

Significance
We uncovered a selective autophagic mechanism that regulates c-Myc protein stability. The E3 ubiquitin ligase TRIM21 directly interacts with and ubiquitinates c-Myc at lysine 148 (K148) via K63-linkage, enabling c-Myc to be targeted to the autophagy machinery for degradation. Our findings also provide a unique insight into the interplay between KRAS and c-Myc. KRAS/MT-driven MAPK signaling results in the p-TRIM21 at the T396 residue by ERK2. This phosphorylation disrupts the interaction between TRIM21 and c-Myc, preventing c-Myc from being targeted for autophagic degradation. Our findings confirmed that KRAS/MT confers resistance to regorafenib treatment in CRC. The combination of vilazodone with regorafenib may represent a promising treatment approach for regorafenib-resistant colorectal cancer tumors with KRAS/MT.
Keywords: TRIM21, MYC, KRAS, regorafenib, drug resistance
Abstract
Kirsten rat sarcoma virus (KRAS) mutation is associated with malignant tumor transformation and drug resistance. However, the development of clinically effective targeted therapies for KRAS-mutant cancer has proven to be a formidable challenge. Here, we report that tripartite motif-containing protein 21 (TRIM21) functions as a target of extracellular signal-regulated kinase 2 (ERK2) in KRAS-mutant colorectal cancer (CRC), contributing to regorafenib therapy resistance. Mechanistically, TRIM21 directly interacts with and ubiquitinates v-myc avian myelocytomatosis viral oncogene homolog (c-Myc) at lysine 148 (K148) via K63-linkage, enabling c-Myc to be targeted to the autophagy machinery for degradation, ultimately resulting in the downregulation of enolase 2 expression and inhibition of glycolysis. However, mutant KRAS (KRAS/MT)-driven mitogen-activated protein kinase (MAPK) signaling leads to the phosphorylation of TRIM21 (p-TRIM21) at Threonine 396 (T396) by ERK2, disrupting the interaction between TRIM21 and c-Myc and thereby preventing c-Myc from targeting autophagy for degradation. This enhances glycolysis and contributes to regorafenib resistance. Clinically, high p-TRIM21 (T396) is associated with an unfavorable prognosis. Targeting TRIM21 to disrupt KRAS/MT-driven phosphorylation using the antidepressant vilazodone shows potential for enhancing the efficacy of regorafenib in treating KRAS-mutant CRC in preclinical models. These findings are instrumental for KRAS-mutant CRC treatment aiming at activating TRIM21-mediated selective autophagic degradation of c-Myc.
Colorectal cancer (CRC) ranks as the third most frequently diagnosed and the third most lethal cancer globally (1). Oncogenic Kirsten rat sarcoma virus (KRAS) mutations, such as KRASG12D, KRASG12V, and KRASG12C, are found in approximately 40% of human CRC cases and play a crucial role in disease progression and responses to targeted therapies (2–8). Nevertheless, there is currently no clinically effective anti-KRAS therapy available. Various studies have demonstrated the crucial role of the oncogene MYC as a mediator of KRAS function, facilitating KRAS-driven oncogenesis (9–14). Consequently, targeting MYC could present a promising therapeutic strategy for addressing KRAS-mutant-driven CRC. However, like KRAS, MYC has been commonly regarded as “undruggable” (15). Current therapeutic strategies for targeting MYC have largely focused on indirect approaches, such as inhibiting MYC transcription and/or translation, as well as destabilizing MYC (16–19). Despite these efforts, there is still much to be explored regarding the molecular mechanisms that mediate the cross talk between KRAS mutations and MYC signaling in CRC tumorigenesis and progression, as well as how to simultaneously manipulate them for therapeutic purposes.
Tripartite motif-containing protein 21 (TRIM21), a member of the TRIM protein family of RING E3 ubiquitin ligases, exerts pleiotropic effects on the development and progression of cancers (20–22). For example, TRIM21 inhibits cancer progression by destabilizing mutant p53 accumulation (23). Additionally, TRIM21 is suggested to regulate tumor immunity through the degradation of voltage-dependent anion-selective channel protein 2 and integrin-associated protein CD47 (24, 25). Recent studies, including ours, have revealed the important roles of TRIM21 in CRC tumorigenesis and progression (26, 27). However, whether TRIM21 is regulated in KRAS-mutated CRC, which might provide an alternative therapeutic strategy to treat KRAS-mutated CRC, remains largely unexplored.
In this study, we have uncovered that TRIM21 functions as a target of ERK2 in KRAS-mutant CRC. Our findings suggest that targeting the inhibition of KRAS-mutant-driven TRIM21 phosphorylation using the antidepressant medication vilazodone could provide a different therapeutic strategy to sensitize regorafenib in the treatment of KRAS-mutated CRC.
Results
TRIM21 Destabilizes the c-Myc Protein.
To delve into the crucial regulatory mechanisms of TRIM21 in CRC, we employed global RNA-sequencing (RNA-seq) analysis to pinpoint the pathways influenced by TRIM21. We identified the top 3,000 differentially expressed genes ranked according to B-statistics of the limma method (SI Appendix, Fig. S1 A and B). Notably, we found an overlap of 258 down-regulated genes and 250 up-regulated genes, with several c-Myc target genes included in the list of co-up-regulated genes (SI Appendix, Fig. S1 C and D). Gene ontology and pathway enrichment analyses indicated the involvement of the coregulated genes in MYC signaling within both the co-up-regulated and co-down-regulated gene groups (SI Appendix, Fig. S1E). Consistently, gene set enrichment analysis (GSEA) of the The Cancer Genome Atlas-Colon Adenocarcinoma dataset revealed significant enrichment of genes down-regulated by MYC in patients with high TRIM21 expression (SI Appendix, Fig. S1F). We further performed proteomic analysis, which revealed that silencing of TRIM21 led to the upregulation of 265 proteins, including c-Myc, and the downregulation of 273 proteins, including TRIM21 (Fig. 1A and Dataset S1). Analysis of biological processes and pathways indicated that the up-regulated proteins were significantly enriched in the biological functions related to cell proliferation, protein stability, cell division, and enhanced MYC signaling (Fig. 1B). Collectively, these findings suggest that TRIM21 may play a role in regulating MYC signaling, which is considered a critical factor in CRC tumorigenesis.
Fig. 1.

TRIM21 inhibits c-Myc signaling by destabilizing the c-Myc protein. (A) Volcano plot illustrating the differentially expressed proteins identified through proteomic analysis in HCT116 cells (shRNA/TRIM21 versus shRNA/Control). (B) Enrichment analysis of biological processes and pathways for the up-regulated proteins in HCT116 cells (shRNA/TRIM21 versus shRNA/Control). (C and D) WB analysis of c-Myc expression in the CRC cells overexpressing TRIM21 (C) or stably silencing TRIM21 (D). (E) Representative images (Top) and correlation analysis (Bottom) of the indicated protein in 150 cases of CRC tissues. (F and G) The effect of overexpressing (F) or silencing (G) TRIM21 on the half-life of c-Myc protein was analyzed in HCT116 or RKO cells, respectively, treated with cycloheximide (CHX).
We then investigated whether TRIM21 modulates c-Myc expression. Our findings revealed a significant decrease in c-Myc protein levels upon TRIM21 overexpression (Fig. 1C), while the protein levels of c-Myc increased in TRIM21-silenced CRC cells (Fig. 1D). Furthermore, a notable negative correlation between TRIM21 and c-Myc was clinically observed in a CRC cohort (n = 150) (Fig. 1E). Notably, TRIM21 did not affect the messenger ribonucleic acid (mRNA) levels of c-Myc (SI Appendix, Fig. S1 G and H), suggesting that TRIM21 may regulate c-Myc expression at the protein level. Therefore, overexpression of TRIM21 decreased the half-life of c-Myc (Fig. 1F), and downregulation of TRIM21 expression prolonged it (Fig. 1G). Functionally, CRC patient-derived organoids (PDOs) with silenced TRIM21 expression exhibited enhanced organoid generation, which was greatly inhibited by silencing c-Myc gene (SI Appendix, Fig. S1 I and J). Together, these results establish the regulatory link between TRIM21 and c-Myc in CRC.
TRIM21 Interacts with c-Myc to Catalyze the K63-Linked Ubiquitination of c-Myc at Lysine 148.
