

Abstract
The 14α-demethylation step is critical in eukaryotic sterol biosynthesis, catalyzed by cytochrome P450 (P450) Family 51 enzymes, e.g. with lanosterol in mammals. This conserved 3-step reaction terminates in a C-C cleavage step that generates formic acid, the nature of which has been controversial. Proposed mechanisms involve roles of P450 Compound 0 (ferric peroxide anion, FeO2¯) or Compound I (perferryl oxygen, FeO3+) reacting with either the aldehyde or its hydrate, respectively. Analysis of 18O incorporation into formic acid from 18O2 provides a means of distinguishing the two mechanisms. Human P450 51A1 incorporated 88% 18O (one atom) into formic acid, consistent with a major but not exclusive FeO2¯ mechanism. Two P450 51 orthologs from amoeba and yeast showed similar results, while two orthologs from pathogenic trypanosomes showed roughly equal contributions of both mechanisms. An X-ray crystal structure of the human enzyme showed the aldehyde oxygen 3.5 Å away from the heme iron. Experiments with human P450 51A1 and H218O yielded primarily one 18O atom but 14% of the formic acid product with two 18O atoms, indicative of a minor contribution of a Compound I mechanism. LC-MS evidence for a Compound 0-derived Baeyer-Villiger reaction product (a 14α-formyl ester) was also found.
Keywords: cytochrome P450, sterol biosynthesis, enzyme mechanism, mass spectrometry, oxidation mechanism
Graphical Abstract
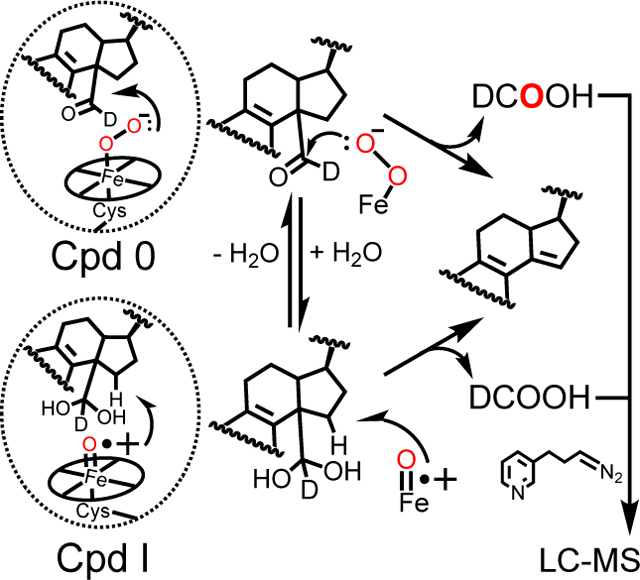
P450 51 enzymes demethylate sterols, releasing formic acid (DCOOH). Analysis of the DCOOH byproduct provides for the mechanistic discrimination of the contribution(s) of Compound 0 (Cpd 0) and Compound I (Cpd I) to catalysis. When enzyme reactions were run under 18O2 (red), >50% of the DCOOH yielded contained one atom of 18O (for all P450 51 enzymes tested), indicative of the major contribution of Cpd 0 in 24,25-dihydrolanosterol C-C cleavage.
Introduction
Cytochrome P450 (P450 or CYP) enzymes catalyze oxidation and reductions of many molecules,[1] particularly drugs, steroids, pesticides, fatty acids, fat-soluble vitamins, and chemical carcinogens.[2] In addition to the extensive involvement of P450s in intermediary steroid metabolism,[3] these enzymes also play a critical role in the biosynthesis of cholesterol in mammals and in the synthesis of similar essential sterols in microorganisms and plants – a role fulfilled by the Family 51 enzymes.[4] Accordingly, inhibition of the Family 51 P450s is a common means of treating a number of diseases caused by fungi and parasites.[5]
The general P450 mechanism (Scheme 1) includes both Compound 0 (ferric peroxide anion, FeO2¯) and its product Compound I (perferryl oxygen, FeO3+) in the catalytic cycle, and the question of which catalytic oxygen species is operative in P450 has long been a source of scientific interest.[6] Two seminal advances towards the rather ubiquitous role of the FeO3+ entity termed Compound I were made by Groves in 1978[7] and Green in 2010,[8] and many P450 oxidations can be rationalized in the context of Compound I chemistry.[9] In fact, some of the alternative explanations for hydroxylation and epoxidation reactions[10] have not held up to further mechanistic scrutiny.[11] However, there are still a number of observed P450 reactions that are difficult to explain with only Compound I mechanisms, and the literature contains some credible evidence for so-called Compound 0 (FeO2¯)-based reactions (Scheme 1).[12]
Scheme 1.
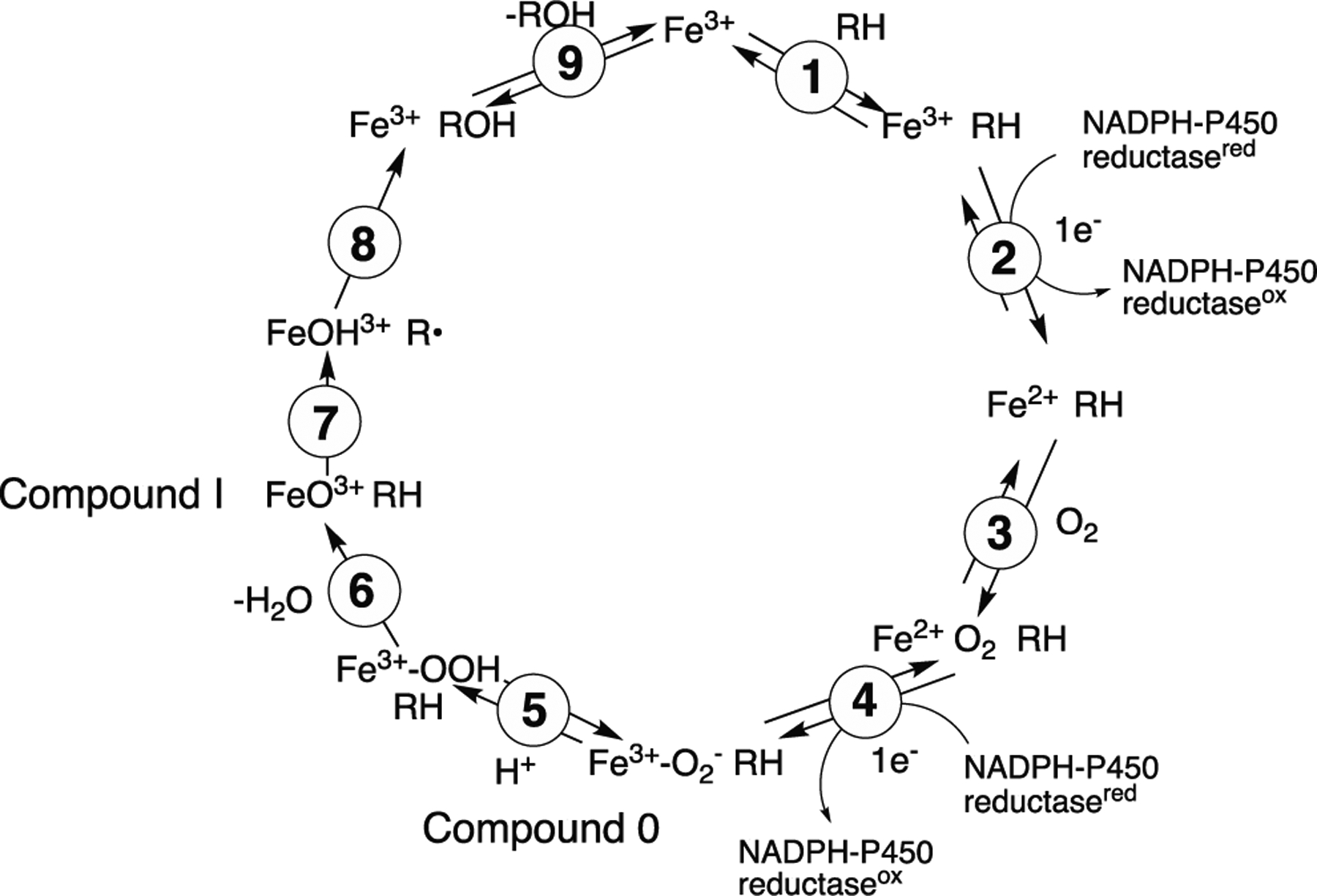
P450 catalytic cycle.
We have recently reviewed the Compound 0 vs. Compound I question within the major P450 enzymes involved in C-C bond cleavage of steroids.[9, 13] With P450 11A1 (cholesterol side chain cleavage enzyme), Compound I has been prepared and shown to be catalytically competent in the reaction.[14] The lyase reaction of P450 17A1 is controversial, with evidence supporting both Compound I and Compound 0 mechanisms.[13, 15] Our own work with P450 19A1 (steroid aromatase) favors a Compound I mechanism.[16]
A variety of approaches have been employed in attempts to distinguish Compound I and Compound 0 (including protonated Compound 0) mechanisms. The list includes the use of biomimetic models,[17] theoretical considerations,[18] oxygen surrogates,[19] site-directed mutagenesis,[20] diagnostic substrates,[21] kinetic solvent isotope effects (KSIE),[22] synthetic formation of Compound 0 or Compound I and use in a reaction,[8, 14] and 18O labeling.[16, 23] Each of these approaches has deficiencies, as well as some attractions. One of the shortcomings of almost all of these approaches is that the answers are interpreted as one-or-the-other mechanism, with no provision for considering a mixed mechanism.
P450 Family 51 enzymes all catalyze a 3-step 14α-demethylation reaction with sterols (Scheme 2). The first two steps involve a carbon hydroxylation and a formal 2-electron oxidation of the carbinol to an aldehyde, both of which are common P450 reactions, generally attributed to a Compound I (FeO3+) mechanism.[24] The third step is more unusual, involving C-C bond cleavage and release of the 14α-carbon atom (C32) as formic acid, where mechanisms involving both P450 Compound 0 and Compound I as the oxidant have been proposed.[12c, 18b, 20b, 25] Fischer et al.[12c] proposed a Compound 0 mechanism involving a Baeyer-Villiger intermediate. Shyadehi et al.[25a] later proposed a Compound 0 mechanism for a yeast (Candida albicans) P450 51 enzyme but without a Baeyer-Villiger rearrangement. At least one theoretical study has been published in favor of a Compound 0 mechanism[25b] but a more recent one is strongly in favor of a Compound I mechanism.[18b]
Scheme 2.
