

Abstract
Bilateral alternation of muscle contractions requires reciprocal inhibition between the two sides of the hindbrain and spinal cord, and disruption of this inhibition should lead to simultaneous activation of bilateral muscles. At 1 day after fertilization, wild-type zebrafish respond to mechanosensory stimulation with multiple fast alternating trunk contractions, whereas bandoneon (beo) mutants contract trunk muscles on both sides simultaneously. Similar simultaneous contractions are observed in wild-type embryos treated with strychnine, a blocker of the inhibitory glycine receptor (GlyR). This result suggests that glycinergic synaptic transmission is defective in beo mutants. Muscle voltage recordings confirmed that muscles on both sides of the trunk in beo are likely to receive simultaneous synaptic input from the CNS. Recordings from motor neurons revealed that glycinergic synaptic transmission was missing in beo mutants. Furthermore, immunostaining with an antibody against GlyR showed clusters in wild-type neurons but not in beo neurons. These data suggest that the failure of GlyRs to aggregate at synaptic sites causes impairment of glycinergic transmission and abnormal behavior in beo mutants. Indeed, mutations in the GlyR β-subunit, which are thought to be required for proper localization of GlyRs, were identified as the basis for the beo mutation. These data demonstrate that GlyRβ is essential for physiologically relevant clustering of GlyRs in vivo. Because GlyR mutations in humans lead to hyperekplexia, a motor disorder characterized by startle responses, the zebrafish beo mutant should be a useful animal model for this condition.
Keywords: channel, synapse, hyperekplexia, strychnine
Zebrafish embryos display three stereotyped behaviors by 36 h postfertilization (hpf) (1, 2). The earliest behavior consists of spontaneous, alternating coiling of the tail. This slow coiling behavior is independent of sensory stimulation and starts at 17 hpf and declines by 26 hpf. After 21 hpf embryos start to respond to mechanosensory stimulation with the two or three rapid trunk contractions that constitute the escape response. After 26 hpf, mechanosensory stimulation starts to initiate swimming episodes. The frequency of muscle contractions during swimming increases from 7 Hz at 26 hpf to 30 Hz at 36 hpf, the latter being comparable with the frequency of swimming in adult zebrafish (3).
The large-scale Tübingen mutagenesis screen isolated 63 zebrafish mutants with abnormal touch responses (4). Amongst them, mutations in seven genes, including accordion, zieharmonika/ache, and bandoneon (beo) were classified as accordion-type mutants. All mutations in this class displayed apparent simultaneous muscle contractions in both sides of the trunk, resulting in the shortening of the trunk in response to touch. Because this class of mutants was phenocopied by exposing wild-type animals to strychnine, a glycine receptor (GlyR) blocker, accordion-type mutants were predicted to have defects in inhibitory synaptic transmission within the CNS (4). However, the first accordion-type mutations that have been molecularly identified, accordion and zieharmonika/ache, were genes encoding an ATPase Ca2+ pump (5, 6) and acetylcholine esterase (7, 8), respectively, which were required by muscles. A dominant mutation, nic1dbn12, which carried a gain-of-function mutation in the α-subunit of the muscle nicotinic acetylcholine receptor, also displayed accordion-like phenotype due to the hypercontraction of trunk muscles (9).
The neural circuits in the hindbrain and spinal cord that mediate the earliest behaviors exhibited by vertebrate embryos have been extensively studied in lamprey, frog, and fish (10-12). The generation of reliable alternating activity requires a mechanism for reciprocal inhibition, which is mediated by glycinergic transmission (10-13). In mammals, the GlyRs in the adult spinal cord consist of a pentameric complex composed of three ligand-binding α1-subunits and two β-subunits, whereas fetal GlyRs are homomers composed of five α2-subunits (refs. 14 and 15; reviewed in ref. 16). In zebrafish, GlyR cDNAs encoding the α1-subunit and a β-subunit have been cloned, and their expression patterns were described mainly in the adult CNS (17, 18). The β-subunit interacts with gephyrin, a cytoplasmic tubulin-binding protein found in postsynaptic densities, to localize GlyRs to the synapse (19-23).
In this paper, we use in vivo electrophysiology to show that the accordion-type phenotype of beo embryos results from mechanosensory stimulation-induced simultaneous, bilateral activation of the trunk muscles. Furthermore, the coactivation of the muscles stems from a lack of glycinergic synaptic transmission due to an absence of GlyR clustering. The underlying basis for the beo phenotype are a putative null mutation and two missense mutations in the glrb2 gene that encodes the β-subunit of the GlyR. Mutations in the α1- or β-subunit of the GlyR cause an inherited human disorder known as hyperekplexia, which is characterized by exaggerated startle responses, neonatal hypertonia, and excessive falling in response to sudden acoustic or tactile stimuli (24-26). Because the beo embryos are accessible at stages that exhibit mutant phenotype, these zebrafish mutations may be useful as an animal model for hyperekplexia.
Materials and Methods
Recording from Muscle and Motoneurons. The dissection protocols for in vivo patch recordings have been described in refs. 3 and 27. Strychnine was applied at 5 μM. For the miniature currents, 1 μM tetrodotoxin (TTX; Sigma), 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 50 μM d-2-amino-5-phosphonovaleric acid (APV), and 5 μM strychnine were applied in the bath to block the voltage-gated sodium channel, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, NMDA receptor, and glycine receptor, respectively.