To investigate the potential mechanism by which TRIM21 regulates c-Myc protein stability, we conducted tests to determine whether there was a direct physical interaction between the two proteins. We confirmed the endogenous and exogenous interactions between TRIM21 and c-Myc through coimmunoprecipitation (Co-IP) analysis in RKO and HEK293T cells, respectively (Fig. 2 A and B). Additionally, proximity ligation assay (PLA) demonstrated the association between TRIM21 and c-Myc (Fig. 2C). Furthermore, the in vitro His-pull-down assay provided further evidence of a direct binding between TRIM21 and c-Myc (Fig. 2D). Mapping of the interacting domains revealed that the region 267 to 475 aa, covering the SPRY-associated domain, SPIa and the ryanodine receptor domain (PRY-SPRY) region in TRIM21, was responsible for the association with c-Myc (SI Appendix, Fig. S2 A and B), while the Leucine zipper (LZ) domain of c-Myc was found to be indispensable for its interaction with TRIM21 (SI Appendix, Fig. S2 C and D). Computational modeling of their interaction showed that the TRIM21-c-Myc complex can interact through the PRY-SPRY domain of TRIM21 and the LZ domain of c-Myc. Furthermore, specific residues, including M417, S432, S434, and Y421 in TRIM21 and R424, K428, and H429 in c-Myc, were identified as crucial for maintaining the structural stability of the TRIM21-c-Myc heterodimer (SI Appendix, Fig. S2E). Notably, an alanine scan revealed that replacement of these residues with alanine in TRIM21 significantly impaired the stability of the complex (SI Appendix, Fig. S2F), indicating that these residues, particularly M417, S432, S434, and Y421 in TRIM21, are essential for its ability to bind c-Myc. Additionally, a mutant TRIM21 (TRIM21/MT) was constructed with M417, S432, S434, and Y421 replaced by alanine to further verify the importance of these residues in TRIM21’s interaction with c-Myc. Co-IP assays demonstrated minimal binding ability between TRIM21/MT and c-Myc compared to the wild-type TRIM21 (TRIM21/WT) (SI Appendix, Fig. S2G).
Fig. 2.
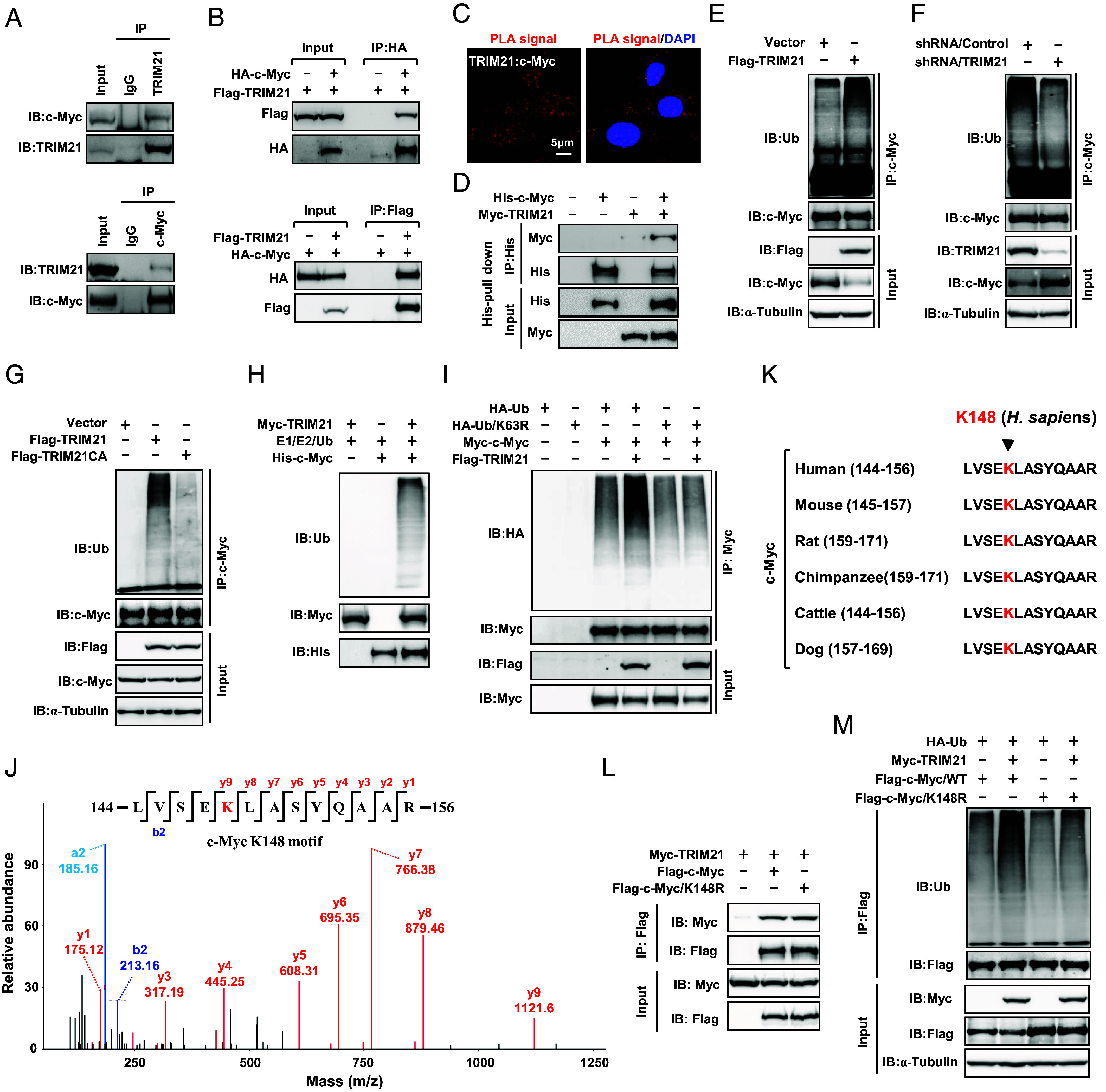
TRIM21 interacts with c-Myc to catalyze K63-linked ubiquitination at lysine 148. (A and B) Co-IP analysis of the interaction between TRIM21 and c-Myc at both the endogenous levels (A) and exogenous levels (B). (C) Confocal microscope images show PLA spots (red) in RKO cells, with each spot representing a single protein interaction. Nuclei were stained with DAPI (blue). (D) His pull-down assays were conducted using purified TRIM21 and c-Myc proteins. (E and F) Ubiquitination assays of endogenous c-Myc were performed using lysates from TRIM21-overexpressing (E) or TRIM21-silencing (F) RKO cells. (G) Ubiquitination assays of endogenous c-Myc in the lysates from RKO cells, overexpressing TRIM21 or the TRIM21CA mutant. (H) In vitro ubiquitination assays were performed by incubating Myc-TRIM21 and His-c-Myc in the presence of E1, E2, and ubiquitin. (I) Ubiquitination assays of exogenous c-Myc were conducted using lysates from HEK293T cells transfected with HA-Ub or HA-Ub/K63R. (J) An illustration is presented to depict c-Myc ubiquitination at K148 identified by Mass-spectrometry (MS). (K) An illustration displays the alignment of various c-Myc amino acid sequences, with the location of K148 depicted in red in the indicated species. (L) Co-IP analysis was performed to assess the interaction between TRIM21 and c-Myc in RKO cells expressing the indicated constructs. (M) Ubiquitination assays of exogenous c-Myc were carried out using lysates from RKO cells expressing the indicated constructs.
As TRIM21 functions as an E3 ubiquitin ligase, we proceeded to investigate its impact on c-Myc ubiquitination. Overexpression of TRIM21 significantly enhanced c-Myc ubiquitination (Fig. 2E), while knockdown of TRIM21 resulted in reduced c-Myc ubiquitination (Fig. 2F). Additionally, the overexpression of a catalytically inactive mutant, TRIM21CA, negated its ability to enhance c-Myc ubiquitination (Fig. 2G). The in vitro ubiquitination assay further confirmed the ubiquitination of c-Myc by TRIM21 (Fig. 2H). Interestingly, introduction of the Lys63-only ubiquitin mutant (Ub/K63R) was unable to support c-Myc ubiquitination (Fig. 2I), indicating that TRIM21 mediates the K63 ubiquitination of c-Myc. Furthermore, the TRIM21/MT was unable to ubiquitinate c-Myc or induce c-Myc degradation (SI Appendix, Fig. S2H), suggesting that the interaction between TRIM21 and c-Myc is essential for the ubiquitination and subsequent degradation of c-Myc.
To identify potential c-Myc ubiquitination sites, we performed mass-spectrometry and identified Lys 148 (K148) as a putative c-Myc ubiquitination site (Fig. 2J). Furthermore, sequence comparison revealed conservation of the K148 site in c-Myc across several animal species (Fig. 2K). In order to validate this site, we generated a c-Myc mutant (c-Myc/K148R) with arginine substitutions for the lysine residue. Our findings indicated that while the c-Myc/K148R mutant retained its ability to interact with TRIM21 (Fig. 2L), the mutation significantly impaired its ubiquitination by TRIM21 (Fig. 2M). Collectively, these results indicate that TRIM21, an E3 ubiquitin ligase, targets c-Myc for K63-linked polyubiquitination at lysine 148.
TRIM21 Recruits Autophagy Machinery to Facilitate the K63-Linked Ubiquitination of c-Myc for Autophagic Degradation.
The findings above suggest that TRIM21-induced c-Myc ubiquitination is attributed to K63 ubiquitination, which does not mark substrates for proteasomal degradation. As depicted in Fig. 3A, treatment with the proteasome inhibitor MG132 increased total c-Myc levels in the cell but did not alter the inhibitory effect of TRIM21 on c-Myc expression. However, the autophagy inhibitor chloroquine (CQ) reinstated c-Myc expression in the TRIM21-overexpressed CRC cells. Similarly, inhibiting autophagy by silencing ATG5 or ATG7, two key autophagy genes, notably hindered the suppressive effect of TRIM21 on c-Myc expression (Fig. 3B). These results indicate that c-Myc may be targeted for TRIM21-mediated autophagic degradation.
