

ABSTRACT
Background and Aim
Pseudomyxoma peritonei (PMP) is an unusual condition with unique behaviour caused by a mucinous neoplasm, usually arising from the appendix. The aim of this study was to evaluate the prevalence of genomic alterations in clinical specimens of PMP using a targeted assay and correlate the findings with clinical, pathological and outcome data. Sequencing data from 223 patients were analysed.
Results
The median follow‐up interval was 48 months. The primary neoplasm was appendiceal in 216 patients, ovarian in 4, urachal in 2 and renal in one. We confirmed common mutations in GNAS and KRAS (42% each) with significant co‐occurrence of variants in these genes. TP53 mutations were found in 8%. Other mutations were rare but included novel mutations in BAP1 and ERBB4. Of 17 patients with acellular peritoneal mucin, 6 (35%) were positive for DNA mutations. The non‐appendiceal cases generally showed a similar mutational landscape to the appendiceal lesions with GNAS and KRAS commonly mutated, although one urachal lesion showed multi‐hit TP53 mutation without variants in either GNAS or KRAS. Survival was significantly associated with the grade of the primary neoplasm, the grade of the peritoneal disease, the completeness of cytoreduction score and with mutation in either GNAS, KRAS or both. The hazard ratio (HR) associated with mutation in GNAS and/or KRAS was 1.87 (p = 0.004).
Conclusions
Survival outcome was more closely associated with the grade of the peritoneal disease than with the grade of the primary neoplasm. Our findings support the developing concept that mutational analysis may provide prognostic information in patients with PMP.
Abbreviations
- AM
acellular mucin
- CC
completeness of cytoreduction
- G
grade
- HAMN
high‐grade appendiceal neoplasm
- HG
high grade
- HGSC
high‐grade with signet‐ring cells
- HR
hazard ratio
- LAMN
low‐grade appendiceal neoplasm
- LG
low grade
- MAC
mucinous adenocarcinoma
- NGS
next‐generation sequencing
- PMP
pseudomyxoma peritonei
- PSOGI
Peritoneal Surface Oncology Group International
- SRC
mucinous adenocarcinoma with signet‐ring cells
- VAF
variant allele frequency
- WHO
World Health Organisation
1. Introduction
Pseudomyxoma peritonei (PMP) is a syndrome characterised by the accumulation of mucinous ascites and tumours within the abdominopelvic cavity [1, 2]. The incidence of PMP is about 2–3 per million per year [3, 4]. Typical features include abundant mucinous ascites, peritoneal implants of mucinous material, thickening of the omentum (‘omental cake’) and Krukenberg tumours of the ovaries. PMP has unusual features not shared with other types of neoplasm. In particular, it spreads widely through the peritoneal cavity by following the physiological flow of peritoneal fluid, a process that has been named the redistribution phenomenon, and it tends to push its way into underlying organs rather than frank tumour infiltration. Furthermore, at least in its low‐grade form, PMP very rarely metastasises to lymph nodes or distant organs [5, 6]. There is a spectrum of diseases varying from indolent low‐grade to more aggressive variants that are more invasive. Nevertheless, PMP is a malignant condition in all grades: if untreated, it grows relentlessly and ultimately causes death, usually through intestinal obstruction. For this reason, many authors prefer the term ‘mucinous carcinoma peritonei’ to PMP [7, 8].
By far the most common cause of PMP is a primary mucinous neoplasm of the appendix. There are four main types of mucinous appendiceal neoplasm according to the Peritoneal Surface Oncology Group International (PSOGI) classification: low‐grade appendiceal mucinous neoplasm (LAMN), high‐grade appendiceal mucinous neoplasm (HAMN), mucinous adenocarcinoma and mucinous adenocarcinoma with signet ring cells [7]. When one of these tumours ruptures, neoplastic cells are released into the peritoneal cavity where they can continue to grow and produce PMP. On rare occasions, PMP can develop from other primary tumours, for example, mucinous tumours of the urachus, ovarian teratomas, intraductal papillary mucinous neoplasms of the pancreas and mucinous neoplasms arising in retrorectal hamartomas [2, 9, 10].
The histological grade (G) of PMP is an independent predictor of prognosis [11]. There are three grades of PMP in the World Health Organization (WHO) classification: G1 is low grade, G2 is high grade, and G3 is high grade with signet ring cells [12, 13]. Tumours with signet‐ring cells are designated separately because of their worse prognosis [14]. In PMP of appendiceal origin, the grades of the appendiceal primary and the peritoneal disease are usually the same, but in 4%–5% of patients, there is discordance in grade. In these cases, it has been found that the grade of the PMP is more closely associated with prognosis, although the evidence is relatively scanty [15].
In about 10%–15% of patients with PMP syndrome, the intra‐abdominal mucin contains no neoplastic cells when it is examined histologically [16, 17]. This acellular mucin is not graded in the WHO classification. In the majority of such cases, the disease does not progress [18].
Appendiceal neoplasia and PMP are genetically distinct from colorectal carcinoma [19, 20]. Studies have shown the most frequent mutations in PMP to be in KRAS and GNAS, with frequencies of about 70% and 50%, respectively [21, 22, 23]. Most KRAS mutations are found in codon 12 (G12D, G12C and G12V) and in codon 13 (G13D) [24]. TP53 is mutated in some cases, but this finding is usually confined to high‐grade PMP [25, 26]. Other genes exhibiting mutation in PMP include FAT3/4, TGFBR1/2, RNF43, PIK3CA, CTNNB1, AKT1, ATM, SMAD4, SMAD2, ARID1A/B, ARID2, RBM10, BRCA2, MML, MLL2/3 and CDKN2A [21, 24, 27, 28, 29, 30]. BRAF mutations occur occasionally but are uncommon [31]. Studies using whole‐exome sequencing also identified mutations in SMAD3, PRKACA, PRKAR1A, TCHH, HERC2, SPEG, TGFBR2, TTN, MUC17, PMEPA1, AHNAK, AHNAK2, APOB, FCGPB, HRNR and OBSCN [20, 32]. One study demonstrated amplification of MYC in a subset of patients [30]. In contrast to colorectal carcinoma, it is unusual to find either APC mutations or microsatellite instability in PMP [22, 28, 31, 33]. Furthermore, on the rare occasions that microsatellite instability is found in appendiceal neoplasia, it is not generally associated with MLH1 hypermethylation [34].
A study of appendiceal adenocarcinomas found that neoplasms that were KRAS mutant and GNAS wild type were associated with better overall survival than neoplasms with mutations in both genes [19], and a study of PMP of appendiceal origin found a mutation in any one of TP53, SMAD4, ATM, CDKN2A, PIK3CA or PTEN was associated with worse overall survival [28]. However, in general, there is little consensus in the literature concerning the prognostic implications of the various genetic mutations, partly because many studies include relatively few patients [21, 24].
The optimal treatment of PMP includes complete tumour removal by radical cytoreductive surgery combined with hyperthermic intraperitoneal chemotherapy, with overall 10‐year survival of over 50% [35, 36]. Occasional patients with non‐resectable recurrent disease may undergo intestinal transplantation [37]. Indicators of likely clinical outcome include the extent of intraperitoneal disease, the histological grade, preoperative serum concentrations of the tumour markers CA125, CA19‐9 and CEA and completeness of tumour removal [38, 39]. However, these clinical and pathological parameters do not accurately predict outcomes in all cases, and a better knowledge of genetic changes in PMP will contribute to the search for improved biomarkers of prognosis [32]. Furthermore, because of the unique behaviour of PMP, an increased understanding of its genetic landscape could shed light on mechanisms of metastasis in general and have implications beyond the treatment of this rare condition [40].
