

Abstract
Bacterial conjugation is an example of macromolecular trafficking between cells, based on the translocation of single-stranded DNA across membranes through a type IV secretion system. TrwBΔN70 is the soluble domain of TrwB, an essential integral membrane protein that couples the relaxosome (a nucleoprotein complex) to the DNA transport apparatus in plasmid R388 conjugation. TrwBΔN70 crystallographic structure revealed a hexamer with six equivalent subunits and a central channel. In this work, we characterize a DNA-dependent ATPase activity for TrwBΔN70. The protein displays positive cooperativity for ATP hydrolysis, with at least three catalytic sites involved. The activity is sensitive to pH and salt concentration, being more active at low pH values. The effective oligonucleotide size required for activation of the ATPase function is between 40 and 45 nucleotides, and the same length is required for the formation of high-molecular-weight TrwBΔN70–DNA complexes, as observed by gel filtration chromatography. A mutation in a tryptophan residue (W216A), placed in the central pore formed by the hexameric structure, resulted in a protein that did not hydrolyze ATP. In addition, it exerted a dominant negative effect, both on R388 conjugation frequency and ATP hydrolysis, underscoring the multimeric state of the protein. ATP hydrolysis was not coupled to a DNA unwinding activity under the tested conditions, which included forked DNA substrates. These results, together with TrwB structural similarity to F1-ATPase, lead us to propose a mechanism for TrwB as a DNA-translocating motor.
Keywords: molecular motor, DNA transfer
Conjugative plasmids provide a main route for acquisition of new genetic information in bacteria. Bacterial conjugation systems (including the related Vir system that Agrobacterium tumefaciens uses to transfer DNA to plants) show similarities to both DNA replication and protein transport systems (1, 2). The translocated substrate is a nucleoprotein particle that crosses the bacterial envelope into other bacterial or eukaryotic cells, crossing the kingdom boundaries. A two-step mechanism for DNA transport was proposed (2), in which conjugation is visualized as a DNA rolling-circle replication process linked to a type IV protein secretion system. Conjugation is encoded by two gene clusters in most conjugative plasmids. One cluster (dtr) encodes the DNA transfer replication proteins, and the other (mpf) codes for the proteins that assemble the type IV protein secretion system channel. R388 is a 34-kb plasmid, which displays the simplest known dtr region, encoding only three proteins (TrwA, TrwB, and TrwC). The mpf region codes for 11 proteins (TrwD–N) involved in the formation of an extracellular pilus and a membrane-associated complex that translocates the nucleoprotein substrate across membranes.
TrwB, an integral membrane and DNA-binding protein, is a key player in R388 conjugation (3–6). It has a counterpart in all conjugative systems, and it is thought to be responsible for recruiting the relaxosome DNA–protein complex and transferring a single DNA strand during cell mating. Its sequence includes conserved Walker motifs and binds both single- and double-stranded DNA nonspecifically (6). The atomic structure of TrwBΔN70, the soluble fraction of TrwB, shows a molecule consisting of two domains: a nucleotide-binding domain, reminiscent of RecA and DNA ring helicases, and an “all α-domain” with partial structural similarity to TraM, a DNA-binding protein of the F plasmid dtr system (7, 8). Six equivalent protein monomers associate to form a quaternary structure strikingly similar to F1-ATPase, leaving a central channel of 20 Å. The nucleotide-binding site is essential for conjugation, because a point mutation affecting the Walker A motif (K136T) abolished DNA transfer (6).
In this work, we characterize a TrwBΔN70 DNA-dependent ATPase activity. Hydrolysis has a nonlinear dependence on ATP concentration, showing positive cooperativity. The isolation of high-molecular-weight complexes by gel filtration data underscores TrwBΔN70 assembly into oligomeric DNA–protein complexes, required for ATPase activity. The crystallographic structure of TrwBΔN70 reveals important similarities with a disparate group of proteins, involved in different aspects of DNA processing such as helicases (9, 10), chromosome segregation (11), as well as in transforming energy, as F1-ATPase (12). TrwBΔN70 structural data, together with the described DNA dependent ATPase activity, suggests mechanistic similarities between these enzymes. Such a relationship provides insight into how TrwB might function as a DNA pump.