Fig. 3.

TRIM21 promotes c-Myc targeting for autolysosomal degradation. (A) WB analysis of c-Myc expression in RKO cells transfected with TRIM21 and treated with 10 μM MG132 or 10 mM CQ for 6 h prior to harvest. (B) WB analysis of c-Myc expression in RKO cells with stable silencing ATG5 or ATG7. (C) Co-IP analysis of the interaction between TRIM21 and c-Myc, TRIM21 and Beclin1, or TRIM21 and p62 in RKO cells. (D) Co-IP analysis was conducted to assess the interaction between TRIM21 or its TRIM21/MT (1 to 267) with c-Myc or p62 in RKO cells. (E) The PLA was used to detect the interaction between TRIM21 or its TRIM21/MT (1 to 267) and c-Myc in RKO cells. Confocal microscope images display PLA spots (in red), with each spot representing a single protein interaction. Nuclei were stained with DAPI (in blue). (F and G) The PLA was employed to detect the interaction between p62 and c-Myc in RKO cells, with TRIM21-silenced (F) or overexpressed (G). Each data point represents the number of interacting signals observed per cell. (H and I) The PLA was employed to detect the interaction between LC3B and c-Myc in RKO cells, with TRIM21-silenced (H) or overexpressed (I), during growth in normal medium with or without fetal bovine serum (FBS). (J) Co-IP analysis was performed to assess the interaction between p62 and c-Myc or its mutant c-Myc/K148R in RKO cells expressing the indicated constructs. (K and L) The PLA was used to detect the interaction between p62 and c-Myc or its mutant c-Myc/K148R (K), as well as between LC3B and c-Myc or its mutant c-Myc/K148R (L) in TRIM21 stably overexpressed HCT116 cells during growth in normal medium (RPMI-1640) without FBS.
We next investigated how TRIM21-mediated autophagic degradation of c-Myc. Our results revealed that TRIM21 promoted autophagy induction in CRC. As shown in SI Appendix, Fig. S3 A–D, overexpression of TRIM21 led to an increase in LC3B II levels and LC3B puncta, while silencing of TRIM21 resulted in a reduction of these markers. This is consistent with previous observations indicating that a subset of TRIM proteins, including TRIM21, serve as specialized receptors for highly specific autophagy (28). Indeed, in CRC cells, TRIM21 was capable of binding both the autophagic regulator Beclin 1 and the autophagic receptor p62 (Fig. 3C). Given that the PRY-SPRY domain of TRIM21 is responsible for binding its cargoes, such as IRF3 and CDK2 (28, 29), to the autophagic machinery for degradation, we examined whether this domain is also crucial for the autophagic degradation of c-Myc. Our results revealed that the deletion of the PRY-SPRY domain of TRIM21 [TRIM21 (1 to 267)] did not affect its ability to interact with Beclin 1; however, the interaction between TRIM21 and c-Myc was abolished when the PRY-SPRY domain of TRIM21 was deleted (Fig. 3D, IP). Consequently, the deletion of the PRY-SPRY domain of TRIM21 failed to induce the degradation of c-Myc protein compared to the wild-type TRIM21 (TRIM21/WT) (Fig. 3D, Input). Furthermore, consistent with these results, PLA experiments also supported that the PRY-SPRY domain of TRIM21 is critical for binding its cargo, c-Myc (Fig. 3E).
To further verify that TRIM21 targets c-Myc for autophagic degradation, we conducted several PLA experiments to assess the binding affinity of c-Myc with the autophagy machinery. Our findings revealed that silencing of TRIM21 in CRC cells significantly disrupted the interaction of c-Myc with the autophagic receptor p62 (Fig. 3F). Conversely, Overexpression of TRIM21 in CRC cells exerted the opposite effects (Fig. 3G and SI Appendix, Fig. S3E). These results were further supported by the observation that, under both basal conditions and during serum starvation, silencing of TRIM21 hindered the colocalization of c-Myc and LC3B (Fig. 3H), an autophagy marker, while overexpressing TRIM21 enhanced their colocalization (Fig. 3I and SI Appendix, Fig. S3F). Furthermore, our results showed that reexpression of wild-type c-Myc (c-Myc/WT), but not the c-Myc ubiquitination resistant mutant c-Myc/K148R, in c-Myc-depleted CRC cells, was able to restore TRIM21-induced interaction of c-Myc with p62 (Fig. 3J). These findings were further confirmed by PLA experiments, which revealed that overexpression of TRIM21 did not enhance the interaction between c-Myc/K148R and p62 (Fig. 3K) or promote the colocalization of c-Myc/K148R and LC3B (Fig. 3L) compared to the wild-type c-Myc (c-Myc/WT). Collectively, these results suggest that the ubiquitination of c-Myc at K148 by TRIM21 is required for TRIM21 to enable c-Myc to be targeted to the autophagy machinery for degradation.
Downregulation of TRIM21 Promotes Enolase 2 (ENO2) Expression and Enhances Glycolysis through a c-Myc-Dependent Pathway.
Increased glycolytic activity provides cancer cells with the energy and building blocks necessary to support their rapid growth and proliferation (30, 31). Interestingly, GSEA revealed that CRC cells with silenced TRIM21 exhibited enhanced expression of glycolysis-related genes (Fig. 4A). We identified three up-regulated glycolysis-related genes, namely ENO2, aldolase C, and phosphoglycerate kinase 1, from RNA-seq data (Fig. 4B). Among these up-regulated genes, ENO2 exhibited the most significant upregulation in both RKO cells and HCT116 cells (Fig. 4B). As a result, we proceeded to investigate the regulatory role of TRIM21 in ENO2. Consistent with the aforementioned findings from RNA-seq data, the silencing of TRIM21 in both RKO cells and HCT116 cells significantly enhanced the expression of ENO2 protein (Fig. 4C). Conversely, stable expression of TRIM21 in CRC cells markedly inhibited ENO2 mRNA and protein levels (Fig. 4 D and E and SI Appendix, Fig. S4 A and B). Furthermore, immunohistochemistry (IHC) staining detected a significant negative correlation between TRIM21 and ENO2 in a CRC cohort (n = 150) (Fig. 4F). Collectively, these results demonstrate that TRIM21 negatively regulates ENO2 expression.
Fig. 4.
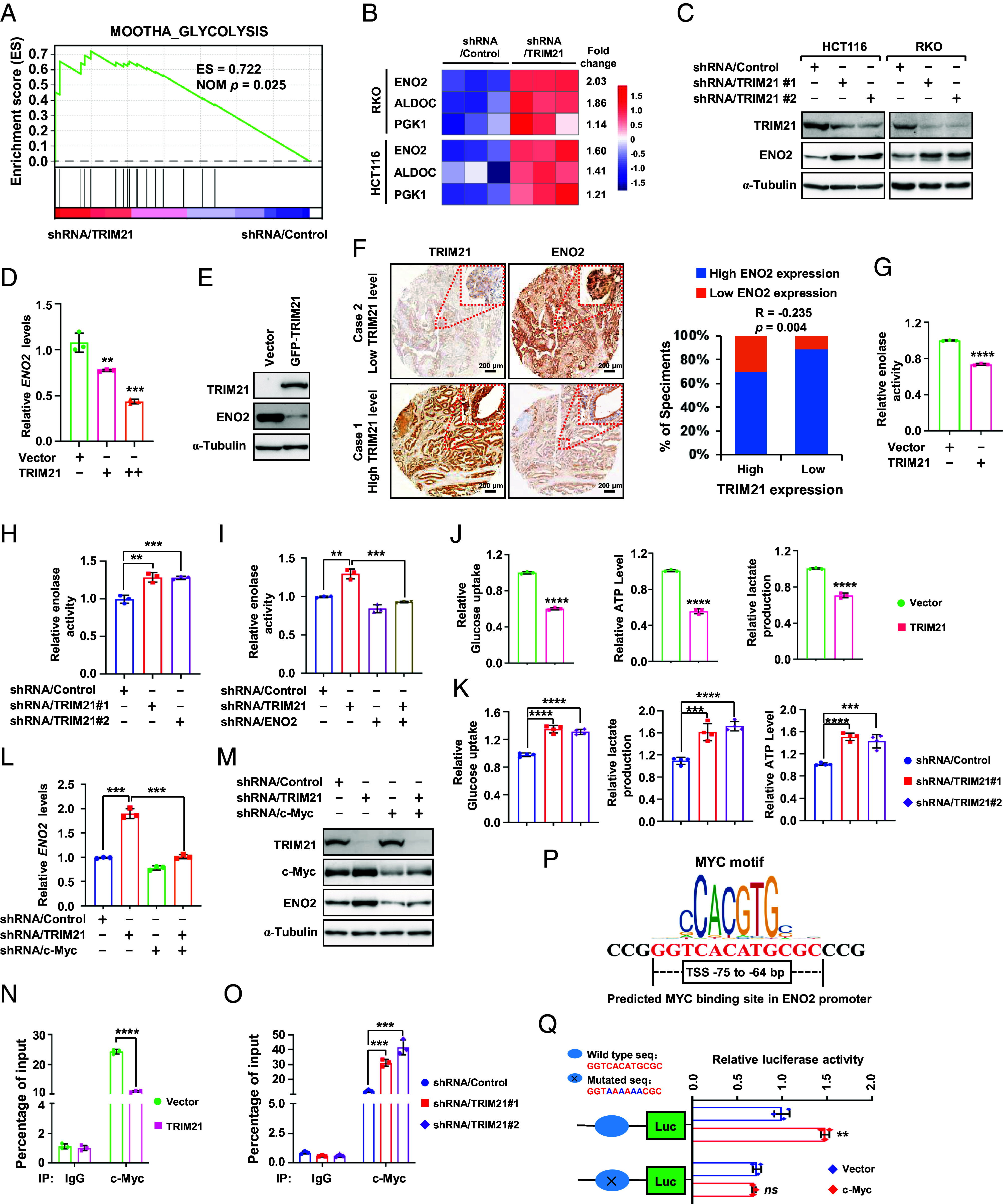
TRIM21 suppresses glycolysis by regulating c-Myc-mediated ENO2 expression. (A) GSEA of glycolysis-associated genes in HCT116 cells stably expressing shRNA/TRIM21 or shRNA/control. (B) A heatmap illustrates the expression of specific glycolysis-related genes. (C) WB analysis of the indicated protein in HCT116 and RKO cells. (D and E) qPCR (D) and WB (E) analysis were carried out to assess ENO2 expression in TRIM21-overexpressing HCT116 cells. (F) Representative images (Left) and correlation analysis (Right) of TRIM21 and ENO2 protein levels in 150 cases of CRC tissues. (G and H) Enolase activity was determined using ELISA in TRIM21-overexpressing (G) or –silencing (H) HCT116 cells. (I) Enolase activity was determined using ELISA in HCT116 cells coexpressing shRNA/TRIM21 and shRNA/ENO2. (J and K) ELISA analysis of glucose uptake, lactate production, and ATP levels in TRIM21-overexpressing (J) or –silencing (K) HCT116 cells. (L and M) qPCR (L) and WB (M) were used to measure ENO2 levels in HCT116 cells coexpressing shRNA/TRIM21 and shRNA/c-Myc. (N and O) ChIP assays were performed to assess the ability of c-Myc to bind the Eno2 promoter in TRIM21-overexpressing HCT116 cells (N) and TRIM21-silencing RKO cells (O). (P) An illustration showing the predicted MYC binding site in the Eno2 promoter. (Q) Mutation of the potential MYC binding site in Eno2 promotor and analysis of the promoter activity after transfection of c-Myc.
