

Abstract
Omaveloxolone is a nuclear factor (erythroid‐derived 2)‐like 2 activator approved in the United States and the European Union for the treatment of patients with Friedreich ataxia aged ≥16 years, with a recommended dosage of 150 mg orally once daily on an empty stomach. The effect of the US Food and Drug Administration (FDA) high‐fat breakfast on the pharmacokinetic profile of omaveloxolone observed in study 408‐C‐1703 (NCT03664453) deviated from the usual linear correlation between fed/fasted maximum plasma concentration (C max) and area under the concentration–time curve (AUC) ratios reported for various oral drugs across 323 food effect studies. Here, physiologically based biopharmaceutics modeling (PBBM) was implemented to predict and explain the effect of the FDA high‐fat breakfast on a 150‐mg dose of omaveloxolone. The model was developed and validated based on dissolution and pharmacokinetic data available across dose‐ranging, food effect, and drug–drug interaction clinical studies. PBBM predictions support clinical observations of the unique effect of a high‐fat meal on omaveloxolone pharmacokinetic profile, in which the C max increased by 350% with only a 15% increase in the AUC. Key parameters influencing omaveloxolone pharmacokinetics in the fasted state based on a parameter sensitivity analysis included bile salt solubilization, CYP3A4 activity, drug substance particle size distribution, and permeability. Mechanistically, in vivo omaveloxolone absorption was solubility and dissolution rate limited. However, in the fed state, higher bile salt solubilization led to more rapid dissolution with predominant absorption in the upper gastrointestinal tract, resulting in increased susceptibility to first‐pass gut extraction; this accounts for the lack of correlation between C max and AUC for omaveloxolone.

Study highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Omaveloxolone is approved in patients with Friedreich ataxia aged ≥16 years at an oral dosage of 150 mg once daily on an empty stomach. Food effect on omaveloxolone pharmacokinetics observed clinically deviates from the usual correlation between fed/fasted maximum plasma concentration (C max) and area under the concentration–time curve (AUC) ratios reported for other oral drugs across food effect studies. Physiologically based biopharmaceutics modeling (PBBM) can predict and explain food effect through integration of drug physiochemical properties, formulation, and metabolism data.
WHAT QUESTION DID THIS STUDY ADDRESS?
A PBBM was developed and validated to predict and explain the effect of the US Food and Drug Administration high‐fat meal on the pharmacokinetics of omaveloxolone 150 mg.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Consistent with clinical findings, the PBBM predicts the unique pharmacokinetic profile for omaveloxolone administered after a high‐fat meal, for which the AUC was only modestly increased (+15%) despite a substantial rise (+350%) in the C max. In vivo, omaveloxolone absorption was solubility and dissolution rate limited in the fasted state. However, in the fed state, bile salt solubilization led to more rapid drug dissolution with predominant absorption in the upper gastrointestinal tract, resulting in increased susceptibility to CYP3A4‐mediated first‐pass gut extraction; this mechanistically explains the lack of correlation between C max and AUC for omaveloxolone.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Omaveloxolone pharmacokinetics vary in the presence of a high‐fat meal versus the fasted state, reinforcing the importance of physician and patient education on proper administration and dosing. The PBBM shows potential in accurately predicting food‐related impacts on drug pharmacokinetics even for outlier trends and potentially reduces the need for clinical pharmacokinetic evaluation.
INTRODUCTION
Friedreich ataxia (FA) is a progressive, autosomal recessive neurodegenerative disorder characterized by difficulty with ambulation, coordination, and speech. 1 , 2 In FA, a biallelic trinucleotide (GAA) repeat expansion in the first intron of the frataxin gene leads to impaired transcription and reduced amounts of functional frataxin protein. 1 , 2 Frataxin deficiency results in dysregulation of antioxidant defenses, mitochondrial dysfunction, and impaired nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2) signaling. 3 , 4 , 5
Omaveloxolone, an Nrf2 activator, has been shown to improve mitochondrial function, restore redox balance, and reduce inflammation in FA models. 6 , 7 In a registrational phase II trial (MOXIe; NCT02255435), omaveloxolone significantly improved neurological function versus placebo, with an acceptable safety profile. 8 , 9 Omaveloxolone was approved in the United States and the European Union for the treatment of FA in patients aged ≥16 years. 10 The recommended dosage is 150 mg administered orally once daily (QD) in the form of three 50‐mg capsules or the entire capsule contents sprinkled on and mixed in 2 tablespoons (30 mL) of applesauce, on an empty stomach at least 1 h before (United States and European Union) or 2 h after (European Union) eating. 11 , 12
Food–drug interactions can potentially affect the release (from a given formulation), solubility, dissolution, absorption, first‐pass metabolism, and/or elimination of an oral drug and possibly impact efficacy and safety. 13 Understanding the differences in the pharmacokinetics (PK) of omaveloxolone in various prandial states and the underlying mechanisms explaining these phenomena is essential. Physiologically based biopharmaceutics modeling (PBBM) is an evolving tool that has been widely applied to predict the absorption and PK of oral drug products (DP). 14 These models integrate the physicochemical and biopharmaceutics properties of the drug substance (DS), formulation characteristics, and system physiological parameters. 14 By using (biopredictive) dissolution testing as a key input, PBBM enables manufacturing flexibility by delineating a safe space for DP critical quality attributes. 15 Through virtual population studies, PBBM has served as an alternative to clinical PK trials that evaluate food effects, drug–drug interaction (DDI), or formulation changes. 15
Here, a PBBM was developed to predict and explain the effect of a high‐fat meal on the PK of a 150‐mg dose of omaveloxolone. The model was validated against PK data from dose‐ranging, food effect, and DDI clinical studies.
MATERIALS AND METHODS
Overview of modeling strategy
The modeling strategy is illustrated in Figure 1. A physiologically based PK (PBPK) modeling absorption baseline model was first established using intravenous and oral data in monkeys to derive relevant PK distribution parameters and then applied to humans. The in vivo capsule opening time and dissolution were based on the DS and DP performances; these together with other biopharmaceutical drug properties such as solubility, passive permeability, and the effect of systemic drug transporter P‐glycoprotein (P‐gP) on drug efflux were integrated into the PBBM. Metabolic clearance was specified based on in vitro data and verified with DDI studies. The determination of solubility, precipitation rate, dissolution, permeability, drug efflux, distribution, metabolism, and elimination are described in the Supplementary Materials and Methods in Data S1 (including Figure S1).
FIGURE 1.