Immunostaining, in Situ Hybridization, and Acridine Orange Labeling. Immunostaining and in situ hybridization were done as described in refs. 5 and 28. Anti-slow muscle fiber (F59, Devlopmental Studies Hybridoma Bank, Iowa City, IA) at a concentration of 1:50 and anti-GlyRα (mAb4a, Synaptic Systems, Goettingen, Germany) at a concentration of 1:50 (29) were used for primary antibodies. Alexa Fluor 488-conjugated anti-mouse IgG (Molecular Probes) at a concentration of 1:2,000, biotinylated anti-mouse IgG (Vector Laboratories) at a concentration of 1:250, and Qdot 605 streptavidin (Quantum Dot, Hayward, CA) at a concentration of 1:1,000 were used for visualization. The color of anti-GlyRα staining was false-colored green to make the labeling more apparent in the micrographs. For in situ hybridization, glra1 (GenBank accession no. NM_131402) and glrb2 (GenBank accession no. AB_195560) probes covering all coding region was used. Acridine orange (Sigma) staining was performed as described in ref. 30.
Additional Details. Details for animals, behavioral assay, recording from muscle and motor neurons, meiotic physical mapping, cloning, knock down, mRNA rescue, mutagenesis of glrb2, RT-PCR, and Western blotting are provided in Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results
beo Mutants Display Dorsal Bend in Response to Touch. As part of an ongoing N-ethyl-N-nitrosourea mutagenesis screen, we isolated a new allele (mi106a) of beo that showed indistinguishable phenotype from a Tübingen mutant beotp221 (4). We performed phenotypic analysis on beotp221. The three early behaviors of beo embryos were analyzed to better clarify the nature of the mutation. beo mutants displayed normal spontaneous coiling at a frequency (0.27 ± 0.11 Hz, n = 10) at 22 hpf that was comparable with that of wild-type siblings (0.23 ± 0.08 Hz, n = 10). This finding indicates that the mutation does not perturb spontaneous coiling. However, when beo embryos are touched with forceps at 24 hpf, they appear to simultaneously contract the trunk muscles on both sides, resulting in the shortening of the body and a dorsal flexure of the trunk (Fig. 1B and Movie 1, which is published as supporting information on the PNAS web site) much like accordion embryos (5). In comparison, 24 hpf wild-type siblings respond to touch with two or three fast, alternating contractions of the trunk (Fig. 1A and Movie 2, which is published as supporting information on the PNAS web site). Unlike the relatively long lasting response of accordion mutants (≈5 s), the bilateral response in beo mutants is fast and is over within 1 s of the touch, much like the escape response of wild-type siblings (wild-type: 0.91 ± 0.12 s, n = 10; beo: 0.86 ± 0.10 s, n = 10). Interestingly, wild-type embryos treated with 70 μM strychnine exhibited apparent, fast, bilateral contractions that were indistinguishable from beo mutants (data not shown). At 48 hpf, mutants failed to initiate swimming in response to touch and exhibited a response similar to the bilateral contractions observed at 24 hpf (data not shown). Thus, beo mutants exhibit normal spontaneous coiling, but they have defective touch responses and lack the ability to swim.
Fig. 1.

beo embryos exhibit aberrant touch responses and morphological defects. (A) Frames from a movie showing a wild-type sibling (24 hpf) respond to mechanosensory stimulation with two alternating contractions of the trunk. The time of each frame is shown on the upper right corner of each frame. (B) A beo embryo (24 hpf) responds to touch with a strong bilateral trunk contraction that causes the trunk to bend dorsally. (C) At 48 hpf, beo mutants are shorter in length compared with wild-type siblings. (D) The normal pattern of trunk slow-twitch muscle fibers labeled with monoclonal antibody F59 seen in a side view of a wild-type sibling (48 hpf). (E) The trunk slow-twitch fibers are disarrayed in a beo mutant (48 hpf). (F) Micrograph showing the notochord in a wild-type sibling (48 hpf). (G) The notochord of a beo mutant (48 hpf) exhibits defects.
In addition to abnormal behavior, beo embryos exhibited morphological defects at 48 hpf. The head-to-tail length of beo embryos at rest was 15% shorter (2.58 ± 0.14 mm, n = 10) than that of wild-type siblings (3.03 ± 0.08 mm, n = 10; Student's t test, P < 0.01) (Fig. 1C). Slow-twitch muscle fibers in beo were disturbed (Fig. 1 D and E), but acridine orange labeling showed no significant increase in cell death (data not shown). Disruption of the notochord was also observed in beo (Fig. 1 F and G). These morphological defects were presumably secondary effects due to the mechanical stress caused by simultaneous, bilateral muscle contractions. Supporting this idea, suppression of motor behavior by tricaine (ethyl 3-aminobenzoate methanesulfonate), a weak Na+ channel inhibitor, or N-benzyl-p-toluene sulfonamide, a specific inhibitor for muscle myosin, abolishes these morphological defects in beo (data not shown). As with the behavior, wild-type embryos treated with 70 μM strychnine showed these morphological defects that were indistinguishable from beo mutants (data not shown). The beo larvae died at 7-10 days postfertilization (dpf), possibly from an inability to swim and feed effectively.
Muscle in beo Receive Abnormal Input in Response to Touch. One way to explain the abnormal behavior in beo is that trunk muscles on both sides seem to contract at the same time despite normal alternating output from motor neurons because muscles contract for longer than normal, as was the case with accordion, zieharmonika/ache, and nic1dbn12 mutants, in which all of the defects were due to muscle defects (5-9). Alternatively, the output from motor neurons on the two sides could coincide, thereby causing simultaneous contraction on both sides of the trunk. To distinguish these two possibilities, we recorded from muscle cells and applied mechanosensory stimulation to the side of the yolk that was either ipsilateral or contralateral to the muscle (Fig. 2A). In wild-type siblings, the response to contralateral stimulation (latency to 50% of peak depolarization: 21.1 ± 1.7 ms, n = 5) (Fig. 2 B and C) preceded that to ipsilateral stimulation (latency to 50% of peak depolarization: 44.9 ± 2.4 ms, n = 5; Student's t test, P < 0.01) by ≈25 ms, as shown in ref. 31. In contrast, the latencies of response to ipsilateral (31.9 ± 1.9 ms, n = 4) and contralateral (30.0 ± 1.5 ms, n = 4) stimulation in beo embryos were comparable. Furthermore, strychnine-treated wild-type embryos exhibited touch response latencies that were similar to that of beo mutants (ipsilateral: 32.9 ± 0.8 ms, n = 3; contralateral: 29.9 ± 0.6 ms, n = 3) (Fig. 2C). These results indicate that the trunk muscles were simultaneously activated by motor neurons on the two sides in beo mutants and in strychnine-treated wild-type embryos. Interestingly, the latency of beo was between that of ipsilateral stimulation and contralateral stimulation in wild-type embryos, suggesting that the absence of beo gene product slows down the response in addition to causing bilateral contractions.