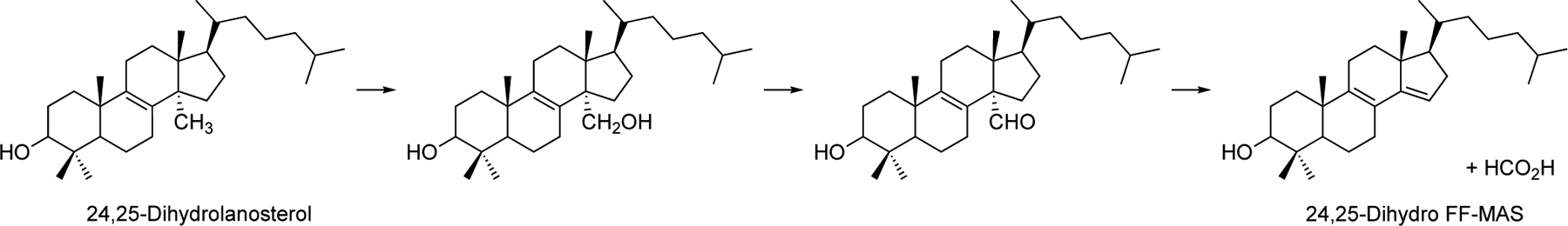
Oxidation of 24,25-dihydrolanosterol to 24,25-dihydro-4.4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol (24,25-dihydro FF-MAS) and formic acid.
On the basis of some mutagenesis and structural information, we proposed that human P450 51A1 uses a Compound I mechanism in catalysis.[20b] A major tenet of the Shyadehi et al.[25a] conclusion about the role of Compound 0 is work on the incorporation of 18O from 18O2 into formic acid in an incubation of a 14α-formyl analogue (Δ7) of 24,25-dihydrolanosterol. However, there are a number of caveats about the results of Shyadehi et al.,[25a] including the utilization of a crude yeast P450 51 microsomal preparation with low enzymatic activity (rate of 0.25 nmol product formed min−1 (nmol enzyme)−1), the use of an artificial Δ7 sterol instead of the natural Δ8 isomer, and analysis using only low resolution MS (which cannot distinguish enzymatically produced DCO2H from the 13C component of HCO2H (H13CO2H)). Although only 62% incorporation of 18O (from 18O2) into formic acid was reported (and only 5–7% of the total formic acid was derived from the substrate), the authors concluded that an exclusive Compound 0 mechanism was operative.[25a]
In considering the significance of this 18O2 incorporation study to the catalytic mechanism of P450 51 enzymes, we synthesized the requisite deuterated 14α-aldehyde derivative of (24,25-dihydro) lanosterol (14α-CDO dihydrolanosterol) and utilized it in 18O incorporation studies using (five) different purified Family 51 P450 enzymes, employed a sensitive derivatization procedure with a formate ester suitable for UPLC-electrospray MS analysis of formic acid, and analyzed the products with HRMS to resolve issues of 13C contribution that we had developed in a similar study with P450 19A1.[16] We also performed complementary labeling studies with H218O, labeling the carbonyl atom of the aldehyde substrate and analyzing the formic acid for 18O content. We conclude that P450 51 enzymes catalyze the last step of sterol 14α-demethylation reactions using both Compound I and Compound 0 mechanisms, the latter also including a Baeyer-Villiger intermediate. Thus, an individual P450 enzyme can catalyze a single reaction via multiple chemical mechanisms.
Results and Discussion
Strategy: 18O Labeling.
The approach to discerning the role of Compound I vs. Compound 0 is shown in Scheme 3. A Compound 0 mechanism incorporates one 18O atom from 18O2 into the product formic acid, and a Compound I mechanism does not. The background level of formic acid in the laboratory environment is problematic for these measurements, necessitating (i) the use of substrate with a deuterated formyl group (DC=O), (ii) chemical measures to reduce the contamination of formic acid in solvents, and (iii) the use of high resolution mass spctrometery (HRMS) (resolution > 56000) for analysis. HRMS can readily distinguish between (derivatized) H13CO2H (m/z 167.0896) and D12CO2H (m/z 167.0925),[16] which is necessary because of the natural abundance of 13C in the endogenous formic acid and its derivatives. HRMS traces of the esters of H13CO2H and D12CO2H in a typical experiment are shown in Figures S3, S4, and S6.
Scheme 3.
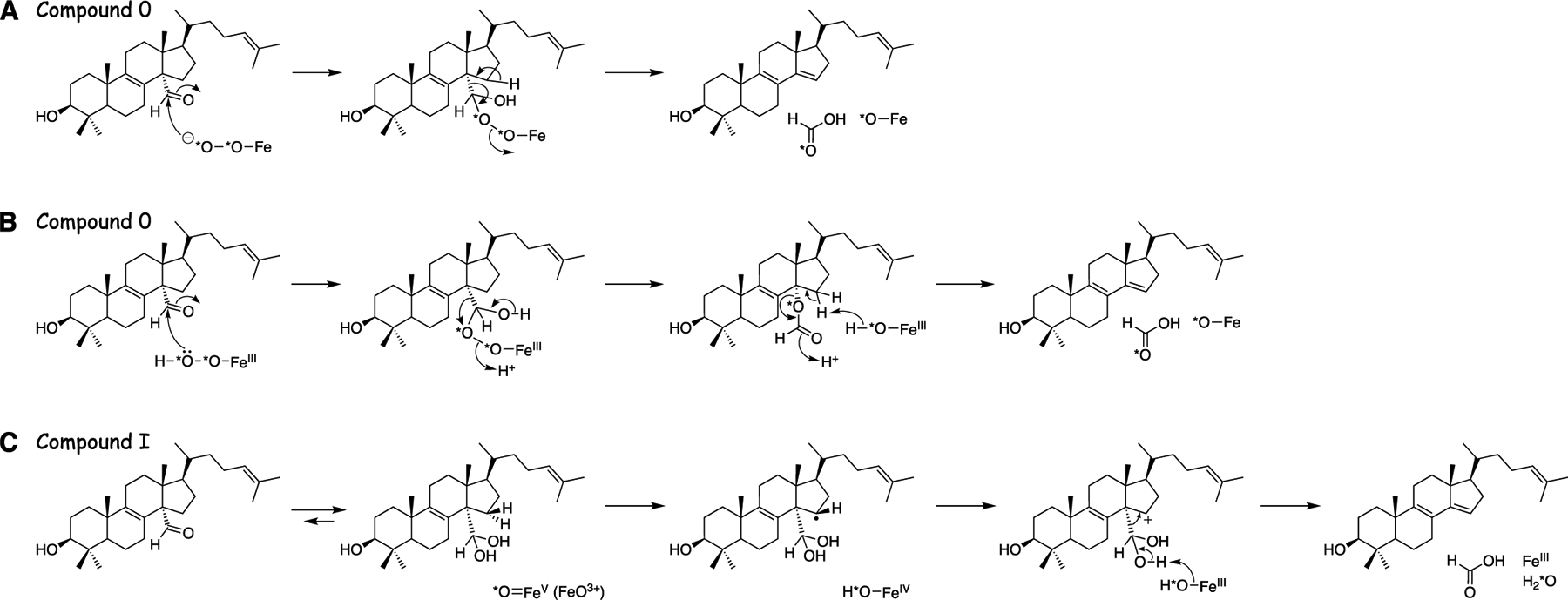
Three proposed mechanisms of oxidation of 14α-CHO dihydrolanosterol to 24,25-dihydro FF-MAS and formic acid.[12c, 18b, 20b, 25] (A) Compound 0. (B) Compound 0 plus Baeyer-Villiger rearrangement. (C) Compound I.
The synthesis of deuterated 14α-formyl 24,25-dihydrolanosterol (14α-CDO dihydrolanosterol) was based on work of Morisaki and associates,[26] which began with commercial 7-dehydrocholesterol and generated the protiated product in 14 steps to give the desired 14α-aldehyde.[27] The partially deuterated aldehyde was generated in three more steps by sequential Dess-Martin periodinane oxidation,[28] reduction with NaBD4, Dess-Martin periodinane oxidation, and hydrolytic cleavage of the 3-O-acetyl group (Figure S1). The deuterium content in the product was ~75% (not 50%), presumably due to a kinetic isotope effect in the Dess-Martin oxidation. The lack of complete substitution is not a requirement in the experimental protocol, in that the formate ester derived from the protium is not used in the mechanistic analysis (i.e., it is identical to the ester derived from endogenous formate contaminant (HCO2H)).
In the course of our studies, we also made several changes to improve the extraction and derivatization of formic acid since our earlier report.[16] The major changes (Figure S2) were (1) the use of tert-butyl methyl ether to extract formic acid instead of CH2Cl2, (2) omitting the MgSO4 drying step, and (3) the inclusion of 10% CH3OH (v/v) in the diazotization step.[29] Extraction of formic acid from the aqueous reaction mixture was found to be inefficient with CH2Cl2. While drying the organic extract with MgSO4 was performed previously based on the premise that the derivatization reaction was sensitive to H2O, the derivatization of acid with the diazo reagent was shown to be improved by the addition of both H2O and CH3OH at 10% (v/v). The drying step routinely reduced formic acid recovery, so the step was omitted. Finally, inclusion of CH3OH (10%, v/v) in the derivatization mixture was found to increase the yield of formic acid recovered as the pyridyl ester, and accordingly this was subsequently included in all derivatizations. Collectively these modifications increased the recovery by three orders of magnitude.