Materials and Methods
Protein Purification. Purification of protein TrwBΔN70 and W216A and K136T mutants was carried out as described in ref. 6 with modifications. Strains used were derivatives of Escherichia coli C41 (13) containing plasmids pSU4637 (6), pSU4639 (6), and pMEC1, respectively (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site). Bacteria were grown in 4 liters of LB medium. After addition of isopropyl β-d-thiogalactoside (Apollo Scientific, Bredbury, U.K.), cells were incubated overnight at 37°C, harvested, and resuspended in 60 ml of buffer A (50 mM Tris·HCl, pH 7.6/1 mM EDTA/5 mM MgCl2/0.001% PMSF) plus 5 mM ATP and 10% sucrose. One tablet of a protease inhibitor mixture (Roche) was added at this step. Lysozyme and benzamidine were added to 0.8 mg/ml and 2.5 mM final concentrations, respectively. After 45 min of incubation on ice, an equal volume of buffer A supplemented with 0.5% (wt/vol) Triton X-100 and 1 M NaCl was added, and the lysate was centrifuged at 138,000 × g for 30 min at 4°C. The supernatant was diluted with buffer B (50 mM Tris·HCl, pH 7.6/0.1 mM EDTA/2 mM MgCl2/0.001% PMSF) to a final concentration of 0.15 M NaCl and adsorbed to a 35-ml phosphocellulose P11 chromatographic column (Whatman), equilibrated with buffer B and 0.15 M NaCl. Bound proteins were eluted with buffer B plus 1 M NaCl. TrwBΔN70-containing fractions were pooled and diluted to 0.15 M NaCl with buffer B. Then, they were loaded onto a 5-ml HiTrap-SP (Amersham Pharmacia) column equilibrated with buffer B plus 0.15 M NaCl. Proteins eluted in a 120-ml salt gradient (0.15–1 M NaCl) were pooled and concentrated in a Centricon YM30 (Amicon) concentrator to 4 ml. Finally, TrwBΔN70 was loaded on a HiLoad 16/60 Superdex 200 column (Amersham Pharmacia) equilibrated with buffer (50 mM Pipes·NaOH, pH 6.0/100 mM NaCl/2 mM MgCl2/0.1 mM EDTA/10% glycerol/0.001% PMSF). Glycerol was added to TrwBΔN70 containing fractions up to 20% final concentration and protein stored at -70°C. Protein concentrations were determined by the method of Bradford (Bio-Rad).
Nucleotide Hydrolysis Assays. ATP hydrolysis was analyzed by three methods. For radiometric analysis (14), TrwBΔN70 protein samples were preincubated for 5 min at 37°C at various ssM13 DNA and salt concentrations, in 10 μl of buffer (50 mM Pipes·NaOH, pH 6.0/100 mM NaCl/2 mM MgCl2/10% glycerol). Reactions were started by addition of ATP/[γ32-P]ATP (Amersham Pharmacia Biosciences) to 2 mM/0.6 μCi (1 Ci = 37 GBq) final concentration. After 10 min, they were stopped by adding 1 μl of 500 mM EDTA (pH 8.0). One-microliter aliquots were spotted onto polyethyleneimine-cellulose plates (Merck), and products were separated by thin-layer chromatography in 0.5 M LiCl/1 M formic acid. To test the effect of pH, samples containing TrwBΔN70 (12 μM) and ssM13 DNA (8 nM) were analyzed in acetic/acetate buffer for a pH range from 5.0 to 5.75, Pipes·NaOH buffer for pH range 6.0–6.5, and Tris·HCl buffer for pH range 7.0–8.0. Radioactive samples were quantified with personal molecular imager software (Bio-Rad).
TrwBΔN70 ATPase activity also was measured by a coupled enzyme assay (15). To analyze ATP concentration dependency, TrwBΔN70 (3 μM) was preincubated with ssM13 DNA (6 nM) for 5 min at 37°C in 150 μl of ATPase assay mixture, consisting of 50 mM Pipes·NaOH (pH 6.2), 75 mM NaCl, 6 mM MgCl2, 10% glycerol, 0.5 mM phosphoenolpyruvate, 0.25 mM NADH, 60 μg/ml pyruvate kinase, and 60 μg/ml lactate dehydrogenase (Roche). Reactions were started by addition of ATP. Activity was measured by the decrease in NADH absorbance at 340 nm for 10 min at 37°C in a UV-1603 spectrophotometer (Shimadzu). The same procedure was repeated at pH 6.5 and 7.5, using Tris·HCl buffer in the latter case. DNA concentration-dependent assays were carried out in the same way, except that ATP (5 mM) and DNA were added first to the assay mixture, and the reaction was started by addition of TrwBΔN70 (3 μM). Oligonucleotides used were 29-mer (A), 36-mer (B), 40-mer (C), 45-mer (D), and 66-mer (E) (sequences are supplied in Table 2, which is published as supporting information on the PNAS web site).
A third colorimetric assay (16) was used to measure hydrolysis of different nucleotides and desoxynucleotides as follows: ATP, dATP, GTP, dGTP, CTP, dCTP, UTP, and dTTP (Sigma). Activity was measured as the amount of inorganic phosphate liberated. TrwBΔN70 (3 μM) was preincubated with ssM13 DNA (6 nM) for 15 min at 37°C in 600 μl of buffer (50 mM Pipes·NaOH, pH 6.0/100 mM NaCl/2 mM MgCl2/10% glycerol). Reactions were started by addition of the respective NTP (5 mM), incubated at 37°C for 10 min, and then stopped by the addition of 400 μl of 10% SDS (wt/vol). A ferrous sulfate–ammonium molybdate solution (500 μl) was added, and the amount of inorganic phosphate was estimated by absorbance at 740 nm.
Preparation of DNA Substrates and Helicase Assay. Circular ssM13DNA was prepared as described in ref. 17. ssDNA concentration of the stock solution was measured by absorbance at 260 nm, using 40 μg·ml-1·A260-1 as the conversion factor. DNA substrates used in helicase assays were generated by annealing radioactive oligonucleotides to ssM13 DNA, as described in Supporting Materials and Methods. Strand-displacement helicase assay was carried out as in ref. 18, with some modifications described in Supporting Materials and Methods.