The aim of this study was to apply next‐generation sequencing (NGS) techniques to clinical specimens of pseudomyxoma peritonei by evaluating the prevalence of genomic alterations with a targeted NGS assay and to correlate the results with clinical, pathological and outcome data. A secondary aim was to compare the survival outcomes in patients with PMP of appendiceal origin according to the grade of the primary tumour compared with the grade of the peritoneal disease.
2. Materials and Methods
2.1. Patient Sample Collection/Criteria
Patients treated at the Peritoneal Malignancy Institute, Basingstoke, UK were recruited to the study. All patients provided written informed consent. All patients had pseudomyxoma peritonei treated by cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. Ethical approval was provided by the National Research Ethics Service, reference 09/H0504/3.
Samples of tumour were taken intra‐operatively during surgery; these samples were snap‐frozen and stored in liquid nitrogen until further processing. Histological examination of all specimens was performed by a pathologist with a special interest in the appendix and PMP. The appendiceal primary tumours were classified according to PSOGI criteria as follows (WHO grade in parentheses): LAMN (G1), HAMN (G2), mucinous adenocarcinoma (G2) and mucinous adenocarcinoma with signet ring cells (G3). The peritoneal disease was classified as acellular mucin (ungraded), low‐grade PMP (G1), high‐grade PMP (G2) and high‐grade PMP with signet ring cells (G3) [7, 41].
Clinical details were retrieved from a prospectively maintained database supplemented by reference to the clinical records if required. The presence of any residual disease at the end of the operation was recorded by the surgeon using the completeness of cytoreduction (CC) score: no visible disease (CC0), nodules of residual tumour less than 0.25 cm diameter (CC1), nodules 0.25–2.5 cm diameter (CC2) and nodules more than 2.5 cm diameter (CC3). For modelling, the CC score was dichotomised into CC0/1 and CC2/3, corresponding to a cutoff value of 0.25 cm diameter, consistent with findings that this is the most clinically significant distinction [35, 42].
2.2. DNA Extraction, Panel Design, Library Preparation and Sequencing
DNA was extracted from fresh frozen samples. Firstly, samples were homogenised using a Precellys 24 in 2 mL CKMix tissue homogenising tubes (Bertin Technologies SAS) at 5000 rpm for two 10 s periods. Following homogenisation, DNA was extracted using the QIAamp DNA Blood Mini kit, automated using the QIASymphony platform (Qiagen). DNA samples were quantified using the Qubit Fluorimeter (Life Technologies).
A custom‐targeted panel was designed for peritoneal malignancies, including those underlying PMP. This panel included 207 key regions across 50 genes frequently mutated across cancers, and the coding regions of six key genes of interest in peritoneal malignancy (BAP1, CDC42, NF2, RNF43, SETDB1 and TRAF7) (Table S1). In addition, 24 SNPs were included to allow verification of sample identity following sequencing [43]. The custom panel was generated using an amplicon‐based approach (YouSeq, Winchester, UK). Capture and library preparation was performed in accordance with manufacturer's instructions. Libraries were paired‐end sequenced (300 cycles) on a NextSeq500. Parallel genotyping of the 24‐sample tracking panel was performed using KASP genotyping (LGC Genomics, UK).
2.3. Bioinformatic and Statistical Analyses
Raw‐sequencing fastq files from 2 runs were merged per sample. Alignment of the merged sequences to the hg38 (release 13) genome was performed using the BWA‐MEM module from the Burrows‐Wheeler Aligner (BWA) software (v.0.7.17) [44] and sorted and indexed using samtools (v.1.16.1). The sorted bams were trimmed using the bamutil (V1.0.14) filter programme to trim ends of reads where there were > 10% mismatches from the reference genome and exclude reads where there were mismatches with a cumulative phred scaled quality of 60.
The trimmed bams were input into Stitcher (v5.2.9) to merge read pairs followed by variant calling and variant quality score recalibration using Pisces (v5.2.9). These variants were then annotated by the Functotator software (v.4.2.2) using base data sources and output as maf format files. These files were analysed in maftools 2.18.0, in Rstudio (R v4.3.2). Variants were filtered for quality score > 100 and variant allele frequency (VAF) ≥ 2.5% (1% for GNAS/KRAS). To exclude known germline variants, all variants were filtered to remove those present in gnomAD at a minor allele frequency of > 0.0001. Manual curation of all mutated genes was carried out using IGV (v. 2.14.1) and mutations were assessed and tagged for exclusion based on a published standard operating procedure [45].
Survival analyses were carried out using the ‘survival’ (3.5‐7), ‘survminer’ (0.4.9) and ‘forestmodel’ (0.6.2) R packages in Rstudio (R v4.3.2).
3. Results
3.1. Clinical Characteristics
Samples from 263 patients were submitted for analysis. In 223 samples, DNA was successfully extracted and good‐quality sequencing data were achieved (Table S2). The demographic and clinical details of these 223 patients are shown in Table 1. Pre‐operative chemotherapy had been received by 36 (16.1%) patients; the others had received no chemotherapy prior to cytoreductive surgery. Follow‐up was available for all patients and the median follow‐up interval was 50 months.
TABLE 1.
Patient/sample demographic and clinical data according to the grade of the peritoneal disease.
