

Abstract
Rotavirus particles are activated for cell entry by trypsin cleavage of the outer capsid spike protein, VP4, into a hemagglutinin, VP8*, and a membrane penetration protein, VP5*. We have purified rhesus rotavirus VP4, expressed in baculovirus-infected insect cells. Purified VP4 is a soluble, elongated monomer, as determined by analytical ultracentrifugation. Trypsin cleaves purified VP4 at a number of sites that are protected on the virion and yields a heterogeneous group of protease-resistant cores of VP5*. The most abundant tryptic VP5* core is trimmed past the N terminus associated with activation for virus entry into cells. Sequential digestion of purified VP4 with chymotrypsin and trypsin generates homogeneous VP8* and VP5* cores (VP8CT and VP5CT, respectively), which have the authentic trypsin cleavages in the activation region. VP8CT is a soluble monomer composed primarily of β-sheets. VP5CT forms sodium dodecyl sulfate-resistant dimers. These results suggest that trypsinization of rotavirus particles triggers a rearrangement in the VP5* region of VP4 to yield the dimeric spikes observed in icosahedral image reconstructions from electron cryomicroscopy of trypsinized rotavirus virions. The solubility of VP5CT and of trypsinized rotavirus particles suggests that the trypsin-triggered conformational change primes VP4 for a subsequent rearrangement that accomplishes membrane penetration. The domains of VP4 defined by protease analysis contain all mapped neutralizing epitopes, sialic acid binding residues, the heptad repeat region, and the membrane permeabilization region. This biochemical analysis of VP4 provides sequence-specific structural information that complements electron cryomicroscopy data and defines targets and strategies for atomic-resolution structural studies.
Dehydrating diarrhea caused by rotavirus infection kills an estimated 600,000 children annually (2). The entry apparatus of rotavirus, consisting of the spike protein, VP4, and the outer capsid glycoprotein, VP7, is an important target of preventive and therapeutic interventions. VP4 and VP7 make up the outer capsid of the triple-layered, nonenveloped, icosahedral rotavirus virion and are the targets of neutralizing and protective antibodies against rotavirus (26). The outer capsid is shed during entry, a poorly understood process that delivers the transcriptionally active, 660-Å double-layered particle across a lipid bilayer and into the cytoplasm.
VP4 has a central role in cell entry by rotavirus. Efficient infectivity of rotavirus in cell culture requires trypsin cleavage of VP4 into two fragments, VP5* and VP8*, both of which remain associated with the virion (7, 12). Activation of rotavirus for membrane interaction and infectivity has been mapped to a specific cleavage site after residue R247 of VP4 (3, 15). The VP8* trypsin cleavage product contains the viral hemagglutinin (HA) (13); the VP5* fragment contains an internal hydrophobic region that has been linked to the ability of activated rotavirus virions to permeabilize membranes (9, 11). VP5* has also been implicated in the binding of sialic acid-independent strains of rotavirus to cells (40).
Image reconstructions from electron cryomicroscopy of trypsinized rotavirus particles demonstrate that VP4 forms dimeric spikes with lobed heads (36, 39). An additional domain of VP4 is buried beneath the VP7 shell and interacts extensively with the underlying VP6 layer (36, 39). Reconstructions of rotavirus virions bound by Fabs recognizing VP8* or VP5* show that a VP8* epitope is located in the heads of the spikes and that an epitope in the internal hydrophobic region of VP5* is located just proximal to the heads (K. A. Dryden, M. Tihova, A. R. Bellamy, H. Greenberg, and M. Yeager, Abstr. Seventh Int. Symp. Double-Stranded RNA Viruses, abstr. P2-15, 2000).
This work describes the purification and characterization of recombinant rhesus rotavirus (RRV) VP4 expressed in insect cells. It describes a proteolysis-triggered conformational change in the VP5* region of VP4 that probably mimics trypsin-induced rearrangements during virus activation. The proteolytic mapping of VP4 provides sequence-specific structural information, which complements electron cryomicroscopy data and defines targets for high-resolution structural studies.
MATERIALS AND METHODS
Antibodies and virus strains.
Monoclonal antibodies HS1 and HS2 were produced as mouse ascites fluids. HS1 recognizes a nonneutralizing epitope on VP8*, and HS2 recognizes a nonneutralizing epitope on VP5* (28). The recombinant baculovirus that expresses RRV VP4 was constructed by Erich Mackow and has been previously described (21).
Insect cell culture and baculovirus cultivation.
The recombinant baculovirus was propagated in Sf9 insect cells with Hinks' TMN FH insect cell medium (JRH Biosciences) supplemented with 10% fetal bovine serum (HyClone) and in Sf900II insect cell medium (Gibco-BRL). Virus titration, amplification, and storage were performed as previously described (38). For production of recombinant VP4, Sf9 insect cells were infected with recombinant baculovirus at a multiplicity of infection of 0.4, maintained in spinner flasks at 28°C, and harvested by centrifugation at 72 to 86 h postinfection.
Purification of VP4.