The sensitive analysis of DCO2H to discriminate the involvement of Compound 0 and Compound I requires nearly complete exchange of the endogenous environment with 18O2. To monitor our reaction vessels for leaks (and ensure the accuracy of our 18O labeling results) we included in each reaction mixture a control incubation of P450 17A1 and progesterone, done in the absence of cytochrome b5 for nearly exclusive formation of its 17α-hydroxy product. Because the environment is replaced with 18O2 and the P450 51 system is mixed with NADPH to start the reaction, the P450 17A1 control reaction is also initiated. The incorporation of 18O into both progesterone to yield 17α−18OH-progesterone by P450 17A1 and DCO2H by P450 51A1 is monitored simultaneously by LC-MS. The P450 17A1 18O incorporation results report on the actual 18O2 content in each reaction vessel and act as a normalization factor for the P450 51 labeling results, allowing us to assess the exact contribution of each mechanism to dihydrolanosterol deformylation. Our previous work with P450 19A1 established minimal (< 6%) DCO2H oxygen exchange with the medium under these conditions.[16]
The 18O labeling approach is limited in its scope, in that only some reactions such as deformylations are amenable. With an α-ketol substrate (α-hydroxyketone), the results are not unambiguous for the interpretation, e.g., P450 17A1 lyase.[9, 13] This approach cannot be applied to many P450 reactions, but it is realistic for the last reaction steps of P450s 19A1 and 51. The approach is technically challenging, due to the need to synthesize deuterated aldehydes (because of background formic acid)[23b] and the requirement to generate an 18O2 atmosphere. However, it is an approach that, when carefully applied, can identify a mixed mechanism, as observed here.
X-ray Crystallography of Human P450 51A1.
The first two oxidations in the demethylation of lanosterol (hydroxylation of C32 and oxidation to an aldehyde) are understood to be mediated by a P450 Compound I mechanism.[24] We had previously reported that the overall three-step reaction was highly kinetically processive, in that lanosterol and reaction intermediates are bound tightly to the enzyme and driven to final product formation.[27] Accordingly, we investigated whether the binding mode of the aldehyde (for C-C cleavage) was similar to the binding mode of lanosterol (for hydroxylation) and crystallized the enzyme in complex with our synthetic aldehyde intermediate for comparison.
Human P450 51A1 was crystallized and the 2.25 Å resolution structure was refined. The overall binding mode of the aldehyde is essentially the same as the binding of lanosterol (Fig. 1A). The protein conformation is also essentially the same (with a closed substrate entry and only minor fluctuations in the positions of some loops, GH in particular, which is known to be the most flexible region in P450 51 structures) (Figure 1B,C). This is in line with the finding that the three-step reaction of sterol 14α-demethylation is a highly kinetically processive process.[27]
Figure 1.
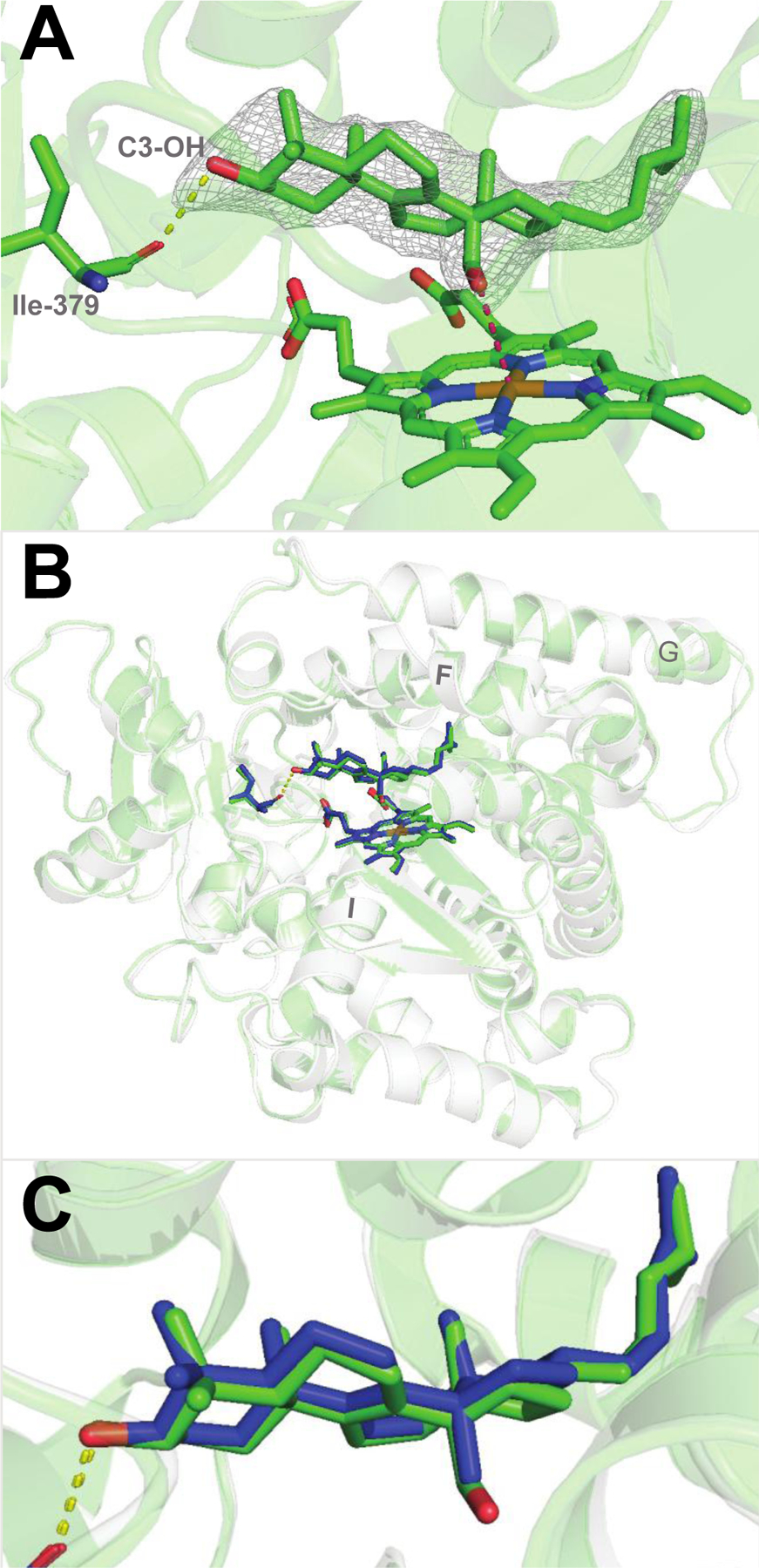
Substrate-bound human P450 51A1. (A) Binding mode of the 14α-aldehyde reaction intermediate inside the enzyme active site (PDB 8SS0, 2.25 Å.). The 2Fo-Fc electron density map within 1.6 Å of the sterol atoms is shown as gray mesh and contoured at 1.5σ. The H-bond between the C3-OH of the sterol molecule and the main chain oxygen of Ile-379 is depicted as yellow dashes. The distance between the aldehyde oxygen and the heme iron is 3.5Å (pink dashes). (B) Superimposition with the lanosterol-bound structure (PDB 6UEZ): the carbon atoms of lanosterol are colored in blue, the ribbon is transparent light-gray, and the rmsd of all Cα-atoms is 0.47 Å. (C) Enlarged view of the superimposed molecules of lanosterol and the 14α-aldehyde intermediate of 24,25-dihydrolanosterol.
18O2 Incorporation into Formic Acid Reveals a Dominant Compound 0 Mechanism.
With an optimized protocol for the extraction and derivatization of formic acid in-hand (see Strategy section), we began by assaying the 24,25-dihydrolanosterol C-C cleavage reaction catalyzed by human P450 51A1. When an incubation of the enzyme and 14α-CDO dihydrolanosterol was performed under an atmosphere of 18O2, >80% of the recovered pyridyl ester of the formate product (DCO2H) contained one atom (m/z 169.0968) of the isotopic label (Figure 2, S3), with the remaining < 20% containing zero atoms (m/z 167.0925). The concomitant P450 17A1 control reaction (included in the P450 51A1 reaction mixture) incorporated >92% of the label (one atom) in the 17α-hydroxylation of progesterone, indicating high fidelity in the replacement of the endogenous atmosphere with 18O2 (see Strategy section, vide supra). When normalized to the control reaction (i.e., P450 17A1), the deformylation of dihydrolanosterol by human P450 51A1 yielded primarily the recovery of one atom of 18O2 in DCO2H (88 ± 3%, n= 5, Figure 2), suggestive of a dominant but not exclusive contribution of the ferric peroxide nucleophile (Compound 0) in facilitating 24,25-dihydrolanosterol C-C cleavage. Because our control experiments established that the chromatographic peaks of interest (Figure 2) are not formed non-enzymatically (e.g., in the absence of NADPH) we were confident in this result, which was reproducible in both technical and biological replicates.
Figure 2.