Results
TrwBΔN70 Is a DNA-Dependent ATPase Active only at Acidic pH Values. TrwBΔN70 protein was purified to homogeneity and analyzed for ATPase activity in the absence and presence of viral (+) strand of M13 phage. ATPase activity was only detected in the presence of ssM13 DNA. Because of previous failures to detect ATPase activity in conjugative coupling proteins (19), we carried out several tests to confirm that the found ATPase activity corresponded to TrwB and not to a contaminating protein. First, ATPase activities of fractions from the last gel filtration chromatography coincided exactly with the TrwBΔN70 absorbance peak (Fig. 1A). Second, mutant proteins TrwBΔN70(W216A) and TrwBΔN70(K136T) purified under exactly the same conditions did not show significant ATPase activity (<1% and 4%, respectively; Fig. 1 B and C). Third, mutant protein TrwBΔN70(W216A) exerted a dominant negative effect on TrwBΔN70 ATP hydrolysis in vitro when added to the assay mixture (see below). As a consequence, we conclude that the observed ATPase activity is an intrinsic activity of TrwB and is not due to a contaminating activity.
Fig. 1.

Correlation of ATPase activity and protein elution profiles in gelfiltration chromatography. For each of the three proteins, the image represents the elution profile obtained in the last purification step (gel-filtration chromatography through a Hi-Load Superdex-200 column). ATP hydrolysis was quantified by radiometric analysis of 1.0 μl of protein samples after a 5-min reaction in the presence of 10 nM M13 ssDNA. Amount of protein loaded was 35.5 mg of WT TrwBΔN70 (A), 38.7 mg of TrwBΔN70(W216A) (B), and 28.2 mg of TrwBΔN70(K136T) (C).
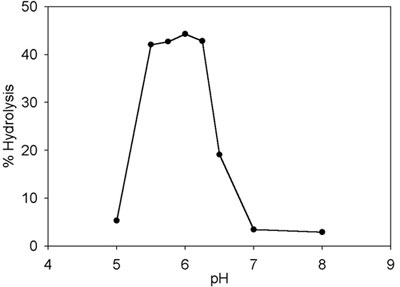
ATP hydrolysis was initially monitored by thin-layer chromatography to characterize the main requirements for the reaction. TrwBΔN70 ATPase activity was sensitive to pH, the optimal range being between 5.25 and 6.25 (see Fig. 7, which is published as supporting information on the PNAS web site). The activity was inhibited at pH ≥ 6.5. This finding is a key result that explains why no ATPase activity was detected in previous works (6, 19). Activity also was stimulated by 50–100 mM NaCl, with significant inhibition occurring at values of >200 mM (see Fig. 8, which is published as supporting information on the PNAS web site). The optimal reaction temperature was in the range of 32–37°C (see Fig. 9, which is published as supporting information on the PNAS web site). At 42°C, the rate of ATP hydrolysis was half the optimal value, and at 50°C the protein was inactive, probably because of thermal denaturation (20).
Given the structural similarity between TrwBΔN70 and hexameric helicases such as T7 DNA helicase, and the preference of the latter for dTTP instead of ATP, we checked TrwBΔN70 NTP specificity for hydrolysis by using a colorimetric assay. Rates of hydrolysis for the eight common NTPs and dNTPs were determined (see Table 3, which is published as supporting information on the PNAS web site). Both, NTPs and dNTPs were hydrolyzed at different rates in the presence of ssM13 DNA, with the exception of dCTP, which was not significantly hydrolyzed. ATP was the preferred nucleoside 5′-triphosphate for hydrolysis.
TrwBΔN70 Displays Positive Cooperativity for ATP Hydrolysis. TrwBΔN70 kinetic parameters for ssDNA-dependent ATP hydrolysis were assayed by a coupled enzyme assay. Fig. 2A shows the effect of ATP concentration on ATPase activity rates, in the presence of 6 nM ssM13 DNA and by using an ATP-regenerating system. The effect of pH variation on TrwBΔN70 ATPase activity is underscored by these curves. ATP hydrolysis rates increased with increasing ATP concentration, until saturation was reached at 2.5 mM ATP. The data indicate strong cooperativity, because deviation from Michaelis–Menten behavior was evident in a standard velocity vs. substrate plot (Fig. 2 A). A commonly used method for determining ATP-induced cooperativity is by calculating the Hill coefficient (21). Assuming the reaction rate is proportional to the fractional saturation of the enzyme, a slope (Hill coefficient) greater than one derived from a plot of the fractional rate [v/(Vmax - v)] vs. the substrate concentration indicates positive cooperativity. Rates of ATP hydrolysis by TrwBΔN70 could not be fit to a simple hyperbolic curve (Michaelis–Menten equation). The data fit a Hill equation, and analysis of the plot demonstrated strong positive cooperativity (Hill coefficients: nH = 2.78 ± 0.05 at pH 6.2 and nH = 3.38 ± 0.11 at pH 6.5) in a range from 0.1 to 3.5 mM ATP (Fig. 2B). The Hill coefficient (nH) gives the lowest estimation of the number of protomers recruited into cooperative binding. Thus, nH can be interpreted as the minimum number of protomers within the oligomeric structure involved in ATP cooperative binding, being at least three for TrwBΔN70.