Recombinant baculovirus-infected cell pellets were lysed by freezing and then thawed in a solution containing 75 mM Tris (pH 8.0), 100 mM NaCl, 2 mM EDTA, 7.5% glycerol, 2.5 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was clarified by centrifugation at 235,400 × g for 2 h. The clarified insect cell lysate was diluted with 19 volumes of 20 mM Tris (pH 8.0)–10 mM NaCl–1 mM EDTA–0.5 mM benzamidine. Proteins in the diluted lysate were bound to Fast-flow DEAE Sepharose (Amersham-Pharmacia Biotech) and eluted with an NaCl gradient. Fractions containing VP4 were identified by Western blotting with monoclonal antibodies HS1 and HS2 and pooled. Pooled fractions were concentrated and exchanged into a buffer containing less than 20 mM NaCl using a Centricon-plus 80 ultrafiltration unit (Amicon, Inc.). The exchanged sample was bound to a 20 HQ anion-exchange column (PerSeptive Biosystems) and eluted with an NaCl gradient. Fractions containing VP4 were pooled and concentrated by ultrafiltration. Finally, VP4 was fractionated on a Hi-Load 16/60 Superdex 200 gel filtration column (Amersham-Pharmacia Biotech) equilibrated in 20 mM Tris (pH 8.0)–100 mM NaCl–1 mM EDTA (TNE). DEAE and Superdex 200 chromatography were performed on an FPLC system (Amersham-Pharmacia Biotech). 20 HQ chromatography was performed on a BioCAD Sprint chromatography system (PerSeptive Biosystems).
Generation of purified VP8CT and VP5CT from VP4.
A 1.1-mg/ml solution of purified VP4 in TNE was made 4.2 μg/ml in TLCK (1-chloro-3-tosylamido-7-amino-2-heptanone)-treated chymotrypsin (Worthington Biochemical) and incubated for 30 min at 37°C. The solution was briefly chilled on ice. TPCK [l-(tosylamido-2-phenyl) chloromethyl ketone]-treated trypsin (Worthington Biochemical) was added to 3.6 μg/ml, and the solution was incubated for an additional hour at room temperature. The digestion was stopped by the addition of PMSF (Sigma) to 1 mM, followed by incubation on ice for 10 min and the addition of benzamidine to 2.5 mM. In some preparations, the sample was passed over a benzamidine-Sepharose column (Amersham-Pharmacia Biotech) prior to the addition of soluble protease inhibitors. The stopped digest was concentrated with a Centricon 10 ultrafiltration unit (Amicon). The resulting fragments were separated by gel filtration using a Superdex 200 HR 10/30 column (Amersham-Pharmacia Biotech) that had been equilibrated in TNE. Fractions in the main peaks containing well-separated VP5* and VP8* protease-resistant cores were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and pooled separately. Stocks of trypsin and chymotrypsin were resuspended from a lyophilized powder to 5 mg/ml in 1 mM HCl and used within 1 week of preparation. Trypsin and chymotrypsin stock solutions were diluted 1:200 in 20 mM Tris-Cl (pH 8.0)–100 mM NaCl–2 mM CaCl2 immediately before use. PMSF was prepared as a 100 mM stock solution in methanol. For small-volume digestions, the PMSF stock was diluted with TNE immediately before addition to each reaction mixture. The above is an optimized protocol. Minor variations used in some digestions are noted in the text and figure legends.
Analytical ultracentrifugation.
Samples for analytical ultracentrifugation were microdialyzed against TNE using a 6,000- to 8,000-molecular-weight (MW) cutoff dialysis membrane (Spectrum Medical Industries). The diasylate was used as a blank and as diluent for centrifugation. Analytical centrifugation was performed using an Optima XL-A analytical ultracentrifuge (Beckman Coulter) with an AN-60 Ti rotor. Absorbance data were obtained at 280 nm with 10 replicates per measurement. Centrifugation speeds were set based on preliminary estimates of molecular masses and standard nomograms (31). The attainment of equilibrium was confirmed by comparing absorbance curves obtained at 6-h intervals.
Equilibrium ultracentrifugation data were fit to the following model:
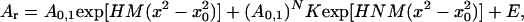 |
where Ar is the absorbance at radius x; A0,1 is the absorbance of the monomer at the reference radius x0; H is a constant to account for the specific volume of the protein, the solvent density, the angular velocity of the rotor, and the temperature; M is the monomer MW; N is the stoichiometry of the association; K is the association constant; and E is the baseline offset. A0,1 is constrained to be greater than 0. Specific volumes of the proteins and the density of TNE at 4°C were calculated based on amino acid composition and published data (24).
The sedimentation coefficient, s, at 4°C in TNE was determined by the slope of a plot of ln(r) versus ω2t, where r is the radial position (in centimeters) of the square root of the second moment of the sedimenting boundary of VP4 as determined by absorbance at 280 nm, ω is the rotor angular velocity (radians per second), and t is the duration of sedimentation (in seconds). The S value was converted to S20,w (S at 20°C in water) using a TNE viscosity of 1.018 cP at 20°C and a VP4-specific volume of 0.729 cm3/g at 20°C (31). The viscosity and specific volume were calculated based on published data (24). For calculation of the frictional ratio (f/f0), the actual (f) and minimal (f0) frictional coefficients were calculated, as described previously (17), based on the S20,w for VP4, an estimate of hydration of 0.3 g of H2O/g of VP4, and the mass spectrometry-determined MW of VP4. The axial ratio was estimated from the frictional ratio based on published graphs (17).