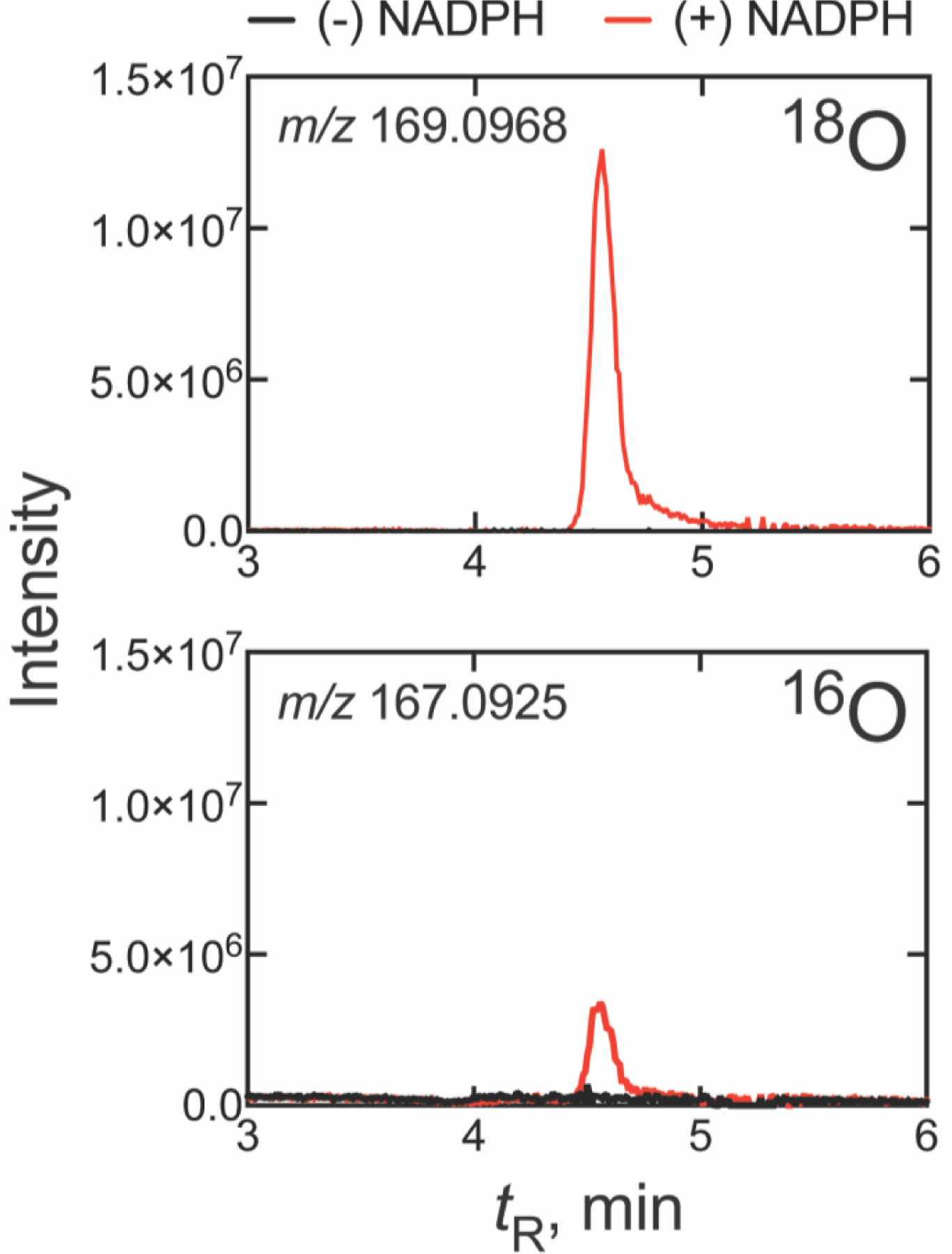
UPLC-HRMS analysis of formic acid formed from 14α-CDO dihydrolanosterol by human P450 51A1 in an atmosphere of 18O2. The two frames show the 18O (m/z 169.0968) and 16O (m/z 167.0925) channels. All m/z values are calculated for the derivatized pyridyl formate esters. (The corresponding raw chromatographic traces are presented in Figure S3).
In light of the dominance of Compound 0 observed in incubations with the human enzyme, we examined whether this extends to other well-studied P450 51 orthologs. Interestingly, we found that P450 51 enzymes from Naegleria fowleri and Candida albicans (Figure 3) showed nearly identical results to the human ortholog, where incubations with 14α-CDO dihydrolanosterol yielded DCO2H that incorporated >82% of the label (one atom), on average, from 18O2 (87 ± 2 and 82 ± 3%, respectively, after normalization, n = 5) (Table 1, Figure 4). In these experiments, the mean label incorporation in the control reaction (P450 17A1) was >91% for both reactions, again ensuring high technical accuracy. As we found with human P450 51A1, the 18O labeling results with P450 51 enzymes from N. fowleri and C. albicans were also suggestive of Compound 0 dominance in 24,25-dihydrolanosterol C-C cleavage.
Figure 3.
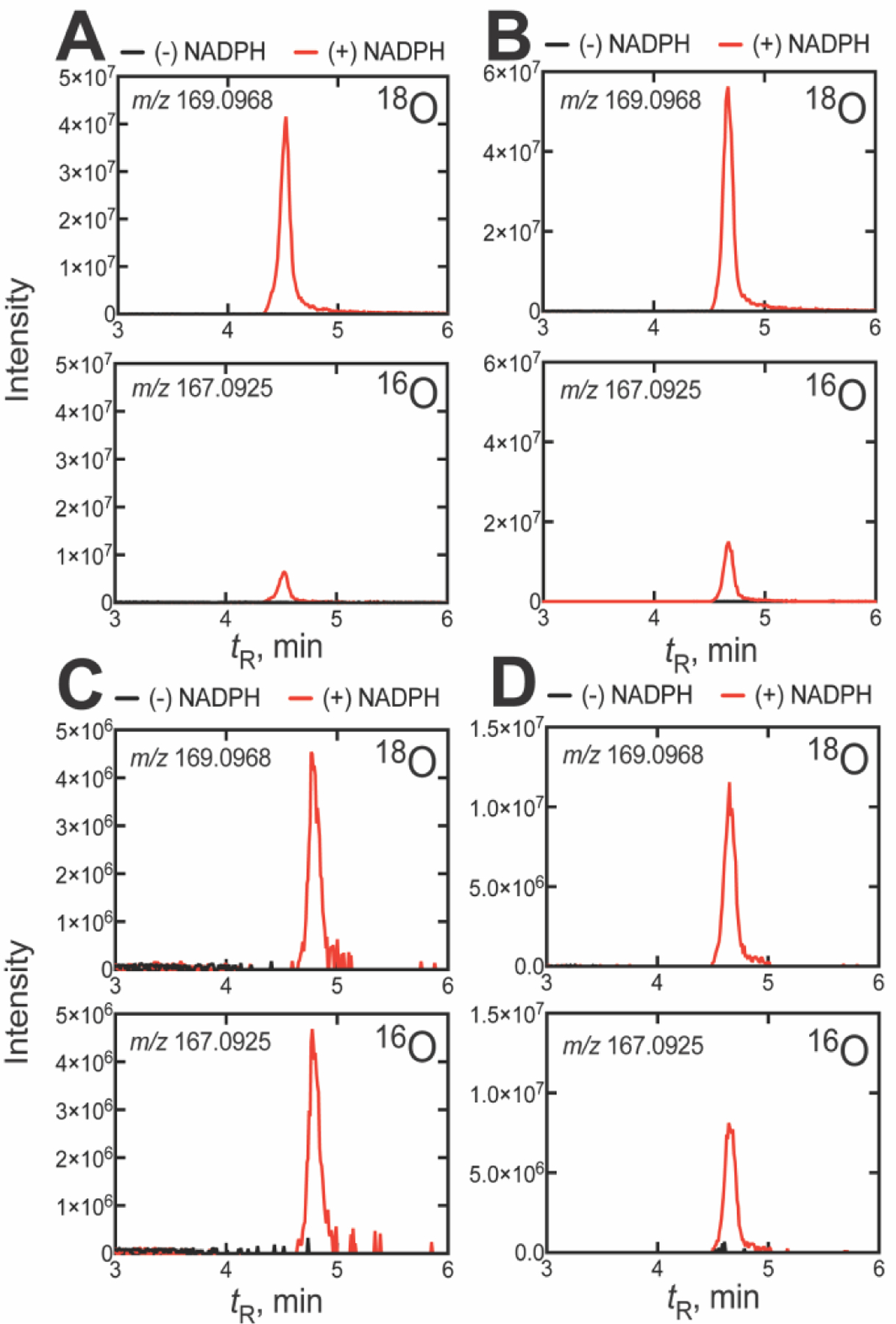
UPLC-HRMS analysis of formic acid formed from 14α-CDO dihydrolanosterol by other P450 51A enzymes in an atmosphere of 18O2. The two frames show the 18O (m/z 169.0968) and 16O (m/z 167.0925) channels for the formic acid derivative formed from the P450 51A enzymes of (A) N. fowleri, (B) C. albicans, (C) T. brucei, and (D) T. cruzi. All m/z values are calculated for the derivatized pyridyl formate esters. (The corresponding raw chromatographic traces for panel C are presented in Figure S4).
Table 1.
Summary of 18O2 incorporation results
| P450 51 enzyme | DCO2H (% 18O) | 17-OH progesterone (% 18O) | % Compound 0 |
|---|---|---|---|
| Human | 82.6 ± 2.0 | 93.6 ± 1.5 | 88.3 ± 3.2 |
| N. fowleri | 79.4 ± 4.0 | 91.4 ± 3.0 | 86.9 ± 1.8 |
| C. albicans | 76.8 ± 3.9 | 93.8 ± 1.2 | 81.9 ± 3.4 |
| T. brucei | 48.0 ± 0.8 | 94.8 ± 0.2 | 50.6 ± 0.9 |
| T. cruzi | 55.3 ± 1.8 | 93.2 ± 0.3 | 59.3 ± 2.0 |
Figure 4.