We then investigated the enolase activity using Enzyme-linked immunosorbent assay (ELISA). The results showed that overexpression of TRIM21 significantly inhibited enolase activity (Fig. 4G and SI Appendix, Fig. S4C), while silencing of TRIM21 had the opposite effect (Fig. 4H). Furthermore, inhibition of ENO2 expression in CRC cells greatly abolished the upregulation of enolase activity induced by the silencing of TRIM21 (Fig. 4I and SI Appendix, Fig. S4D), indicating that ENO2 is responsible for the enhanced enolase activity resulting from TRIM21-silencing. ENO2 serves as an important enzyme in promoting glycolysis. Consequently, overexpression of TRIM21 led to reduced glycolysis as revealed by decreased glucose uptake, adenosine triphosphate (ATP) level, and lactate production (Fig. 4J and SI Appendix, Fig. S4E). Consistent with this, silencing of TRIM21 resulted in enhanced glycolysis, as evidenced by a significant increase in glucose uptake, ATP level, and lactate production compared to the control (Fig. 4K and SI Appendix, Fig. S4F).
The results above suggest that TRIM21 regulates the autophagic degradation of c-Myc. This leads us to contemplate whether TRIM21 governs the expression of ENO2 via c-Myc. In TRIM21-silenced CRC cells, the levels of ENO2 mRNA and protein significantly increased, whereas they showed minimal change in CRC cells with simultaneous silencing of c-Myc (Fig. 4 L and M). Consistently, silencing of TRIM21 in tumor organoids significantly increased ENO2 expression. However, this regulatory effect was entirely abolished by depleting c-Myc expression (SI Appendix, Fig. S4G). These findings indicate that TRIM21 regulates ENO2 expression through a c-Myc-dependent mechanism. Chromatin immunoprecipitation analysis using the c-Myc antibody revealed substantial enrichment of c-Myc in the promoter regions of the ENO2 gene. However, overexpression of TRIM21 significantly reduced this enrichment, while silencing TRIM21 markedly increased it in the promoter region of ENO2 (Fig. 4 N and O). Bioinformatic analysis indicated potential c-Myc binding sites in the ENO2 promoter (Fig. 4P). To validate this hypothesis, we mutated this site and then assessed its luciferase activity. As shown in Fig. 4Q, the luciferase activity induced by c-Myc was significantly impaired compared to that of the wild-type promoter construct. Taken together, our results suggest that ENO2 is a direct target gene of c-Myc.
Deletion of Trim21 Amplifies Colorectal Carcinogenesis in Apcmin/+ Mice and Colitis-Associated Colon Cancer Mice by Strengthening ENO2-Mediated Glycolysis.
To validate the in vivo regulatory roles of TRIM21-mediated c-Myc signaling in CRC development, we first established Azoxymethane (AOM)/Dextran sodium sulfate (DSS)-induced colon cancer model using Trim21 knockout (Trim21−/−) mice (SI Appendix, Fig. S5A). Our results showed that Trim21−/− mice exhibited a higher number of visible colon tumor foci (SI Appendix, Fig. S5B) and demonstrated a significant increase in tumor number and burden (SI Appendix, Fig. S5C). Immunohistochemical (IHC) analysis of the colon tumors using PCNA antibody revealed a marked increase in the percentage of tumor cells with PCNA-positive (PCNA+) nuclear immunostaining in Trim21−/− mice compared with wild-type (Trim21+/+) mice (SI Appendix, Fig. S5 D and E). Although the levels of c-Myc mRNA in the tumors did not show a significant difference (SI Appendix, Fig. S5F), TRIM21 deficiency significantly impaired the K63-linked polyubiquitination of c-Myc protein and led to a notable increase in the total levels of c-Myc protein (SI Appendix, Fig. S5G). Furthermore, qPCR and WB analysis confirmed a significant increase in both ENO2 mRNA and protein levels in the colon tumors of Trim21−/− mice compared with wild-type (Trim21+/+) mice (SI Appendix, Fig. S5 G and H). Consequently, increased enolase activity and glycolysis were observed in the colon tumors from Trim21−/− mice (SI Appendix, Fig. S5 I and J). We further crossed Apcmin/+ mice with Trim21−/− mice to generate Apcmin/+; Trim21−/− mice. Similar to the results obtained from the AOM/DSS-induced inflammatory colon cancer mouse model, deletion of TRIM21 in the Apc-driven mouse model of colon cancer resulted in the development of more visible colon tumor foci (SI Appendix, Fig. S5K), as well as an increase in the total number of tumors and maximal tumor size (SI Appendix, Fig. S5L). Furthermore, TRIM21 deficiency markedly promoted the proliferation of tumor cells, as indicated by the analysis of PCNA in the colon tumor samples (SI Appendix, Fig. S5 M and N). We also investigated the molecular mechanism by which TRIM21 regulates colon tumorigenesis in this mouse model. We found no significant difference in c-Myc mRNA levels in colon tumor samples of Trim21−/− compared to Trim21+/+ mice (SI Appendix, Fig. S5O). However, we observed decreased K63-linked polyubiquitination and increased total levels of c-Myc protein in the colon tumor from Trim21−/− mice (SI Appendix, Fig. S5P). Additionally, we found that the mRNA and protein levels of ENO2, enolase activity, lactate production, and ATP levels were greatly enhanced in colon tumor samples from Trim21−/− mice (SI Appendix, Fig. S5 Q–S). Taken together, the above results establish a functional relationship between TRIM21 and c-Myc signaling in vivo.
We further treated intestinal organoids derived from Apcmin/+; Trim21−/− mice and Apcmin/+; Trim21+/+mice with the ENO2-specific inhibitor POMHEX. The results showed that TRIM21 deficiency enhanced organoid formation; however, the inhibition of ENO2 by POMHEX abrogated organoid formation (SI Appendix, Fig. S5T), suggesting that ENO2 plays a vital role in TRIM21-associated tumor growth. To delve deeper into ENO2's pivotal role in TRIM21-associated colorectal tumorigenesis, we generated mice with conditional deletion of Eno2 in the intestinal epithelium (referred to as Eno2ΔIEC mice) by crossing Eno2-floxed mice with Villin-Cre mice. We then bred Trim21−/− mice with Eno2ΔIEC mice to produce Trim21−/−; Eno2ΔIEC offspring (Fig. 5A). Utilizing the AOM/DSS model for colon cancer induction (Fig. 5B), ENO2 ablation in the intestinal epithelium notably abrogated the onset of colon tumors. As shown in Fig. 5 C–E, Trim21−/−; Eno2fl/fl mice exhibited a pronounced increase in both the quantity and size of colon tumors relative to Trim21+/+; Eno2fl/fl mice. Nonetheless, the absence of ENO2 in Trim21−/−; Eno2ΔIEC mice appeared to partially mitigate these effects. Importantly, ENO2 deletion substantially reduced lactate and ATP levels in the colon tumor tissues from Trim21−/−; Eno2ΔIEC mice compared with Trim21−/−; Eno2fl/fl counterparts (Fig. 5F). Consistently, ENO2 deficiency abrogated organoid growth (Fig. 5G). Collectively, these findings underscore the functional interplay between TRIM21 and ENO2 in governing intestinal tumor development.