CD spectroscopy.
Samples for circular dichroism (CD) spectroscopy were buffer exchanged into 20 mM boric acid-NaOH (pH 8.0)–100 mM sodium fluoride by microdialysis. CD was measured with an Aviv model 62DS CD spectrometer using a 1-mm-path-length quartz cuvette. Measurements were obtained in 0.5-nm intervals from 200 to 250 nm with 12 replicates, a 1-nm bandwidth, and a 0.5-s measurement time at each wavelength. Total ellipticity was converted to mean residue molar ellipticity based on the measured concentration of samples recovered from the CD cuvette (as below). To obtain secondary-structure predictions, a ridge regression algorithm (a variant of the least-squares method) was used to fit the CD data to a linear combination of the CD spectra of 16 proteins with determined structures (30).
Quantitation of protein.
The concentrations of VP4, VP5CT, and VP8CT were determined by spectrophotometry at 280 nm. Absorbances were converted to concentrations using molar extinction coefficients of 95.3 × 103 M−1 cm−1 for VP4, 42.2 × 103 M−1 cm−1 for VP5CT, and 39.6 × 103 M−1 cm−1 for VP8CT. Extinction coefficients were calculated based on the predicted amino acid sequences. For more accurate quantitation of the samples used in CD spectroscopy, the samples were diluted with 3 volumes of 8 M guanidine-HCl prior to spectrophotometry.
Amino acid sequencing.
Protein fragments for N-terminal sequencing were separated by SDS-PAGE and electroblotted onto a Sequi-Blot polyvinylidene difluoride membrane (Bio-Rad). Coomassie blue-stained bands were excised, and the N-terminal sequence was obtained at the Tufts Protein Chemistry Facility (Boston, Mass.).
Mass spectrometry.
Matrix-assisted laser desorption ionization (MALDI)–time of flight mass spectrometry for full-length VP4 and cross-linked VP5CT was performed at the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University using a sinapinic acid matrix. The sample was calibrated using bovine serum albumin as a standard. The accuracy of measurement is estimated to be ±0.2%. MALDI-time of flight mass spectrometry of protease cleavage fragments of VP4 was performed at the Tufts Protein Chemistry Facility.
Chemical cross-linking.
VP5CT at a concentration of 2 mg/ml was exchanged into 20 mM HEPES (pH 7.0)–100 mM NaCl by microdialysis. Freshly prepared 5-mg/ml bis(sulfosuccinimidyl)suberate (BS3; Pierce Endogen) in 5 mM Na-citrate, pH 5.0, was added to a final concentration of 0.83 mg/ml, and the sample was incubated at room temperature for 30 min. A second equal amount of BS3 was added, and the sample was incubated for an additional 30 min. The cross-linking reaction was quenched by adding Tris-Cl, pH 7.5, to a final concentration of 222 mM. The cross-linked material was microdialyzed against 20 mM ammonium bicarbonate, pH 7.7, and divided into aliquots for SDS-PAGE and mass spectrometry.
Calibration of gel filtration columns.
Superdex 200 gel filtration columns were calibrated using a set of globular protein standards (Amersham-Pharmacia Biotech) consisting of ferritin (440 kDa), aldolase (158 kDa), ovalbumin (43 kDa), and RNase A (13.7 kDa). Calibration curves were constructed by plotting KAV versus ln(MW), where KAV equals (Ve − V0)/(Vt − V0). Ve is the measured elution volume of the markers. V0, the void volume of the column, was determined by gel filtration of blue dextran, and Vt, the total column volume, was specified by the manufacturer (Amersham-Pharmacia Biotech).
Molecular biology and electrophoresis.
Plasmid DNA for sequencing was prepared using Qiagen plasmid Midi kits. DNA oligonucleotide primer synthesis and plasmid DNA sequencing were performed by the Howard Hughes Medical Institute Biopolymer Facility (Boston, Mass.). SDS-PAGE, Coomassie blue staining, electroblotting, and Western blotting were performed using established techniques (34). Sample buffer for SDS-PAGE contained 125 mM Tris-Cl (pH. 6.8), 4% SDS, 1% β-mercaptoethanol, 40% glycerol, and bromphenol blue.
Native PAGE and isoelectric focusing (IEF) were performed on a Phast-gel system (Amersham-Pharmacia Biotech). The native polyacrylamide gel buffer contained 112 mM acetate and 112 mM Tris (pH 8.8), and the native running buffer contained 880 mM l-alanine and 250 mM Tris (pH 8.8). Gradient gels for SDS-PAGE were obtained from Bio-Rad, SDS-PAGE standards were obtained from Gibco-BRL, and markers for native gel electrophoresis and IEF were obtained from Amersham-Pharmacia Biotech. The following native gel markers have the indicated molecular masses: catalase, 232 kDa; lactate dehydrogenase, 140 kDa; and bovine serum albumin, 66 kDa. To stain IEF gels with Coomassie blue, 0.1% CuSO4 was added to the staining solution to decrease background (32).