| AM | LG (G1) | HG (G2) | HGSC (G3) | Overall | |
|---|---|---|---|---|---|
| (N = 17) | (N = 150) | (N = 50) | (N = 6) | (N = 223) | |
| Age at surgery | |||||
| Mean (SD) | 61.4 (11.1) | 57.6 (12.0) | 55.6 (13.3) | 50.0 (17.0) | 57.2 (12.5) |
| Median [Min, Max] | 60.0 [43.0, 76.0] | 58.0 [30.0, 85.0] | 55.0 [27.0, 84.0] | 49.0 [32.0, 78.0] | 57.0 [27.0, 85.0] |
| Sex | |||||
| Female | 12 (70.6%) | 104 (69.3%) | 29 (58.0%) | 2 (33.3%) | 147 (65.9%) |
| Male | 5 (29.4%) | 46 (30.7%) | 21 (42.0%) | 4 (66.7%) | 76 (34.1%) |
| Time from first surgery to follow up (months) | |||||
| Mean (SD) | 68.5 (35.9) | 61.9 (33.8) | 46.7 (34.0) | 27.0 (21.1) | 58.1 (34.6) |
| Median [Min, Max] | 53.0 [19.0, 121] | 57.5 [4.00, 145] | 40.0 [4.00, 123] | 26.0 [2.00, 61.0] | 50.0 [2.00, 145] |
| CC score | |||||
| CC0 (no residual disease) | 9 (52.9%) | 33 (22.0%) | 24 (48.0%) | 2 (33.3%) | 68 (30.5%) |
| CC1 (< 0.25 cm) | 8 (47.1%) | 76 (50.7%) | 21 (42.0%) | 2 (33.3%) | 107 (48.0%) |
| CC2 (0.25–2.5 cm) | 0 (0%) | 8 (5.3%) | 1 (2.0%) | 0 (0%) | 9 (4.0%) |
| CC3 (> 2.5 cm) | 0 (0%) | 28 (18.7%) | 4 (8.0%) | 2 (33.3%) | 34 (15.2%) |
| Missing | 0 (0%) | 5 (3.3%) | 0 (0%) | 0 (0%) | 5 (2.2%) |
| Primary site | |||||
| Appendix | 16 (94.1%) | 147 (98.0%) | 47 (94.0%) | 6 (100%) | 216 (96.9%) |
| Ovarian teratoma | 1 (5.9%) | 2 (1.3%) | 1 (2.0%) | 0 (0%) | 4 (1.8%) |
| Urachus | 0 (0%) | 1 (0.7%) | 1 (2.0%) | 0 (0%) | 2 (0.9%) |
| Kidney | 0 (0%) | 0 (0%) | 1 (2.0%) | 0 (0%) | 1 (0.4%) |
| Primary tumour | |||||
| LAMN | 14 (82.4%) | 145 (96.7%) | 3 (6.0%) | 0 (0%) | 162 (72.6%) |
| HAMN | 0 (0%) | 0 (0%) | 6 (12.0%) | 0 (0%) | 6 (2.7%) |
| Appendiceal MAC | 2 (11.8%) | 2 (1.3%) | 36 (72.0%) | 0 (0%) | 40 (17.9%) |
| Appendiceal SRC | 0 (0%) | 0 (0%) | 2 (4.0%) | 6 (100%) | 8 (3.6%) |
| PMP non‐appendiceal | 1 (5.9%) | 3 (2.0%) | 3 (6.0%) | 0 (0%) | 7 (3.1%) |
| Pre‐op CA19.9 (U/mL) | |||||
| Mean (SD) | 48.6 (66.8) | 862 (2570) | 376 (965) | 266 (378) | 679 (2190) |
| Median [Min, Max] | 16.0 [6.00, 251] | 187 [0, 20,400] | 121 [0, 6040] | 48.0 [3.00, 879] | 127 [0, 20,400] |
| Missing | 1 (5.9%) | 5 (3.3%) | 4 (8.0%) | 0 (0%) | 10 (4.5%) |
| Pre‐op CA125 (U/mL) | |||||
| Mean (SD) | 25.1 (20.3) | 58.2 (61.3) | 75.2 (181) | 74.3 (36.9) | 59.9 (100) |
| Median [Min, Max] | 18.0 [3.00, 73.0] | 38.0 [2.00, 403] | 25.3 [4.00, 1260] | 75.5 [15.0, 122] | 34.5 [2.00, 1260] |
| Missing | 0 (0%) | 6 (4.0%) | 1 (2.0%) | 0 (0%) | 7 (3.1%) |
| Pre‐op CEA (ng/mL) | |||||
| Mean (SD) | 25.2 (66.1) | 61.9 (154) | 50.2 (151) | 13.6 (18.6) | 55.0 (147) |
| Median [Min, Max] | 4.70 [0.500, 272] | 21.0 [0.500, 1650] | 9.70 [0.500, 890] | 6.85 [2.30, 51.0] | 15.0 [0.500, 1650] |
| Missing | 0 (0%) | 6 (4.0%) | 1 (2.0%) | 0 (0%) | 7 (3.1%) |
Abbreviations: AM, acellular mucin; HAMN, high‐grade appendiceal neoplasm; HG, high grade; HGSC, high‐grade with signet ring cells; LAMN, low‐grade appendiceal neoplasm; LG, low grade; MAC, mucinous adenocarcinoma; SRC, signet ring cells.
The PMP originated from a mucinous neoplasm of the appendix in 216 (96.9%) patients (Table 2). The other primary tumours were an ovarian teratoma in 4 patients, a mucinous tumour of the urachus in 2 patients and a mucinous tumour of the renal pelvis in one patient. There were 17 (7.6%) patients with histologically acellular mucin; in 14 of them, the primary was an LAMN, in 2, the primary was an appendiceal mucinous adenocarcinoma and in one the primary was an ovarian teratoma. The presence or absence of lymph node metastasis was recorded in 166 (74.4%) patients. Nodal metastasis was present in 18/166 (10.8%) patients; 5 with G1 PMP, 11 with G2 PMP and 2 with G3 PMP. There was discordance between the grade of the appendiceal primary and the grade of the PMP in 7 (3.1%) patients: three G1 primaries were associated with G2 PMP, two G2 primaries were associated with G1 PMP, and two G3 primaries were associated with G2 PMP (Table 2).
TABLE 2.
Grade of pseudomyxoma peritonei according to type of primary neoplasm.
| Peritoneal disease | |||||
|---|---|---|---|---|---|
| AM | LG (G1) | HG (G2) | HGSC (G3) | ||
| Primary tumour | LAMN (G1) | 14 | 145 | 3 | 0 |
| HAMN (G2) | 0 | 0 | 6 | 0 | |
| Appendiceal MAC (G2) | 2 | 2 | 36 | 0 | |
| Appendiceal SRC (G3) | 0 | 0 | 2 | 6 | |
| Non‐appendiceal primary | 1 | 3 | 3 | 0 | |
Abbreviations: AM, acellular mucin; HAMN, high‐grade appendiceal neoplasm; HG, high grade; HGSC, high grade with signet ring cells; LAMN, low‐grade appendiceal neoplasm; LG, low grade; MAC, mucinous adenocarcinoma; SRC, signet ring cells.
3.2. Mutational Landscape
Of the 223 patients successfully sequenced across cancer hotspot regions, we observed good‐quality variants in 126 (56.5%) patients. The most commonly mutated genes were GNAS and KRAS at 42% each (Figure 1). Mutations in KRAS were most commonly seen at the known mutation hotspots in codons 12 and 13, and those in GNAS were at the known hotspot in codon 201 (Figure S2). TP53 mutations were found in 8% and were more common in high‐grade lesions. SMAD4, ERBB4, BAP1 and PIK3CA were detected at rates of 2%–4%. Mutations in APC and BRAF were unusual (1% each). Mutated NRAS was found in only one (0.4%) specimen, a mucinous appendiceal adenocarcinoma. In the 17 specimens consisting of acellular peritoneal mucin, 6 (35%) were positive for mutated DNA (Figure 1B).
FIGURE 1.