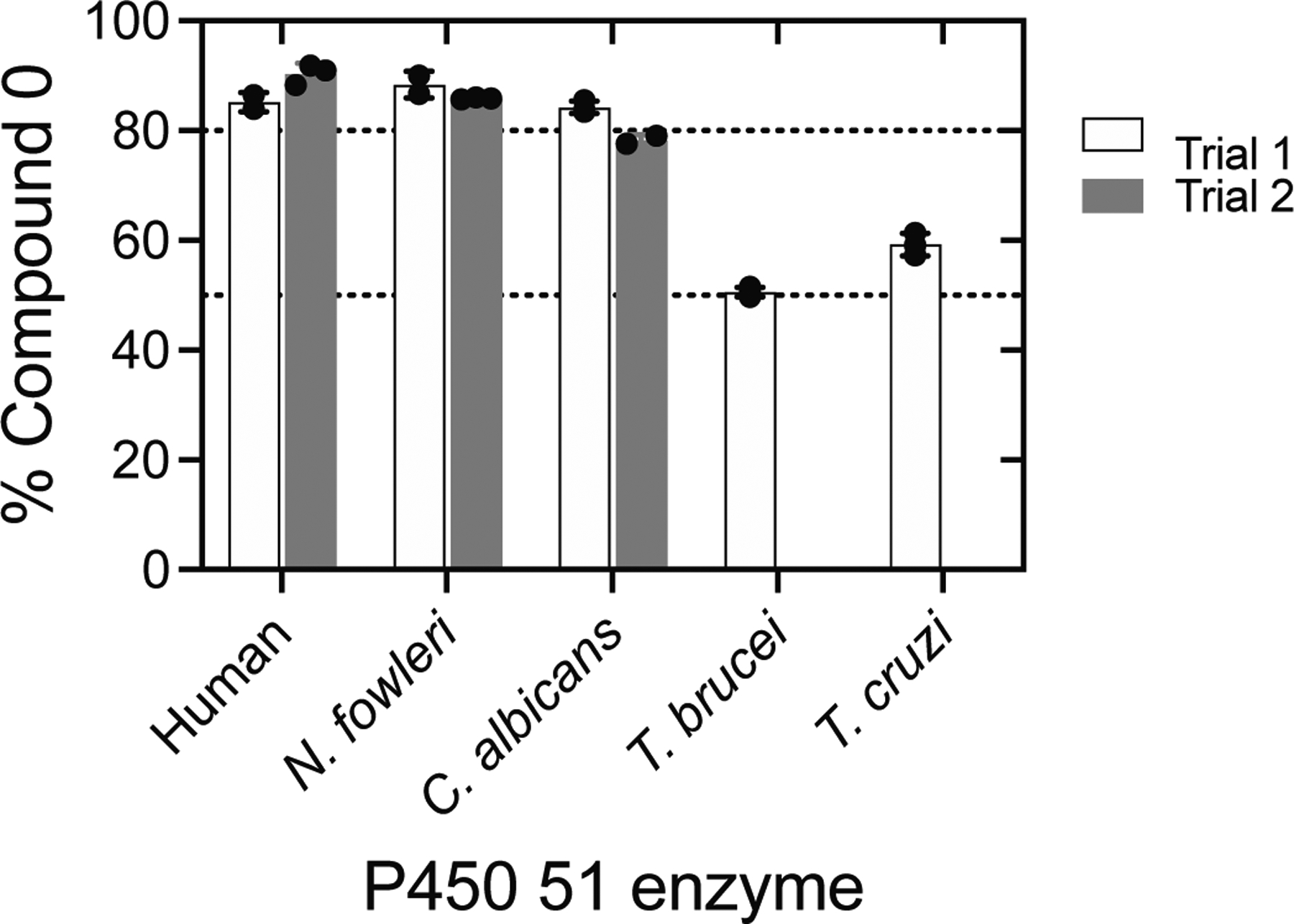
Summary of 18O incorporation from 18O2 into formic acid with five P450 51A enzymes. Results from Figures 2 and 3. All values were normalized for the content of 18O (from 18O2) incorporated into 17α-OH progesterone by P450 17A1. The individual points indicate the results of individual incubations (3), with the means ± SD. Separate trials were run on different days, each with a set of three more enzyme reactions. The stippled lines are set at 50 and 80% for reference.
We then applied the analysis to P450 51 enzymes from Trypanosoma cruzi and Trypanosoma brucei (Figure 3). As lanosterol is not a natural substrate of either enzyme, both orthologs show low rates of 24,25-dihydrolanosterol 14α-demethylation. Accordingly, incubations of trypanosomal P450 51 enzymes were performed with an elevated P450 concentration (1 μM), an extended reaction time (60 min), and modified analysis conditions, all to increase sensitivity (see Experimental Procedures). When we performed the incubations of these orthologs with 14α-CDO dihydrolanosterol, the DCO2H recovered showed lower label incorporation from 18O2 than the other P450 51 enzymes tested (59 ± 2% and 51 ± 1%, (Figure S4), respectively, after normalization, n = 3) (Table 1, Figure 4). Despite this (significant and reproducible) difference, the mean label incorporation in the control reaction (progesterone 17α-hydroxylation by P450 17A1) was >93% for both reactions, and residual substrate (progesterone) was detected after reaction termination, indicating that reduced label incorporation (into DCO2H) could not be attributed to loss of 18O2 in the reaction (e.g., due to gas leaks). For both trypanosomal P450 51 orthologs tested, the 18O labeling results were suggestive of roughly equal contributions of both Compound 0 and Compound I mechanisms in dihydrolanosterol C-C cleavage.
H218O Labeling with Human P450 51A1 –a Minor Contribution of Compound I.
While the incorporation of 18O (from 18O2) into DCO2H (~85%) is unambiguous evidence for the contribution of a P450 Compound 0 mechanism, the lack of complete incorporation (~15%) is only indirect evidence of the role of Compound I. To directly measure the contribution of Compound I to dihydrolanosterol C-C cleavage by human P450 51A1, we employed an alternative approach to the 18O2 experiment by running the enzymatic incubation in H218O. In this experiment, the starting aldehyde substrate is equilibrated with the medium (H218O) prior to the enzymatic incubation (to yield 14α-CD18O dihydrolanosterol, with the carbonyl oxygen exchanged). The action of P450 Compound 0 (in an 16O2 atmosphere) yields one atom of 16O incorporated into DCO2H upon C-C cleavage and recovers the 18O label from the (aldehyde) starting substrate (Scheme 3A). Conversely, P450 Compound I acts on the substrate only after hydration of the 14α-CD18O aldehyde to 14α-CD(18OH)2 (i.e., gem-diol). Upon C-C cleavage, both atoms of 18O present in the gem-diol (derived from the medium) will be recovered in DCO2H. Accordingly, the H218O experiment discriminates between the relative contributions of Compound 0 and Compound I on the basis of incorporation of either one or two atoms of label into DCO2H, respectively (Scheme 3).
When 14α-CD18O dihydrolanosterol was prepared (Figure S5) and incubated with human P450 51A1 (in H218O), the major DCO2H product contained just one atom of 18O label (from the starting substrate) (86 ± 1%, m/z 169.0968 (for the ester derivative), with the minor product incorporating an additional atom from the solvent (H218O, 14 ± 1%, m/z 171.1010) (Figures 5, S6). The finding that the Compound I product (two 18O atoms recovered in DCO2H) was a minor product in the H218O experiment (~15%) is in excellent agreement with our finding that the Compound 0 product (one atom of 18O (from 18O2) recovered in DCO2H) was the major product (~85%, Figure 2, Table 1) in the 18O2 experiment. Together, the 18O2 and H218O labeling experiments track the relative contributions of Compound 0 and Compound I in the mechanism of dihydrolanosterol C-C cleavage by human P450 51A1, independently demonstrating that the enzyme predominantly (≥ 85%) incorporates one atom of O2 from the atmosphere in DCO2H to achieve deformylation of 14α-CDO dihydrolanosterol, i.e. the Compound 0 mechanism predominates but the Compound I mechanism is also operative.
Figure 5.
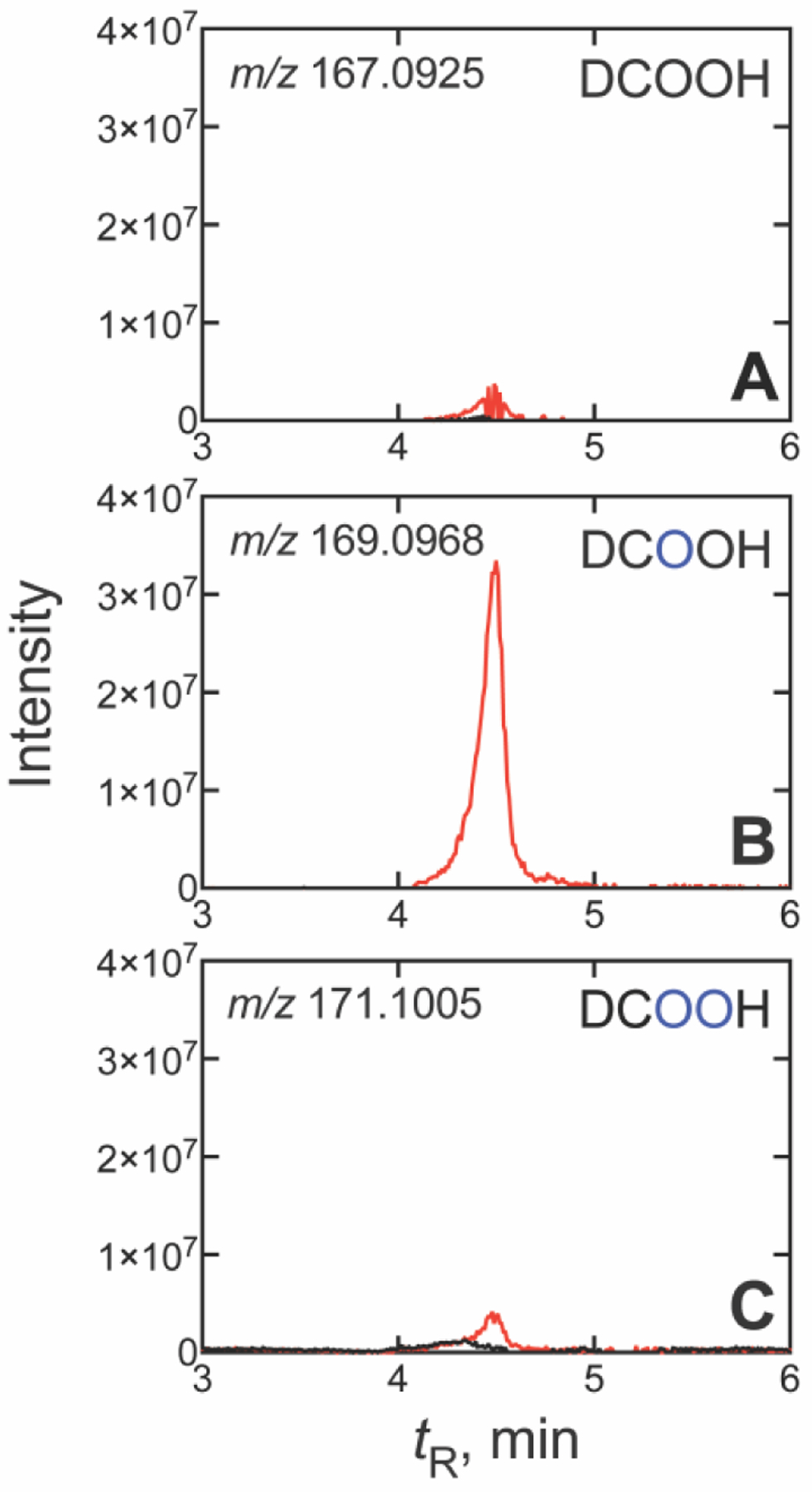
UPLC-HRMS analysis of formic acid formed from 14α-CD18O dihydrolanosterol by human P450 51A1 in H218O. Traces are shown for (A) no 18O (only 16O, m/z 167.0925), (B) one 18O atom (m/z 169.0968), and (C) two 18O atoms (m/z 171.1010). Isotopically labelled oxygen atoms (18O) are indicated in blue, and all m/z values are calculated for the derivatized pyridyl formate esters. (The corresponding raw chromatographic traces are presented in Figure S6).