Computation.
DNA and amino acid sequence data were analyzed using the Lasergene suite of DNAstar sequence analysis software. Ultracentrifugation data were analyzed using Optima XL-A software version 3.0 (Beckman Instruments, Inc.) and Origin version 3.78 (Microcal Software, Inc). CD data were analyzed using CONTIN (30). Graphs were produced using Origin version 5.0 (Microcal Software, Inc.). Chromatograms were traced in Photoshop, version 6.0 (Adobe, Inc.), and scaled in Illustrator, version 8.0 (Adobe, Inc.).
Nucleotide sequence accession number.
The RRV gene segment 4 cDNA sequence was submitted to GenBank and has been assigned accession number AY033150.
RESULTS
Sequencing of cloned RRV gene segment 4.
The clone of RRV gene segment 4 that was used to make the recombinant baculovirus was sequenced. This clone contains 32 nucleotide changes and encodes 11 amino acid changes relative to a published RRV gene segment 4 sequence, which is based on direct sequencing of pooled RRV mRNA (22). Each amino acid change replaces the residue encoded by the published VP4 sequence with a residue found in the corresponding position in VP4 of rotavirus strains SA11 or YM (18, 19). The changes are as follows: S73 to T, Y132 to N, D311 to E, I338 to V, F421 to L, G445 to S, G446 to R, Y454 to N, L468 to F, Y519 to D, and Y690 to F.
Purification of VP4.
VP4 was purified from the supernatant of a freeze-thaw lysate of recombinant baculovirus-infected Sf9 cells using anion-exchange chromatography with DEAE Sepharose and 20 HQ Poros resin followed by gel filtration chromatography through Superdex 200 beads (see Materials and Methods). Gel filtration chromatography (Fig. 1A) yielded a major peak of VP4 with a KAV of 0.326, corresponding to an apparent molecular mass of 111 kDa. A tiny additional peak, with a KAV of 0.198, corresponding to an apparent molecular mass of 249 kDa, was also seen. Although such higher molecular mass peaks also contain VP4, they are very small. Their formation is increased by denaturing procedures such as freezing and thawing of the sample, and they revert to the 111-kDa peak upon repeat gel filtration of stored fractions (data not shown).
FIG. 1.

Purification of VP4. (A) Gel filtration chromatography. Chromatogram of VP4 separated on a Hi-Load 16/60 Superdex 200 gel filtration column as a final purification step. The Vo is 44.1 ml. Solid line, A280; dashed line, calibration curve; circles, elution volumes of MW markers. (B) SDS-PAGE. Coomassie blue-stained SDS-polyacrylamide gel of purified recombinant VP4. Lane MW, molecular mass standards labeled in kilodaltons; lane 4, VP4.
From each liter of starting insect cell culture we obtained up to 1.5 mg of pure VP4. By Coomassie blue stained and SDS-PAGE, VP4 migrated with an apparent molecular mass of 86.1 kDa and no contaminating protein bands were visible (Fig. 1b). MALDI-time of flight mass spectrometry of purified VP4 yielded a molecular mass of 86,307 ± 172 Da. The predicted molecular mass of VP4 based on its amino acid sequence, assuming N acetylation of the initial methionine, is 86,633 Da. Amino acid sequence could not be obtained from the N terminus of the recombinant protein, suggesting that it, like authentic virion-derived VP4, has a blocked N terminus (18). These results, together with analysis of an early trypsin cleavage fragment of VP4 (see below), indicate that purified recombinant VP4 is missing a single residue, L776, from its C terminus, likely due to proteolysis after R775.
Analytical ultracentrifugation of purified VP4.
Equilibrium analytical ultracentrifugation of VP4 was used to determine the mass of VP4 independent of shape and charge and without disturbing noncovalent interactions. Centrifugation of VP4 at three different starting concentrations yielded absorbance-versus-radius distributions that fit the theoretical distribution of an 86.0-kDa monomeric protein (Fig. 2A). Small systematic residuals near the bottom of the cell in the highest-concentration sample (shown in Fig. 2A) gave evidence of a slight tendency to self-associate. Models in which the VP4 monomer was in equilibrium with a dimer (not shown) did not provide a closer fit to the experimental data, suggesting that aggregation accounts for the residuals.
FIG. 2.