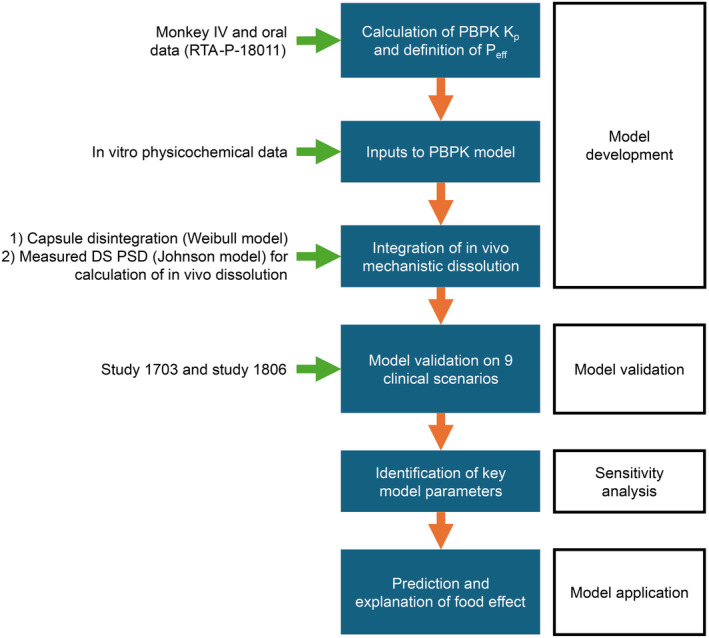
Modeling strategy for omaveloxolone PBBM. DS, drug substance; IV, intravenous; K p, permeability constant; PBBM, physiologically based biopharmaceutics modeling; PBPK, physiologically based pharmacokinetic; P eff, effective permeability; PSD, particle size distribution.
The PBBM was validated using data from nine clinical scenarios from clinical PK studies 408‐C‐1703 (NCT03664453), thereafter referred to as study 1703, and 408‐C‐1806 (NCT04008186), thereafter referred to as study 1806, which tested different doses, prandial states, and DDIs. Parameter sensitivity analyses (PSAs) were run on the validated model to identify the main sources of within‐ and between‐participant variability and the factors limiting omaveloxolone absorption. With the validated model, the effect of a high‐fat meal on the PK of omaveloxolone was evaluated and explained mechanistically. The DDI and PBPKPlus modules of GastroPlus v9.8.2 (GastroPlus; Simulations Plus) and the ADMET Predictor v10.3 (APv10.3; Simulations Plus) were used.
Clinical studies
The average participant demographic information from two selected studies with comprehensive PK data was used for validation of the PBPK absorption baseline model. Study 1703 was a phase I, open‐label, two‐part, food effect (part 1) and dose proportionality (part 2) study of omaveloxolone in healthy adult participants (N = 34). In part 1 (two‐period, fixed‐sequence, randomized crossover design), participants were randomly assigned 1:1 to one of the two treatment sequences (sequence 1: period 1 fed and period 2 fasted; sequence 2: period 1 fasted and period 2 fed), each with a 1‐week washout period. Participants were administered two single doses of omaveloxolone 150 mg at the start of each period. During the fed state, participants were provided the US Food and Drug Administration (FDA) high‐fat standardized breakfast (800–1000 calories, with ≥50% from fat) 16 prior to dosing. In part 2, participants were randomized 1:1 to receive a single dose of either 50‐mg or 100‐mg omaveloxolone in a fasted state; data for omaveloxolone 150 mg (taken in a fasted state) from part 1 were included in part 2 analyses. The design of study 1703 is further detailed in the Supplementary Materials and Methods (Data S1).
Study 1806 was a phase I, open‐label, four‐part, DDI study of omaveloxolone in healthy participants (N = 61). Participants were treated with omaveloxolone 150 mg QD on days 1 and 13, and oral doses of a cytochrome P450 2C8 (CYP2C8) inhibitor gemfibrozil 600 mg twice daily (part 2), a strong CYP3A4 inhibitor itraconazole 200 mg QD (part 3), or a P‐gP inhibitor and a moderate CYP3A4 inhibitor verapamil 120 mg QD (part 4), on days 10 to 18. Primary end points were maximum plasma concentration (C max) and area under the concentration–time curve (AUC) from time 0 extrapolated to infinity (AUC0‐∞), and time to C max (t max); AUC from time 0 to the last quantifiable plasma concentration (AUC0‐t) was a secondary end point.
Model validation
For model validation, the prediction performance indicators were calculated for the PK parameters and profiles as described below.
Average fold error (AFE) is defined by the following equation:
The AFE is an indicator of prediction bias. A method that predicted all actual values with no bias would have a value of 1; underpredictions are indicated by an AFE of <1 and overpredictions by AFE of >1. AFE values generally vary between 0 and infinity; a prediction may be considered satisfactory if the AFE is between 0.8 and 1.2, passable if the AFE is 0.5 to 0.8 or 1.2 to 2, and poor if the AFE is 0 to <0.5 or >2. A satisfactory AFE is needed for model validation.
Absolute average fold error (AAFE) is defined by the following equation:
The AAFE converts negative log fold errors to positive values before averaging them and measures the spread of the predictions. AAFE values vary between 1 and infinity. A method that predicted all actual values perfectly would have a value of 1; one with predictions that were on average twofold off (above 100% or below 50%) would have a value of 2 and so forth. A prediction may be considered satisfactory if the AAFE is <1.2, passable if the AAFE is in the range of 1.2 to 2, and poor if the AAFE is >2. A satisfactory AAFE is needed for model validation.
Average absolute prediction error (AAPE%) is defined by the following equation:
AAPE is the measurement of prediction error scaled to percentage units. It approximates (AAFE −1) × 100. A model is considered satisfactory if the AAPE is <20%, passable if the AAPE is ≥20 to <50%, and poor if the AAPE is ≥50%.
Percent predictions within clinical variability (PPWCV) are defined by following equation:
For each PK sampling time point 1 to n total (apart from pre‐dose), a binary criterion yes or no is determined based on whether the predicted concentration falls within the 95% confidence interval of the measured clinical data. The PPWCV calculated for each PK profile was averaged across all the clinical scenarios tested. The same calculations were performed for PK parameters—the binary criterion for each clinical scenario was based on whether the predicted PK parameter fell within the 95% confidence interval of the measured average value of that parameter. A satisfactory PPWCV is >80%, a passable PPWCV is in the range of ≥65% to 80%, and a poor PPWCV is <65%.