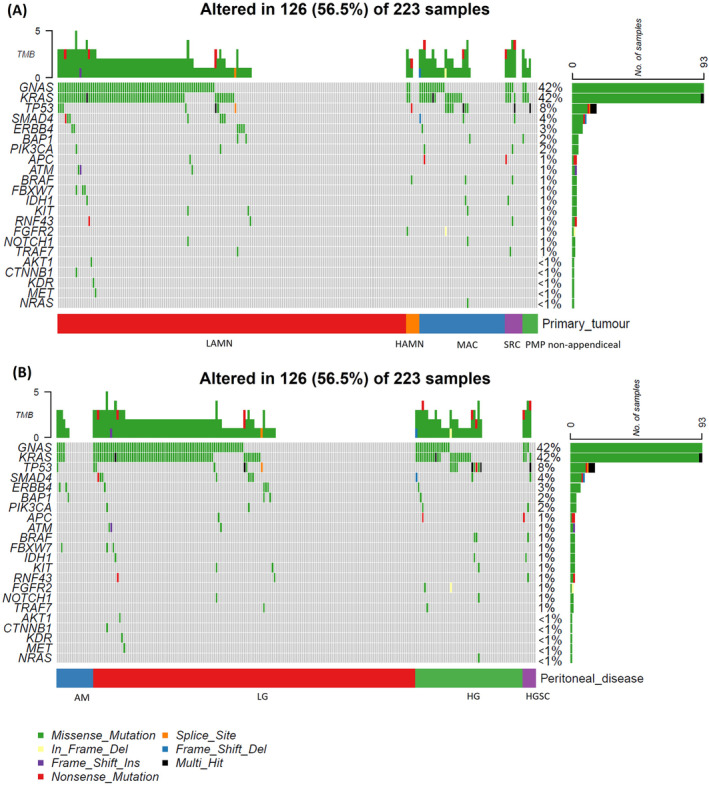
Oncoplot of mutations grouped by (A) primary tumour and (B) by peritoneal disease.
The 7 lesions of non‐appendiceal origin generally showed a similar mutational landscape to the appendiceal lesions, with GNAS and KRAS variants the most frequent, although one patient had no mutation in either gene but did harbour multihit mutation of TP53; this lesion was a mucinous adenocarcinoma of the urachus (Figure 1A). One case of appendiceal mucinous adenocarcinoma also contained multihit mutation of TP53 without mutation in either KRAS or GNAS.
3.3. Mutual Exclusivity Analysis
Mutations in GNAS and KRAS significantly co‐occur (adj. p‐value 2.72 × 10−21, odds ratio 22.3). The most mutually exclusive mutations were between GNAS and TP53 although this was not significant (adj. p‐value 0.9, odds ratio 0.4) (Figure 2). Mutations in NOTCH1 and KIT were also significantly mutually inclusive (adj. p‐value 0.003), but this was based on very few mutations, where two patients with NOTCH1 mutations both also had mutations in KIT.
FIGURE 2.
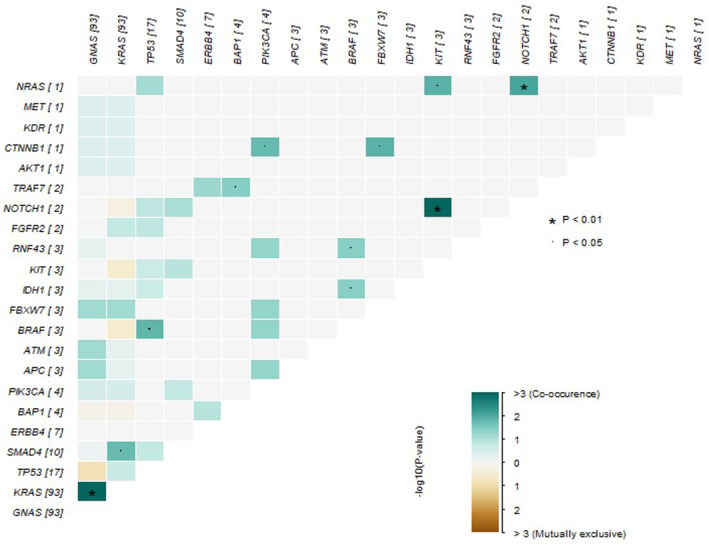
Mutual exclusivity and co‐occurrence of mutations.
3.4. Survival Analyses
The median survival for all patients with PMP of appendiceal origin was 96 months. Overall median survival for the non‐appendiceal group was 51% at 61 months (median survival not reached in this cohort). Modelling survival of patients using key clinical data identified CC score as the factor with most impact. The CC score was available for 218 patients; the median survival of the 175 patients with CC0/1 was 116 months and the median survival of the 43 patients with CC2/3 was 26 months (p = 2.57 × 10−20).
For the appendiceal neoplasms, survival rates differed significantly according to the grade of the primary tumour (Figure 3A). Dichotomising the primary tumours into LAMN (G1) and HAMN/mucinous adenocarcinoma/mucinous adenocarcinoma with signet ring cells (HAMN/MAC/SRC) (G2/3) showed the median overall survival for the LAMN group was 115 months and for the HAMN/MAC/SRC group 42 months (p = 4.59 × 10−6, HR = 4.06). The 5‐year survival was 69% and 38% for the LAMN and HAMN/MAC/SRC groups, respectively. The 5‐year survival for the non‐appendiceal group was 69%. Survival was also significantly different between peritoneal disease types (p = 3.292 × 10−8) (Figure 3B). The median survival for low‐grade PMP (G1) was 105 months, for high‐grade PMP (G2) 42 months and for high‐grade PMP with signet ring cells (G3) 26 months. In the 17 patients with acellular mucin, only three experienced recurrence and the 5‐year survival rate was 88%, compared with an overall 5‐year survival of 59% in the 206 patients with neoplastic cells in the peritoneal mucin (p = 0.035).
FIGURE 3.
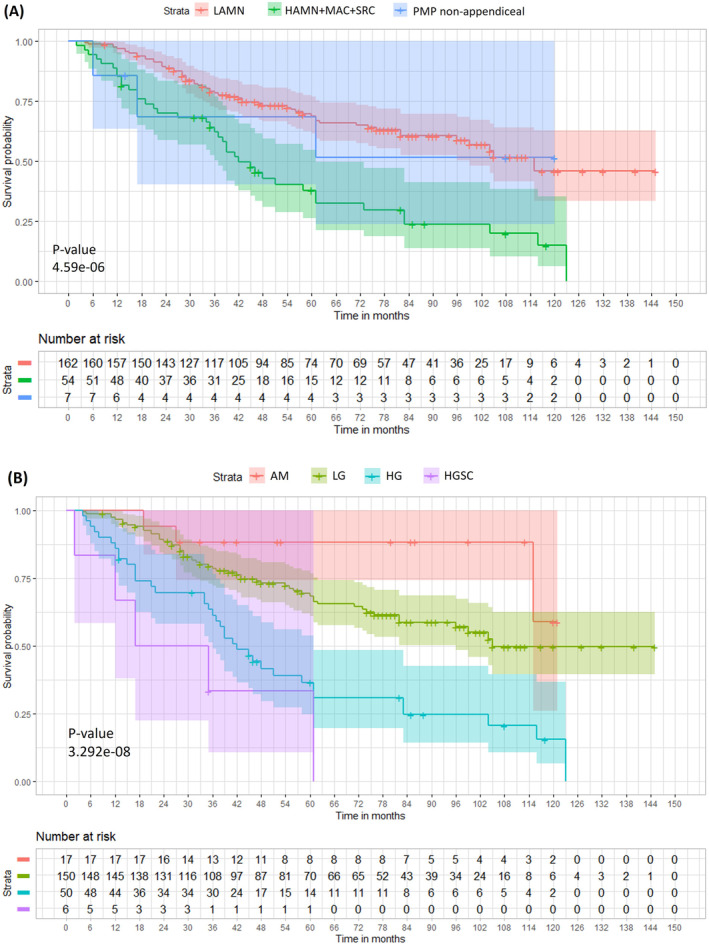
Kaplan–Meier Survival plot, stratified by (A) primary tumour and (B) peritoneal disease.