Analytical ultracentrifugation. Curves were fit and residuals were determined as described in Materials and Methods. (A) Equilibrium sedimentation of VP4. Shown is the A280-versus-radius plot of 453 μg of VP4 per ml in TNE following centrifugation at 9,000 rpm in an AN-60 Ti rotor (Beckman Coulter) at 4°C for 89 h. The curve, fit to data sets of VP4 centrifuged at starting concentrations of 112, 226, and 453 μg/ml, is the theoretical distribution of a 86.0-kDa particle with a partial specific volume of 0.7222 cm3/g in a buffer with a density of 1.005 g/ml using a baseline offset of 0.008 A280 unit. (B) Velocity sedimentation of VP4. Shown are results of a second-moment analysis of the sedimentation of 865 μg of VP4 per ml in TNE at 4°C during centrifugation at 25,000 rpm. The fitted line has a slope of 3.3 s. (C) Equilibrium sedimentation of VP8CT. Shown is an A280-versus-radius plot of 317 μg of VP8CT per ml in TNE following centrifugation at 20,000 rpm at 4°C for 45 h. The curve, fit to data sets of VP8CT centrifuged at starting concentrations of 79, 158, and 317 μg/ml, is the theoretical distribution of a 22.4-kDa particle with a partial specific volume of 0.7158 cm3/g in a buffer with a density of 1.005 g/ml using a baseline offset of −0.003 A280 unit. (D) Equilibrium sedimentation of VP5CT. Shown is a A280-versus-radius plot of 392 μg of VP5CT per ml in TNE following centrifugation at 9,000 rpm at 4°C for 90 h. The curve, fit to data sets of VP5CT centrifuged at starting concentrations of 98, 196, and 392 μg/ml, is the theoretical distribution of a 63.4-kDa particle with a partial specific volume of 0.7215 cm3/g that dimerizes with an association constant of 5.1 in a buffer with a density of 1.005 g/ml using a baseline offset of 0.002 A280 unit.
To obtain information about the shape of purified VP4, a sedimentation coefficient was determined by velocity analytical ultracentrifugation. Second-moment analysis of a sedimenting boundary of VP4 at 20 time points during centrifugation yielded an S of 3.30 s in TNE at 4°C (Fig. 2B). This sedimentation coefficient corresponds to an S20,w of 5.21 s. The sedimentation coefficient and the MW determined by mass spectrometry yield a frictional ratio of 1.21. Assuming that VP4 can be modeled as a smooth prolate ellipsoid, the frictional ratio predicts an axial ratio of approximately 1:3.3. (See Materials and Methods for a description of the above calculations.) Because an irregular contour adds drag, it is likely that this number somewhat overestimates the elongation of VP4.
Trypsin digestion of purified VP4.
A protease analysis of soluble VP4 was undertaken to determine if purified VP4 was cleaved at the sites associated with virus activation, to seek evidence for a protease-induced structural rearrangement, and to obtain protease-resistant cores corresponding to structural domains. Trypsin initially cleaved VP4 to produce a fragment with a mass spectrometry-determined molecular mass of 21.86 kDa (not shown, although the minor boiled VP4 band seen in Fig. 5A results from unintended cleavage at or near this site by trace protease). This cleavage was complete by the time that digestion was stopped by adding PMSF immediately after a 850-μg/ml solution of VP4 was made 2.5 μg/ml in trypsin. Edman degradation showed that trypsin initially cleaved after R582 (N-terminal sequence, SVGSS) to produce a fragment (designated VP5Tx in Fig. 3B) with a predicted C terminus after R775, one residue short of the predicted C terminus at L776. Therefore, while trypsin efficiently cleaves virion-associated VP4 only in the activation region (3), the most trypsin-sensitive site on purified VP4 is within the VP5* region, after R582. Mass spectrometry of freshly purified VP4 indicates that the cleavage after R775 likely occurred prior to intentional trypsin digestion (described above).
FIG. 5.

Electrophoresis of VP4, VP5CT, and VP8CT. (A) Coomassie blue-stained SDS-polyacrylamide gel. Purified proteins were denatured with SDS-PAGE sample buffer containing 1% β-mercaptoethanol and either boiled or not boiled (indicated above brackets) prior to separation on a 4-to-15% polyacrylamide gradient gel. Lane MW, molecular mass standards (in kilodaltons); lanes 4, VP4; lanes 5, VP5CT; lanes 8, VP8CT. (B) Coomassie blue-stained native polyacrylamide gel. Shown are purified proteins in TNE electrophoresed on a 4-to-25% polyacrylamide gradient gel with a discontinuous native buffer system (described in Materials and Methods). Lane M, native PAGE markers; LDH, lactate dehydrogenase; BSA, bovine serum albumin. Samples are labeled as in panel A. (C) Coomassie blue stained IEF gel. Purified proteins in TNE were applied to the cathode end of a polyacrylamide gel containing carrier ampholytes in the pH range 4 to 6.5. Markers were applied either at the cathode end (cat) or in the middle of the gel (mid), as indicated above the lanes. IEF was performed until the markers applied at either location had focused to the same position. The pI of each marker is indicated adjacent to the corresponding band, and the position of the cathode is indicated. Samples are labeled as in panel A.
FIG. 3.