Fig. 2.

TrwBΔN70 ATPase activity displays positive cooperativity. Shown is the effect of ATP concentration on ATP hydrolysis rates at various pH values, as measured by the coupled enzyme method. Reactions contained 3 μM TrwBΔN70 and 6 nM ssM13 DNA. (A) The three curves represent ATPase activity at the following three pH values: pH 6.2 (▪), 6.5 (▴), and 7.5 (•). (B) Hill plot of data obtained at pH 6.2 (▪) and 6.5 (▴). The slope of the lines in the range of 0.1 mM ATP (log[ATP] = -1) to 3.5 mM ATP (log[ATP] = 0.54) was estimated as the apparent Hill coefficient (nH).
TrwBΔN70(W216A) Mutant Protein Does Not Exhibit ATPase Activity but Exerts a Dominant Negative Effect on the ATPase Activity of Protein TrwBΔN70. Mutation K136T, affecting a key residue at the TrwB ATP-binding site, drastically affected DNA transfer in vivo (6). In this work, we analyzed the effect of a mutation in a tryptophan residue (W216A), placed in the internal channel formed by a TrwBΔN70 hexamer. TrwBΔN70(W216A) mutant protein did not show ATPase activity under the tested conditions (Fig. 1B). Moreover, when ATPase activity was tested in the presence of both TrwBΔN70 and TrwBΔN70(W216A) proteins, the activity of the former was inhibited, indicating a clear dominant negative effect. The increase in TrwBΔN70(W216A) concentration correlated with a decrease in the ATPase rate (Fig. 3A). TrwBΔN70 ATPase activity was 50% inhibited when the mutant vs. wild-type (WT) concentration ratio was 0.18, which roughly corresponds to one TrwBΔN70(W216A) protomer per five to six TrwBΔN70 protomers. Less than 3% ATPase activity remained in an equimolar mixture of TrwBΔN70 and W216A mutant, which correlates well with a theoretical value of 1.5% (i.e., 1/26). Fig. 3B shows the theoretical curve obtained assuming that a mutant monomer assembles in a mixed hexamer with WT efficiency and that the resultant multimer containing at least one mutant monomer is completely inactive. Experimental data approached the theoretical curve, indicating that the mutant protein probably incorporates to hexamers with almost WT efficiency and that multimers containing just one mutant monomer are inactive.
Fig. 3.

Dominant negative effect on TrwBΔN70 ATPase activity exerted by TrwBΔN70(W216A) mutant protein. (A) ATPase rates were measured by the enzyme coupled assay by using a constant 1.5 μM TrwBΔN70 protein concentration and increasing concentrations of mutant protein TrwBΔN70(W216A) (▴), shown on the x axis as relative concentrations. Protein samples were first incubated together for 5 min, and then the rest of the reactants were added. The control assay (▪) reproduces the same final protein concentrations but using only TrwBΔN70. One hundred percent hydrolysis corresponds to 700 nM ATP hydrolyzed per min per mg of protein. Relative activity is expressed as percent ATPase rate per milligram of total protein (thus, the value is constant in the control assay). (B) Triangles (▴) represent the same experimental values as in A, except that the x axis indicates absolute concentrations of the added mutant protein. If we assume that the mutant protein assembles into hexamers with WT efficiency but hexamers containing at least one mutant monomer are inactive, the dotted line shows the theoretical curve that represents the relative activity of the resulting mixture [relative activity = [a/(a + b)]6 × 100, where a and b are the concentration of proteins TrwBΔN70 and TrwBΔN70(W216A), respectively].
Mutation W216A also abolished DNA transfer in vivo (Table 1). Without a functional trwB gene, there was no conjugation of plasmid pSU1456 (a R388 mutant with a transposon insertion within trwB). Conjugation was restored when pSU1456 was complemented with a plasmid containing a functional trwB gene (pSU4622) but not when plasmid pMTX611 [coding for the mutant protein TrwB(W216A)] was used as a complementing plasmid. Moreover, a 10,000-fold decrease in the frequency of R388 conjugation was observed when pMTX611 was present in trans in an otherwise isogenic donor strain, indicating that the mutant protein strongly interfered with the function of the WT protein. The W216A mutant exerts a more pronounced dominant negative effect than K136T mutant, which was previously reported to promote a 300-fold decrease in the frequency of R388 conjugation (ref. 6 and Table 1). This difference could explain why a dominant negative phenotype in ATPase activity was not observed in vitro when mutant TrwBΔN70(K136T) protein was added to the reaction sample (data not shown).