Given the consistency of results for a dominant (~85%) role of Compound 0 with a minor (15%) contribution of Compound I (Figures 4 and 5, Table 1) in human P450 51A1, we questioned if structural elements can be identified that govern the mechanistic balance.[30] We had previously mutated salt-bridged residues (Asp-231 and His-314) to alanine in P450 51A1 and analyzed the effects of the resulting double-mutant on sterol demethylation.[20b] These results had been interpreted to support a Compound I mechanism due to a significant (~160-fold) reduction in aldehyde demethylation (providing evidence for a requirement for solvent accessible protons, i.e. in the formation of Compound I).
When we incubated the human P450 51A1 double mutant (D213A/H314A, 1 μM) with 14α-CDO dihydrolanosterol in more H218O experiments, we were unable to reproducibly recover significant amounts of DCOOH from the reaction (as a pyridyl formate ester), likely due to the low rates of sterol demethylation (even from 60-min incubations). We then incubated the single-mutant P450 51A1 D213A, which had shown higher reaction rates (roughly ¼ that of the WT enzyme, starting with lanosterol [31]) with 14α-CDO dihydrolanosterol (50 μM) we observed that 8.1 ± 0.2% (n = 3) of the 18O-labeled DCOOH product was derived from Compound I (2 18O atoms incorporated), representing a reduction of the Compound I product by ~½ compared to the WT enzyme (i.e.,14% (Figure 5) to 8%). However, a ~50% reduction in the Compound I contribution to sterol deformylation does not account for the >4-fold reduction in enzymatic activity observed for the enzyme, [31] and it is likely that the catalytic consequence of this mutation may be the result of unknown structural changes in the enzyme.
Oxygen Surrogates were not Catalytically Competent with P450 51A1 Deformylation.
Historically, one approach commonly employed to discern the roles of Compound 0 and Compound I in P450 reactions has been the use of chemical reagents acting as “oxygen surrogates” (iodosylbenzene (PhI=O), H2O2, cumene hydroperoxide (CuOOH)), in efforts to gauge the dependence of catalysis on either species.[12a, 19] In theory, a reaction that is mediated via a ferric peroxide (Compound 0) mechanism might be supported by the addition of H2O2 (or CuOOH) to ferric P450. Similarly, a reaction mediated by the oxyferryl species Compound I might be supported by the addition of the single-oxygen donor PhI=O to the enzyme. In these experiments, reactions supported by single oxygen donors can rule out a Compound 0 pathway, but reactions supported by 2-oxygen donors (hydroperoxides) cannot definitively rule out a Compound I pathway (i.e., Compound I forms naturally from Compound 0, Scheme 1). (All of these reagents are often (eventually) destructive to the P450 heme, however.)
We examined whether such oxygen surrogates could be used to gauge the dependence of P450 51 catalysis on Compound I. We tested the ability of PhI=O, CuOOH, and H2O2 to support demethylation of 14α-CDO dihydrolanosterol by human P450 51A1. We did not detect any product formation from any of these surrogates at any time point (≤ 5 min) tested but detected typical enzymatic activity (> 20 nmol product min−1 (nmol P450)−1) from the control reaction (supported by NADPH), as expected.[27] Interestingly, we did observe some product formation with both PhI=O and H2O2 (but not CuOOH) when the reaction mixture was reconstituted with NADPH-P450 reductase, although the activity was still < 1% of the control (NADPH) reaction in both cases. Overall, experiments with oxygen surrogates were largely unable to support P450 51A1 deformylation and did not provide a means of addressing the P450 Compound 0 vs. Compound I question.
Kinetic Solvent Isotope Effect (KSIE) Analyses are Unreliable.
Previous cases for discerning the involvement of Compound I vs. Compound 0 in P450 catalysis have also been proposed through the analysis of KSIE data.[22, 32] In the general P450 catalytic cycle (Scheme 1), protonation of Compound 0 (step 5) to yield Compound I ultimately relies on the availability of protons (from the solvent). Substitution of the solvent (H2O) with D2O may – in theory – slow the formation of Compound I and either (i) increase the rate of a Compound 0-driven process (inverse KSIE), or (ii) decrease the rate of a Compound I-driven process (normal KSIE).
While curious as to whether such type of analysis would support our 18O labeling results (e.g. the dominant but not exclulsive role of P450 Compound 0), we also recognize that the use of D2O in enzyme incubations is controversial due to the accompanying (and unknown) structural changes arising from the exchange of labile backbone protons in enzymes,[33] in that a protein prepared in D2O may be chemically, structurally, and functionally distinct from the native enzyme (in H2O). Accordingly, results obtained utilizing each solvent system may not be directly comparable, as is done in the KSIE experiment.
For the sake of comparison to existing studies addressing the P450 Compound 0 vs. Compound I question, we conducted the experiment with human P450 51A1 (and 14α-CDO dihydrolanosterol). Our results indicated a decrease in enzymatic activity from 72 ± 7 min−1 (in 100% H2O, v/v) to 42 ± 6 min−1 (in 80% D2O, v/v) corresponding to a (normal) KSIE (kH/kD) of 1.7 (Figure S7). A normal KSIE has been historically interpreted as a dominant contribution of P450 Compound I, although our result is not a significant KSIE (< 2) and we are inherently skeptical of the use of KSIE data to assess protein function due to the unknown enzyme structural (and potentially functional) changes that occur with deuterium exchange, as noted earlier by others.[33]
A P450 51A1 Baeyer-Villiger Intermediate.
Baeyer-Villiger rearrangements are well-known in chemistry, and a number of flavoproteins use 4a-hydroperoxide intermediates to catalyze such reactions,[9, 34] including mammalian flavin-containing monooxygenase (FMO) enzymes.[35] There has been limited evidence for these reactions in P450 catalysis, likely due in part to the preponderance of Compound I (FeO3+) mechanisms.[9, 24a] Mak and Swinney[22a, 36] proposed Baeyer-Villiger chemistry in the oxidation of progesterone to 17-O-acetyltestosterone by P450 17A1 in hog testicular microsomes, but we were unable to detect this compound in our own work with recombinant human P450 17A1.[15d]
In plants, Arabidopsis P450 85A2 and 85A3 oxidize a 6-keto steroid to brassinolide, a secosteroid with a 7-membered B-ring lactone, suggestive of Baeyer-Villiger chemistry.[37] However, even this transformation can be rationalized in the context of a FeO3+ reaction (Compound I) in the absence of 18O labeling studies.[9] Ortiz de Montellano and associates have identified several oxidations of cholesterol by Mycobacterium tuberculosis P450 125A1, including five deformylation products derived from the C26-aldehyde.[38] Several of these can be rationalized by Compound 0 (FeO2¯) chemistry, and one (M2) was proposed to be generated by 1-electron oxidation of the C25 radical generated by homolytic decomposition of a peroxyhemiacetal intermediate, followed by trapping of a C25 carbocation by the released formic acid.[38] However, the authors indicate that a Baeyer-Villiger rearrangement could also explain their results. Another possibility of a P450 Baeyer-Villiger reaction is that of Sordaria araneosa SdnB.[39]
The best case for Baeyer-Villiger P450 chemistry may be the study of Fischer et al.[12c] on lanosterol 14α-demethylation in rat liver microsomes. While it is generally accepted that the P450 51 reaction culminates in cleavage of the C14-C32 bond of 14α-CHO lanosterol, their group identified a competing reaction facilitated by Baeyer-Villiger type chemistry (Scheme 3C) that yielded a third oxidative intermediate in the reaction series.[12c] The intermediate (a 14α-formyl derivative of lanosterol) was initially identified by radiochromatography as an NADPH-dependent product of lanosterol metabolism in rat liver microsomes, based on chromatography as an early shoulder on the substrate peak when separated using reversed-phase (C18) HPLC. The group found that the chromatographic separation (of substrate and intermediate) was substantially improved when performed with a silica column (normal phase HPLC), and after isolation and some spectroscopic analyses assigned the molecule as 14α-formyloxy-lanost-8-en-3β-ol. To our knowledge, this remains the only report of an additional intermediate of lanosterol metabolism, in that further kinetic establishment of the intermediacy of the Baeyer-Villiger compound and catalytic competence has not been described. Our recent analysis of the kinetics of dihydrolanosterol 14α-demethylation by human P450 51A1[27] did not include this as an intermediate. The compound is reportedly formed in lower abundance relative to the final product[12c] and may have been below the limit of detection in our previous analyses. Additionally, it might not be expected to survive the acidic conditions used in quenching with HCl in single-turnover analysis.[27]
As our 18O labelling data clearly indicated the dominance of P450 Compound 0 (Figure 4), we were interested as to whether the proposed 14α-formyl lanosterol derivative (Baeyer-Villiger intermediate) was also a product under our experimental conditions, namely, utilizing a purified recombinant enzyme system (devoid of esterases in microsomes, which could degrade such an ester intermediate) and on-line LC-MS (which was not available to Fischer et al.[12c]). We performed a 14α-CDO dihydrolanosterol deformylation assay under steady-state conditions[27] and subjected the products of the reaction to reversed-phase (C18) HPLC coupled to electrospray ionization (ESI) MS analysis. Our analysis revealed an NADPH-dependent chromatographic peak (tR 3.05 min) eluting as an early shoulder on the substrate peak (tR 3.51 min), corresponding to the chromatographic migration of the putative Baeyer-Villiger intermediate in the earlier report (Figures 6, S8).[12c] HRMS analysis of this product was also consistent with that of a Baeyer-Villiger intermediate, which was characterized by the fragment ions of m/z 413.3783 due to the in-source loss of DCO2 from the 14α-formyl group and a hydrogen atom from C15, and m/z 395.3672 due to the additional loss of H2O from the 3β-hydroxyl in the ESI source. All detected m/z values were within 5 ppm of theoretical values, giving high confidence in the assignment.