Sequential digestion of VP4 with chymotrypsin and trypsin. (A) Coomassie blue stained SDS-polyacrylamide gel. A 1.1-mg/ml solution of VP4 was either digested with 4.2 μg of chymotrypsin per ml for 30 min at 37°C (lanes marked with +) or incubated on ice for 30 min (lanes marked with −). Trypsin was then added to a concentration of 3.6 μg/ml (except in those samples marked − immediately above the lane), and the samples were incubated at room temperature for an additional 15 to 210 min (indicated by numbers immediately above the lanes). Digestions were stopped by the addition of PMSF to 0.625 mM prior to denaturation by boiling in reducing SDS-PAGE sample buffer and separation on a 4-to-15% polyacrylamide gradient gel. Molecular mass standards are identified in kilodaltons adjacent to marker bands. The gel has a slight distortion that causes bands on the right-hand side to appear to migrate further. (B) Schematic. The scale indicates residue numbers of intact VP4. “VP8*” and “VP5*” designate fragments generated by trypsinization of virions (3). “VP5Ca” and “VP5Cb” designate alternate N termini found on fragments with the electrophoretic mobility of band VP5C in panel A. “VP5Tx” designates the fragment first cleaved from intact VP4 upon exposure to trypsin. Other fragment designations refer to the digestion products labeled in panel A. Amino acids specified by number at the N termini of bars were determined by Edman degradation. Amino acids specified at the C termini of bars are preceded by “∼” if an alternate C terminus is also compatible with the data. Dashed ends of boxes designate C termini estimated from SDS-PAGE data. Bold letters in the amino acid sequence designate residues after which cleavages occur upon trypsinization of virion-associated VP4 (3). (C) Mass spectrometry of VP8CT and VP5CT. A dual digestion of VP4 was performed as described in Materials and Methods, and the products were analyzed by MALDI-time of flight mass spectrometry. The amount of VP5CT in the sample was diminished by its relatively poor elution from a benzamidine-Sepharose column. The range of the mass spectrum containing the single ionization peaks for VP8CT and VP5CT is displayed.
More thorough trypsin digestion of VP4 generated protease-resistant cores of VP8* and VP5*. Trypsin cleaved after K29 (N-terminal sequence, TQNVT) to produce VP8T, which has an apparent molecular mass of 23.0 kDa by SDS-PAGE (Fig. 3A and B). Trypsin digestion produced heterogeneous VP5* cores with apparent molecular masses of 32.0, 29.6, and 27.6 kDa by SDS-PAGE (VP5Ta, VP5Tb, and VP5Tc, respectively) (Fig. 3A and B). This pattern was highly reproducible, and the larger fragments did not chase into the smaller fragments with longer digestion. N-terminal sequencing showed that trypsin cleavage after R247 had produced VP5Ta (N-terminal sequence, AQAN) and that cleavage after K258 had produced VP5Tc (N-terminal sequence, TSLW). VP5Tb contained a mixture of both of the above-described N termini, suggesting that it may represent two comigrating fragments. Thus, VP5Tc, the most abundant VP5* tryptic core, lacked the N terminus at A248 that is linked to virus activation for cell entry (3).
Chymotrypsin digestion of purified VP4.
Digestion of VP4 with chymotrypsin also produced cores of VP8* and VP5* (VP8C and VP5C) (Fig. 3A). The VP8C N terminus was A46 (N-terminal sequence, APVNW), and its mass spectrometry-determined molecular mass was 21.32 kDa, predicting a C terminus at L236 (Fig. 3B). Depending on the time of digestion (30 versus 210 min), VP5C had an N terminus at S239 (N-terminal sequence, SARN; VP5Ca in Fig. 3B) or at R247 (N-terminal sequence, RAQAN; VP5Cb in Fig. 3B). VP5Cb produced a broad and weak mass spectrometry peak with a maximum between 31.44 and 31.82 kDa, predicting a C terminus (or C termini) between L525 and F528. Therefore, unlike trypsin, chymotrypsin cut in the activation region of purified VP4 without trimming the N termini of the resulting VP5* cores beyond A248.
Trypsin cleavage of chymotryptic VP4 fragments.
Trypsin cleaved the chymotryptic VP4 cores to form VP8CT and VP5CT (Fig. 3). The products of digestion with chymotrypsin for 30 min at 37°C followed by combined digestion with trypsin and chymotrypsin for 1 h at room temperature were subjected to further analysis. The VP8CT N terminus was at A46 (N-terminal sequence, APVN), demonstrating that trypsin had left the N terminus of VP8C intact. Although VP8CT had a slightly greater apparent molecular mass than that of VP8C by SDS-PAGE (Fig. 3A), mass spectrometry showed its true molecular mass to be 20.88 kDa (Fig. 3C), approximately 440 Da smaller than the molecular mass of VP8C. The anomalous SDS-PAGE mobility of VP8CT relative to that of VP8C was highly reproducible and probably reflects anomalous SDS binding or a retained secondary structure. The N-terminal sequencing and mass spectrometry data show that trypsin cleaved the 5 C-terminal residues from VP8C to yield VP8CT, which is predicted to have the authentic VP8* C terminus at R231 (3).
Edman degradation of VP5CT revealed a single N terminus at A248 (N-terminal sequence, AQAN) (Fig. 3B). VP5CT produced a broad and relatively weak mass spectrometry peak with a maximum at 31.73 kDa (Fig. 3C), predicting that it shares the VP5C C terminus (or C termini) between L525 and F528. Therefore, trypsin had trimmed 9 N-terminal residues from VP5Ca or 1 N-terminal residue from VP5Cb to yield VP5CT, which has the single authentic, entry-associated VP5* N terminus at A248 (Fig. 3). These data indicate that the potential trypsin cleavage site after K258 is more resistant to trypsin cleavage in VP5C than in intact purified VP4. Because VP8CT and VP5CT are relatively (though not completely) homogeneous and have the entry-associated trypsin cleavages in the activation region, these fragments were purified and analyzed further.