Fig. 5.
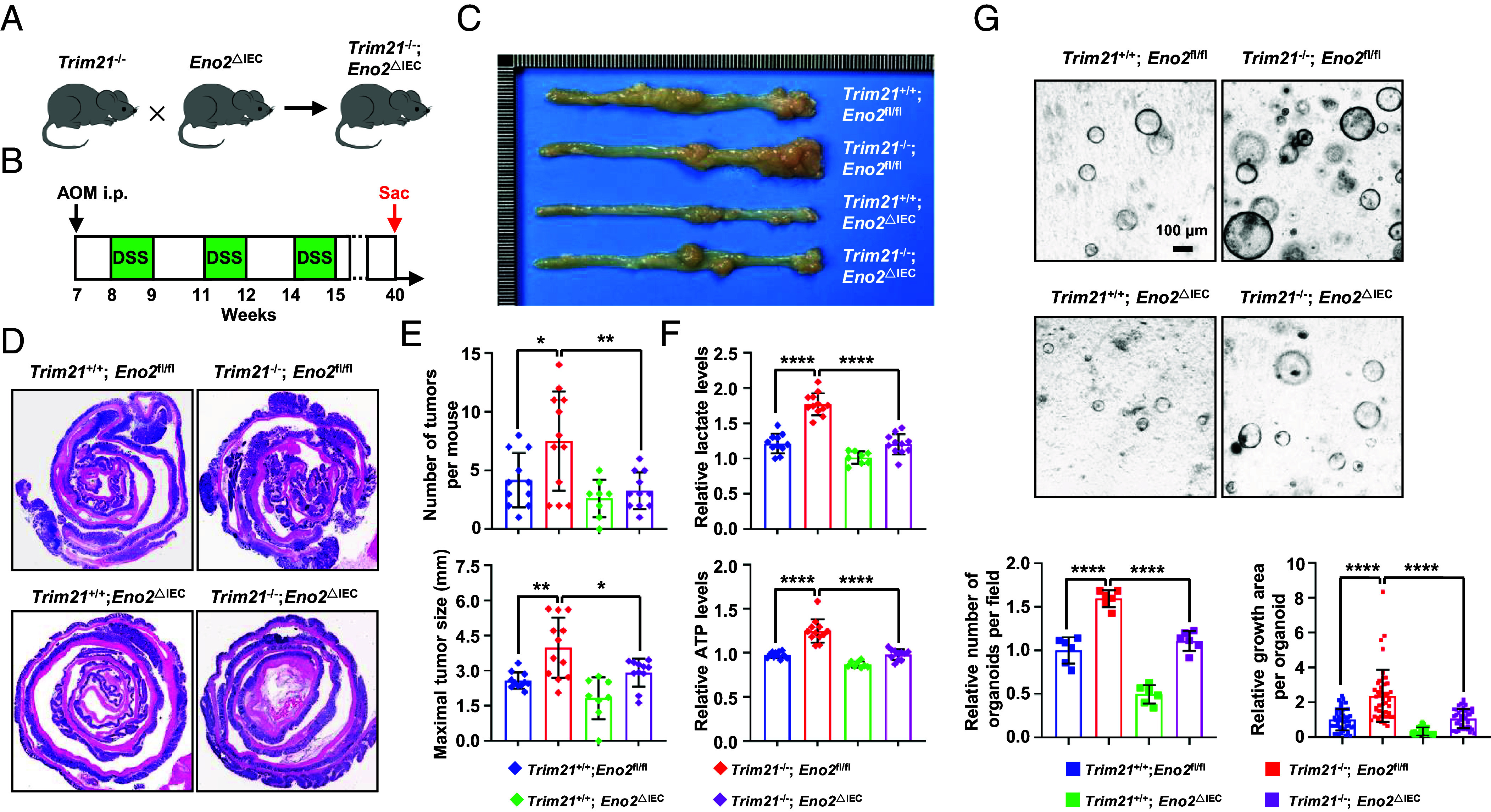
ENO2 plays a crucial role in TRIM21-associated tumor growth in the CRC mouse model. (A) The schematic shows the strategy to generate Trim21−/−; Eno2ΔIEC mice. (B) Design of the experimental AOM/DSS model. (C–E) Representative photographs (C) and H&E staining (D) of the colon, as well as the colon tumor number (E, Top) and tumor burden (E, Bottom) from Trim21+/+; Eno2fl/fl (n = 11), Trim21−/−; Eno2fl/fl (n = 12), Trim21+/+; Eno2ΔIEC (n = 8), and Trim21−/−; Eno2ΔIEC (n = 11) mice. (F) ELISA analysis of lactate (Top) and ATP levels (Bottom) in colon tumors of Trim21+/+; Eno2fl/fl (n = 11), Trim21−/−; Eno2fl/fl (n = 12), Trim21+/+; Eno2ΔIEC (n = 8), and Trim21−/−; Eno2ΔIEC (n = 11) mice. (G) Representative images (Top) and quantification (Bottom) of the indicated organoids formed by the colonic tumor from AOM/DSS-induced Trim21+/+; Eno2fl/fl, Trim21−/−; Eno2fl/fl, Trim21+/+; Eno2ΔIEC, and Trim21−/−; Eno2ΔIEC mice.
KRAS-Mutant Activates EKR2 to Phosphorylate TRIM21 at Threonine 396 (T396).
To elucidate the molecular impact of Trim21 deficiency in mice, we analyzed the gene expression profiles of colon tumor tissues from AOM/DSS-treated Trim21+/+ and Trim21−/− mice using RNA-seq. Our analysis identified 1,748 genes with increased expression and 1,252 genes with decreased expression as a result of Trim21 loss according to B-statistics of the limma method (SI Appendix, Fig. S6A). Notably, gene ontology and pathway enrichment analyses of the differentially expressed genes indicated a significant enrichment of targets related to KRAS and the KRAS oncogenic signaling pathway (SI Appendix, Fig. S6B). Considering the prevalence of KRAS mutations in CRC, which are pivotal in CRC pathogenesis through their activation of the Ras/Raf/MEK/ERK signaling cascade (32, 33), we further probed into the potential involvement of TRIM21 in the oncogenic signaling mediated by KRAS mutations. In CRC cells expressing KRAS/G12D, one of the most common mutations in CRC, we noted a significant increase in the phosphorylation status of the TRIM21 protein (Fig. 6A). Pretreatment of these cells with PD98059, an inhibitor of ERK’s upstream pathways, effectively prevented the phosphorylation of TRIM21 (p-TRIM21) induced by KRAS/G12D; in contrast, inhibition of p38 with SB203580 or of JNK with SP600125 did not produce this effect (Fig. 6B), suggesting that ERK is involved in TRIM21 phosphorylation. Further supporting this, the presence of KRAS/G12D was found to enhance the interaction between TRIM21 and ERK2, but not ERK1 (SI Appendix, Fig. S6 C and D). Moreover, silencing of ERK2, but not ERK1, led to the suppression of TRIM21 phosphorylation induced by KRAS/G12D (Fig. 6C), thereby establishing ERK2 as an upstream kinase regulating TRIM21.
Fig. 6.
