

Abstract
Objective:
A de novo antineoplastic drug was planned to suppress and modulate the Head, Neck, and Oral Cancer.
Methods:
Using the computational software tools including molecular docking, molecular dynamics (MD), and post-molecular dynamics bond contact analyses, it has been shown that the new drug called ‘’Innovative Head, Neck, and Oral Cancer Suppressor’’, or simply abbreviated as “IHNOCS” is very effective in terms of suppressing and co-modulating TGF-β and KRTAP2-3 together.
Result:
The drug suppresses the KRTAP2-3 protein activity while also holding onto TGF-β and modulating it to slow down and halt the metastasis.
Conclusion:
We have effectively created a novel medication using principles of theoretical chemistry, biochemistry, pharmaceutical chemistry and organic chemistry and organic chemistry to inhibit Head, Neck, and Oral Cancer. This medication should further undergo experimental testing in various stages, including in vitro, in vivo, and human clinical phases. It exhibits significant effectiveness in inhibiting the progression of cancer by simultaneously targeting TGF-β and KRTAP2-3, thereby impeding metastasis and suppressing the disease.
Key Words: Head- Neck, Oral Cancer, De novo in silico Drug Design- KRTAP 2-3, TGF-β, Molecular Docking
Introduction
Head and neck cancer is squamous cell carcinoma originating from the epithelium in certain parts of the throat and head, and is called head and neck cancer. Head and neck cancer accounts for approximately 10% of all cancer types [1]. In the data discussed in 2020, 19.3 million new cancer cases occurred worldwide. The data of some regions called head and neck cancer are listed as follows; 604,100 cases in the esophagus, 377,713 cases in the lip and oral cavity, 184,615 cases in the larynx, 53,583 cases in the salivary glands [2].
One can think right away the method of reducing the metastasis of head, neck and oral cancer is via suppressing the Transforming Growth Factor β (TGF-β) according to the vast scientific data and literature. However, when TGF-β was suppressed, the metastasis diminishes rapidly while enhancing the proliferation since TGF-β is known to possess high motility and metastatic potential to cancer cells [3–6]. It should be noted that in the scientific literature, the motility of cancerous cells increases with the expression of Keratin-associated protein 2-3 (KRTAP2-3) since it’s the regulator and mediator of the cancer promoting effect of TGF-β [7]. Thus, TGF-β induced KRTAP2-3 is overexpressed in migrated cancerous cells, and somehow if it can be suppressed, the metastasis would slow down to a halt.Designing a new drug which will be named as ‘’Innovative Head, Neck, and Oral Cancer Suppressor’’, or simply abbreviated as “IHNOCS” in its patent for the argument’s sake that can both diminish the activity of TGF-β and suppress the KRTAP2-3 protein as co-modulating both of them via in silico studies is the idea in this research to effectively the solve the problem. The goal here is to complete the research with comprehensive computational simulation tools to illustrate efficiently how the drug matches the functional groups of TGF-β and KRTAP2-3 forming a triple complex to modulate TGF-β and suppress KRTAP2-3. Computational chemistry has reached a spectacular point that studying these kinds of bonding affinities via molecular docking, molecular dynamics (MD) and post-molecular dynamics bonding contact analyses is a crucial methodology to precisely compute and estimate the affinity and tendency of the mode of binding as shown in the scientific literature since when these tools come together, they lead to 100% verification by in vitro experiments [8–11].
This such precision and accuracy of the computational simulation tools once more allow us to study TGF-β’s Hydrogen binding to our de novo drug. TGF-β should not be suppressed totally since this would lead to the cancerous cell proliferation while the metastasis and migration stopping. However, when TGF-β’s activity is diminished by a designed drug and allowing it to be modulated via Hydrogen bonding rather than very strong covalent bonding, both metastasis and the cell proliferation effect of TGF-β will diminish by the help of reversible competitive inhibition. Forming a triple complex of “drug - TGF-β - KRTAP2-3” would lead to further diminish in metastasis, leading to almost stopping the cancerous cell migration. Using this hypothesis, the computed and simulated drug has revealed the capabilities of hitherto unknown mechanisms as a brand-new antineoplastic drug that can be studied in further in vitro, in vivo and human phase stage studies to shed light onto a brighter future to eliminate Head, Neck, and Oral cancer.