Separation of VP8CT and VP5CT by gel filtration chromatography.
Gel filtration chromatography of VP4 fragments generated by sequential digestion with chymotrypsin and trypsin produced two major peaks (Fig. 4A). The major peak, designated “8” in Fig. 4A, had a KAV of 0.502, corresponding to an apparent molecular mass of 27.5 kDa. SDS-PAGE showed that peak 8 fractions contained VP8CT (Fig. 4B). The major peak designated “5” in Fig. 4A had a KAV of 0.348, corresponding to an apparent molecular mass of 90.1 kDa. SDS-PAGE showed that peak 5 fractions contained VP5CT (Fig. 4B). A minor peak eluting before peak 5 had a KAV of 0.225, corresponding to an apparent molecular mass of 231 kDa, and also contained VP5CT by SDS-PAGE (Fig. 4B). Thus, VP5CT formed two peaks with unexpectedly high apparent molecular masses, suggesting the presence of more than one oligomeric form. The main VP5CT peak was substantially broader than the VP8CT peak. The ratio of height to width at half-height was 2.4 × 10−3 A280 units/ml for VP5CT and 1.3 × 10−3 A280 units/ml for VP8CT (measured on a chromatogram where both peaks are fully recorded). The broadening of the VP5CT main peak suggests heterogeneity of VP5CT particle size or a nonideal interaction of VP5CT with the size exclusion medium. Gel filtration chromatography cleanly purified VP5CT and VP8CT from each other (Fig. 5b).
FIG. 4.

Purification of VP5CT and VP8CT. (A) Gel filtration chromatography. Shown is a chromatogram of VP5CT and VP8CT, produced by sequential digestion of VP4 with chymotrypsin and trypsin (protocol is described in Materials and Methods), separated on a Superdex 200 HR 10/30 column. The Vo is 8.43 ml. Solid line, A280; dashed line, calibration curve; circles, elution volumes of MW markers; peak 5, main VP5CT peak; peak 8, VP8CT peak. Fractions are numbered above the abscissa. (B) Coomassie blue-stained SDS-polyacrylamide gel. The fractions labeled in panel A were analyzed on a reducing 4-to-15% polyacrylamide gradient gel. Fraction numbers are indicated immediately above the lanes. Fractions pooled for further analysis of VP5CT and VP8CT are indicated above the brackets. Molecular mass standards are identified in kilodaltons adjacent to the marker bands.
Gel filtration chromatography of the VP4 fragments produced by digestion with chymotrypsin alone produced a similar chromatogram (not shown) in which VP5C formed a peak with a KAV of 0.414, corresponding to an apparent molecular mass of 74.9 kDa. The high apparent molecular mass of VP5C suggests that it, like VP5CT, had oligomerized.
Electrophoretic analyses of VP4, VP8CT, and VP5CT.
Electrophoretic experiments were undertaken to investigate the oligomeric state of VP5CT (Fig. 5). Reducing SDS-PAGE of boiled VP4, VP5CT, and VP8CT yielded major bands with apparent molecular masses of 86.0, 32.7, and 22.6 kDa, respectively (Fig. 5A). These apparent molecular masses are within 2 kDa of the molecular masses determined by mass spectrometry. Reducing SDS-PAGE of unboiled VP4 and VP8 produced major bands with electrophoretic mobilities indistinguishable from those seen with boiled samples, but reducing SDS-PAGE of unboiled VP5CT yielded a major band with an apparent molecular mass of 57.0 kDa (Fig. 5A). Reduction of VP5CT by sample buffer containing 5% β-mercaptoethanol instead of 1% β-mercaptoethanol gave the same result (not shown). Minor bands of unboiled VP5CT had apparent molecular masses of 29.0 and 32.7 kDa, suggesting that boiling had relatively little effect on the SDS-PAGE mobility of monomeric VP5CT. These data indicate that VP5CT forms a dimer that is disrupted by boiling in the presence of SDS and β-mercaptoethanol but not by exposure to SDS and β-mercaptoethanol at room temperature.
Other minor bands in Fig. 5A bear mentioning. SDS-PAGE of boiled or unboiled VP4 produced a minor band with an apparent molecular mass of 70.9 kDa. This band probably results from unintended cleavage of soluble VP4 by traces of protease at a highly sensitive site approximately 22 kDa from the VP4 C terminus (described above). A more slowly migrating band of unboiled VP8CT has an apparent molecular mass of 27.2 kDa, indicating retention of SDS-resistant secondary structure.