We compared survival outcomes of patients with PMP of appendiceal origin when classified according to the WHO grade of the appendiceal primary and according to the WHO grade of the PMP. Patients with acellular peritoneal mucin were excluded. The HR for primary tumour grade was 2.82 (p = 9.96 × 10−9) while the HR for peritoneal disease grade was higher, 3.82 (p = 2.91 × 10−9). In the Cox model with both variables included, PMP grade was more closely associated with survival and the primary tumour grade made no further contribution (p = 0.0002, Figure S5). These findings show that prognosis is more closely associated with the grade of the PMP than the grade of the appendiceal primary.
Comparing survival rates in patients with and without KRAS and GNAS mutations, individuals harbouring a KRAS variant had poorer survival than those without (p = 0.006, HR = 1.72). Similarly, those carrying a GNAS variant had poorer survival than those without (p = 0.048, HR = 1.48). The next most frequently mutated gene was TP53 (mutated in 17); although there was a trend towards poorer survival in the mutated cases this was not significant (p = 0.061). There was no significant association between CC score and genetic mutation in our data.
As mutations in GNAS and KRAS genes frequently co‐occur in the same patients, we merged these to create a subset of patients with KRAS and/or GNAS mutations. This subset had significantly poorer survival than those without mutations at either gene (p = 0.001) (Figure S3). When modelling carriage of KRAS and/or GNAS mutational status (excluding cases with acellular peritoneal mucin), genetic variation in these genes significantly contributed to hazard (p = 0.004, HR = 1.87) compared to cases with no mutation in either gene after accounting for CC scoring and peritoneal disease grade (Figure 4B). However, our data indicate that they represent a lesser hazard than peritoneal disease grade.
FIGURE 4.
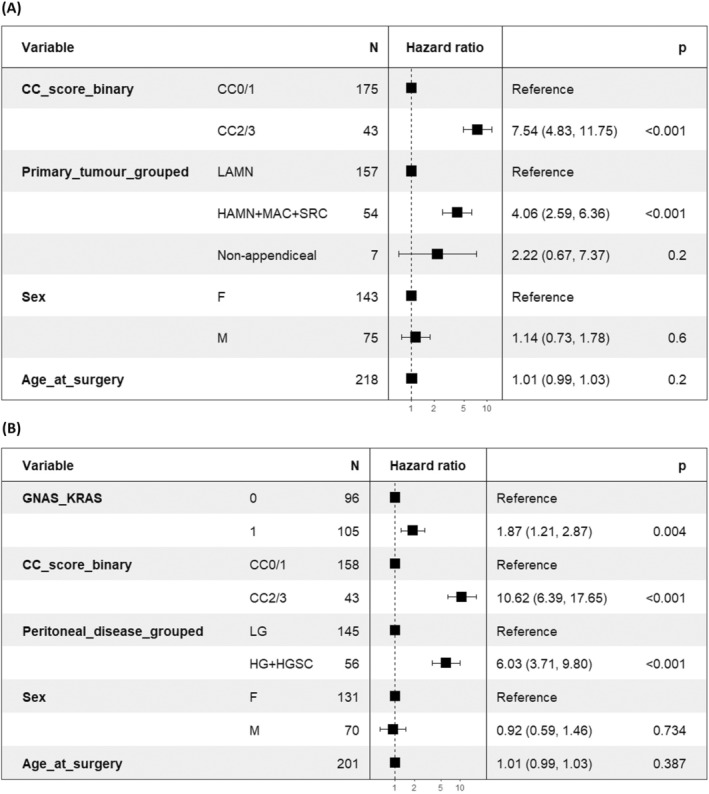
Cox proportional hazards multivariable model forest plot(s) (A) including CC score and primary tumour and (B) peritoneal disease type with acellular mucin (AM) excluded.
4. Discussion
As expected, the most common mutations were found in KRAS and GNAS, although at lower frequencies than in some other studies (Figure 1) [20, 21]. Other mutations in our series have generally been reported previously, although unexpected findings were mutated BAP1 in 2% of patients and mutated ERBB4 in 3%. BAP1 is commonly mutated in other neoplasms, such as malignant mesothelioma and melanoma [46, 47]. ERBB4, which encodes the tyrosine kinase receptor HER4, has varied effects on cancer development, but mutations in this gene have been implicated in several neoplasms, including melanoma, lung carcinoma and stomach carcinoma [48]. Of the 4 cases with BAP1 mutation, 3 showed no variants in either KRAS or GNAS, and of the 7 cases with ERBB4 mutation, 4 (all LAMNs) likewise showed no variants in either KRAS or GNAS (Figure 1A). These findings raise the possibility of novel genetic pathways in PMP that may not involve KRAS or GNAS. Of note in this respect, the targeted hotspot panel was not sufficient to analyse amplifications. Another limitation of the study was a lack of matched normal tissue, which is advantageous in confirming somatic mutation status.
A previous study of mucinous appendiceal neoplasms found wild‐type GNAS in 5/6 high‐grade tumours, raising the possibility that high‐grade lesions may not arise from low‐grade tumours [20]. We found GNAS mutations in 18/54 (33%) of high‐grade lesions and 73/162 (45%) of low‐grade lesions (Figure 1A); this difference was not significant in our data (p = 0.18). For patients with PMP, a potential implication of wild‐type KRAS is the possible use of drugs such as cetuximab in treatment. A retrospective study of patients with appendiceal adenocarcinoma showed no survival benefit from cetuximab or panitumumab [49]. However, in a xenograft mouse model of high‐grade PMP, the KRAS G12D inhibitor MRTX1133 was associated with a marked reduction of tumour growth [50]. It could be hypothesised that sequencing of percutaneous or laparoscopic biopsies might identify patients with PMP who could benefit from cetuximab or panitumumab prior to surgery to improve CC0 rates, and further studies in this area are indicated.
Other candidates for targeted therapy are neoplasms with BRAF mutations. In a study of patient‐derived organoid and xenograft mouse models of PMP, the BRAF V600E inhibitor encorafenib reduced cell viability and tumour growth [51]. Although BRAF mutation in appendiceal neoplasia is rare, the subset of patients harbouring such a mutation might benefit from this treatment and further research is needed.
The low frequency of APC and BRAF mutation in our series is in keeping with the findings of others and supports the concept that these genes are mutated rarely in pseudomyxoma peritonei, in contrast to colorectal carcinomas [20, 22, 28, 31, 33]. This emphasises the fact that colorectal and appendiceal neoplasms are genetically distinct.
In the 17 specimens consisting of acellular peritoneal mucin, 6 (35%) were positive for mutated DNA (Figure 1B). PMP with acellular mucin has an excellent prognosis [18]. In some cases, it may be that the mucin does contain neoplastic cells, but at such a low concentration that they are missed by routine histopathological assessment. Alternatively, the mucin may be genuinely acellular because the neoplastic cells do not survive after extrusion from the appendix. The detection of mutated DNA in histologically acellular mucin could be consistent with either scenario. In a study of three patients with acellular peritoneal mucin associated with LAMNs harbouring mutated KRAS, the KRAS mutation was detected in cell‐free DNA in the mucin from all three patients [17].