Table 1. Conjugation frequencies of R388 and trwB mutants in the presence or absence of complementing proteins.
| Plasmids in donor | Variant of TrwB protein in donor | Transfer frequency, transconjugants/donor |
|---|---|---|
| pSU1456 | None | <10-7 |
| pSU1456 + pSU4622 | TrwB | 2 × 10-2 |
| pSU1456 + pSU4632 | TrwB(K136T) | <10-7 |
| pSU1456 + pMTX611 | TrwB(W216A) | <10-7 |
| R388 | TrwB | 5 × 10-1 |
| R388 + pSU4622 | TrwB + TrwB | 7 × 10-2 |
| R388 + pSU4632 | TrwB + TrwB(K136T) | 3 × 10-4 |
| R388 + pMTX611 | TrwB + TrwB(W216A) | 8 × 10-6 |
Derivatives of E. coli K12 strain DH5α carrying the plasmids shown in the first column were mated with strain UB1637 as described in ref. 6. Transconjugants were selected on plates containing streptomycin and trimethoprim. All plasmids were described in ref. 6, except pMTX611, which is described in Supporting Materials and Methods. Values are the mean of six experiments.
Minimal Oligonucleotide Length Required to Promote ATPase Activity. Fig. 4 shows that, in the absence of DNA, TrwBΔN70 ATPase activity was low (30 nmol/min per mg). Addition of oligonucleotides or viral M13 (+) strand lead to efficient ATP hydrolysis, with Vmax of ≈700 nmol/min per mg. We investigated the minimal length of ssDNA needed for activating TrwBΔN70 ATPase. Twenty-nine-, 36-, 40-, 45-, and 66-mer oligonucleotides were used as substrates for the ATPase reaction. No ATP hydrolysis was observed for the 29-, 36-, and 40-mer substrates. Increasing the length of the DNA to 45 or 66 nt resulted in a large increase in ATP hydrolysis, reaching the rate observed for ssM13 DNA (80% and 90% of Vmax, respectively; Fig. 4). The optimal ATPase activity was obtained at a DNA concentration of 52 μM in nucleotides, which reflects an optimal relationship of ≈17 nt per TrwBΔN70 monomer. Because the assayed oligonucleotides were not related in sequence (see Table 2), results suggest that TrwBΔN70 ATPase activity is largely sequence unspecific.
Fig. 4.

Effect of oligonucleotide size and concentration on TrwBΔN70 ATP hydrolysis. Reactions contained 3 μM TrwBΔN70, 5 mM ATP, and oligonucleotides of different sizes as represented in the image as follows: 29-mer (▵), 36-mer (○), 40-mer (▪), 45-mer (▴), 66-mer (□), and M13 ssDNA (•). Activities were measured by using the coupled enzyme assay. Each symbol represents the average of three different experiments.
Strand Displacement Helicase Assays. To test whether TrwBΔN70 ATP hydrolysis was coupled to a DNA unwinding activity, we used an electrophoretic helicase assay. TrwBΔN70 was tested for its ability to displace an oligonucleotide of either 90- or 63-nt long from a partial DNA duplex. Three different heteroduplexes were prepared by annealing oligonucleotides of different lengths to ssM13 DNA. Because some ring helicases require two noncomplementary ssDNA tails at one end of a duplex DNA (a DNA fork) to initiate DNA strand separation, we also prepared two different forked DNA substrates by annealing a 63-mer with a 18-base noncomplementary 3′ tail or a 63-mer with a 18-base noncomplementary 5′ tail to ssM13 DNA. These lengths were shown to provide productive DNA substrates for hexameric helicases that require a DNA fork (22). Whereas TrwC protein (18), used as a positive control in these experiments, was effective in unwinding all three types of DNA substrates, TrwBΔN70 was defective in helicase activity under all tested conditions (data not shown).
DNA Promotes TrwBΔN70 Oligomerization. In gel filtration experiments, TrwBΔN70 exists as a monomer in solution, as shown from its elution from a Superdex 200 column as a single peak with an estimated molecular mass of 50 kDa (peak 1, Fig. 5A). Incubation of the protein with oligonucleotides of different lengths (29-, 36-, or 40-mer) lead to the formation of a second complex, which eluted with a molecular mass of ≈100 kDa (peak 2, Fig. 5B). The complex contains DNA, as shown by a correlated increase in peak 2 absorbance at 260 nm, so we estimate it to be a complex of one monomer protein with one oligonucleotide molecule. Peak 1 corresponds to free protein, because it shows a relative lack of absorbance at 260 nm. Strikingly, when TrwBΔN70 protein was incubated with longer nucleotides (45- or 66-mer), a third fast-migrating complex appeared, with an estimated molecular mass of 440 kDa, also correlated with high absorbance at 260 nm. This DNA–protein complex could correspond to a hexameric TrwBΔN70–DNA complex (peak 3, Fig. 5C). Therefore, there is a correlation between the conditions that allow the formation of the large complex and ATPase activity (Fig. 4). Finally, gel filtration experiments carried out in the presence of ATP, but in the absence of a DNA substrate, did not affect the monomeric state of the protein.
Fig. 5.