Parameter sensitivity analyses
The PSA for C max, AUC0‐t, and other PK parameters (Supplementary Materials and Methods) was based on a range of selected DP properties and physiological parameters that could impact omaveloxolone absorption or metabolism by affecting capsule opening time, size of the DS (controlling in vivo dissolution), first‐pass gut and liver extraction, and metabolic elimination in vivo (Table S1). The analysis was performed using omaveloxolone 150 mg (target dose) in the fasted state (for increased sensitivity with a lower fraction absorbed vs. the fed state) on a representative population based on the MOXIe registrational study cohort with FA (average age: 26 years; average weight: 69 kg). 10
Simulation design
The PBBM was built using default values based on human fasted or fed physiologies. Since omaveloxolone is not ionized in the physiological pH range, adjustment of surface solubility was not needed. For PBBM validation, populations representative of the clinical trials were created based on the average height and weight of the cohorts. The advanced compartmental and transit model physiologies were adjusted for body weight. All the doses and prandial states tested in the clinical trials were reproduced in the PBBM. The default optimum log D model SA/V 6.1 was applied to calculate absorption scaling factors. Since omaveloxolone is lipophilic with log p > 5, the amount of absorption scaling factors was increased in the cecum and colon.
To predict food effect in study 1703, the FDA high‐fat breakfast option was selected with default zero‐order gastric emptying. The gastric emptying time was increased from 3.51 to 5 h to match the time needed for the intragastric volume to fall below 100 mL after a high‐fat meal intake. 17
Because CYP3A4, but not CYP2C8, is largely involved in omaveloxolone metabolism, 18 DDI is expected for co‐administration of omaveloxolone with CYP3A4 inhibitors. Hence, two DDI simulations based on parts 3 and 4 of study 1806 were performed (Supplementary Materials and Methods, Figures S2‐S5). The DDI module of GastroPlus was used, and simpler modeling options based on reported values for inhibition were also tested for model validation. To account for itraconazole 200 mg co‐administration outside of the DDI module, the maximum rate of reaction (V max) in the gut and liver for CYP3A4 was reduced by a factor of 3.9. 19 , 20 The V max for P‐gp was set to 0 for verapamil co‐administration.
Ethics statement
The clinical studies were designed and monitored in accordance with sponsor procedures, which complied with the ethical principles of good clinical practice and with the Declaration of Helsinki. Each trial was approved by the institutional review board or independent ethics committee at the respective centers. All patients provided written informed consent.
RESULTS
Modeling parameters
The main physicochemical and biopharmaceutical properties of omaveloxolone used for model parameterization are shown in Table 1. The derivation of these parameters (solubility and precipitation [Figure S6], dissolution, permeability [Figures S7 and S8], P‐gp efflux, and metabolism and elimination [Figure S9]) is shown in the Supplementary Results.
TABLE 1.
Main physicochemical and biopharmaceutics properties of omaveloxolone used in the model.
| Parameter | Value for crystalline drug | Value for amorphous drug | Reference/rationale |
|---|---|---|---|
| Molecular weight | 554.7 | From structure | |
| Compound nature | Crystalline | Amorphous | From FDA, chemistry review for omaveloxolone (NDA 216718) 41 |
| pKa | 7.26 (A) | Measured | |
| Log P | >5 | Measured | |
| True density (g/mL) | 1.2 | Not reported. Default value used | |
| Intrinsic solubility (μg/mL) | 0.154 | Approx. 1.1 | Measured |
| Precipitation time (s) | NA | 25,167 | Does not play a role in drug exposure per sensitivity analysis |
| In vitro degradation rate (min−1) | 0 | No measured chemical degradation for up to 24 h in biorelevant media | |
| FaSSIF‐V2 solubility (μg/mL) | 1.4 (24 h) | 14 | Measured. The value for the amorphous drug was taken as 10 times that for the crystalline drug (see page 11 of Supplementary Materials [Supplementary Results–Solubility and precipitation]) |
| FeSSIF solubility (μg/mL) | 9 (24 h) | 90 | Measured. The value for the amorphous drug was taken as 10 times that for the crystalline drug (see Supplementary Results–Solubility and precipitation) |
| Bile salt solubilization ratio | 7.3 × 105 | 9.34 × 105 | Fitted from solubility data in GastroPlus |
| Exponent for bile salt solubilization | 1.3039 | 1.2813 | Fitted from solubility data in GastroPlus |
| fu plasma (%) | 3.1 | From FDA, clinical pharmacology review for omaveloxolone (NDA 216718) 18 | |
| B/P ratio | 0.70 | ||
| DS particle size diameter (μm) | Not reported. 50 μm radius for model use on crystalline content |
D (v,0.1) = measured D (v,0.5) = measured D (v,0.9) = measured |
Measured. Typical D (v,0.9) < 30 μm for amorphous drug |
| Jejunum P eff (10−4 cm/s) | 3 | Fitted to oral monkey data from RTA‐P‐18011. Predicted to be 3.841 using APv10.3 | |
| Formulation type | Suspension (if precipitate) or same as amorphous or IR capsule (when spiked) | 50‐mg immediate‐release capsules | The recommended dose is 150 mg once daily |
| Model for dissolution | Based on Johnson model using DS PSD, adjusted for bile salt effect on solubility and diffusion. Dissolution model employed a “controlled‐release undissolved” formulation type using measured dissolution with a Weibull fit of the data | Based on mechanism of release and measured DS PSD (see Supplementary Materials and Methods–Integration of DP dissolution) | |
| Model for distribution | Full PBPK model with Lukacova parameters for K p calculation | Based on best fit to monkey IV data | |
| V max of CYP3A4 in the gut (mg/s) | 0.5 | Converted to in vivo V max from intrinsic HLM clearance and adjusted to PK data from study 1806 | |
| V max of CYP3A4 in the PBPK model (mg/s/mg‐enz) | 0.015 | Converted to in vivo V max from intrinsic HLM clearance and adjusted to PK data from study 1806 | |
| K m of CYP3A4 (μg/mL) | 1 | Rounded from 0.694 μg/mL; converted from CYP3A4 measured K m in study RTA408‐P‐1212 | |
| V max of P‐gp (mg/s) | 2.523 × 10−4 | Converted from Caco‐2‐derived data | |
| K m of P‐gp (μg/mL) | 2.9401 | Converted from Caco‐2‐derived data | |
Abbreviations: B/P, blood:plasma concentration ratio; CYP3A4, cytochrome P450 3A4; D (v,0.1/0.5/0.9), 10%/50%/90% of particles are smaller than the stated size; DP, drug product; DS, drug substance; FaSSIF‐V2, fasted‐state simulated intestinal fluid V2; FDA, US Food and Drug Administration; FeSSIF, fed‐state simulated intestinal fluid; fu, fraction unbound; HLM, human liver microsome; IR, immediate‐release; IV, intravenous; K m, Michaelis–Menten constant; K p, permeability constant; log P; partition coefficient/lipophilicity; NDA, new drug application; PBPK, physiologically based pharmacokinetic; P eff, effective permeability; P‐gp, P‐glycoprotein; PK, pharmacokinetic; pKa, acid‐dissociation constant; PSD, particle size distribution; V max, maximum rate of reaction.