There is little consistent evidence in the literature about the prognostic impact of genomic alterations in PMP [19, 21, 24]. Our data showed poorer survival was associated with either KRAS mutation (p = 0.006, HR = 1.72) or GNAS mutation (p = 0.048, HR = 1.48). Furthermore, the subset of patients with KRAS and/or GNAS mutations had significantly worse survival than those without mutations at either of these established genes (p = 0.001) (Figure S3). TP53 variants were associated with a trend towards poorer survival, consistent with the findings of others [28]. However, this was not statistically significant in our data (p = 0.061). Nevertheless, TP53 mutations were more likely to be found in high‐grade lesions: they were identified in 9/54 (17%) of high‐grade neoplasms and 7/162 (4%) of low‐grade neoplasms (p = 0.007) (Figure 1A), in keeping with other studies [20, 25, 31].
The associations between the various clinical features and survival in our results were generally as expected in the light of previous studies [11, 35, 36]. The CC score influenced survival more than any other factor in our data, and the appendiceal neoplasms showed better overall survival in the LAMN (G1) subgroup compared with the HAMN/MAC/SRC (G2/G3) subgroup. The non‐appendiceal subgroup demonstrated an intermediate survival in this small set of patients (n = 7). The grade of the pseudomyxoma peritonei was also significantly associated with survival, and patients with acellular mucin had the best outcome. Survival was more closely associated with the grade of the peritoneal disease than the grade of the primary tumour in cases of PMP of appendiceal origin (Figure S5). This finding supports the practice of grading the primary and peritoneal disease separately in patients with PMP [15].
Our results demonstrate the importance of KRAS and GNAS variants in the oncogenesis of PMP and are consistent with the conclusions of others that TP53 appears to be associated with high grade disease. The relative rarity of mutations in other genes suggests we need to look beyond the genes analysed in panels such as the one used in our study to identify other genes that may be commonly involved. The starting point for many studies of the genetics and pathology of appendiceal mucinous neoplasms and PMP is the assumption that they are similar to colorectal carcinoma, but there are many important clinical, pathological and genetic differences between them [19]. We speculate that widening the range of genes studied by NGS sequencing may not only improve prognostic data but also provide insights into the unique behaviour of these tumours. A possible pointer in this direction is our documentation of novel variants in BAP1 and ERBB4. We have also demonstrated that meaningful genetic information can be obtained from PMP that is histologically acellular. Further work in the genetics of PMP may enlighten the whole field of peritoneal metastatic disease, a frequent and clinically difficult condition in patients with common primary cancers of gastrointestinal origin.
Author Contributions
Jane Gibson: formal analysis (equal), writing – review and editing (equal). Norman John Carr: conceptualization (equal), data curation (equal), funding acquisition (equal), resources (equal), writing – original draft (equal), writing – review and editing (equal). Alex Mirnezami: conceptualization (equal), data curation (equal), funding acquisition (equal), resources (equal), writing – review and editing (equal). Brendan John Moran: data curation (equal), resources (equal), writing – review and editing (equal). Sarah Ennis: conceptualization (equal), funding acquisition (equal), supervision (equal), writing – review and editing (equal). Alexios Tzivanakis: data curation (equal), resources (equal), writing – review and editing (equal). Konstantinos Boukas: investigation (equal), writing – review and editing (equal). Amatta Mirandari: formal analysis (equal), writing – review and editing (equal). Reuben J. Pengelly: formal analysis (equal), writing – review and editing (equal). Sophia Stanford: data curation (equal), project administration (equal), resources (equal), writing – review and editing (equal). Sanjeev Paul Dayal: data curation (equal), resources (equal), writing – review and editing (equal). Faheez Mohamed: data curation (equal), resources (equal), writing – review and editing (equal). Thomas Desmond Cecil: data curation (equal), resources (equal), writing – review and editing (equal).
Ethics Statement
Ethical approval was provided by the National Research Ethics Service, reference 09/H0504/3.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1.
Figure S1.
Acknowledgements
We wish to thank Emily Arbuthnot for clinical data coordination and administration; Stephen Doherty from Wessex Medical Laboratory Services (Southampton) for technical support, and YouSeq for targeted panel design. The authors acknowledge the use of the IRIDIS High Performance Computing Facility and associated support services at the University of Southampton, in the completion of this work. The authors acknowledge the Faculty of Medicine BIO‐R, at the University of Southampton for their support and assistance in this work.
This research study is funded by Mesothelioma, UK and the Peritoneal Malignancy Institute Basingstoke. The collection of patient samples was supported by the Pelican Cancer Foundation.
Funding: This research study is funded by Mesothelioma, UK and the Peritoneal Malignancy Institute Basingstoke. The collection of patient samples was supported by the Pelican Cancer Foundation.
Norman John Carr and Sarah Ennis contributed equally.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Moran B. J. and Cecil T. D., “The Etiology, Clinical Presentation, and Management of Pseudomyxoma Peritonei,” Surgical Oncology Clinics of North America 12, no. 3 (2003): 585–603, 10.1016/s1055-3207(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 2. Carr N. J., “New Insights in the Pathology of Peritoneal Surface Malignancy,” Journal of Gastrointestinal Oncology 12, no. Suppl 1 (2021): S216–S229, 10.21037/jgo-2020-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smeenk R. M., van Velthuysen M. L., Verwaal V. J., and Zoetmulder F. A., “Appendiceal Neoplasms and Pseudomyxoma Peritonei: A Population Based Study,” European Journal of Surgical Oncology 34, no. 2 (2008): 196–201, 10.1016/j.ejso.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 4. Patrick‐Brown T., Carr N. J., Swanson D. M., Larsen S., Mohamed F., and Flatmark K., “Estimating the Prevalence of Pseudomyxoma Peritonei in Europe Using a Novel Statistical Method,” Annals of Surgical Oncology 28, no. 1 (2021): 252–257, 10.1245/s10434-020-08655-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sugarbaker P. H., “Pseudomyxoma Peritonei. A Cancer Whose Biology Is Characterized by a Redistribution Phenomenon,” Annals of Surgery 219, no. 2 (1994): 109–111, 10.1097/00000658-199402000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jarvinen P. and Lepisto A., “Clinical Presentation of Pseudomyxoma Peritonei,” Scandinavian Journal of Surgery 99, no. 4 (2010): 213–216, 10.1177/145749691009900406. [DOI] [PubMed] [Google Scholar]