Fig. 2.

Voltage response of beo muscles after mechanosensory stimulation is abnormal. (A) Schematic diagram of the experimental method. Embryos (48 hpf) were pinned on a dish, and their skin was peeled off to allow access to the muscle cells for electrophysiological recordings. Responses were evoked by mechanosensory stimulation delivered by a puff of bath solution on either side of the yolk ipsilateral or contralateral to the recorded muscle. (B) Superimposed traces of voltage responses of muscles evoked by mechanosensory stimulation. Arrows indicate the time of stimulation. The latency of the muscle response to contralateral stimulation was shorter than that to ipsilateral stimulation in wild-type siblings, whereas the latency to ipsilateral and contralateral stimulation was the same in beo embryos. (C) Histograms showing that the latency to half-maximal amplitude of the first depolarization was shorter to contralateral stimulation compared with ipsilateral stimulation in wild-type siblings and equal in duration to contralateral and ipsilateral stimulation in beo embryos and strychnine-treated wild-type embryos. Note that the latency in beo and strychnine-treated wild-type embryos is in between that of contralateral and ipsilateral stimulation in untreated wild-type embryos. (D) The long-lasting rhythmic depolarization recorded in a muscle after ipsilateral mechanosensory stimulation of a wild-type sibling. (E) The short and large response devoid of rhythmicity after ipsilateral stimulation in a strychnine-treated wild-type embryo. (F) The short and large response devoid of rhythmicity after ipsilateral stimulation in a beo embryo.
To further characterize the output from the CNS, we examined the pattern of activity after mechanosensory stimulation. At 48 hpf, muscle recordings from wild-type embryos showed sustained episodes of rhythmic depolarizations after mechanosensory stimulation (Fig. 2D). The frequency of this rhythmic activity (28.9 ± 4.1 Hz, n = 10) was within the normal range of swimming (3). The duration of the muscle response in wild-type siblings was 2.53 ± 1.2 s with a peak depolarization of 7.0 ± 1.8 mV (n = 10). When strychnine was added to the bath solution, the pattern of activity was dramatically altered so that the responses were shorter in duration (0.51 ± 0.06 s, n = 6; Student's t test, P < 0.01), greater in peak depolarization (12.9 ± 2.5 mV; Student's t test, P < 0.01), and completely devoid of rhythmic activity (Fig. 2E). Similarly, muscle recordings from beo embryos showed a single peak of depolarization without rhythmicity that was shorter in duration (0.50 ± 0.05 s, n = 6; Student's t test, P < 0.01) and larger in amplitude (13.2 ± 2.4 mV, n = 6; Student's t test, P < 0.01) than in wild-type embryos without strychnine (Fig. 2F). Thus, the similarity of defective behavior and muscle physiology in beo mutants and strychnine-treated wild-type embryos is consistent with the hypothesis that glycinergic transmission is aberrant in beo embryos.
Glycinergic Synaptic Transmission Is Aberrant in beo Mutants. To see whether glycinergic transmission is defective in mutants, we measured spontaneous glycinergic synaptic currents in motor neurons at 48 hpf. Spontaneous synaptic currents in the presence of TTX were less frequent in beo (0.30 ± 0.05 Hz, n = 4) (Fig. 3D) compared with wild-type embryos (0.64 ± 0.06 Hz, n = 4) (Fig. 3A). Application of blockers for the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (CNQX) and NMDA receptor (APV) removed a subset of the spontaneous currents in wild-type (0.33 ± 0.07 Hz, n = 10) (Fig. 3B). The remaining events were abolished by the addition of 5 μM strychnine (0 Hz, n = 6) (Fig. 3C), suggesting that they were glycinergic currents. In beo, spontaneous currents were completely eliminated by blocking glutamate receptors with CNQX and APV (0 Hz, n = 11) (Fig. 3E), and further application of strychnine had no effect (0 Hz, n = 11) (Fig. 3F). Thus, the frequency of glutamatergic currents in wild-type motor neurons (0.31 Hz) was comparable to that in mutant motor neurons (0.30 Hz). These results indicate that spontaneous glutamatergic synaptic currents were unperturbed and that spontaneous glycinergic synaptic currents were missing in beo mutants.
Fig. 3.

Glycinergic synaptic transmission is missing in beo embryos because of a defect in GlyR clustering. (A) Spontaneous synaptic currents recorded in the presence of TTX in a wild-type motor neuron. (B) Nonglutamatergic spontaneous currents in the wild-type motor neuron after block of NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors with CNQX and APV, respectively. (C) The nonglutamatergic spontaneous currents are eliminated by strychnine in the wild-type motor neuron, showing that they are glycinergic currents. (D) Spontaneous synaptic currents recorded in the presence of TTX in a beo motor neuron. (E) Nonglutamatergic spontaneous currents in the presence of CNQX and APV are absent in the beo motor neuron, demonstrating that spontaneous glycinergic synaptic currents are missing in beo.(F) No further effect of strychnine on the beo motor neuron. (G and H) A puff of exogenous glycine induces a current in a wild-type motor neuron (G) and a smaller current in a beo motor neuron (H). (I) Cross sections of the spinal cord outlined by the square in the schematic illustration. (J) Labeling of GlyRα with mAb4a in wild-type marks clusters of GlyRs that are associated with the surface of presumptive neurons, which is outlined with dashed lines. (K) No clusters of GlyRs were labeled by mAb4a in the beo spinal cord. (L) Western blots labeled with mAb4a showing that GlyRα is expressed at levels comparable in wild-type siblings and beo. Blotting with antiacetylated tubulin is control.