Model validation
The PBPK absorption model and PBBM were validated across nine distinct clinical scenarios in studies 1703 and 1806. Profile predictions are shown in Figures S9 (right panel), S10, and S11; calculations of model prediction performance indicators are shown in Table 2, Tables S2‐S4.
TABLE 2.
Prediction performance indicators for profile prediction.
| AFE | AAFE | AAPE (%) | % Profile within clinical variability | |
|---|---|---|---|---|
| Study dose , (average) | 1.05 | 1.36 | 20.1 | 98 |
| 1703–50 mg (fasted) | 0.96 | 1.16 | 10.9 | 100 |
| 1703–100 mg (fasted) | 0.97 | 1.26 | 16.7 | 100 |
| 1703–150 mg (fasted) | 1.05 | 1.35 | 23.0 | 100 |
| 1703–150 mg (fed) | 1.62 | 1.90 | 47.69 | 88 |
| 1806–150 mg (part 2) | 1.06 | 1.25 | 17.1 | 100 |
| 1806–150 mg (part 3) | 0.80 | 1.38 | 15.3 | 100 |
| 1806–150 mg (part 4) | 1.06 | 1.26 | 16.2 | 100 |
| 1806–150 mg (part 3 + itraconazole) | 1.18 | 1.27 | 13.2 | 95 |
| 1806–150 mg (part 4 + verapamil) | 0.71 | 1.43 | 20.6 | 100 |
Note: Bold numbers on the top line represent average values of the simulated clinical scenarios.
Abbreviations: AAFE, absolute average fold error; AAPE, average absolute prediction error; AFE, average fold error.
All predicted PK parameters over the nine clinical scenarios were within the observed clinical variability (Table 2, Figures S9 [right panel], S10, and S11). The use of the GastroPlus “controlled release undissolved dispersed” model in conjunction with the DS particle size distribution (PSD) allowed the separation of in vivo drug release (capsule opening) from in vivo dissolution of the capsule contents. This approach provided a satisfactory PK profile prediction, with 98% of the predicted concentrations within the clinical variability at each data point. All model prediction performance indicators on AUC and C max were satisfactory based on predefined criteria, and the PBBM was considered validated.
Parameter sensitivity analyses
The PSA revealed that the main parameters influencing the PK of omaveloxolone 150 mg were bile salt solubilization, V max of CYP3A4, DS PSD, drug permeability, and volume in the small intestine (Figure 2, Supplementary Results, Figure S12). The model's sensitivity to the DS PSD confirmed the decision to use the measured DS batch particle size for the analysis. Precipitation time, transit times, and P‐gp function did not have a substantial effect on omaveloxolone PK. In particular, the stomach transit time from 0.25 to 0.5 h or Weibull lag from 0.12 to 0.5 h did not affect omaveloxolone PK. The main between‐participant sources of variability were drug permeability and CYP3A4 expression and function. The main within‐participant sources of variability were differences in bile salt concentration during the day in the fasted and fed states and the type of food.
FIGURE 2.
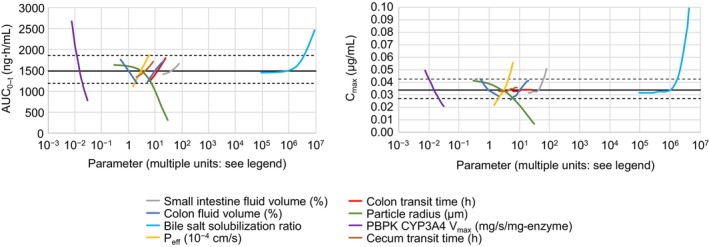
PSA results showing the main parameters that affect AUC0‐t (left panel) and C max (right panel). In each panel, the first eight most influential parameters are displayed for clarity. AUC0‐t, area under the curve from time 0 to the last quantifiable plasma concentration; C max, maximum plasma concentration; CYP3A4, cytochrome P450 3A4; PBPK, physiologically based pharmacokinetic; P eff, effective permeability; PSA, parameter sensitivity analysis; V max, maximum rate of reaction.
Prediction of food effect
The effects of a high‐fat meal on omaveloxolone 150 mg in vivo dissolution, first‐pass extraction, and PK are shown in Figure 3.
FIGURE 3.
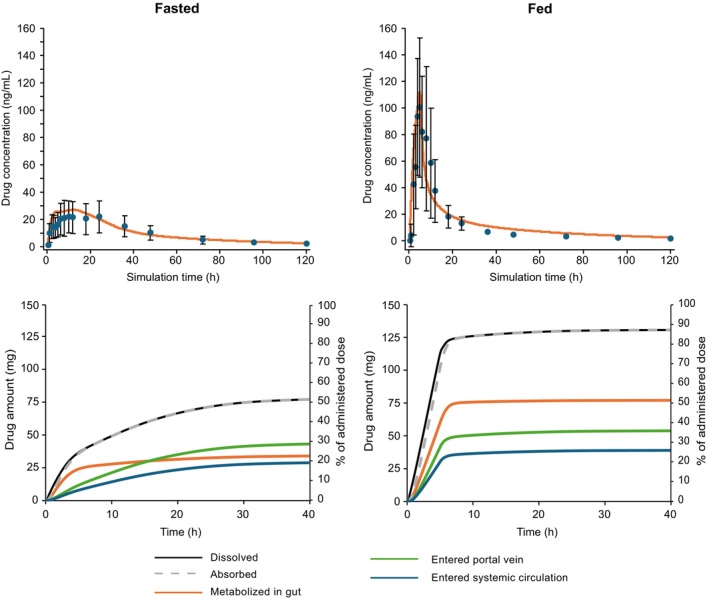
PBBM prediction of omaveloxolone PK profiles (upper panels) and the amount and percentage of drug dissolved, absorbed, metabolized, and entered into the portal vein or the systemic circulation (lower panels) following a single dose of omaveloxolone 150 mg in the fasted (left panels) and fed (right panels) states. In the top panel, the symbols represent the (arithmetic) mean of measured omaveloxolone concentrations and the error bars represent ± standard deviation. PBBM, physiologically based biopharmaceutics modeling; PK, pharmacokinetic.