- 7. Carr N. J., Cecil T. D., Mohamed F., et al., “A Consensus for Classification and Pathologic Reporting of Pseudomyxoma Peritonei and Associated Appendiceal Neoplasia: The Results of the Peritoneal Surface Oncology Group International (PSOGI) Modified Delphi Process,” American Journal of Surgical Pathology 40, no. 1 (2016): 14–26, 10.1097/PAS.0000000000000535. [DOI] [PubMed] [Google Scholar]
- 8. Bradley R. F., JHt S., Russell G. B., Levine E. A., and Geisinger K. R., “Pseudomyxoma Peritonei of Appendiceal Origin: A Clinicopathologic Analysis of 101 Patients Uniformly Treated at a Single Institution, With Literature Review,” American Journal of Surgical Pathology 30, no. 5 (2006): 551–559, 10.1097/01.pas.0000202039.74837.7d. [DOI] [PubMed] [Google Scholar]
- 9. Yoshizaki Y., Gohda Y., Inagaki F., et al., “A Case of Pseudomyxoma Peritonei Arising From a Perforated Intraductal Papillary Mucinous Neoplasm That Underwent Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy,” Clinical Journal of Gastroenterology 17, no. 1 (2024): 188–197, 10.1007/s12328-023-01890-y. [DOI] [PubMed] [Google Scholar]
- 10. Taguchi A., Rokutan H., Oda K., et al., “Genetic Diagnosis of Pseudomyxoma Peritonei Originating From Mucinous Borderline Tumor Inside an Ovarian Teratoma,” BioMed Central Medical Genomics 15, no. 1 (2022): 51, 10.1186/s12920-022-01188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang Y., Alzahrani N. A., Chua T. C., and Morris D. L., “Histological Subtype Remains a Significant Prognostic Factor for Survival Outcomes in Patients With Appendiceal Mucinous Neoplasm With Peritoneal Dissemination,” Diseases of the Colon and Rectum 60, no. 4 (2017): 360–367, 10.1097/DCR.0000000000000719. [DOI] [PubMed] [Google Scholar]
- 12. Misdraji J., Carr N., and Pai R., Appendiceal Mucinous Neoplasm, Digestive System Tumours (Geneva, Switzerland: World Health Organization Classification of Tumours; 5th. International Agency for Research on Cancer, World Health Organization, 2019). [Google Scholar]
- 13. Misdraji J., Carr N., and Pai R., Appendiceal Adenocarcinoma, Digestive System Tumours (Geneva, Switzerland: World Health Organization classification of tumours; 5th. International Agency for Research on Cancer, World Health Organization, 2019). [Google Scholar]
- 14. Shamavonian R., Lansom J. D., Karpes J. B., Alzahrani N. A., and Morris D. L., “Impact of Signet Ring Cells on Overall Survival in Peritoneal Disseminated Appendix Cancer Treated With Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy,” European Journal of Surgical Oncology 47, no. 1 (2021): 194–198, 10.1016/j.ejso.2020.11.134. [DOI] [PubMed] [Google Scholar]
- 15. Memon A. A., Godbole C., Cecil T., et al., “Overall Survival Is More Closely Associated With Peritoneal Than Primary Appendiceal Pathological Grade in Pseudomyxoma Peritonei With Discordant Pathology,” Annals of Surgical Oncology 29, no. 4 (2022): 2607–2613, 10.1245/s10434-021-10994-z. [DOI] [PubMed] [Google Scholar]
- 16. Al‐Azzawi M., Misdraji J., van Velthuysen M. F., et al., “Acellular Mucin in Pseudomyxoma Peritonei of Appendiceal Origin: What Is Adequate Sampling for Histopathology?,” Journal of Clinical Pathology 73, no. 4 (2020): 220–222, 10.1136/jclinpath-2019-206213. [DOI] [PubMed] [Google Scholar]
- 17. Garcia‐Olmo D., Olmedillas‐Lopez S., Cortes‐Guiral D., et al., “The Role of Mucin Cell‐Free DNA Detection as a New Marker for the Study of Acellular Pseudomyxoma Peritonei of Appendicular Origin by Liquid Biopsy,” Therapeutic Advances in Medical Oncology 12 (2020): 1758835920928233, 10.1177/1758835920928233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roxburgh C. S., Fenig Y. M., Cercek A., et al., “Outcomes of Low‐Grade Appendiceal Mucinous Neoplasms With Remote Acellular Mucinous Peritoneal Deposits,” Annals of Surgical Oncology 26, no. 1 (2019): 118–124, 10.1245/s10434-018-7003-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foote M. B., Walch H., Chatila W., et al., “Molecular Classification of Appendiceal Adenocarcinoma,” Journal of Clinical Oncology 41, no. 8 (2023): 1553–1564, 10.1200/JCO.22.01392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alakus H., Babicky M. L., Ghosh P., et al., “Genome‐Wide Mutational Landscape of Mucinous Carcinomatosis Peritonei of Appendiceal Origin,” Genome Medicine 6, no. 5 (2014): 43, 10.1186/gm559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lund‐Andersen C., Torgunrud A., Fleten K. G., and Flatmark K., “Omics Analyses in Peritoneal Metastasis‐Utility in the Management of Peritoneal Metastases From Colorectal Cancer and Pseudomyxoma Peritonei: A Narrative Review,” Journal of Gastrointestinal Oncology 12, no. Suppl 1 (2021): S191–S203, 10.21037/jgo-20-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stein A., Strong E., Clark Gamblin T., et al., “Molecular and Genetic Markers in Appendiceal Mucinous Tumors: A Systematic Review,” Annals of Surgical Oncology 27, no. 1 (2020): 85–97, 10.1245/s10434-019-07879-7. [DOI] [PubMed] [Google Scholar]
- 23. Pietrantonio F., Perrone F., Mennitto A., et al., “Toward the Molecular Dissection of Peritoneal Pseudomyxoma,” Annals of Oncology 27, no. 11 (2016): 2097–2103, 10.1093/annonc/mdw314. [DOI] [PubMed] [Google Scholar]
- 24. Murage N. W., Ahmed N. M., Underwood T. J., Walters Z. S., and Breininger S. P., “The Genetic Profile and Molecular Subtypes of Human Pseudomyxoma Peritonei and Appendiceal Mucinous Neoplasms: A Systematic Review,” Cancer Metastasis Reviews 42, no. 1 (2023): 335–359, 10.1007/s10555-023-10088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu X., Salhab M., Tomaszewicz K., et al., “Heterogeneous Mutational Profile and Prognosis Conferred by TP53 Mutations in Appendiceal Mucinous Neoplasms,” Human Pathology 85 (2019): 260–269, 10.1016/j.humpath.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 26. Noguchi R., Yano H., Gohda Y., et al., “Molecular Profiles of High‐Grade and Low‐Grade Pseudomyxoma Peritonei,” Cancer Medicine 4, no. 12 (2015): 1809–1816, 10.1002/cam4.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pengelly R. J., Rowaiye B., Pickard K., et al., “Analysis of Mutation and Loss of Heterozygosity by Whole‐Exome Sequencing Yields Insights Into Pseudomyxoma Peritonei,” Journal of Molecular Diagnostics 20, no. 5 (2018): 635–642, 10.1016/j.jmoldx.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 28. Wald A. I., Pingpank J. F., Ongchin M., et al., “Targeted Next‐Generation Sequencing Improves the Prognostication of Patients With Disseminated Appendiceal Mucinous Neoplasms (Pseudomyxoma Peritonei),” Annals of Surgical Oncology 30, no. 12 (2023): 7517–7526, 10.1245/s10434-023-13721-y. [DOI] [PubMed] [Google Scholar]