The conclusion that beo mutations lack spontaneous glycinergic synaptic currents was corroborated by examination of the durations of the currents, which varied depending on whether they were glycinergic or glutamatergic. The duration when the currents were >50% of the peak currents was measured as the duration of spontaneous synaptic currents. The frequency histogram of the duration appeared to exhibit two populations of synaptic currents in wild-type embryos: a fast current with a peak at 0.8 ms and a slower current with a peak at 2.8 ms (Fig. 5, which is published as supporting information on the PNAS web site). Pharmacological removal of glutamatergic currents with CNQX and APV in wild-type siblings left the slow population, suggesting that the glycinergic synaptic currents are longer in duration than the glutamatergic ones. In beo, the frequency histogram of the duration showed only a fast population of glutamatergic synaptic currents. The removal of the glutamatergic synaptic currents with CNQX and APV abolished all spontaneous synaptic events in beo, demonstrating the lack of spontaneous glycinergic synaptic currents in beo mutants.
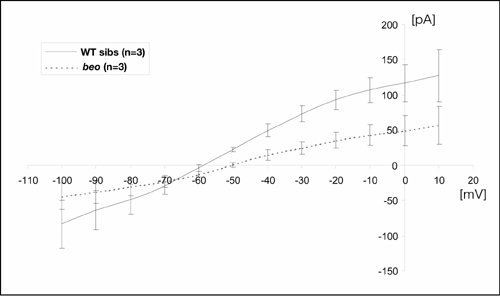
The absence of spontaneous glycinergic synaptic currents in beo mutants could result from the absence of functional GlyRs or the failure to aggregate GlyRs at synapses. To distinguish between these two possibilities, the sensitivity of motor neurons to exogenous glycine was assayed. Puffing glycine (100 mM, 20 ms) onto motor neurons in vivo revealed that wild-type (Fig. 3G) and beo (Fig. 3H) motor neurons responded to exogenous glycine. However, the amplitude of the glycine response in wild-type siblings (96.1 ± 6.8 pA, n = 3) was consistently larger than that in beo mutants (38.3 ± 18.3 pA, n = 3; Student's t test, P < 0.01). Glycine-induced currents were also measured by comparing strychnine-sensitive whole-cell currents during voltage steps in the presence of bath-applied glycine (Fig. 6, which is published as supporting information on the PNAS web site). These experiments showed that the strychnine-sensitive conductance in beo motor neurons (1.5 nA/V, n = 3) was about half of that in wild-type motor neurons (2.8 nA/V, n = 3). These results demonstrate that functional GlyRs are expressed on the cell surface of motor neurons in beo and suggest that the defect is a lack of GlyRs clustering at synapses.
To test whether there is a lack of GlyR aggregation at synapses within the CNS in beo, localization of GlyRs was assayed by immunolabeling with an antibody (mAb4a) against an epitope that is common to all GlyR α-subunits (29). In wild-type embryos, clusters of GlyRs were found associated with the plasma membranes of cell bodies in the spinal cord, including the ventrolateral portion that contains the cell bodies of motor neurons (Fig. 3 I and J). In contrast, no clusters of GlyRs were visible in beo spinal cord (Fig. 3K). In these sections, it was not possible to determine whether there was increased labeling throughout the plasma membrane in the mutants. Furthermore, Western blotting with mAb4a showed that GlyRα was expressed comparably in wild-type and beo embryos (Fig. 3L). These results along with the electrophysiological evidence suggest that GlyRs are expressed on the surface of neurons but are not clustered at presumptive synaptic sites in beo mutants.
beo Encodes for a β-Subunit of the GlyR and Is Expressed Early in the Hindbrain and Spinal Cord. Because the β-subunit of the GlyR encoded by the glrb gene is essential for the clustering of GlyRs in mammals (16, 21), we focused on glrb as a candidate for the beo gene. Genomic sequences encoding glrb were identified from the ongoing Danio rerio sequencing project (available at www.sanger.ac.uk/Projects/D_rerio). Two positive genomic fragments that represented two β-subunit genes were found. One β-subunit gene in linkage group (LG)1 was reported in ref. 18. This gene was termed glrb1, and the second β-subunit found in genomic fragment Scaffold1268 was termed glrb2. The glrb2 gene was physically mapped by using the LN54 radiation hybrid panel and found to be located between two microsatellites, Z21080 and Z8801, in LG14 (Fig. 7A, which is published as supporting information on the PNAS web site). Furthermore, bulk segregant analysis by meiotic mapping showed that these two markers in LG14 were linked to the beo mutation. To see whether the glrb2 gene was the beo gene, glrb2 cDNA was cloned and sequenced from wild type and three alleles of the beo mutation. Wild-type glrb2 encodes 494 amino acid residues (GenBank accession no. AB195560) (Fig. 7 B and C) that contain a signal peptide, four transmembrane domains (M1-M4) and a gephyrin-binding domain). In beotp221, Tyr-101 in the N terminus extracellular domain was changed to a stop codon to generate a severely truncated protein. Thus, there appears to be a null mutation of the glrb2 gene in beotp221. Missense mutations were found in the other alleles of beo; Leu-277 in M1 was changed to Arg in beotw38f, and Arg-297 in the cytoplasmic loop between M1 and M2 was changed to His in beomi106a.