The fraction of dose absorbed (Fa) for omaveloxolone and the fraction reaching the portal vein after passing through the gut wall without metabolism (Fa × fraction of drug escaping first‐pass gut metabolism [Fg]) in the fasted and fed states are shown in Figure 4. In both prandial states, the in vivo dissolution of omaveloxolone was limited by solubility in all compartments. However, the Fa of omaveloxolone increased from 52% in the fasted to 87% in the fed state, attributed to the drug's lipophilicity and strong affinity to bile salt micelles, along with higher concentration of micelles after a high‐fat meal. The resulting increased solubility and faster dissolution contributed to the majority of absorption occurring in the upper gastrointestinal (GI) tract in the fed state. In the fasted state, omaveloxolone absorption occurred along the GI tract, with the highest fraction absorbed in the cecum and colon as shown by the secondary peaks in the concentration–time plots (Figure S12). As there is a lower expression of CYP3A4 in the lower versus upper GI tract, the fraction of omaveloxolone lost by first‐pass gut extraction was limited in the fasted versus fed state. Fa × Fg was 36% in the fed state versus 29% in the fasted state (Figure 4), and hepatic extraction was 33% and 28%, respectively. These phenomena explain the negligible increase in AUC (+15%) despite a large increase in C max (+350%) in the fed state for omaveloxolone 150 mg observed in the food effect study 1703.
FIGURE 4.
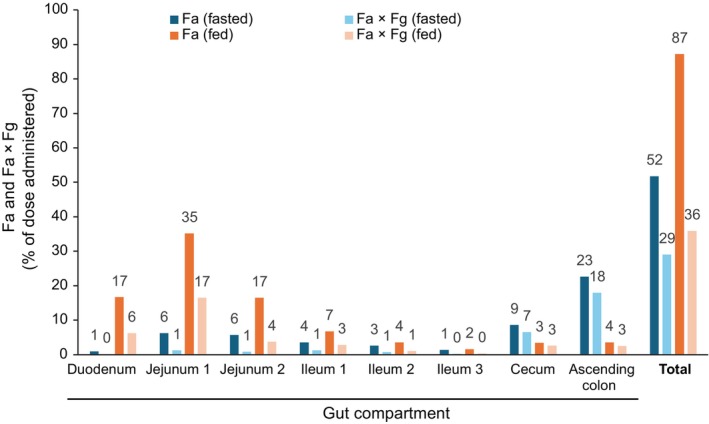
PBBM prediction of the fraction absorbed (Fa) and the fraction reaching the portal vein after passing through the gut wall without metabolism (Fa × Fg) at each gut compartment for a single dose of omaveloxolone 150 mg administered in the fasted and fed states. Data labels shown are rounded to whole numbers. PBBM, physiologically based biopharmaceutics modeling.
Food effect and dose proportionality results from study 1703
Findings from study 1703 corroborated the results of the PBBM analysis (detailed in the Supplementary Results, Tables S5–S9, Figure S14).
DISCUSSION
Omaveloxolone is currently indicated to be taken on an empty stomach with capsules either swallowed whole or contents sprinkled on and mixed in 2 tablespoons of applesauce for patients with swallowing difficulties. Omaveloxolone displays a unique PK profile in the presence of standardized FDA high‐fat breakfast: AUC was only modestly increased (+15%) despite a substantial rise in C max (+350%). The PBBM developed for omaveloxolone provides a mechanistic explanation of this unique food effect.
The PBBM, informed by the DS and DP characteristics and drug metabolism, was validated across the doses, prandial states, and DDI scenarios studied. Consistent with permeability and CYP3A4 activity identified as main between‐participant sources of variability from the PSA in this study, Avdeef et al. 21 reported a 60% variability in permeability data obtained from humans across drugs with low and high permeability; furthermore, Lown et al. 22 reported high interparticipant variability in gut CYP3A4 expression, at 11‐fold based on protein, 8‐fold based on mRNA, and 6‐fold based on catalytic activity. These sources of variability should not exist for cross‐over trials. Difference in bile salt concentration was identified as a main within‐participant source of variability in this study, in line with a previous report. 23 The variability in bile salt lumen concentration has also been reported to span several logs, even in the fasted state. 24 The within‐participant variability during the day was reported to be high even in the fasted state, covering 2 log differences. 25
Based on the PBBM prediction, omaveloxolone absorption was mainly limited by drug solubility along the GI tract in the fasted state (Figure 3). Omaveloxolone belongs to Biopharmaceutics Classification System class 4 DS, which is characterized by low permeability, restricting absorption in the GI tract, and continuous drug dissolution. This underscores the importance of permeability as a limiting factor for AUC and C max in the fasted state (Figure 2). The Fa of omaveloxolone in the fasted state as predicted by the PBBM was approximately 52%. Consistently, based on the human absorption, metabolism, and excretion study (Study 1805), omaveloxolone was the most abundant component (approximately 40%) in the feces of participants who were administered a 150‐mg dose of [14C]‐omaveloxolone, indicating an Fa of up to 60%. 18 Under fed conditions, omaveloxolone absorption occurred more rapidly due to the drug's affinity for bile salts, which resulted in faster dissolution in the fed state versus the fasted state.
The PBBM prediction is corroborated by the individual fasted‐ and fed‐state omaveloxolone PK profiles in Study 1703 (Figure S13). Under the fasted state, in addition to an initial rise in omaveloxolone plasma concentration, multiple peaks were observed in most individual PK profiles, mainly occurring after 4 h (when the drug should have reached the lower intestine in humans), suggesting that absorption predominantly occurred in the colon. This supports the model prediction that drug dissolution and solubility were the rate‐limiting factors for absorption in the fasted state. Under fed conditions, there was a notably faster drug absorption with t max more frequently observed before omaveloxolone reached the colon (Figure S13), indicating that drug absorption predominantly occurred in the upper segments of the GI tract. The faster (within‐participant) absorption in the fed state also suggested that higher concentrations of bile salts in the GI tract lumen could partially overcome the limited solubility (and therefore in vivo dissolution) of omaveloxolone.
The uniqueness of the food effect observed with omaveloxolone, compared with other medications, is shown by the correlation between fed/fasted C max and AUC ratios from 323 food effect studies encompassing a range of compounds and formulations. 26 , 27 , 28 , 29 , 30 , 31 The linear correlation between log(C max ratio) and log(AUC ratio) for investigated drugs that were proposed by Omachi et al. 29 was applied to the dataset of the present study (Figure 5a). Based on the linear regression, when log(AUC ratio) = 0 (i.e., AUC ratio = 1), the corresponding log(C max ratio) is expected to be slightly negative at −0.0651 (i.e., C max ratio = 0.86 on average). This lower C max ratio to AUC ratio (<1) is likely related to prolonged gastric emptying in the fed state, which delays the passage of drug to the small intestine. C max and AUC (ratios) are usually expected to be highly correlated since the AUC is calculated from the integration of concentration–time profiles. In most cases, drugs with solubility‐limited absorption and without significant first‐pass extraction have increased solubility due to the presence of higher amounts of bile salts in the GI tract lumen under the fed state. This leads to increased drug concentration in close proximity to the absorptive surface of the intestine and a larger amount of drug reaching the systemic circulation (i.e., higher AUC) versus in the fasted state.