Materials and Methods
Geometric Optimization
To identify the active sites of a molecule and explore its interactions with receptors, it is crucial to accurately determine its optimal geometric structure. In our current investigation, we designed a ligand to inhibit the TGF-β - KRTAP2-3, drawing upon the extensive pharmaceutical chemistry expertise of our research group. The resulting organic chemical structures, in their most stable molecular geometries, were subjected to analysis using the Gaussian 09 program [12] with density functional theory (DFT)/B3LYP functional [13] and utilizing the 6-31G(d,p) principle. This process led to the formation of the most stable molecular structures of IHNOCS - TGF-β - KRTAP2-3 intended for further computational and simulation-based research, as depicted in Figure 1 (patented). For molecular docking and molecular dynamics computations, as well as post-processing of output files, Gauss View 6.0 and Avogadro 1.95 software [14] were employed to prepare input files.
Figure 1.
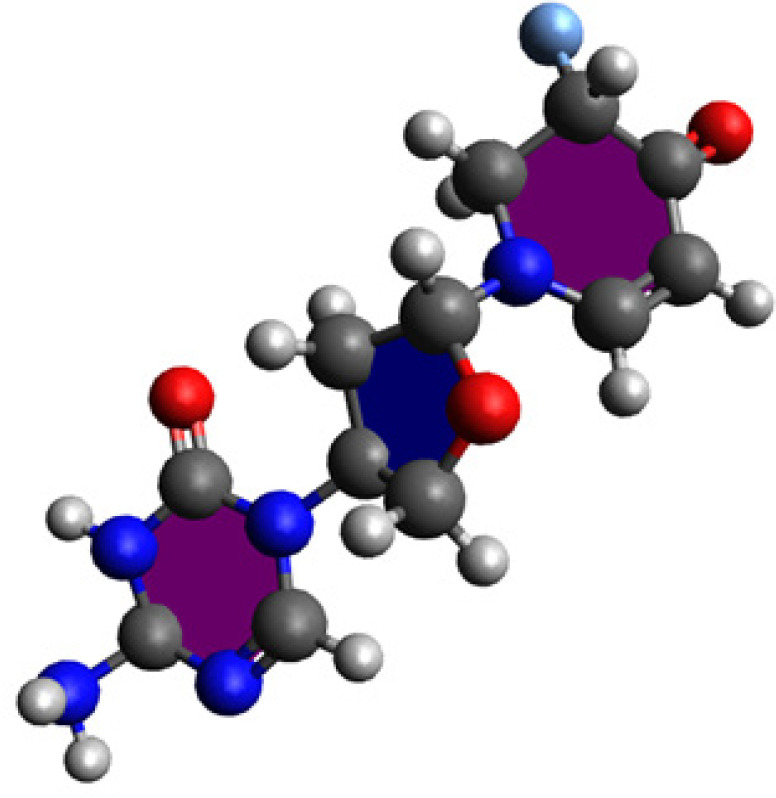
The Designed Chemical Structure of IHNOCS at pH 5.0 Medium for Cancerous Cell Cystoplasm.
Molecular Docking Procedure
The molecular docking simulations were carried out using AutoDock Vina 1.1.2 and PyRx 0.8 software [15]. These programs are widely recognized for their high precision and accuracy in biochemical docking simulations. A total of 1000 poses were generated, with 100 poses for each simulation. The simulations involved the newly designed drug IHNOCS and its interaction with the receptor structure of TGF-β, which had been optimized and designed as an organic molecule. Additionally, the KRTAP2-3 protein, derived from the expression of the NCBI gene 100288323, was also studied.
The optimization of the drug and protein structures was performed using Gauss View 6.0 and Avogadro 1.95 software. The simulations demonstrated the interactions and binding of the drug to the receptor. The docking scores obtained in kcal/mol represented the Gibbs free binding energy.
From all the simulations, the docking pose with the most accurate and favorable binding energy, identified within the best-clustered data, was selected as the initial structure and input file for the subsequent molecular dynamics (MD) simulations. Each MD simulation was conducted with different seed numbers.