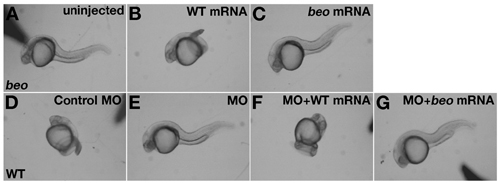
The molecular identification of beo as the glrb2 gene was confirmed by mutant rescue and antisense phenocopy. We injected wild-type glrb2 mRNA or glrb2 mRNA carrying the beomi106a point mutation into recently fertilized embryos of beo heterozygous carriers. Nearly all of the progeny injected with wild-type mRNA displayed normal escape responses after touch at 24 hpf (113 of 123 embryos, 92%) (Fig. 8B, which is published as supporting information on the PNAS web site), whereas ≈25% of uninjected progeny (40 of 153 embryos, 26.1%) (Fig. 8A) and beo mRNA-injected progeny (41 of 147 embryos, 27.9%) (Fig. 8C) showed the mutant response. To knockdown synthesis of GlyRβ2, antisense morpholino oligonucleotides (MO) against glrb2 mRNA was injected into recently fertilized wild-type embryos. An overwhelming number of the antisense MO-injected wild-type embryos displayed the beo behavior after touch (84 of 95 embryos, 88.4%) (Fig. 8E), whereas none of the control MO-injected wild-type embryos exhibited aberrant responses (0 of 65 embryos, 0%) (Fig. 8D). A wild-type glrb2 mRNA was engineered with differences in the 5′-untranslated region, which should make it immune to the antisense glrb2 MO. When the antisense MO was coinjected with the engineered wild-type glrb2 mRNA into wild-type embryos, they responded normally to touch (73 of 81 embryos, 90.1%) (Fig. 8F). In contrast, embryos coinjected with antisense MO and beomi106a mRNA exhibited the beo response (73 of 84 embryos, 86.9%) (Fig. 8G). Thus, mRNA rescue and antisense knockdown confirm that glrb2, encoding for the β2-subunit of GlyR, is the gene responsible for the beo phenotype.
Because beo mutants showed abnormal escape responses at 24 hpf, expression of glrb2 along with glra1 and glrb1 was assayed with RT-PCR at different stages. glra1 and glrb2 were expressed starting at 1 day of development, whereas glrb1 begins expression at 3 days (Fig. 4A). This finding suggests that the β2- but not the β1-subunit is active in GlyRs before day 3. RT-PCR also found that glrb1 expression was unchanged in beo mutants; i.e., it was not observed before day 3 and not increased at later stages (data not shown). Thus, glrb1 does not compensate for the lack of glrb2 in beo mutants. These data suggest that the lack of the GlyRβ2 during the first 3 days of development is responsible for the failure of GlyRs to aggregate at synapses (see above).
Fig. 4.

Temporal and spatial expression of GlyR genes. (A) RT-PCR shows that glra1 and glrb2 are expressed starting on 1 dpf, whereas glrb1 expression starts at 3 dpf. (B-G) Whole-mount in situ hybridization with a glra1 (B, D, and F) or glrb2 (C, E, and G) probe in 24-hpf embryos. Lateral views show expression of glra1 and glrb2 in putative neurons in the hindbrain (B and C, respectively) and spinal cord (F and G, respectively). Dorsal views show the bilaterally symmetric, presumptive hindbrain neurons expressing glra1 (D) and glrb2 (E).
Because the hindbrain and spinal cord are necessary and sufficient for the escape response and swimming (2, 32, 33) and beo mutants are defective in these behaviors, glrb2 should be expressed in these regions of the CNS. The patterns of expression of the glra1 and glrb2 genes were indistinguishable at 24 and 48 hpf. At 24 hpf, both genes were expressed in repeating bilateral clusters of cells in the hindbrain (Fig. 4 B-E), which was similar to glra1 expression in Xenopus (34, 35) and glra4a in zebrafish (36). The neurons in the hindbrain are likely reticulospinal neurons, such as the Mauthner cells, that are essential for the escape response in frog and fish (11, 32, 33). glra1 and glrb2 were also extensively expressed by neurons in the spinal cord (Fig. 4 F and G). By 48 hpf, expressions of glra1 and glrb2 had extended to many other presumptive neurons in the hindbrain and spinal cord (data not shown). These data suggest that the GlyRβ2 normally serves to localize GlyRs to synapses within the hindbrain and spinal cord.
Discussion
beo Mutants Simultaneously and Bilaterally Contract Trunk Muscles. The Tübingen screen found 7 accordion-type mutations that caused bilateral contractions of the trunk muscle (4). Two of these mutations, accordion and zieharmonika/ache, exhibited muscle defects with mutations in muscle ATPase Ca2+ pump and acetylcholine esterase, respectively (5-8). The present study demonstrates that one of the remaining five mutations, beo, is due to a defect in the CNS that causes the simultaneous activation of axial muscles on both sides of the trunk. In wild-type embryos, muscles contralateral to the side receiving mechanosensory stimulation received motor input 25 ms before ipsilateral muscles. This was similar to the latency difference between the two sides reported in ref. 31. In contrast, contralateral and ipsilateral muscles were activated simultaneously in beo embryos. Similar simultaneous activation of muscles on the two sides of the trunk examined in strychnine-treated wild-type embryos suggested that glycinergic synaptic transmission was defective in mutants. Indeed, glycinergic synaptic transmission mediates reciprocal inhibition between the two sides of the spinal cord to ensure antiphasic activation of motor neurons on the two sides in lamprey, Xenopus, and fish (10-12). Because the beo gene encodes for the GlyR β2-subunit of the GlyR (see below), beo provides genetic evidence for the importance of glycinergic transmission during alternating contractions of antagonistic muscle groups.