Gel-filtration chromatography of TrwBΔN70 and TrwBΔN70–DNA complexes. Protein samples (60 μM) were preincubated with oligonucleotides of different lengths (10 μM) and chromatographed at 20°C at a flow rate of 30 μl/min on a Superdex-200 PC 3.2/30 column on a Amersham Pharmacia SMART system. The column was equilibrated in buffer containing 50 mM Pipes·NaOH (pH 6.0), 100 mM NaCl, 2 mM MgCl2, 0.1 mM EDTA, and 10% glycerol. Absorbance was monitored at 280 nm (black) and 260 nm (red). Ve represents the elution volume. Elution profiles are as follows: TrwBΔN70 (A); TrwBΔN70 preincubated with oligonucleotides 29-, 36-, or 40-mer (B); and TrwBΔN70 preincubated with oligonucleotides 45- or 66-mer (C). Peaks were interpreted as follows: (1) free protein (relative lack of absorbance at 260; apparent molecular mass = 50 kDa), (2) slow complex (contains protein and DNA; apparent molecular mass = 100 kDa), and (3) fast complex (contains protein and DNA; apparent molecular mass = 440 kDa).
Interestingly, the TrwBΔN70(W216A) mutant protein showed the same oligomerization properties as the WT protein (data not shown), indicating that the mutant protein assembles normally as a complex in vitro, and probably exerts its dominant effect by forming unproductive heterohexamers. Therefore, W216A mutation does not affect oligomerization but inactivates TrwB ATPase function.
Discussion
TrwBΔN70, the soluble fraction of the conjugative coupling protein TrwB, displays a robust ATPase activity that requires DNA for activation. The activity was affected by pH and salt concentration, with significant inhibition occurring at 200 mM salt and pH values of >6.5. Both factors, pH and salt concentration, are probably affecting electrostatic interactions between protein and DNA and contribute to stability of the DNA–protein complex. Data analysis showed positive cooperativity for ATP hydrolysis in the presence of DNA (Hill coefficients of 2.78 and 3.38 at pH 6.2 and 6.5, respectively). Hill coefficient can be interpreted as the minimum number of protomers within the oligomeric structure that bind ATP cooperatively. By analogy to what happens in F1-ATPase (23), this result implies that a minimum of three ATP molecules bind cooperatively, suggesting a cooperative process in which TrwB would have at least three catalytic sites. The existence of an oligomeric state is underscored by gel filtration data (Fig. 5), which show the formation of a high-molecular-weight complex when TrwB binds oligonucleotide DNA in a productive manner. Under purification conditions, TrwBΔN70 protein exists as a monomer in solution. Gel-filtration experiments carried out in the presence of ATP (and in the absence of DNA) did not affect the monomeric state of the protein, indicating that it is DNA, and not ATP, that is the key factor that promotes TrwBΔN70 oligomerization. This result is consistent with previous electron microscopy observations that protein complexes were only formed in the presence of DNA. Moreover, TrwBΔN70 ssDNA binding activity was ATP-independent (6).
The minimal oligonucleotide size required for the formation of a high-molecular-weight complex was between 40 and 45 nt. The same DNA substrate length was required to observe an ATPase activity, which suggests that formation of an oligomeric assembly of TrwB on ssDNA is a prerequisite for activation of TrwBΔN70 ATPase function. TrwBΔN70 crystallographic structure unveiled a hexamer (7), and native TrwB, including the transmembrane domain, appears in both monomeric and hexameric forms in solution (5). Given the estimated molecular mass of the TrwBΔN70–DNA complex in gel-filtration chromatography (≈440 kDa), it is most likely that the protein assembles on DNA as a hexamer. If we assume a distance of 7 Å between successive phosphate residues in an extended ssDNA structure, only ≈15 nt would be needed to expand the 10-nm-long channel traversing a TrwBΔN70 hexamer. The requirement for a DNA substrate of >40 nt might indicate the minimal length needed to trigger the translocating activity along ssDNA.
TrwB ATPase activity displayed an interesting dependence on the absolute concentration of ssDNA. Hydrolysis rates reached a maximum for a particular ssDNA and protein concentration and, at higher ssDNA concentrations, started to decline (Fig. 4). This result can be explained if at low concentrations of ssDNA, successive protomers are recruited to the same ssDNA, constituting the active oligomeric structure. As ssDNA concentration is increased, an increasing proportion of monomers would bind to separate ssDNA molecules, where they cannot productively interact to assemble into hexamers. Similar effects were reported for at least two other ATPases, human RAD51 protein, a member of the RecA family (24), and simian virus 40 T-antigen hexamers (25).
The atomic structure of TrwBΔN70 hexamer reveals a conspicuous ring formed by six tryptophan residues in the central channel, suggesting a possible role in DNA binding and translocation. Mutation of that residue (W216A) was deleterious for DNA conjugation. Moreover, a 10,000-fold decrease in the frequency of R388 conjugation was observed when both TrwB and TrwB(W216A) proteins were expressed in the same donor cell (Table 1). The dominant negative phenotype also was obser ved in experiments in vitro (Fig. 3). Protein TrwBΔN70(W216A) not only failed to hydrolyze ATP but produced a rapid decrease of TrwBΔN70 ATPase activity when added to the assay mixture together with the native protein. An explanation for this result could be an heterogeneous assembly of TrwBΔN70 and TrwBΔN70(W216A) monomers into inactive hexameric complexes. A mechanism based on sequential hydrolysis in such a structure would be defective in ATP hydrolysis (see below). Interestingly, TrwBΔN70(W216A) protein is able to oligomerize in the presence of a productive DNA substrate (gel filtration data), suggesting that it exerts its dominant effect by forming unproductive heterohexamers. Therefore, W216A mutation is affecting not oligomerization but ATPase activity, although the mutation is far from the ATP binding site. Because this ATPase activity is still DNA-dependent, W216 might play a key role in DNA translocation through the central pore of the hexamer.