FIGURE 5.
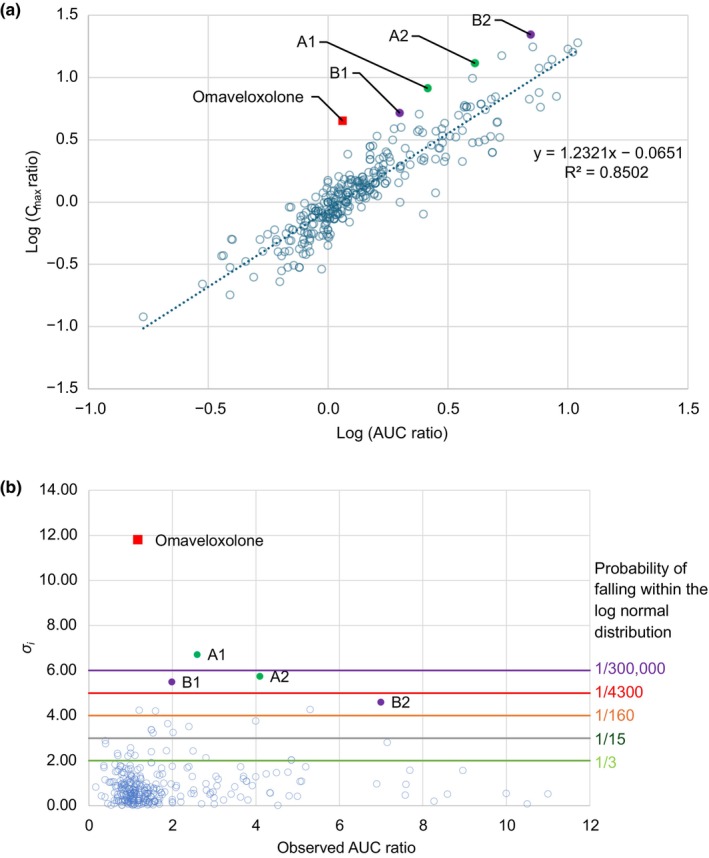
(a) Correlation between fed/fasted C max ratio versus AUC ratio. (b) σ i values for each observed C max ratio versus AUC ratio for omaveloxolone and 323 food effect studies. The solid square symbol shows omaveloxolone. A1: CPD3 100 mg administered with a high‐fat meal 38 ; A2: CPD3 50 mg administered with a high‐fat meal 38 ; B1: progesterone—Utrogestran 200 mg administered with a 688‐calorie meal containing 34% fat content 39 ; B2: progesterone—Utrogestran 100 mg administered with an 890‐calorie meal containing 55% fat content. 27 σ i , mean of sigma; AUC, area under the curve; C max, maximum plasma concentration; CPD3, adenosine monophosphate deaminase inhibitor.
There are, however, physiological factors and methodological issues that can explain deviations in the correlation between C max and AUC. PK sampling frequency could miss the C max during clinical food effect studies, as the same sampling time points are typically used for the fasted and fed states. In some studies, PK sampling was not frequent enough to capture C max in the fed state due to prolonged gastric emptying, which may lead to underestimation of both C max and AUC and a false‐negative finding of food effect. 32 In addition, in both prandial states, gastric emptying of solid phases that comprise the drug might not be happening all at once, and gastric retention could account for a lower C max compared to single‐phase emptying. Partial gastric emptying has been reported in the fasted and fed states for various drugs. 32 , 33 , 34 , 35 , 36 , 37 If first‐pass extraction is not limiting drug absorption in a saturable way, a lower C max due to partial gastric emptying could be associated with the same AUC as when gastric emptying happens in a single phase, leading to decorrelation of C max and AUC.
Taken together, a correlation between drug‐fed/fasted C max and AUC ratios is expected and justifiable. The presence of outliers in this correlation is particularly intriguing, primarily to understand the mechanism of food effect and to assess the predictive capability of mechanistic models. The correlation depicted in Figure 5a could prognosticate measured fed/fasted C max ratios based on corresponding fed/fasted AUC ratios. The statistics associated with using the correlation to predict measured C max ratios would lead to an AFE of 1.00, an AAFE of 1.29, and an AAPE of 27%. The relative standard deviation for the prediction of C max based on this correlation was assumed to be 29% from the AAFE value. Assuming that C max ratio observations were randomly spread around the predicted C max ratios with a log‐normal distribution, the probability of observing any C max ratio value is calculated by means of sigma (σ i ) derivation.
In the above equation, the value of σ i can help estimate the probability of observing a C max ratio (O i) around the prediction P i . For σ i = 2, the chance to observe (O i ) based on P i and the spread of the data approximates 1 of 3; for σ i = 3, the chance is 1 of 15; and for σ i = 4, the chance is 1 of 160. σ i can be used to identify outliers to the correlation between C max ratio and observed AUC ratio. The σ i value for omaveloxolone and the 323 food effects studies from the literature are shown in Figure 5b. The C max ratio versus AUC ratio correlation established is good and roughly what would be expected from a log‐normal distribution with 69%, 91%, and 95% of the observed C max ratio within 1, 2, and 3 σ i of the predictions, respectively. The unique nature of the food effect on omaveloxolone was evident as the σ i value for omaveloxolone was largest at 11.7 (close to 12) away from the prediction, while the second largest outlier σ i value was only at approximately 7. Interestingly, the other four outliers (besides omaveloxolone) in the C max ratio versus AUC ratio correlation in Figure 5b corresponded to only two drugs (CPD3 and progesterone). 27 , 38 , 39 These two drugs show similarities with omaveloxolone (e.g., lipophilicity [log P] was reported to be 6.6 and 3.9, respectively). 38 , 40 Both CPD3 and progesterone showed secondary absorption phases based on average PK profiles in the fasted state, which was not observed or occurred to a much lower extent in the fed state. 27 , 40 Although the detailed evaluation of these effects goes beyond the scope of this article, the effect of food on the PK of CPD3 was correctly predicted using GastroPlus. 40
CONCLUSIONS
A PBBM was successfully developed and validated for omaveloxolone. Drug absorption for omaveloxolone is solubility and dissolution rate limited. In the fasted state, omaveloxolone is incompletely absorbed, with absorption predominantly occurring in the lower segment of the GI tract where the CYP3A4‐mediated first‐pass gut extraction is low. In the fed state, the higher amount of bile salt micelles present led to an increased solubility of omaveloxolone attributed to its weakly acidic and highly lipophilic nature, thereby accelerating in vivo dissolution, resulting in predominant absorption in the upper GI tract. In this region, omaveloxolone is subjected to a more substantial first‐pass gut extraction, causing a notable transient surge in C max without a correspondingly large increase in AUC. This food effect on the PK of omaveloxolone deviates from that of other drugs for which the fed/fasted ratios for C max and AUC are generally well correlated. These findings point to the impact of fed versus fasted condition on the PK profile of omaveloxolone; hence, reinforcing the importance of physician and patient education on administration and dosing compliance.