A recent study by Masino and Fetcho (37) used extracellular motor root recordings and found that fictive swimming activity in beo mutants was disorganized but still exhibited side-to-side alternation. There are two potential reasons for the disparity with our results, which show complete absence of alternating activity. First, the allele of beo analyzed in their experiment (beota86d) may have been a weak allele. There are seven different alleles of beo (4). We examined beotp221, beotw38f, and beomi106a with the phenotypic analysis done with beotp221, which contained a stop codon at the position of amino acid 101 of the predicted 494-aa protein. This fact suggested that the beotp221 product was severely truncated and likely to be nonfunctional. Second, there might be some behavioral recovery from the early severe beo phenotype. Masino and Fetcho (37) assayed motor roots at 4-6 dpf, whereas we examined output from motor neurons at 2 dpf.
In beo embryos, the ipsilateral and contralateral muscles not only respond simultaneously but also show shorter and larger depolarizations lacking rhythmicity. This result suggests that glycinergic transmission is necessary for generation of the sustained rhythmic drive for swimming. Interestingly, the response to contralateral stimulation of beo mutants is delayed by 10 ms compared with that of wild-type siblings. The increased latency in mutants further suggests that glycinergic transmission can also accelerate the responses to stimuli in wild-type embryos. Although it is unclear how this acceleration occurs, one possibility is that glycinergic transmission is excitatory at early stages and that removal of the excitation can slow down the response rate of early neural circuits in beo mutants. In fact, glycinergic transmission is known to be excitatory at early stages in other organisms because of chloride ion levels that make the equilibrium potential for chloride ion with respect to the resting potential (38), which may also be the case in early zebrafish neurons.
beo Encodes the β-Subunit of the GlyR That Is Required for Synaptic Clustering of GlyRs. We identified mutations in the glrb2 gene of beo mutants and confirmed that glrb2 mutation gave rise to the mutant phenotype by mRNA rescue and MO knockdown. In beotp221, a stop codon was located in the N-terminal extracellular domain, which was 5′ to the transmembrane domains, suggesting that this allele carried a null mutation. In beomi106a, an Arg-297-His mutation was found in the loop between M1 and M2. GlyRβ has a sequence similarity of ≈47% with GlyRα1, with transmembrane domains and loops between them being well conserved. A human glra1 mutation, which carries a homologous mutation in the residue corresponding to Arg-297 in GlyRβ2 (Arg-252-His in human GlyRα1), was reported to cause hyperekplexia (39). Functional analysis of GlyRα1 carrying the Arg-252-His mutation revealed that this mutation likely accelerated degradation of GlyRα1 (40). Thus, it is possible that beomi106a is a hypomorph of GlyRβ2. In beotw38f, Leu-277 in M1 was changed to Arg. M1 is highly conserved, and a missense mutation in M1 of glra1 was reported to be responsible for hyperekplexia because of the reduced expression of GlyRα1 at the cell surface (41). Thus, beotw38f may also be a hypomorph of GlyRβ2. GlyRs normally associate with gephyrin via direct binding to GlyRβ to aggregate GlyRs at synaptic sites (19-23). Indeed, the mouse mutation spastic is a GlyRβ hypomorph in which GlyRs and glycinergic inhibitory postsynaptic currents are dramatically decreased (42-45). Whether GlyRs are clustered in spastic mice, however, has not been reported.
In this study, we showed that beo embryos exhibited defects in glycinergic synaptic transmission. First, electrophysiological recordings from strychnine-treated wild-type muscles showed that blocking glycinergic transmission phenocopied the beo mutation, suggesting that the output from the CNS was aberrant in beo muscles because of a defect in glycinergic transmission. Second, motor neurons in beo embryos can respond to exogenous glycine, showing that they express functional GlyRs at the cell surface. Third, spontaneous glycinergic currents were completely absent in beo motor neurons, indicating that glycinergic transmission was impaired in beo. Fourth, immunostaining with an antibody against GlyRα revealed that GlyRs were not clustered in the beo spinal cord. Taken together, these results indicate that a mutation of the glrb2 gene leads to impairment of clustering by GlyRs and defective function of neural circuits, leading to aberrant behavior starting with one of the earliest behaviors emitted by embryos. This study directly demonstrates that GlyRβ is required for physiologically relevant clustering of GlyRs in the CNS.
Western blotting analysis suggested that the amount of GlyRα was not diminished in beo embryos despite the fact that they were not clustered in mutants. However, the response of beo motor neurons to exogenous glycine suggested a decrease in responsiveness to glycine. In fact, coexpression of GlyRα1 with GlyRβ significantly increased glycine-gated whole-cell currents compared with expression of GlyRα1 alone in HEK-293 cells (46). The decreased response of beo motor neurons to exogenous glycine might be due to differences in the properties of GlyRs made up of α1- and β-subunits versus those made up of homomeric α1-subunits (46, 47). GlyRs made up of α1- and β-subunits have a lower single channel conductance but a lower Hill coefficient and EC50 compared with GlyRs made up of homomeric α1-subunits. Another possibility is that surface expression of GlyRs is reduced in the absence of GlyRβ, much like the β-, γ-, and δ-subunits of the acetylcholine receptor, which are retained in intracellular compartments in the absence of the α-subunit (48).
beo Mutants Are an Animal Model for Hyperekplexia. Hyperekplexia is a rare neurological disorder in humans that is characterized by exaggerated startle responses to unexpected acoustic or tactile stimuli (24). Various autosomal dominant and recessive mutations in the glra1 gene have been identified as responsible for hyperekplexia (16, 25). Furthermore, a human patient carrying missense and nonsense mutations in glrb also exhibits hyperekplexia (26). Mice harboring mutations in glra1 also exhibit a similar startle disease (49), with spastic mice carrying an insertion of the long interspersed nuclear element retrotransposon in an intron of the glrb gene (43, 44). Because spastic mice are hypomorphs for glrb expression (50), the beotp221 mutation, which is likely a null allele, might be useful as a system to study synaptic clustering of GlyRs due to the putative absence of GlyRβ.