Given the DNA dependency for ATP hydrolysis, we checked whether such an activity was coupled to DNA unwinding. Whereas the DNA helicase TrwC, used as a control in our experiments, was effective in unwinding all types of DNA substrates (including forked DNA substrates), TrwBΔN70 did not show detectable helicase activity under the tested conditions. Although our experiments do not rule out rigorously that ATP hydrolysis by TrwBΔN70 is coupled to DNA unwinding, a helicase activity might not be necessary for TrwB function, because TrwB may act on a preexisting ssDNA strand produced by the initial shooting step in the conjugation process (2). According to this view, TrwB function would be to pump the single DNA strand already threaded on the type IV protein secretion system channel, by using energy derived from ATP hydrolysis.
Our work provides important insights into the mechanism of DNA translocation between conjugating cells. Available structural and biochemical data allow us to propose a mechanism for conjugative DNA translocation that shows remarkable similarity to the F1-ATPase mechanism. TrwBΔN70 and F1-ATPase share striking structural similarities (Fig. 6). F1-ATPase is a α3β3 hexamer with a central coiled-coil γ-subunit. It contains six ATP-binding sites, of which only three are catalytic (23). We propose that the γ-subunit of F1-ATPase is a functional analog of the DNA strand that occupies TrwB central channel. The so-called “binding change mechanism” followed by F1-ATPase shows cooperative catalysis among three active sites, alternating between ATP-bound, ADP-bound, and empty conformations, as shown by structural analysis (12). Although the structures of TrwB and ring helicases display six identical subunits, noncatalytic sites might result from induced asymmetry after DNA binding. Results presented here are consistent with this hypothesis, because TrwBΔN70 shows positive cooperativity for ATP hydrolysis, with at least three putative sites for ATP binding. It can be expected that TrwB complexes with bound DNA will show evidence of this asymmetry.
Fig. 6.

Structural comparison between TrwB and F1-ATPase. TrwB and F1-ATPase structures are shown in A and B, respectively, represented in opposite orientation with respect to the inner membrane. For clarity, only α-, β-, and γ-subunits are represented in F1-ATPase. Note also that the transmembrane α-helices in the N-terminal domain of TrwB are missing in this representation. β-strands and α-helices are colored in blue and red, respectively, with the exception of the γ-subunit in F1-ATPase, where the coiled-coil structure is colored in green. In A, the DNA is represented in green, showing the 5′ and 3′ ends. Following the mechanism proposed in this work, TrwB would bind the 5′-DNA strand through the central channel, and it would exclude the 3′-DNA strand (that may or may not loop around the perimeter of the hexamer). Strand separation might not be achieved by TrwB directly but may occur in a previous step, perhaps associated with TrwC helicase. Blue arrows represent the postulated opening and closing of the catalytic subunits coupled to ATP hydrolysis. TrwB and F1-ATPase structures (Protein Data Bank ID codes 1E9R and 1E1Q, respectively) were generated with bobscript (28). The DNA drawing was created as a separate image and superposed on TrwB structure.
Fig. 6 emphasizes that TrwBΔN70 is anchored to the bacterial membrane in opposite orientation to that of F1-ATPase. The “all-α domain” in TrwB is orientated toward the cytoplasm, probably to contact DNA, whereas in F1-ATPase it is adjacent to the membrane to contact the coiled-coil structure of the γ-subunit. The cytoplasmic channel entrance in the TrwBΔN70 hexamer is plugged by a ring of asparagine residues and restricted to ≈8 Å in diameter. This region is the narrowest part of the channel, which is otherwise ≈20-Å wide along its whole length (7). TrwBΔN70 structure represents a symmetrical conformation of the enzyme. By analogy to F1-ATPase, we propose that DNA binding could promote an asymmetrical conformation, as the γ-subunit does in F1-ATPase. In perfect agreement with this interpretation, the atomic structure of the thermophilic Bacillus PS3 α3β3 complex (without the γ-subunit) shows an exact threefold symmetry (that is, equivalent to a TrwB symmetric hexamer) (26). If our interpretation proves to be correct, TrwB would couple sequential ATP hydrolysis to DNA translocation through the central pore, following a mechanism similar to the binding change mechanism of F1-ATPase. Cycles of ATP hydrolysis would allow protomers within the TrwB hexamer to alternate between ATP-bound, ADP-bound, and empty conformational states. Cyclic conformational changes of the ring subunits would pump the DNA strand through the central channel. Therefore, we propose a model in which TrwB operates as a molecular motor, translocating DNA by a mechanism similar to that used by F1-ATPase. A somehow similar mechanism has been proposed for DNA movement across T7 gene4 hexameric helicase (10, 27).
Supplementary Material
Acknowledgments
This work was supported by Ministerio de Educación y Ciencia (Spain) Grant BMC2002-00379 (to F.d.l.C.). I.T. received a fellowship from the Basque Government.