Figure 6. Reversed phase (C18) UPLC-HRMS analysis of Baeyer-Villiger intermediate.
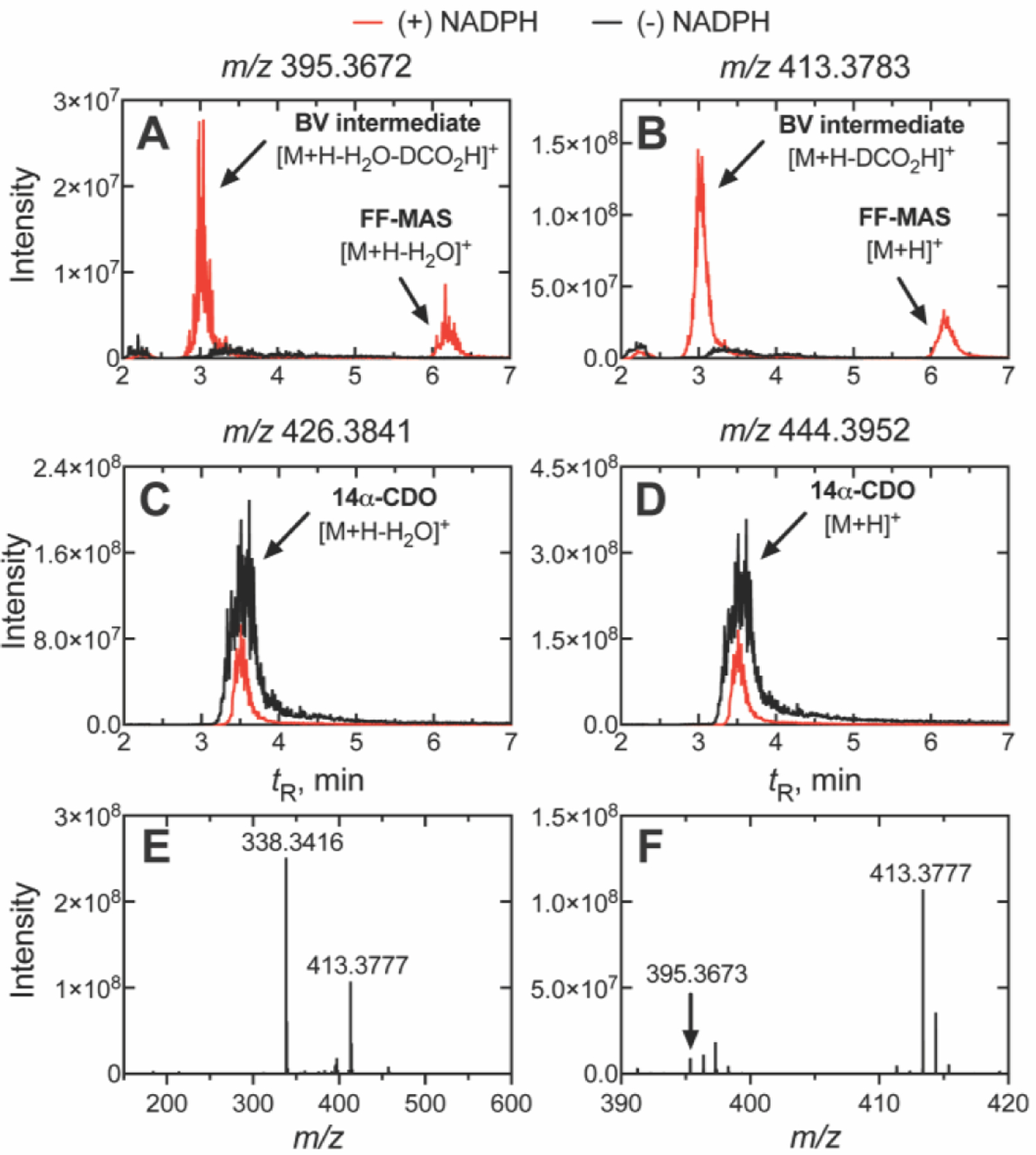
Steady-state incubations of P450 51A1 and 14α-CDO dihydrolanosterol were run for 2.5 min and the products were extracted and analyzed via (reversed phase) LC-ESI-HRMS. The extracted m/z values of (A) 395.3672 and (B) 413.3783 (utilizing a ± 5 ppm mass window) are fragment ions of the suspected Baeyer-Villiger (BV) intermediate corresponding to the [M+H-H2O-DCO2-H]+ and [M+H-DCO2-H]+ ions, respectively. Depletion of the aldehyde starting substrate (14α-CDO dihydrolanosterol) was observed with extracted m/z values of (C) 426.3841 and (D) 444.3952 corresponding to the intact [M+H]+ and [M+H-H2O]+ ions. The full mass spectrum (m/z 150–600) of the intermediate (tR 3.05 min, Panels A and B) showed the major [M+H- DCO2-H]+ fragment ion (m/z 413.3777) of the intermediate (Panel E), while the minor [M+H-H2O- DCO2-H]+ fragment ion (m/z 395.3673) was observable when the trace was magnified (m/z 390–420, Panel F). The Baeyer-Villiger (BV) intermediate (tR 3.05 min) eluted as an early shoulder of the substrate peak (14α-CDO dihydrolanosterol, tR 3.51 min) while FF-MAS eluted later (tR 6.17 min). similar to the results reported by Fischer et al.[12c] (The corresponding raw chromatographic traces are presented in Figure S8). (The ion m/z 338.3416 in Panel E is from a closely eluting peak (ΔtR 0.1 min) and could not be identified; it is not a fragment of m/z 413.3777).
We then applied the sample to a normal phase HPLC system coupled to atmospheric pressure chemical ionization (APCI) MS, adapting the conditions of Fischer et. al.[12c] to determine if the chromatographic retention of our suspected intermediate–like that described in their report–would subsequently elute later than the substrate and product. We observed a chromatographic peak (Figure S9; tR 5.38 min) with ions of m/z 395.3672 and 413.3783 now eluting significantly after both the final product (dihydro FF-MAS, tR 1.41 min) and substrate (14α-CDO dihydrolanosterol, tR 2.22 min). The formation of this peak was NADPH-dependent, reproducible, and appeared to increase with increasing reaction time.
We believe that this intermediate compound is the same one observed by Fischer et al.,[12c] who reported IR and 1H NMR spectra consistent with the proposed structure (i.e., IR 1717 cm−1, NMR δ 7.86) but were also unable to obtain mass spectra of the intact molecule. Despite numerous attempts utilizing both normal and reversed phase chromatography systems coupled to either APCI or ESI HRMS, under various ionization conditions, we were also unable to obtain the parent ion. Fischer et al.[12c] noted that this product (which was acid-labile) decomposed when isolated and added back to rat liver microsomes, but the roles of microsomal esterases[40] cannot be dismissed. Based on the chromatographic behavior on both normal and reversed phase columns (in comparison to the original report) and HRMS analysis of the molecule, we assign the product as the 14α-formyloxy-lanost-8-en-3β-ol intermediate proposed by Fischer et al.[12c]
Conclusion
Our 18O2 analyses indicate that all of the P450 51 reactions examined operate with a mixed reaction mechanism with participation of both Compound 0 (FeO2¯) and Compound I (FeO3+) mechanisms, the former accounting for ~85% of the reaction with all enzymes tested (Figure 4), with the exception of the T. cruzi and T. brucei enzymes. The ~85% values (Figure 4) are based on internal standard P450 17A1 progesterone 17α-hydroxylation, and the lack of complete incorporation is not due to leaks in the glassware or other artifacts. The contribution (14%) of the FeO3+ pathway is supported by our results with reactions done in H218O, labeling the carbonyl oxygen atom (Figure 5, Table 1). Although we provide evidence for the involvement of a Baeyer-Villiger rearrangement in the Compound 0 pathway (Figure 6), we cannot conclude what fraction of the reaction proceeds through this pathway as opposed to a direct elimination (Scheme 3B).
Supplementary Material
Acknowledgements
We thank L. Liu for the preparation of enzymes. We acknowledge Dr. M. W. Calcutt, Dr. S. Chetyrkin, and B. Hachey of the Vanderbilt University Mass Spectrometry Core for technical assistance. We also thank W. Boeglin for assistance with the normal-phase LC-MS analysis and K. Trisler for assistance in preparation of the manuscript. This study was supported in part by United States Public Health Service (USPHS) grants R01 GM118122 (to F. P. G.) and R01 GM067871 (to G. I. L.). This material is also based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1937963 (K. D. M.). We also acknowledge USPHS supplement 3R01 GM134548-03W1 for the normal phase HPLC equipment. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Institutes of Health or the National Science Foundation.