Zebrafish beo mutants and patients with hyperekplexia share the most critical feature of an exaggerated startle response due to impaired glycinergic synaptic transmission caused by mutations in GlyR genes. Because zebrafish embryos are readily accessible to molecular, genetic, pharmacological, and physiological interventions as well as in vivo visualization of fluorescent-tagged molecules, beo mutants could serve as an attractive model for hyperekplexia. Additionally, there are four other mutations of the accordion-type that remain to be characterized (4). It will be interesting to see whether any of these genes may be associated with synaptic clustering of GlyRs and could potentially be candidates for additional hyperekplexia genes.
Supplementary Material
Acknowledgments
We thank S. M. Sprague and A. Nagashima for fish care and R. I. Hume for helpful discussion and generous use of electrophysiology equipment. This work was supported by grants from the National Institute of Neurological Disorders and Stroke (to J.Y.K.). L.S.-A. was partly supported by Fond de Recherche en Santé du Québec. G.B.D. was supported by a Postdoctoral Fellowship from the National Science Foundation. H.H. was supported by a Long-Term Fellowship from the Human Frontier Science Program.
Author contributions: H.H. and J.Y.K. designed research; H.H., L.S.-A., G.B.D., W.W.C., and W.Z. performed research; H.H. and M.G. contributed new reagents/analytic tools; H.H. analyzed data; and H.H. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: beo, bandoneon; GlyR, glycine receptor; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; APV, d-2-amino-5-phosphonovaleric acid; hpf, hours postfertilization; MO, morpholino oligonucleotide; dpf, days postfertilization; TTX, tetrodotoxin.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AB195560).
References
- 1.Eaton, R. C. & Farley, R. D. (1973) Copeia 4, 673-682. [Google Scholar]
- 2.Saint-Amant, L. & Drapeau, P. (1998) J. Neurobiol. 37, 622-632. [DOI] [PubMed] [Google Scholar]
- 3.Buss, R. R. & Drapeau, P. (2001) J. Neurophysiol. 86, 197-210. [DOI] [PubMed] [Google Scholar]
- 4.Granato, M., van Eeden, F. J. M., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C.-P., Jiang, Y.-J., et al. (1996) Development (Cambridge, U.K.) 123, 399-413. [DOI] [PubMed] [Google Scholar]
- 5.Hirata, H., Saint-Amant, L., Waterbury, J., Cui, W., Zhou, W., Li, Q., Goldman, D., Granato, M. & Kuwada, J. Y. (2004) Development (Cambridge, U.K.) 131, 5457-5468. [DOI] [PubMed] [Google Scholar]
- 6.Gleason, M. R., Armisen, R., Verdecia, M. A., Sirotkin, H., Brehm, P. & Mandel, G. (2004) Dev. Biol. 276, 441-451. [DOI] [PubMed] [Google Scholar]
- 7.Behra, M., Cousin, X., Bertrand, C., Vonesch, J.-L., Biellmann, D., Chatonnet, A. & Strähle, U. (2002) Nat. Neurosci. 5, 111-118.11753420 [Google Scholar]
- 8.Downes, G. B. & Granato, M. (2004) Dev. Biol. 270, 232-245. [DOI] [PubMed] [Google Scholar]
- 9.Lefebvre, J. L., Ono, F., Puglielli, C., Seidner, G., Franzini-Armstrong, C., Brehm, P. & Granato, M. (2004) Development (Cambridge, U.K.) 131, 2605-2618. [DOI] [PubMed] [Google Scholar]
- 10.Grillner, S. (2003) Nat. Rev. Neurosci. 4, 573-586. [DOI] [PubMed] [Google Scholar]
- 11.Roberts, A. (2000) Brain Res. Bull. 53, 585-593. [DOI] [PubMed] [Google Scholar]
- 12.Fetcho, J. R. (1992) Brain Behav. Evol. 40, 82-97. [DOI] [PubMed] [Google Scholar]
- 13.Kiehn, O. & Kullander, K. (2004) Neuron 41, 317-321. [DOI] [PubMed] [Google Scholar]
- 14.Langosch, D., Thomas, L. & Betz, H. (1988) Proc. Natl. Acad. Sci. USA 85, 7394-7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoch, W., Betz, H. & Becker, C.-M. (1989) Neuron 3, 339-348. [DOI] [PubMed] [Google Scholar]
- 16.Lynch, J. W. (2004) Physiol. Rev. 84, 1051-1095. [DOI] [PubMed] [Google Scholar]
- 17.David-Watine, B., Goblet, C., de Saint Jan, D., Fucile, S., Devignot, V., Bregestovski, P. & Korn, H. (1999) Neuroscience 90, 303-317. [DOI] [PubMed] [Google Scholar]
- 18.Imboden, M., Devignot, V., Korn, H. & Goblet, C. (2001) Neuroscience 103, 811-830. [DOI] [PubMed] [Google Scholar]
- 19.Kirsch, J., Wolters, I., Triller, A. & Betz, H. (1993) Nature 366, 745-748. [DOI] [PubMed] [Google Scholar]