- 29. Munari G., Businello G., Mattiolo P., et al., “Molecular Profiling of Appendiceal Serrated Lesions, Polyps and Mucinous Neoplasms: A Single‐Centre Experience,” Journal of Cancer Research and Clinical Oncology 147, no. 7 (2021): 1897–1904, 10.1007/s00432-021-03589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ang C. S., Shen J. P., Hardy‐Abeloos C. J., et al., “Genomic Landscape of Appendiceal Neoplasms,” JCO Precision Oncology 2 (2018): 1–18, 10.1200/PO.17.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gleeson E. M., Feldman R., Mapow B. L., et al., “Appendix‐Derived Pseudomyxoma Peritonei (PMP): Molecular Profiling Toward Treatment of a Rare Malignancy,” American Journal of Clinical Oncology 41, no. 8 (2018): 777–783, 10.1097/COC.0000000000000376. [DOI] [PubMed] [Google Scholar]
- 32. Lin Y. L., Zhu J. Q., Ma R. Q., et al., “Whole‐Exome Sequencing Identifies Mutation Profile and Mutation Signature‐Based Clustering Associated With Prognosis in Appendiceal Pseudomyxoma Peritonei,” Molecular Cancer Research 22, no. 1 (2024): 70–81, 10.1158/1541-7786.MCR-22-0801. [DOI] [PubMed] [Google Scholar]
- 33. Nummela P., Saarinen L., Thiel A., et al., “Genomic Profile of Pseudomyxoma Peritonei Analyzed Using Next‐Generation Sequencing and Immunohistochemistry,” International Journal of Cancer 136, no. 5 (2015): E282–E289, 10.1002/ijc.29245. [DOI] [PubMed] [Google Scholar]
- 34. Taggart M. W., Galbincea J., Mansfield P. F., et al., “High‐Level Microsatellite Instability in Appendiceal Carcinomas,” American Journal of Surgical Pathology 37, no. 8 (2013): 1192–1200, 10.1097/PAS.0b013e318282649b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chua T. C., Moran B. J., Sugarbaker P. H., et al., “Early‐ and Long‐Term Outcome Data of Patients With Pseudomyxoma Peritonei From Appendiceal Origin Treated by a Strategy of Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy,” Journal of Clinical Oncology 30, no. 20 (2012): 2449–2456, 10.1200/JCO.2011.39.7166. [DOI] [PubMed] [Google Scholar]
- 36. Govaerts K., Lurvink R. J., De Hingh I., et al., “Appendiceal Tumours and Pseudomyxoma Peritonei: Literature Review With PSOGI/EURACAN Clinical Practice Guidelines for Diagnosis and Treatment,” European Journal of Surgical Oncology 47, no. 1 (2021): 11–35, 10.1016/j.ejso.2020.02.012. [DOI] [PubMed] [Google Scholar]
- 37. Reddy S., Punjala S. R., Allan P., et al., “First Report With Medium‐Term Follow‐Up of Intestinal Transplantation for Advanced and Recurrent Nonresectable Pseudomyxoma Peritonei,” Annals of Surgery 277, no. 5 (2023): 835–840, 10.1097/SLA.0000000000005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kusamura S., Baratti D., Hutanu I., et al., “The Role of Baseline Inflammatory‐Based Scores and Serum Tumor Markers to Risk Stratify Pseudomyxoma Peritonei Patients Treated With Cytoreduction (CRS) and Hyperthermic Intraperitoneal Chemotherapy (HIPEC),” European Journal of Surgical Oncology 41, no. 8 (2015): 1097–1105, 10.1016/j.ejso.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 39. Taflampas P., Dayal S., Chandrakumaran K., Mohamed F., Cecil T. D., and Moran B. J., “Pre‐Operative Tumour Marker Status Predicts Recurrence and Survival After Complete Cytoreduction and Hyperthermic Intraperitoneal Chemotherapy for Appendiceal Pseudomyxoma Peritonei: Analysis of 519 Patients,” European Journal of Surgical Oncology 40, no. 5 (2014): 515–520, 10.1016/j.ejso.2013.12.021. [DOI] [PubMed] [Google Scholar]
- 40. Pretzsch E., Neumann J., Niess H., et al., “Comparative Transcriptomic Analyses Reveal Activation of the Epithelial‐Mesenchymal Transition Program in Non‐Metastasizing Low Grade Pseudomyxoma Peritonei,” Pathology, Research and Practice 254 (2024): 155129, 10.1016/j.prp.2024.155129. [DOI] [PubMed] [Google Scholar]
- 41. Carr N. J., Bibeau F., Bradley R. F., et al., “The Histopathological Classification, Diagnosis and Differential Diagnosis of Mucinous Appendiceal Neoplasms, Appendiceal Adenocarcinomas and Pseudomyxoma Peritonei,” Histopathology 71, no. 6 (2017): 847–858, 10.1111/his.13324. [DOI] [PubMed] [Google Scholar]
- 42. Arjona‐Sanchez A., Munoz‐Casares F. C., Casado‐Adam A., et al., “Outcome of Patients With Aggressive Pseudomyxoma Peritonei Treated by Cytoreductive Surgery and Intraperitoneal Chemotherapy,” World Journal of Surgery 37, no. 6 (2013): 1263–1270, 10.1007/s00268-013-2000-2. [DOI] [PubMed] [Google Scholar]
- 43. Pengelly R. J., Gibson J., Andreoletti G., Collins A., Mattocks C. J., and Ennis S., “A SNP Profiling Panel for Sample Tracking in Whole‐Exome Sequencing Studies,” Genome Medicine 5, no. 9 (2013): 89, 10.1186/gm492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H. and Durbin R., “Fast and Accurate Long‐Read Alignment With Burrows‐Wheeler Transform,” Bioinformatics 26, no. 5 (2010): 589–595, 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barnell E. K., Ronning P., Campbell K. M., et al., “Standard Operating Procedure for Somatic Variant Refinement of Sequencing Data With Paired Tumor and Normal Samples,” Genetics in Medicine 21, no. 4 (2019): 972–981, 10.1038/s41436-018-0278-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo G., Chmielecki J., Goparaju C., et al., “Whole‐Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A, and CUL1 in Malignant Pleural Mesothelioma,” Cancer Research 75, no. 2 (2015): 264–269, 10.1158/0008-5472.CAN-14-1008. [DOI] [PubMed] [Google Scholar]
- 47. Murali R., Wiesner T., and Scolyer R. A., “Tumours Associated With BAP1 Mutations,” Pathology 45, no. 2 (2013): 116–126, 10.1097/PAT.0b013e32835d0efb. [DOI] [PubMed] [Google Scholar]
- 48. El‐Gamal M. I., Mewafi N. H., Abdelmotteleb N. E., et al., “A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors,” Molecules 26, no. 23 (2021): 7376, 10.3390/molecules26237376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raghav K. P., Shetty A. V., Kazmi S. M., et al., “Impact of Molecular Alterations and Targeted Therapy in Appendiceal Adenocarcinomas,” Oncologist 18, no. 12 (2013): 1270–1277, 10.1634/theoncologist.2013-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vazquez‐Borrego M. C., Granados‐Rodriguez M., Bura F. I., et al., “Antitumor Effect of a Small‐Molecule Inhibitor of KRAS(G12D) in Xenograft Models of Mucinous Appendicular Neoplasms,” Experimental Hematology & Oncology 12, no. 1 (2023): 102, 10.1186/s40164-023-00465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martinez‐Quintanilla J., Cabot D., Sabia D., et al., “Precision Oncology and Systemic Targeted Therapy in Pseudomyxoma Peritonei,” Clinical Cancer Research 30, no. 18 (2024): 4082–4099, 10.1158/1078-0432.CCR-23-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Figure S1.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.