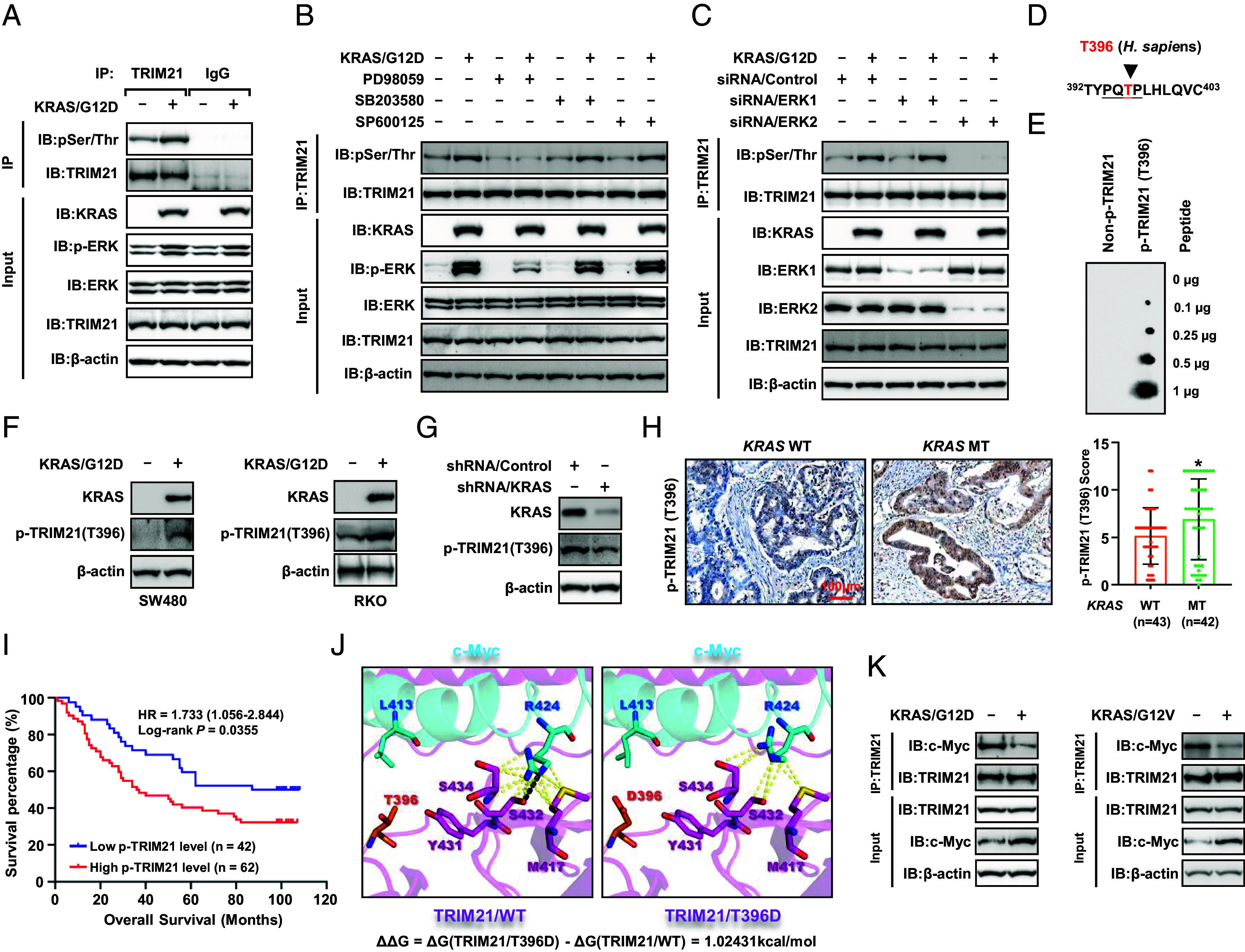
TRIM21 phosphorylation triggered by mutant KRAS enhances c-Myc expression. (A) RKO cells were transfected with KRAS/G12D, and whole-cell extracts were collected for immunoprecipitation with an anti-TRIM21 antibody, followed by WB analysis of TRIM21 phosphorylation using an anti-phospho-Ser/Thr antibody. (B) RKO cells transfected with KRAS/G12D were treated with various kinase inhibitors (PD98059, 10 μM; SB203580, 10 μM; SP600125, 10 μM), and the p-TRIM21 was determined. (C) The p-TRIM21 was analyzed in the ERK1 or ERK2 knockdown RKO cells transfected with KRAS/G12D. (D) An illustration displays that TRIM21 contains a phosphorylation consensus motif of ERK (P-X-S/T-P), 394PQTP397. (E) The dot-blot assay was performed to validate the antibody's specificity for T396-specific phosphorylation [p-TRIM21 (T396)]. (F and G) WB analysis of p-TRIM21 (T396) levels in the CRC cells overexpressing KRAS/G12D (F) or stably silencing KRAS (G). (H) IHC staining of p-TRIM21 (T396) in CRC tissues with and without mutant KRAS (Left) and Boxplot shows the p-TRIM21 (T396) levels in the CRC tissues (Right). (I) Kaplan–Meier survival curve of patients with CRC expressing high (n = 62) and low (n = 42) levels of p-TRIM21 (T396). (J) Interaction diagram between wild-type TRIM21 and c-Myc (Left). Interaction diagram following the mutation of TRIM21’s T396 to D396 (Right), illustrating the alterations in interactions with residue R424 on c-Myc resulting from the mutation of TRIM21’s T396 to D396. The hydrogen bond interaction is indicated by a black dashed line, while the nonpolar interactions are depicted by yellow dashed lines. (K) Co-IP analysis was performed to assess the interaction between TRIM21 and c-Myc in RKO cells transfected with KRAS/G12D (Left) or KRAS/G12V (Right).
By performing a sequence alignment analysis, we found that the TRIM21 contains a phosphorylation consensus motif of ERK (P-X-S/T-P), 394PQTP397 (Fig. 6D). We thus generated a mutant form of TRIM21 (TRIM21/T396A) with threonine residue substituted with alanine. The Co-IP assay showed that the KRAS/G12D mutation induced phosphorylation of wild-type TRIM21 (TRIM21/WT), but not the TRIM21/T396A mutant (SI Appendix, Fig. S6E), indicating that T396 is a major ERK phosphorylation site upon KRAS-mutant. To facilitate the specific recognition of TRIM21 T396 phosphorylation, we generated a T396-specific phosphorylation antibody, p-TRIM21 (T396), which specifically recognized TRIM21 T396-specific phosphorylation peptide using a dot-blot assay (Fig. 6E). We found that TRIM21 T396 phosphorylation markedly increases in response to KRAS/G12D (Fig. 6F), and that the TRIM21/T396A mutation eliminates the signal detectable by the p-TRIM21 (T396) antibody (SI Appendix, Fig. S6F). Furthermore, silencing of KRAS in the KRAS-mutant HCT116 cell line markedly reduced TRIM21 T396 phosphorylation (Fig. 6G). Next, we performed IHC to examine TRIM21 T396 phosphorylation levels in a CRC cohort (n = 85). The results showed that p-TRIM21 (T396) levels were significantly elevated in CRC tissues with the KRAS mutation compared to those with wild-type KRAS (KRAS/WT) (Fig. 6H). Kaplan–Meier survival analysis revealed that patients with high levels of p-TRIM21 (T396) expression had significantly reduced survival compared to those with low expression levels (Fig. 6I). These findings underscore the importance of TRIM21 T396 phosphorylation as a prognostic indicator for the overall survival of CRC patients.
KRAS-Mutant-Triggered p-TRIM21 Hampers the Autophagic Degradation of c-Myc and Enhances Glycolysis.
Since TRIM21 promotes the autophagic degradation of c-Myc and inhibits glycolysis, we investigated whether p-TRIM21 regulates this process. Computational modeling demonstrated that the TRIM21/T396D mutant, designed to mimic phosphorylation, markedly destabilizes the TRIM21-c-Myc complex (Fig. 6J). Co-IP experiments confirmed that the expression of KRAS/G12D or KRAS/G12V in RKO cells significantly disrupted the interaction between TRIM21 and c-Myc (Fig. 6K). Similarly, PLA experiments revealed that the KRAS-mutant inhibited the association of TRIM21 with c-Myc in the intestine tumor tissues from Apcmin/+; KrasG12D; Tp53+/− mice (SI Appendix, Fig. S6G). Additionally, wild-type TRIM21 (TRIM21/WT) markedly promoted the K63-linked polyubiquitination of c-Myc, an effect that could be reversed by KRAS/G12D. However, KRAS/G12D did not have a similar effect in TRIM21/T396A-expressing CRC cells (SI Appendix, Fig. S6H). Consequently, KRAS/G12D significantly impaired the interaction between c-Myc and p62 and hindered the association of c-Myc with LC3B in TRIM21-depleted RKO cells with reexpression of wild-type TRIM21 (TRIM21/WT) but not in the cells with TRIM21/T396A reexpression (SI Appendix, Fig. S6I). Furthermore, KRAS/G12D enhanced enolase activity (SI Appendix, Fig. S6J) and glycolysis (SI Appendix, Fig. S6K) in TRIM21/WT-expressing CRC cells, but not in TRIM21/T396A-expressing CRC cells. Collectively, these results suggest that the KRAS-mutant inhibits the autophagic degradation of c-Myc and enhances glycolysis by phosphorylating TRIM21 at T396.
Inhibition of TRIM21 Phosphorylation Enhances the Efficacy of Regorafenib Therapy in KRAS-Mutant CRC.
Regorafenib, an oral multikinase inhibitor, has shown promising therapeutic efficacy in CRC (34, 35). Retrospective analysis has indicated a less significant benefit for the majority of CRC patients suffering from KRAS-mutant when undergoing regorafenib treatment (35–37). However, direct experimental evidence is lacking, and the underlying molecular mechanisms remain unclear. To address this gap, we conducted an initial half-maximal growth-inhibitory concentration (IC50) analysis in CRC cells. The results showed that regorafenib inhibits the growth of SW620, DLD-1, HCT116, and SW480 cells, which harbor mutant KRAS (KRAS/MT), at nearly twice the concentration required for HT-29 and RKO cells with KRAS/WT (SI Appendix, Fig. S7A). Down-regulating KRAS expression in the KRAS-mutant CRC cells significantly lowered the IC50 value (SI Appendix, Fig. S7B), whereas introducing ectopic KRAS/G12D in HT-29 cells with KRAS/WT substantially raised the IC50 value (SI Appendix, Fig. S7C). These results suggest that KRAS mutation may lead to unsatisfactory therapeutic efficacy. To further verify this, we established CRC PDOs from different patients with CRCs (n = 48). Among these, 35.4% of PDOs (n = 17) exhibited a proliferative growth pattern after passaging (Fig. 7A and SI Appendix, Table S1). Consistent with the results in cell lines, the IC50 of the majority of PDOs harboring KRAS/MT is higher than those PDOs with KRAS/WT (Fig. 7B). Notably, inhibition of KRAS expression in the PDOs with KRAS/MT significantly improved the sensitivity to regorafenib therapy (SI Appendix, Fig. S7D). Furthermore, we employed mouse intestinal organoids from Apcmin/+; KrasWT; Tp53+/− mice and Apcmin/+; KrasG12D; Tp53+/− mice to evaluate the effect of regorafenib treatment. Our results showed that regorafenib inhibited the growth of intestinal organoids derived from Apcmin/+; KrasG12D; Tp53+/− mice at a concentration nearly four times higher than that needed for organoids derived from Apcmin/+; KrasWT; Tp53+/− mice (SI Appendix, Fig. S7E). Collectively, these findings suggest that KRAS/MT confers resistance to regorafenib treatment in CRC.