In silico PBPK modeling and PBBM tools offer promising platforms that integrate drug dissolution, formulation characteristics, precipitation, degradation, first‐pass extraction, and metabolism. These tools accurately forecast the impact of food on oral drugs, such as omaveloxolone, potentially reducing the need for clinical evaluations. Notably, the accurate prediction of a 12‐σ outlier event of C max versus AUC ratio in this case study bolsters the credibility of such models for the prediction of food effects.
AUTHOR CONTRIBUTIONS
X.J.H.P., S.M.H., H.Z., D.W., L.Q.S., and S.S.‐S. wrote the manuscript; X.J.H.P., S.M.H., H.Z., and D.W. designed the research; X.J.H.P., H.Z., and D.W. performed the research; X.J.H.P., S.M.H., H.Z., D.W., L.Q.S., and S.S.‐S. analyzed the data; and H.Z. contributed new reagents/analytical tools.
FUNDING INFORMATION
This study was funded by Reata Pharmaceuticals, Inc.; Reata was acquired by Biogen in 2023.
CONFLICT OF INTEREST STATEMENT
Xavier J.H. Pepin and Sandra Suarez‐Sharp are employees of and hold stock in Simulations Plus, which was commissioned by Reata Pharmaceuticals to conduct the study; Reata was acquired by Biogen in 2023. Scott M. Hynes was an employee and may have held stock in Biogen at the time of development of this publication. Hamim Zahir and Lois Q. Semmens are employees of and may hold stock in Biogen. Deborah Walker was an employee of and held stock and/or stock options in Reata at the time the study was conducted.
Supporting information
Data S1.
ACKNOWLEDGMENTS
Medical writing support was provided by Qing Yun Chong, PhD, CMPP, of Nucleus Global, in accordance with Good Publication Practice (GPP 2022) guidelines, and funded by Biogen.
Pepin XJH, Hynes SM, Zahir H, Walker D, Semmens LQ, Suarez‐Sharp S. Understanding the mechanisms of food effect on omaveloxolone pharmacokinetics through physiologically based biopharmaceutics modeling. CPT Pharmacometrics Syst Pharmacol. 2024;13:1771‐1783. doi: 10.1002/psp4.13221
Scott M. Hynes and Deborah Walker: Employees at the time of development of this publication.
Reata Pharmaceuticals, Inc., was acquired by Biogen in 2023.
DATA AVAILABILITY STATEMENT
Individual participant data collected during the trial may be shared after anonymization and upon approval of the research proposal. Biogen commits to sharing patient‐level data, study‐level data, CSRs, and protocols with qualified scientific researchers who provide a methodologically sound proposal. Biogen reviews all data requests internally based on the review criteria and in accordance with our Clinical Trial Transparency and Data Sharing Policy. Deidentified data and documents will be shared under agreements that further protect against participant reidentification. To request access to data, please visit https://vivli.org/.
REFERENCES
- 1. Lynch DR, Farmer JM, Balcer LJ, Wilson RB. Friedreich ataxia: effects of genetic understanding on clinical evaluation and therapy. Arch Neurol. 2002;59:743‐747. [DOI] [PubMed] [Google Scholar]
- 2. Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65:1296‐1303. [DOI] [PubMed] [Google Scholar]
- 3. D'Oria V, Petrini S, Travaglini L, et al. Frataxin deficiency leads to reduced expression and impaired translocation of NF‐E2‐related factor (Nrf2) in cultured motor neurons. Int J Mol Sci. 2013;14:7853‐7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paupe V, Dassa EP, Goncalves S, et al. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One. 2009;4:e4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shan Y, Schoenfeld RA, Hayashi G, et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich's ataxia YG8R mouse model. Antioxid Redox Signal. 2013;19:1481‐1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abeti R, Baccaro A, Esteras N, Giunti P. Novel Nrf2‐inducer prevents mitochondrial defects and oxidative stress in Friedreich's ataxia models. Front Cell Neurosci. 2018;12:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Probst BL, Trevino I, McCauley L, et al. RTA 408, a novel synthetic triterpenoid with broad anticancer and anti‐inflammatory activity. PLoS One. 2015;10:e0122942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lynch DR, Chin MP, Delatycki MB, et al. Safety and efficacy of omaveloxolone in Friedreich ataxia (MOXIe study). Ann Neurol. 2021;89:212‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynch DR, Farmer J, Hauser L, et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2019;6:15‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lynch DR, Chin MP, Boesch S, et al. Efficacy of omaveloxolone in Friedreich's ataxia: delayed‐start analysis of the MOXIe extension. Mov Disord. 2023;38:313‐320. [DOI] [PubMed] [Google Scholar]
- 11. Reata Pharmaceuticals, Inc . SKYCLARYS® highlights of prescribing information. US Food and Drug Administration. 2024. Accessed April 15, 2024. https://www.skyclarys.com/docs/skyclarys_us_prescribing_information/
- 12. Reata Ireland Limited . SKYCLARYS summary of product characteristics. European Medicines Agency. 2024. Accessed February 26, 2024. https://www.ema.europa.eu/en/documents/product‐information/skyclarys‐epar‐product‐information_en.pdf
- 13. Koziolek M, Alcaro S, Augustijns P, et al. The mechanisms of pharmacokinetic food‐drug interactions—a perspective from the UNGAP group. Eur J Pharm Sci. 2019;134:31‐59. [DOI] [PubMed] [Google Scholar]
- 14. Wu D, Li M. Current state and challenges of physiologically based biopharmaceutics modeling (PBBM) in oral drug product development. Pharm Res. 2023;40:321‐336. [DOI] [PubMed] [Google Scholar]
- 15. US Food and Drug Administration . The use of physiologically based pharmacokinetic analyses—biopharmaceutics applications for oral drug product development, manufacturing changes, and controls. 2002. Accessed February 26, 2024. https://www.fda.gov/media/142500/download
- 16. US Food and Drug Administration . Assessing the effects of food on drugs in INDs and NDAs—clinical pharmacology considerations guidance for industry. 2022. Accessed February 26, 2024. https://www.fda.gov/media/121313/download