Molecular Dynamics (MD) Simulations
The initial structures for the MD simulations were selected from the docking poses with the most favorable binding energy methodology [6,16]. Schrödinger’s Maestro Desmond Program [17] was utilized for running the molecular dynamics (MD) simulations, each spanning 50 ns with 5000 poses at 10 ps intervals. To ensure accuracy, each MD simulation was repeated three times with different seed numbers, confirming the correctness of the simulation parameters and the structures of the complexes formed by the IHNOCS (ligand) with TGF-β and KRTAP2-3 (receptors).
During the MD simulations, the dynamic properties of the drug-receptor complexes were assessed over time. The simulation area was defined by a grid box measuring 110 × 110 × 110 Å3 with a spacing of 0.5 Å, offering wider simulation coverage. TIP3P-type water molecules were included within the box, and 0.15 M NaCl ions were added to neutralize the system.
The temperature and pressure conditions were set as follows
NPT at 310 K with Nose-Hoover temperature coupling [18] and a constant pressure of 1.01 bar via Martyna Tobias−Klein pressure coupling [19]. The systems were not constrained, and the default fitting for OPLS 3.0 standards provided the initial velocity values for the forcefield calculations.
Post Molecular Dynamics Characterizations
The MD trajectory datasets were employed for “Hydrogen contact mapping analyses” allowing a detailed examination of the binding effects of the complexes and the assessment of the drug’s effectiveness on an atom-by-atom basis. To achieve this, the “Event Analysis” modules within Schrödinger’s Maestro Desmond Program were utilized. These obtained results were then compared to the inhibition constant values to gain insights into the drug’s performance.
Results
To achieve the goal of above-mentioned hypothesis, one should know the pharmaceutical chemistry tendencies of functional groups, its effects on various amino acids and its bioactivity features. The effectiveness of interactions appears to depend on various factors, including the affinity of the drug’s external groups to protein, and the topology of the binding.
In Figure 1, the organic chemical structure of the designed drug can be seen at pH 5.0 for the cancerous cell medium. By simulating and computing the theoretical stability and binding energies of IHNOCS docked onto TGF-β and KRTAP2-3, Figure 2 illustrates one of the main poses of the IHNOCS, taken under 3D video within Schrödinger’s Maestro Desmond MD software, where the MD run had 5000 frames.
Figure 2.
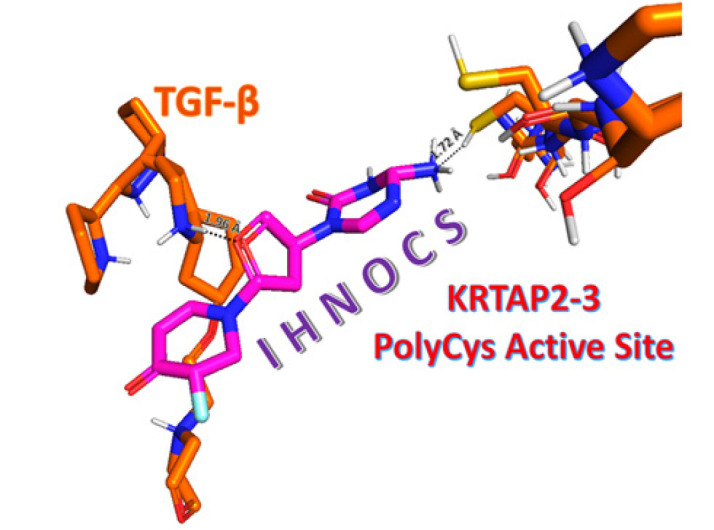
The 3-D Structure of IHNOCS Bound to TGF-β and KRTAP2-3 for Suppression at pH 5.0 Medium Simulating Cancerous Cell Cytoplasm.
After the triple complex equilibrium and stabilization occur approximately 15 - 20 nanoseconds in Figure 3, which is basically after the 2000th frame, IHNOCS formed the stable complex with TGF-β and KRTAP2-3. Along with the MD results in Schrödinger’s Desmond and according to the cluster analyses of 100 posed trial from the molecular docking studies in Autodock Vina, it forms strong H-bonds with TGF-β - KRTAP2-3 and the obtained docking energies are around -11.2 kcal/mol.
Figure 3.