Fig. 7.

Inhibition of TRIM21 phosphorylation sensitizes regorafenib therapy in CRC. (A) Graphical representation of a human PDO and PDX model. (B) Representative images depicting CRC PDOs with or without KRAS/MT in response to regorafenib treatment (Left). Regression curves and IC50 values were depicted in the graph (Right). (C) CRC PDOs carrying KRAS/WT or KRAS/MT were treated with control vehicle, vilazodone (10 μM), regorafenib (5 μM), or a combination of treatments. Representative images of CRC PDOs (Left) and quantification (Right) are presented. (D) A schematic representation of the treatment plan for NOD/SCID mice bearing subcutaneous CRC tumors. (E) Depiction of tumor images (Left), tumor weight (Middle), and tumor growth curve (Right). (F) IHC staining and quantification of p-TRIM21 (T396) and PCNA in CRC PDX tumors in each group.
Our finding that KRAS/MT hampers the autophagic degradation of c-Myc and enhances ENO2-mediated glycolysis, we thus tested whether inhibiting glycolysis could enhance the therapeutic effectiveness of regorafenib in an AOM/DSS-induced mouse colon cancer model (SI Appendix, Fig. S7F). In keeping with our findings above, the deficiency of ENO2 in Trim21+/+; Eno2ΔIEC and Trim21−/−; Eno2ΔIEC mice showed an inhibitory effect on tumor growth compared to the Trim21+/+; Eno2fl/fl and Trim21−/−; Eno2fl/fl mice (SI Appendix, Fig. S7 G and H). Furthermore, tumors from Trim21+/+; Eno2ΔIEC and Trim21−/−; Eno2ΔIEC mice exhibited lower enolase activity, ATP, and lactate production than those from Trim21+/+; Eno2fl/fl and Trim21−/−; Eno2fl/fl mice (SI Appendix, Fig. S7 I–K). Significantly, treatment with regorafenib in Trim21+/+; Eno2ΔIEC and Trim21−/−; Eno2ΔIEC mice had an additive effect in suppressing tumor growth compared to Trim21+/+; Eno2fl/fl and Trim21−/−; Eno2fl/fl mice (SI Appendix, Fig. S7 G and H). These results strongly suggest that the inhibition of ENO2 expression cooperates with regorafenib to suppress the growth of CRC.
Our recent study has identified that vilazodone, an antidepressant, directly binds to TRIM21 to exert effective antimetastatic action both in in vitro and in vivo (27). Interestingly, our findings further showed that vilazodone significantly impaired the KRAS/G12D-induced association between TRIM21 and ERK2 (SI Appendix, Fig. S7L). Importantly, vilazodone effectively inhibits the KRAS/G12D-induced p-TRIM21 at T396 and suppresses the expression of c-Myc and ENO2 induced by KRAS/G12D (SI Appendix, Fig. S7M). Additionally, vilazodone treatment reversed the increase in enolase activity and glycolysis induced by KRAS/G12D (SI Appendix, Fig. S7 N and O). Overall, these results suggest that vilazodone inhibits the KRAS-mutant-driven c-Myc/ENO2/glycolysis signaling axis through targeting TRIM21. Analysis of CRC tissues used to establish the PDOs mentioned above revealed heightened levels of p-TRIM21 (T396), c-Myc, and ENO2 proteins (SI Appendix, Fig. S7 P–R), along with increased enolase activity (SI Appendix, Fig. S7S), lactate and ATP (SI Appendix, Fig. S7T) in KRAS-mutant CRC tissues compared to those with KRAS/WT. Given these results, we hypothesized that the combined use of vilazodone and regorafenib may have an additional impact on tumor growth. To test this hypothesis, we treated mouse intestinal organoids from Apcmin/+; KrasWT; Tp53+/− mice and Apcmin/+; KrasG12D; Tp53+/− mice with vilazodone, regorafenib, either alone or in combination. We observed that the combined treatment significantly inhibited the growth of organoids compared to treatment with either agent alone, particularly in intestinal organoids derived from Apcmin/+; KrasG12D; Tp53+/− mice in comparison to those from Apcmin/+; KrasWT; Tp53+/− mice (SI Appendix, Fig. S7U). Consistently, Treatment with vilazodone or regorafenib alone led to a moderate reduction in the growth of PDOs, and the combined treatment resulted in the lowest cell viability in PDOs carrying KRAS/MT compared to those with KRAS/WT (Fig. 7C and SI Appendix, Fig. S7V). Aligned with the PDO results, the combined treatment of vilazodone and regorafenib significantly restrained tumor growth compared to regorafenib monotherapy in models of CRC patient-derived xenografts (PDXs) (Fig. 7D). Importantly, this combined treatment had the most pronounced effect on reducing tumor growth in PDXs with KRAS/MT compared to those with KRAS/WT (Fig. 7E). Immunohistochemistry (IHC) analysis revealed that the expression of p-TRIM21 (T396) decreased in PDX tumors with KRAS/MT in mice treated with vilazodone alone or in combination with regorafenib, compared with the vehicle-treated and regorafenib-treated groups (Fig. 7F). Additionally, the combination treatment of vilazodone and regorafenib also led to a significant reduction in the number of PCNA+ proliferating tumor cells (Fig. 7F). Collectively, the data suggest that the combination of vilazodone with regorafenib may represent a promising treatment approach for regorafenib-resistant CRC tumors with KRAS/MT.
Discussion
The Kirsten rat sarcoma (KRAS) oncogene is frequently mutated in CRC and is generally associated with a poor prognosis (3, 38). In this study, we uncovered an underappreciated function of KRAS in its interplay with the oncoprotein c-Myc in CRC. We found that KRAS inhibits the TRIM21-triggered selective autophagic degradation of c-Myc by activating ERK2-mediated TRIM21 phosphorylation. Consequently, this process enhances glycolysis and confers resistance to regorafenib therapy. These findings offer unique insights for potential treatments targeting KRAS-mutant CRC.
Selective autophagy is a specialized form of autophagy that targets specific cellular components for degradation. This process plays a crucial role in maintaining cellular homeostasis and can significantly influence tumorigenesis and progression in various ways (39, 40). For example, Flightless-I acts as a checkpoint protein in selective autophagy by interacting with p62, which inhibits the recognition of LC3, thereby promoting tumorigenesis in breast cancer (41). Our previous study also demonstrated that TRAF6 regulates β-catenin protein levels by activating the selective autophagic degradation pathway for β-catenin, thereby helping to prevent the metastasis of primary tumor cells (42). c-Myc plays a central role in nearly every aspect of the oncogenic process, controlling processes such as proliferation, apoptosis, and differentiation (9, 43, 44). The stability of the c-Myc protein is closely regulated by the ubiquitin–proteasome system. Various ubiquitinating enzymes and deubiquitinating enzymes, including Fbw7, USP28, and USP36, have been identified to regulate c-Myc protein stability (45–48). Our previous research highlighted the involvement of the E3 ubiquitin ligase TRAF6 in regulating c-Myc protein abundance in hepatocellular carcinoma (19). However, whether c-Myc is involved in selective autophagy has not been reported. An important finding here is that we have uncovered a selective autophagic mechanism that regulates c-Myc protein stability. We presented evidence that TRIM21 directly interacts with and ubiquitinates c-Myc with K63-linked ubiquitin at the K148 residue, which promotes the targeting of c-Myc to the autophagic machinery for degradation. However, KRAS inhibits this regulatory process. This is due to KRAS/MT-driven MAPK signaling, which leads to the p-TRIM21 at the T396 residue by ERK2. This phosphorylation disrupts the interaction between TRIM21 and c-Myc, preventing c-Myc from being targeted for autophagic degradation. Our present study has further revealed that the expression of p-TRIM21 (T396) is significantly higher in KRAS-mutant CRC patients than in CRC patients with KRAS/WT. Moreover, elevated levels of p-TRIM21 (T396) are correlated with poor prognosis. Although the regulatory mechanism for TRIM21-triggered selective autophagic degradation of c-Myc is not available in KRAS-mutant CRC, which accounts for approximately 40% of all CRC cases (3), these findings provide a different therapeutic strategy for the treatment of KRAS-mutant CRC. Our model showed that blocking TRIM21 phosphorylation with the antidepressant vilazodone, recently identified as TRIM21's ligand by our team (27), could functionally restore TRIM21 to trigger selective autophagic degradation of c-Myc in KRAS-mutant CRC.