- 20.Kirsch, J. & Betz, H. (1995) J. Neurosci. 15, 4148-4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer, G., Kirsch, J., Betz, H. & Langosch, D. (1995) Neuron 15, 563-572. [DOI] [PubMed] [Google Scholar]
- 22.Feng, G., Tintrup, H., Kirsch, J., Nichol, M. C., Kuhse, J., Betz, H. & Sanes, J. R. (1998) Science 282, 1321-1324. [DOI] [PubMed] [Google Scholar]
- 23.Sola, M., Bavro, V. N., Timmins, J., Franz, T., Ricard-Blum, S., Schoehn, G., Ruigrok, R. W. H., Paarmann, I., Saiyed, T., O'Sullivan, G. A., et al. (2004) EMBO J. 23, 2510-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andermann, F. & Andermann, E. (1988) Brain Dev. 10, 213-222. [DOI] [PubMed] [Google Scholar]
- 25.Shiang, R., Ryan, S. G., Zhu, Y.-Z., Hahn, A. F., O'Connell, P. & Wasmuth, J. J. (1993) Nat. Genet. 5, 351-358. [DOI] [PubMed] [Google Scholar]
- 26.Rees, M. I., Lewis, T. M., Kwok, J. B. J., Mortier, G. R., Govaert, P., Snell, R. G., Schofield, P. R. & Owen, M. J. (2002) Hum. Mol. Genet. 11, 853-860. [DOI] [PubMed] [Google Scholar]
- 27.Cui, W. W., Saint-Amant, L. & Kuwada, J. Y. (2004) J. Neurophysiol. 92, 2898-2908. [DOI] [PubMed] [Google Scholar]
- 28.Hirata, H., Yoshiura, S., Ohtsuka, T., Bessho, Y., Harada, T., Yoshikawa, K. & Kageyama R. (2002) Science 298, 840-843. [DOI] [PubMed] [Google Scholar]
- 29.Schröder, S., Hoch, W., Becker, C.-M., Grenningloh, G. & Betz, H. (1991) Biochemistry 30, 42-47. [DOI] [PubMed] [Google Scholar]
- 30.Seiler, C. & Nicolson, T. (1999) J. Neurobiol. 41, 424-434. [PubMed] [Google Scholar]
- 31.Buss, R. R. & Drapeau, P. (2002) J. Neurophysiol. 87, 1244-1251. [DOI] [PubMed] [Google Scholar]
- 32.Liu, K. S. & Fetcho, J. R. (1999) Neuron 23, 325-335. [DOI] [PubMed] [Google Scholar]
- 33.Gahtan, E., Sankrithi, N., Campos, J. B. & O'Malley, D. M. (2002) J. Neurophysiol. 87, 608-614. [DOI] [PubMed] [Google Scholar]
- 34.Dale, N., Ottersen, O. P., Roberts, A. & Storm-Mathisen, J. (1986) Nature 324, 255-257. [DOI] [PubMed] [Google Scholar]
- 35.Roberts, A., Dale, N., Ottersen, O. P. & Storm-Mathisen, J. (1988) Development (Cambridge, U.K.) 103, 447-461. [DOI] [PubMed] [Google Scholar]
- 36.Imboden, M., Devignot, V. & Goblet, C. (2001) Dev. Genes Evol. 211, 415-422. [DOI] [PubMed] [Google Scholar]
- 37.Masino, M. A. & Fetcho, J. R. (2005) J. Neurophysiol., 93, 3177-3188. [DOI] [PubMed] [Google Scholar]
- 38.Ben-Ari, Y. (2002) Nat. Rev. Neurosci. 3, 728-739. [DOI] [PubMed] [Google Scholar]
- 39.Vergouwe, M. N., Tijssen, M. A. J., Peters, A. C. B., Wielaard, R. & Frants, R. R. (1999) Ann. Neurol. 46, 634-638. [DOI] [PubMed] [Google Scholar]
- 40.Rea, R., Tijssen, M. A., Herd, C., Frants, R. R. & Kullmann, D. M. (2002) Eur. J. Neurosci. 16, 186-196. [DOI] [PubMed] [Google Scholar]
- 41.Humeny, A., Bonk, T., Becker, K., Jafari-Boroujerdi, M., Stephani, U., Reuter, K. & Becker, C.-M. (2002) Eur. J. Hum. Genet. 10, 188-196. [DOI] [PubMed] [Google Scholar]
- 42.Becker, C.-M., Schmieden, V., Tarroni, P., Strasser, U. & Betz, H. (1992) Neuron 8, 283-289. [DOI] [PubMed] [Google Scholar]
- 43.Kingsmore, S. F., Giros, B., Suh, D., Bieniarz, M., Caron, M. G. & Seldin, M. F. (1994) Nat. Genet. 7, 136-141. [DOI] [PubMed] [Google Scholar]
- 44.Mülhardt, C., Fischer, M., Gass, P., Simon-Chazottes, D., Guénet, J.-L., Kuhse, J., Betz, H. & Becker, C.-M. (1994) Neuron 13, 1003-1015. [DOI] [PubMed] [Google Scholar]
- 45.von Wegerer, J., Becker, K., Glockenhammer, D., Becker, C.-M., Zeilhofer, H. U. & Swandulla, D. (2003) Neurosci. Lett. 345, 45-48. [DOI] [PubMed] [Google Scholar]
- 46.Bormann, J., Rundström, N., Betz, H. & Langosch, D. (1993) EMBO J. 12, 3729-3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Legendre, P. (1997) J. Neurophysiol. 77, 2400-2415. [DOI] [PubMed] [Google Scholar]
- 48.Claudio, T., Paulson, H. L., Green, W. N., Ross, A. F., Hartman, D. S. & Hayden, D. (1989) J. Cell Biol. 108, 2277-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryan, S. G., Buckwalter, M. S., Lynch, J. W., Handford, C. A., Segura, L., Shiang, R., Wasmuth, J. J., Camper, S. A., Schofield, P. & O'Connell, P. (1994) Nat. Genet. 7, 131-135. [DOI] [PubMed] [Google Scholar]
- 50.Hartenstein, B., Schenkel, J., Kuhse, J., Besenbeck, B., Kling, C., Becker, C.-M., Betz, H. & Weiher, H. (1996) EMBO J. 15, 1275-1782. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}