The Root Mean Square Deviation Plots of All Atoms of IHNOCS - TGF-β - KRTAP2-3 Complex in Blue and Non-Ligand Bound KRTAP2-3 in Red, Respectively.
As can be seen in Figure 3, the dynamic properties of the IHNOCS and KRTAP2-3 protein’s PolyCysteine active site as well as along with the TGF-β were investigated via post-MD analysis technique of RMSD.
The post-MD simulation trajectories yielded us that for the KRTAP2-3 protein with no ligand bound system reached equilibrium without any problem shown in red while the “TGF-β-KRTAP2-3-IHNOCS” complex in blue reaches better stability towards the end of 50 nanoseconds MD. This proves that the MD run occurred and ended without any stability problem and the complex is more stable than the KRTAP2-3 protein itself.
In Table 1, after the MD’s 3D motion trajectory analyses, a further study was researched to calculate all of the possible interactions between the functional groups on drugs and the amino acids. This is a very complex affinity and binding study where all of the hydrogen bonds among thousands of atoms were examined from the matrix data of trajectory files of the MD study. Thus, the post-MD Hydrogen contact mapping analysis was done using matrix matches for each atom manually and a machine learning pre-coded script to find the regioselectivity and probability of tendencies of functional groups on drugs towards the amino acids of KRTAP2-3 protein. When it was completed, cysteine was seen to be having a major regioselectivity (approximately 50%) by the drug.
Table 1.
The Post-Molecular Dynamics Hydrogen Bond Contact Analyses of IHNOCS.
| Protein domain | % of H-Bonds Drug-Amino acid |
Binding Energy (kcal/mol) |
H-Bond Distance (Å) |
|---|---|---|---|
| Proline | 12.1 | -8.4 | 1.94 |
| Cysteine | 51.3 | -11.2 | 1.72 |
| Arginine | 16.1 | -10.3 | 2.11 |
| The rest | 20.5 | - | - |
Discussion
The drug aligns and maneuvers itself into the active site groove of the KRTAP2-3 protein, and strong hydrogen forces (1.72 Å) take place there so that IHNOCS suppresses the protein activity of KRTAP2-3 while also holding onto TGF-β and modulating it.
In conclusion, all in all, we have successfully developed a new medicinal what was designed with pharmaceutical organic chemistry for the suppression of Head, Neck and Oral Cancer that can lead to in vitro, in vivo, and human phase stages. The drugs’ inhibition is significantly effective while co-modulating TGF-β and KRTAP2-3 to halt the metastasis and suppress the cancer.
Acknowledgements
Funding statement
This study was funded by Kocaeli Health and Technology University supercomputer infrastructure and Assistant Professor Soykan Agar at the faculty of Pharmacy. Further software support was achieved by Istanbul Technical University High Performance Computing Center. This study has also been funded by Molecular Cancer Research Association (MOKAD) at Istinye University under the oncology and medical biochemistry team of Prof. Engin Ulukaya (M.D. Ph.D.). The patent application was done for the drug molecule and its mechanism what was shown in this research article via Technology Transfer Office of Istinye University with the application number of P23-0898 and it was accepted in the first stage.
Availability of data (if apply to your research)
It can be shared with open access in case of journal asks of us.
If it was approved by any scientific Body/ if it is part of an approved student thesis
It has passed the preliminary process for the acceptance of ‘’Turkish Patent Office’’.
Any conflict of interest
Authors Assistant Professor Soykan AGAR (Ph.D.), Mohaddeseh Mokhtari (Pharmacy Undergraduate Assistant), Muhammed Yanik (Graduate Software System Manager), Barbaros Akkurt (Post-Doc Teaching Fellow, Ph.D.) and Professor Engin Ulukaya (M.D., Ph.D.) declare that there is no conflict of interest for this research paper and its datasets.
How the ethical issue was handled (name the ethical committee that approved the research)
All in silico data was studied and represented with honest work and since it was an in silico simulation study, not an in vitro experimental study, there was no such need for ethical committee approval for this research paper which is compatible with the laws of national ethical committee.