Metabolic reprogramming is a critical factor contributing to therapeutic resistance in cancer (49). For instance, phosphoglycerate dehydrogenase, the initial enzyme in the serine synthesis pathway, plays a pivotal role in sorafenib resistance (50). Additionally, upregulation of glucose transporter 3 is associated with metabolic reprogramming and resistance to antiangiogenic therapy (51). Our study reveals that directly inhibiting the glycolytic enzyme ENO2 expression in Eno2ΔIEC mice or targeting TRIM21 with the antidepressant vilazodone to inhibit c-Myc-mediated ENO2 expression and glycolysis in CRC PDOs and PDXs harboring KRAS/MT significantly sensitizes regorafenib therapy. These findings align with recent studies demonstrating that pharmacological inhibition of enolase activity by AP-III-a4 or POMHEX enhances the effectiveness of antiangiogenic drugs (52), underscoring the importance of metabolic reprogramming in therapeutic resistance across various cancer types.
In summary, our study has uncovered an autophagy-mediated mechanism for the degradation of the c-Myc protein and has elucidated the interplay between KRAS and c-Myc signaling in KRAS-mutant CRC.
Materials and Methods
The use of human CRC tissues in the study was approved by Soochow University for Biomedical Research Ethics Committee, and all of the patients provided informed consent. The animal experiments were conducted in compliance with the guidelines of the Animal Care and Use Committee of Soochow University. Patient-derived colon cancer organoids, immunofluorescence microscopy, PLA, western immunoblotting, immunoprecipitation, ubiquitination assay, qPCR analysis, pull-down assay, and RNA-seq analysis were performed as previously described (22, 27, 53). Additional materials and methods are available in SI Appendix, SI Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82022050, 82372662, 81972601, 82002520, 82302967, and 82373142), the Natural Science Foundation of Jiangsu Province (BE2023703), the Science and Technology Foundation of Suzhou (SZM2022014), and Suzhou Medical Key Supported Discipline (SZFCXK20241). This work was also supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author contributions
H.W. designed research; W.-L.Y., L.H., X.-Q.Y., S.W., W.-J.G., Y.Y., X.-S.H., F.L., X.G., Y.-X.L., G.H., X.-M.L., W.-Y.S., K.H., Y.-Y.W., W.-X.W., J.-H.L., Y.S., and C.-J.Q. performed research; W.-L.Y., L.H., X.-Q.Y., S.W., Y.Y., X.-S.H., F.L., X.G., Y.-X.L., G.H., X.-M.L., W.-Y.S., K.H., Y.-Y.W., W.-X.W., J.-H.L., Y.S., C.-J.Q., and H.W. analyzed data; and S.W. and H.W. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Yu Song, Email: sy5449@163.com.
Chen-Jiang Qu, Email: qcj6019@suda.edu.cn.
Hua Wu, Email: wuhua@suda.edu.cn.
Data, Materials, and Software Availability
RNA-seq results have already been uploaded to the GEO database, and the reference numbers were GSE254956, GSE254960, and GSE211267 (27), respectively. All other study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Siegel R. L., Miller K. D., Fuchs H. E., Jemal A., Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022). [DOI] [PubMed] [Google Scholar]
- 2.N. Cancer Genome Atlas, Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore A. R., Rosenberg S. C., McCormick F., Malek S., RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug. Discov. 19, 533–552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood L. D., et al. , The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D., RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun C., et al. , Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 7, 86–93 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Liao W., et al. , KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 35, 559–572.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu W. H., et al. , Oncogenic KRAS drives lipofibrogenesis to promote angiogenesis and colon cancer progression. Cancer Discov. 13, 2652–2673 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang C. V., MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuzyk A., Mai S., c-MYC-induced genomic instability. Cold Spring Harb. Perspect. Med. 4, a014373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soucek L., et al. , Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes. Dev. 27, 504–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walz S., et al. , Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaseva A. V., et al. , KRAS suppression-induced degradation of MYC is antagonized by a MEK5-ERK5 compensatory mechanism. Cancer Cell 34, 807–822.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes T. K., et al. , Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 29, 75–89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang C. V., Reddy E. P., Shokat K. M., Soucek L., Drugging the "undruggable" cancer targets. Nat. Rev. Cancer 17, 502–508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delmore J. E., et al. , BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavana O., et al. , HAUSP deubiquitinates and stabilizes N-Myc in neuroblastoma. Nat. Med. 22, 1180–1186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto T., et al. , Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Wu H., et al. , Tumor necrosis factor receptor-associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-Myc gene expression and protein stability. Hepatology 71, 148–163 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Alomari M., TRIM21–A potential novel therapeutic target in cancer. Pharmacol. Res. 165, 105443 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Benton D., Chernoff J., TRIMming away colon cancer: TRIM21-mediated ubiquitination as an activator of the Hippo tumor suppressor pathway. Cell Chem. Biol. 30, 699–701 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Wan S., et al. , SPARC stabilizes ApoE to induce cholesterol-dependent invasion and sorafenib resistance in hepatocellular carcinoma. Cancer Res. 84, 1872–1888 (2024). [DOI] [PubMed] [Google Scholar]
- 23.Liu J., et al. , The ubiquitin ligase TRIM21 regulates mutant p53 accumulation and gain of function in cancer. J. Clin. Invest. 133, e164354 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J. Y., et al. , TRIM21 inhibits irradiation-induced mitochondrial DNA release and impairs antitumour immunity in nasopharyngeal carcinoma tumour models. Nat. Commun. 14, 865 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du L., et al. , EGFR-induced and c-Src-mediated CD47 phosphorylation inhibits TRIM21-dependent polyubiquitylation and degradation of CD47 to promote tumor immune evasion. Adv. Sci. (Weinh) 10, e2206380 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou G., et al. , TRIM21 is decreased in colitis-associated cancer and negatively regulates epithelial carcinogenesis. Inflamm. Bowel. Dis. 27, 458–468 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Liu Y. X., et al. , TRIM21 is a druggable target for the treatment of metastatic colorectal cancer through ubiquitination and activation of MST2. Cell Chem. Biol. 30, 709–725.e6 (2023). [DOI] [PubMed] [Google Scholar]
- 28.Kimura T., et al. , TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 210, 973–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J., et al. , Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun. 13, 2835 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D., Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Lobel G. P., Jiang Y., Simon M. C., Tumor microenvironmental nutrients, cellular responses, and cancer. Cell Chem. Biol. 30, 1015–1032 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samatar A. A., Poulikakos P. I., Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug. Discov. 13, 928–942 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Drosten M., Barbacid M., Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 37, 543–550 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Arai H., et al. , Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat. Rev. 81, 101912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grothey A., et al. , Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Tabernero J., et al. , Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: A retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 16, 937–948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camaj P., et al. , KRAS exon 2 mutations influence activity of regorafenib in an SW48-based disease model of colorectal cancer. Future Oncol. 11, 1919–1929 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Henry J. T., et al. , Comprehensive clinical and molecular characterization of KRAS (G12C)-mutant colorectal cancer. JCO Precis Oncol. 5, PO.20.00256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dikic I., Johansen T., Kirkin V., Selective autophagy in cancer development and therapy. Cancer Res. 70, 3431–3434 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Debnath J., Gammoh N., Ryan K. M., Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Bio. 24, 560–575 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He J. P., et al. , Flightless-I blocks p62-mediated recognition of LC3 to impede selective autophagy and promote breast cancer progression. Cancer Res. 78, 4853–4864 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Wu H., et al. , TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/β-catenin degradation and is targeted for GSK3B/GSK3β-mediated phosphorylation and degradation. Autophagy 15, 1506–1522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhanasekaran R., et al. , The MYC oncogene–The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 19, 23–36 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun L., et al. , Rapamycin targets STAT3 and impacts c-Myc to suppress tumor growth. Cell Chem. Biol. 29, 373–385.e6 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Yada M., et al. , Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Popov N., et al. , The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 9, 765–774 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Sun X. X., et al. , The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. U.S.A. 112, 3734–3739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruiz E. J., et al. , USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. Elife 10, e71596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jimenez-Valerio G., Casanovas O., Angiogenesis and metabolism: Entwined for therapy resistance. Trends Cancer 3, 10–18 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Wei L., et al. , Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 10, 4681 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuang R., et al. , GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2, e88815 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang C., et al. , ENO2-derived phosphoenolpyruvate functions as an endogenous inhibitor of HDAC1 and confers resistance to antiangiogenic therapy. Nat. Metab. 5, 1765–1786 (2023). [DOI] [PubMed] [Google Scholar]
- 53.Li X. M., et al. , Histone lactylation inhibits RARy expression in macrophages to promote colorectal tumorigenesis through activation of TRAF6-IL-6-STAT3 signaling. Cell Rep. 43, 113688 (2024). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
RNA-seq results have already been uploaded to the GEO database, and the reference numbers were GSE254956, GSE254960, and GSE211267 (27), respectively. All other study data are included in the article and/or supporting information.