Author contributions: F.d.l.C. and E.C. designed research; I.T. and S.Z. performed research; I.T., F.d.l.C., and E.C. analyzed data; and F.d.l.C. and E.C. wrote the paper.
References
- 1.Cascales, E. & Christie, P. J. (2003) Nat. Rev. Microbiol. 1, 137-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Llosa, M., Gomis-Ruth, F. X., Coll, M. & de la Cruz, F. (2002) Mol. Microbiol. 45, 1-8. [DOI] [PubMed] [Google Scholar]
- 3.Cabezon, E., Sastre, J. I. & de la Cruz, F. (1997) Mol. Gen. Genet. 254, 400-406. [DOI] [PubMed] [Google Scholar]
- 4.Llosa, M., Zunzunegui, S. & de la Cruz, F. (2003) Proc. Natl. Acad. Sci. USA 100, 10465-10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hormaeche, I., Alkorta, I., Moro, F., Valpuesta, J. M., Goni, F. M. & de la Cruz, F. (2002) J. Biol. Chem. 277, 46456-46462. [DOI] [PubMed] [Google Scholar]
- 6.Moncalian, G., Cabezon, E., Alkorta, I., Valle, M., Moro, F., Valpuesta, J. M., Goni, F. M. & de La Cruz, F. (1999) J. Biol. Chem. 274, 36117-36124. [DOI] [PubMed] [Google Scholar]
- 7.Gomis-Ruth, F. X., Moncalian, G., Perez-Luque, R., Gonzalez, A., Cabezon, E., de la Cruz, F. & Coll, M. (2001) Nature 409, 637-641. [DOI] [PubMed] [Google Scholar]
- 8.Gomis-Ruth, F. X., Moncalian, G., de la Cruz, F. & Coll, M. (2002) J. Biol. Chem. 277, 7556-7566. [DOI] [PubMed] [Google Scholar]
- 9.Yu, X. & Egelman, E. H. (1997) Nat. Struct. Biol. 4, 101-104. [DOI] [PubMed] [Google Scholar]
- 10.Singleton, M. R., Sawaya, M. R., Ellenberger, T. & Wigley, D. B. (2000) Cell 101, 589-600. [DOI] [PubMed] [Google Scholar]
- 11.Errington, J., Bath, J. & Wu, L. J. (2001) Nat. Rev. Mol. Cell. Biol. 2, 538-545. [DOI] [PubMed] [Google Scholar]
- 12.Abrahams, J. P., Leslie, A. G., Lutter, R. & Walker, J. E. (1994) Nature 370, 621-628. [DOI] [PubMed] [Google Scholar]
- 13.Miroux, B. & Walker, J. E. (1996) J. Mol. Biol. 260, 289-298. [DOI] [PubMed] [Google Scholar]
- 14.Weinstock, G. M., McEntee, K. & Lehman, I. R. (1981) J. Biol. Chem. 256, 8829-8834. [PubMed] [Google Scholar]
- 15.Kreuzer, K. N. & Jongeneel, C. V. (1983) Methods Enzymol. 100, 144-160. [DOI] [PubMed] [Google Scholar]
- 16.Taussky, H. H. & Shorr, E. (1953) J. Biol. Chem. 202, 675-685. [PubMed] [Google Scholar]
- 17.Sambrook, J. & Russell, D. W. (2001) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Woodbury, N.Y.).
- 18.Grandoso, G., Llosa, M., Zabala, J. C. & de la Cruz, F. (1994) Eur. J. Biochem. 226, 403-412. [DOI] [PubMed] [Google Scholar]
- 19.Schroder, G. & Lanka, E. (2003) J. Bacteriol. 185, 4371-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hormaeche, I., Iloro, I., Arrondo, J. L., Goni, F. M., de la Cruz, F. & Alkorta, I. (2004) J. Biol. Chem. 279, 10955-10961. [DOI] [PubMed] [Google Scholar]
- 21.Segel, I. H. (1976) Biochemical Calculations: How to Solve Mathematical Problems in General Biochemistry (Wiley, New York).
- 22.Ahnert, P. & Patel, S. S. (1997) J. Biol. Chem. 272, 32267-32273. [DOI] [PubMed] [Google Scholar]
- 23.Boyer, P. D. (1993) Biochim. Biophys. Acta 1140, 215-250. [DOI] [PubMed] [Google Scholar]
- 24.Tombline, G. & Fishel, R. (2002) J. Biol. Chem. 277, 14417-14425. [DOI] [PubMed] [Google Scholar]
- 25.Alexandrov, A. I., Botchan, M. R. & Cozzarelli, N. R. (2002) J. Biol. Chem. 277, 44886-44897. [DOI] [PubMed] [Google Scholar]
- 26.Shirakihara, Y., Leslie, A. G., Abrahams, J. P., Walker, J. E., Ueda, T., Sekimoto, Y., Kambara, M., Saika, K., Kagawa, Y. & Yoshida, M. (1997) Structure (London) 5, 825-836. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani, M. M., Washington, M. T., Moore, K. C. & Patel, S. S. (1997) Proc. Natl. Acad. Sci. USA 94, 5012-5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esnouf, R. M. (1997) J. Mol. Graphics Model 15, 132-134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}