Author Contribution Statement
Soykan Agar: Writing the original and final drafts, experimenting in silico simulations, supervision; Mohaddeseh Mokhtari: Research paper formatting, checking the final draft; Muhammed Yanik: As a software system manager, checking the simulational software progress and mise-en-page of the paper; Barbaros Akkurt: Mise-en-page and reference proofreading; Engin Ulukaya: Supervision.
References
- 1.Gormley M, Creaney G, Schache A, Ingarfield K, Conway DI. Reviewing the epidemiology of head and neck cancer: definitions, trends and risk factors. Br Dent J. 2022;233:780–6. doi: 10.1038/s41415-022-5166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clin. 2021;71:209–49. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 3.Bierie B, Moses H. TGF-β and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Bian Y, Terse A, Du J, Hall B, Molinolo A, Zhang P, et al. Progressive Tumor Formation in Mice with Conditional Deletion of TGF-β Signaling in Head and Neck Epithelia Is Associated with Activation of the PI3K/Akt Pathway. Cancer Res. 2009;69:5918–26. doi: 10.1158/0008-5472.CAN-08-4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellomo C, Caja L, Moustakas A. Transforming growth factor β as regulator of cancer stemness and metastasis. Br J Cancer. 2016;115:761–9. doi: 10.1038/bjc.2016.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi K, Podyma-Inoue KA, Saito M, Sakakitani S, Sugauchi A, Iida K, et al. TGF-β generates a population of cancer cells residing in G1 phase with high motility and metastatic potential via KRTAP2-3. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111411. [DOI] [PubMed] [Google Scholar]
- 8.Şenel P, Agar S, Sayin VO, Altay F, Yurtsever M, Gölcü A. Elucidation of binding interactions and mechanism of Fludarabine with dsDNA via multispectroscopic and molecular docking studies. J Pharm Biomed Anal. 2020;179:112994. doi: 10.1016/j.jpba.2019.112994. [DOI] [PubMed] [Google Scholar]
- 9.Şenel P, Agar S, İş YS, Altay F, Gölcü A, Yurtsever M. Deciphering the mechanism and binding interactions of Pemetrexed with dsDNA with DNA-targeted chemotherapeutics via spectroscopic, analytical, and simulation studies. J Pharm Biomed Anal. 2022;209:114490. doi: 10.1016/j.jpba.2021.114490. [DOI] [PubMed] [Google Scholar]
- 10.Cheraghi S, Şenel P, Dogan Topal B, Agar S, Majidian M, Yurtsever M, et al. Elucidation of DNA-Eltrombopag Binding: Electrochemical, Spectroscopic and Molecular Docking Techniques. Biosensors. 2023;13 doi: 10.3390/bios13030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Şenel P, Agar S, Yurtsever M, Gölcü A. Voltammetric quantification, spectroscopic, and DFT studies on the binding of the antineoplastic drug Azacitidine with DNA. J Pharm Biomed Anal. 2024;237:115746. doi: 10.1016/j.jpba.2023.115746. [DOI] [PubMed] [Google Scholar]
- 12.Frisch MJ. gaussian 09, Revision d. 01. Wallingford CT: Gaussian. Inc; 2009. p. 201. [Google Scholar]
- 13.Becke A. Density-Functional Thermochemistry III The Role of Exact Exchange. J Chem Phys. 1993;98:5648–52. [Google Scholar]
- 14.Dennington R, Keith TA. GaussView V. 6.1. Semichem Inc., Shawnee Mission, KS; 2016. [Google Scholar]
- 15.Gaillard T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J Chem Inf Model. 2018;58:1697–706. doi: 10.1021/acs.jcim.8b00312. [DOI] [PubMed] [Google Scholar]
- 16.Agar S, Akkurt B, Ulukaya E. The Inhibition Mechanism of Pancreatic Ductal Adenocarcinoma via LXR Receptors: A Multifaceted Approach Integrating Molecular Docking, Molecular Dynamics and Post-MD Inter-Molecular Contact Analysis. Asian Pac J Cancer Prev. 2023;24:4103–9. doi: 10.31557/APJCP.2023.24.12.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmond DE. Shaw Research. New York: 2017. [Google Scholar]
- 18.Evans DJ, Holian BL. The Nose–Hoover thermostat. J Chem Phys. 1985;83:4069–74. [Google Scholar]
- 19.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–89. [Google Scholar]