Footnotes
Supporting Information
The authors have cited references within the Supporting Information.[1–14]
References
- [1].Rendic S, Guengerich FP, Chem. Res. Toxicol 2015, 28, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ortiz de Montellano PR, Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed., Springer, New York, 2015. [Google Scholar]
- [3].Auchus RJ, Miller WL, in Cytochrome P450: Structure, Mechanism, and Biochemistry, Vol. 2, 4th ed. (Ed.: Ortiz de Montellano PR), Springer, New York, 2015, pp. 851–879. [Google Scholar]
- [4].McLean KJ, Leys D, Munro AW, in Cytochrome P450:Structure, Mechanism, and Biochemistry, 4th ed. (Ed.: Ortiz de Montellano PR), Springer, New York, 2015, pp. 261–407. [Google Scholar]
- [5].Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI, J. Biol. Chem 2015, 290, 23916–23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ullrich V, Angew. Chem., Int. Ed 1972, 11, 701–712. [DOI] [PubMed] [Google Scholar]
- [7].Groves JT, McClusky GA, White RE, Coon MJ, Biochem. Biophys. Res. Commun 1978, 81, 154–160. [DOI] [PubMed] [Google Scholar]
- [8].Rittle J, Green MT, Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- [9].Guengerich FP, Yoshimoto FK, Chem. Rev 2018, 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Newcomb M, Le Tadic-Biadatti MH, Chestney DL, Roberts ES, Hollenberg PF, J. Am. Chem. Soc 1995, 117, 12085–12091; [Google Scholar]; b) Newcomb M, Shen R, Choi S, Toy PH, Hollenberg PF, Vaz ADN, Coon MJ, J. Am. Chem. Soc 2000, 122, 2677–2686; [Google Scholar]; c) Jin S, Makris TM, Bryson TA, Sligar SG, Dawson JH, J. Am. Chem. Soc 2003, 125, 3406–3407. [DOI] [PubMed] [Google Scholar]
- [11].a) Auclair K, Hu Z, Little DM, Ortiz de Montellano PR, Groves JT, J. Am. Chem. Soc 2002, 124, 6020–6027; [DOI] [PubMed] [Google Scholar]; b) Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT, J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Vaz ADN, Roberts ES, Coon MJ, J. Am. Chem. Soc 1991, 113, 5886–5887; [Google Scholar]; b) Roberts ES, Vaz ADN, Coon MJ, Proc. Natl. Acad. Sci. USA 1991, 88, 8963–8966; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fischer RT, Trzaskos JM, Magolda RL, Ko SS, Brosz CS, Larsen B, J. Biol. Chem 1991, 266, 6124–6132. [PubMed] [Google Scholar]
- [13].Guengerich FP, Tateishi Y, McCarty KD, Med. Chem. Res 2023, 32, 1263–1677. [Google Scholar]
- [14].Davydov R, Strushkevich N, Smil D, Yantsevich A, Gilep A, Usanov S, Hoffman BM, Biochemistry 2015, 54, 7089–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Miller SL, Wright JN, Corina DL, Akhtar M, J. Chem. Soc., Chem. Commun 1991, 157–159; [Google Scholar]; b) Mak PJ, Gregory MC, Denisov IG, Sligar SG, Kincaid JR, Proc. Natl. Acad. Sci. USA 2015, 112, 15856–15861; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gregory M, Mak PJ, Sligar SG, Kincaid JR, Angew. Chem., Int. Ed 2013, 52, 5342–5345; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yoshimoto FK, Gonzalez E, Auchus RJ, Guengerich FP, J. Biol. Chem 2016, 291, 17143–17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yoshimoto FK, Guengerich FP, J. Am. Chem. Soc 2014, 136, 15016–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Cole PA, Robinson CH, J. Am. Chem. Soc 1988, 110, 1284–1285; [Google Scholar]; b) Watanabe Y, Ishimura Y, J. Am. Chem. Soc 1989, 111, 8047–8049. [Google Scholar]
- [18].a) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W, Chem. Rev 2010, 110, 949–1017; [DOI] [PubMed] [Google Scholar]; b) Kalita S, Shaik S, Dubey KD, ACS Catal. 2022, 12, 5673–5683. [Google Scholar]
- [19].a) Lichtenberger F, Nastainczyk W, Ullrich V, Biochem. Biophys. Res. Commun 1976, 70, 939–946; [DOI] [PubMed] [Google Scholar]; b) White RE, Coon MJ, Annu. Rev. Biochem 1980, 49, 315–356; [DOI] [PubMed] [Google Scholar]; c) Guengerich FP, Vaz AD, Raner GN, Pernecky SJ, Coon MJ, Mol. Pharmacol 1997, 51, 147–151. [DOI] [PubMed] [Google Scholar]
- [20].a) Mak PJ, Duggal R, Denisov IG, Gregory MC, Sligar SG, Kincaid JR, J. Am. Chem. Soc 2018, 140, 7324–7331; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hargrove TY, Wawrzak Z, Guengerich FP, Lepesheva GI, J. Biol. Chem 2020, 295, 9998–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Cryle MJ, De Voss JJ, Angew. Chem., Int. Ed 2006, 45, 8221–8223; [DOI] [PubMed] [Google Scholar]; b) Podgorski MN, Coleman T, Churchman LR, Bruning JB, De Voss JJ, Bell SG, Chem. Eur. J 2022, 28, e202202428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Swinney DC, Mak AY, Biochem. 1994, 33, 2185–2190; [DOI] [PubMed] [Google Scholar]; b) Gregory MC, Denisov IG, Grinkova YV, Khatri Y, Sligar SG, J. Am. Chem. Soc 2013, 135, 16245–16247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Akhtar M, Corina D, Pratt J, Smith T, J. Chem. Soc., Chem. Commun 1976, 854–856; [Google Scholar]; b) Akhtar M, Calder MR, Corina DL, Wright JN, Biochem. J 1982, 201, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Ortiz de Montellano PR, in Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed. (Ed.: Ortiz de Montellano PR), Springer, New York, 2015, pp. 111–176; [Google Scholar]; b) Guengerich FP, ACS Catal. 2018, 8, 10964–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Shyadehi AZ, Lamb DC, Kelly SL, Kelly DE, Schunck WH, Wright JN, Corina D, Akhtar M, J. Biol. Chem 1996, 271, 12445–12450; [DOI] [PubMed] [Google Scholar]; b) Sen K, Hackett JC, J. Am. Chem. Soc 2010, 132, 10293–10305. [DOI] [PubMed] [Google Scholar]
- [26].a) Morisaki M, Igata T, Yamamoto S, Chem. Pharm. Bull 2000, 48, 1474–1479; [DOI] [PubMed] [Google Scholar]; b) Araki S, Eguchi S, Morisaki M, Chem. Pharm. Bull 1990, 38, 1796–1797; [Google Scholar]; c) Takano Y, Morisaki M, Chem. Pharm. Bull 1991, 39, 1647–1648. [Google Scholar]
- [27].McCarty KD, Sullivan ME, Tateishi Y, Hargrove TY, Lepesheva GI, Guengerich FP, J. Biol. Chem 2023, 299, 104841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dess DB, Martin JC, J. Org. Chem 1983, 48, 4155–4156. [Google Scholar]
- [29].a) Knapp DR, Handbook of Analytical Derivatization Reactions, Wiley; New York, 1979; [Google Scholar]; b) Schlenk H, Gellerman JL, Anal. Chem 1960, 32, 1412–1414; [Google Scholar]; c) Vorbeck ML, Mattick LR, Lee FA, Pederson CS, Anal. Chem 1961, 33, 1512–1514. [Google Scholar]
- [30].Gotoh O, J. Biol. Chem 1992, 267, 83–90. [PubMed] [Google Scholar]
- [31].Hargrove TY, Lamb DC, Smith JA, Wawrzak Z, Kelly SL, Lepesheva GI, Sci. Rep 2022, 12, 16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Khatri Y, Luthra A, Duggal R, Sligar SG, FEBS Lett. 2014, 588, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Jencks WP, Catalysis in Chemistry and Enzymology, McGraw-Hill, New York, 1969, p. 276; [Google Scholar]; b) Kresge AJ, J. Am. Chem. Soc 1973, 95, 3065–3067; [Google Scholar]; c) Walsh C, Enzymatic Reaction Mechanisms, W. H. Freeman, San Francisco, 1979, pp. 121–122; [Google Scholar]; d) Abeles RH, Frey PA, Jencks WP, Biochemistry, Jones and Bartlett, New York, 1992, pp. 113–115; [Google Scholar]; e) Fersht A, Structure and Mechanism in Protein Science, Freeman, New York, 1999, p. 99; [Google Scholar]; f) Hermans J Jr., Scheraga HA, Biochim. Biophys. Acta 1959, 36, 534–535. [DOI] [PubMed] [Google Scholar]
- [34].Walsh CT, Chen YCJ, Angew. Chem., Int. Ed. Engl 1988, 27, 333–343. [Google Scholar]
- [35].Fiorentini F, Geier M, Binda C, Winkler M, Faber K, Hall M, Mattevi A, ACS Chem. Biol 2016, 11, 1039–1048. [DOI] [PubMed] [Google Scholar]
- [36].Mak AY, Swinney DC, J. Am. Chem. Soc 1992, 114, 8309–8310. [Google Scholar]
- [37].a) Nomura T, Kushiro T, Yokota T, Kamiya Y, Bishop GJ, Yamaguchi S, J. Biol. Chem 2005, 280, 17873–17879; [DOI] [PubMed] [Google Scholar]; b) Kim TW, Hwang JY, Kim YS, Joo SH, Chang SC, Lee JS, Takatsuto S, Kim SK, Plant Cell 2005, 17, 2397–2412; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mizutani M, Sato F, Arch. Biochem. Biophys 2011, 507, 194–203. [DOI] [PubMed] [Google Scholar]
- [38].Sivaramakrishnan S, Ouellet H, Matsumura H, Guan S, Moenne-Loccoz P, Burlingame AL, Ortiz de Montellano PR, J. Am. Chem. Soc 2012, 134, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen QB, Yuan GY, Yuan T, Zeng HT, Zou ZR, Tu ZC, Gao J, Zou Y, J. Am. Chem. Soc 2022, 144, 3580–3589. [DOI] [PubMed] [Google Scholar]
- [40].a) Hosokawa M, Maki T, Satoh T, Mol. Pharmacol 1987, 31, 579–584; [PubMed] [Google Scholar]; b) Satoh T, Hosokawa M, Annu. Rev. Pharmacol 1998, 38, 257–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.