- 17. Koziolek M, Grimm M, Garbacz G, Kühn JP, Weitschies W. Intragastric volume changes after intake of a high‐caloric, high‐fat standard breakfast in healthy human subjects investigated by MRI. Mol Pharm. 2014;11:1632‐1639. [DOI] [PubMed] [Google Scholar]
- 18. US Food and Drug Administration . Center for Drug Evaluation and Research. 216718Orig1s000 clinical review. 2023. Accessed April 19, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/216718Orig1s000MedR.pdf
- 19. Templeton I, Peng CC, Thummel KE, Davis C, Kunze KL, Isoherranen N. Accurate prediction of dose‐dependent CYP3A4 inhibition by itraconazole and its metabolites from in vitro inhibition data. Clin Pharmacol Ther. 2010;88:499‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Templeton IE, Thummel KE, Kharasch ED, et al. Contribution of itraconazole metabolites to inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2008;83:77‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Avdeef A, Tam KY. How well can the Caco‐2/Madin‐Darby canine kidney models predict effective human jejunal permeability? J Med Chem. 2010;53:3566‐3584. [DOI] [PubMed] [Google Scholar]
- 22. Lown KS, Kolars JC, Thummel KE, et al. Interpatient heterogeneity in expression of CYP3A4 and CYP3A5 in small bowel. Lack of prediction by the erythromycin breath test. Drug Metab Dispos. 1994;22:947‐955. [PubMed] [Google Scholar]
- 23. Marciani L, Cox EF, Hoad CL, et al. Effects of various food ingredients on gall bladder emptying. Eur J Clin Nutr. 2013;67:1182‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riethorst D, Mols R, Duchateau G, Tack J, Brouwers J, Augustijns P. Characterization of human duodenal fluids in fasted and fed state conditions. J Pharm Sci. 2016;105:673‐681. [DOI] [PubMed] [Google Scholar]
- 25. Pyper K, Brouwers J, Augustijns P, et al. Multidimensional analysis of human intestinal fluid composition. Eur J Pharm Biopharm. 2020;153:226‐240. [DOI] [PubMed] [Google Scholar]
- 26. Singh BN, Malhotra BK. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet. 2004;43:1127‐1156. [DOI] [PubMed] [Google Scholar]
- 27. Qin H, Zheng L, Fang Y, et al. Pharmacokinetics, safety and bioequivalence of two formulations of progesterone soft capsule in healthy Chinese postmenopausal females: impacts of a high‐fat meal. Basic Clin Pharmacol Toxicol. 2022;130:268‐276. [DOI] [PubMed] [Google Scholar]
- 28. Emami Riedmaier A, DeMent K, Huckle J, et al. Use of PBPK modeling for predicting drug‐food interactions: an industry perspective. AAPS J. 2020;22:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Omachi F, Kaneko M, Iijima R, Watanabe M, Itagaki F. Relationship between the effects of food on the pharmacokinetics of oral antineoplastic drugs and their physicochemical properties. J Pharm Health Care Sci. 2019;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kesisoglou F, Basu S, Belubbi T, et al. Streamlining food effect assessment—are repeated food effect studies needed? An IQ analysis. AAPS J. 2023;25:60. [DOI] [PubMed] [Google Scholar]
- 31. Li M, Zhao P, Pan Y, Wagner C. Predictive performance of physiologically based pharmacokinetic models for the effect of food on oral drug absorption: current status. CPT Pharmacometrics Syst Pharmacol. 2017;7:82‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andreas CJ, Pepin X, Markopoulos C, Vertzoni M, Reppas C, Dressman JB. Mechanistic investigation of the negative food effect of modified release zolpidem. Eur J Pharm Sci. 2017;102:284‐298. [DOI] [PubMed] [Google Scholar]
- 33. Pepin XJ, Flanagan TR, Holt DJ, Eidelman A, Treacy D, Rowlings CE. Justification of drug product dissolution rate and drug substance particle size specifications based on absorption PBPK modeling for lesinurad immediate release tablets. Mol Pharm. 2016;13:3256‐3269. [DOI] [PubMed] [Google Scholar]
- 34. Lipka E, Lee ID, Langguth P, Spahn‐Langguth H, Mutschler E, Amidon GL. Celiprolol double‐peak occurrence and gastric motility: nonlinear mixed effects modeling of bioavailability data obtained in dogs. J Pharmacokinet Biopharm. 1995;23:267‐286. [DOI] [PubMed] [Google Scholar]
- 35. Li M, Zhang X, Wu D, et al. Understanding in vivo dissolution of immediate release (IR) solid oral drug products containing weak acid BCS class 2 (BCS class 2a) drugs. AAPS J. 2021;23:113. [DOI] [PubMed] [Google Scholar]
- 36. Langguth P, Lee KM, Spahn‐Langguth H, Amidon GL. Variable gastric emptying and discontinuities in drug absorption profiles: dependence of rates and extent of cimetidine absorption on motility phase and pH. Biopharm Drug Dispos. 1994;15:719‐746. [DOI] [PubMed] [Google Scholar]
- 37. Weitschies W, Wedemeyer RS, Kosch O, et al. Impact of the intragastric location of extended release tablets on food interactions. J Control Release. 2005;108:375‐385. [DOI] [PubMed] [Google Scholar]
- 38. Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45:1213‐1226. [DOI] [PubMed] [Google Scholar]
- 39. Simon JA, Robinson DE, Andrews MC, et al. The absorption of oral micronized progesterone: the effect of food, dose proportionality, and comparison with intramuscular progesterone. Fertil Steril. 1993;60:26‐33. [PubMed] [Google Scholar]
- 40. Vinarov Z, Dobreva P, Tcholakova S. Effect of surfactant molecular structure on progesterone solubilization. J Drug Deliv Sci Technol. 2018;43:44‐49. [Google Scholar]
- 41. Center for Drug Evaluation and Research. US Food and Drug Administration . 216718Orig1s000 product quality review. 2023. Accessed May 2, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/216718Orig1s000ChemR.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
Individual participant data collected during the trial may be shared after anonymization and upon approval of the research proposal. Biogen commits to sharing patient‐level data, study‐level data, CSRs, and protocols with qualified scientific researchers who provide a methodologically sound proposal. Biogen reviews all data requests internally based on the review criteria and in accordance with our Clinical Trial Transparency and Data Sharing Policy. Deidentified data and documents will be shared under agreements that further protect against participant reidentification. To request access to data, please visit https://vivli.org/.