VP4 and VP8CT produced tight, single bands by native gel electrophoresis (Fig. 5B) and IEF (Fig. 5C), indicating homogeneity. VP4 and VP8CT focused at pIs of 5.0 and 4.3, respectively (Fig. 5C). In contrast, VP5CT formed a diffuse, single band by native gel electrophoresis (Fig. 5B) and a smear between two diffuse bands with pIs of 4.9 and 5.5 by IEF (Fig. 5C), indicating heterogeneity. The dispersion of VP5CT by IEF is not due to an irreversible pH-induced conformational change, as diluting VP5CT with 3 volumes of 100 mM sodium citrate, pH 5.0, did not alter its subsequent migration by native gel electrophoresis (not shown). A heterogeneous C terminus between L525 and F528 might account for the broad mass spectrometry peak of VP5CT (Fig. 3C). In this case, the presence or absence of a charged residue, D526, may cause VP5CT to form distinct bands by IEF and closely comigrating bands by native PAGE. The presence of a smear between two maxima by IEF suggests, however, that a reversible change in VP5CT, such as a dynamic association between VP5CT dimers or conformational flexibility within dimers, accounts for the results.
Chemical cross-linking of VP5CT.
VP5CT was cross-linked chemically by incubation with BS3, boiled in reducing sample buffer, and separated by SDS-PAGE (Fig. 6). Monomeric cross-linked VP5CT comigrated with the untreated VP5CT at an apparent molecular mass of 31.2 kDa. Major cross-linked species migrated with apparent molecular masses of 65.2 and 137.2 kDa, indicating that a significant amount of VP5CT had been cross-linked into dimers and tetramers. Faint bands with apparent molecular masses of 95.2 and 177.2 kDa may represent small amounts of trimer (or incompletely cross-linked tetramer) and hexamer, respectively. A contaminant band can be seen in both sample lanes at approximately 43 kDa, and a faint haze in the untreated VP5CT lane at approximately 65 kDa may represent a non-cross-linked dimer. Mass spectrometry of the cross-linked material detected 31.27-, 63.04-, 95.10-, and 126.8-kDa species. The cross-linking data are most compatible with V5CT dimers self-associating under nondissociating conditions into tetramers and, possibly, higher-order complexes.
FIG. 6.

Coomassie blue-stained SDS-polyacrylamide gel of chemically cross-linked VP5CT. VP5CT, untreated or cross-linked with BS3 (indicated above lanes), was boiled in reducing SDS-PAGE sample buffer and separated on a 4-to-15% polyacrylamide gradient gel. Molecular mass standards are identified in kilodaltons adjacent to the marker bands.
Analytical ultracentrifugation of VP8CT and VP5CT.
The oligomeric states of VP8CT and VP5CT were further assessed by equilibrium analytical ultracentrifugation. Centrifugation of VP8CT at three different starting concentrations yielded absorbance-versus-radius distributions that fit the theoretical distribution of a nonassociating monomeric protein with a molecular mass of 22.4 kDa (Fig. 2C). Residuals showed a small, random scatter.
Equilibrium ultracentrifugation of VP5CT at three different starting concentrations yielded absorbance-versus-radius distributions that fit the theoretical distribution of a 31.7-kDa protein that forms very tightly associated dimers that further pair to form tetramers with a dimer-dimer association constant of 5.1 (Fig. 2D). This model is consistent with the electrophoretic and cross-linking data presented above, and the residuals show a small, random scatter. Despite the very close fit of this model to the data, the theoretical distribution of a 31.8-kDa protein that trimerizes with an association constant of 3.2 × 105 also gives small residuals, though with a more systematic error (not shown).
CD spectroscopy.
The secondary structure compositions of VP4, VP5CT, and VP8CT were assessed by CD spectroscopy. The theoretical spectra (Fig. 7) of proteins with the compositions given in Table 1 closely match the observed spectra (Fig. 7). The CD data indicate that VP4 and VP5CT have mixed structures but that VP8CT is predominantly composed of a β-sheet. VP4 appears to have a higher proportion of α-helix than does either VP8CT or VP5CT (Table 1). Consistent with this observation, sequence-based structure predictions suggest that the portions of VP4 that are missing from both VP5CT and VP8CT (Fig. 3B) are rich in α-helix (19).
FIG. 7.

CD spectroscopy. In each plot, the experimentally measured mean residue molar ellipticity is depicted by open circles (measured); the ellipticity of a model protein with the composition indi- cated in Table 1 is depicted by filled circles (fit). The fit composition was determined using CONTIN (30). (A) VP4; (B) VP5CT; (C) VP8CT. deg, degree.
TABLE 1.
CD spectroscopy-based prediction of secondary-structure composition
| Structure | % in:
|
||
|---|---|---|---|
| VP4 | VP5CT | VP8CT | |
| α-Helix | 22 | 17 | 7 |
| β-Sheet | 45 | 30 | 59 |
| β-Turn | 9 | 16 | 4 |
| Remainder | 24 | 37 | 30 |
DISCUSSION
A number of viral structural proteins undergo entry-associated conformational changes. Influenza virus HA undergoes a dramatic pH-triggered conformational change to extend a fusion domain from a buried position to a viral-membrane-distal position (5). A drop in pH also triggers the tick-borne encephalitis virus E protein to rearrange from a homodimer to a homotrimer (1). After binding to its cell receptor, poliovirus loses VP4, an internal capsid protein generated by autolytic cleavage, and exposes a hydrophobic region of VP1 (16). The capsids of some nonenveloped plant viruses, such as tomato bushy stunt virus, expand in low-calcium environments (33).
Rotavirus undergoes at least one major entry-related conformational transition: uncoating upon calcium chelation in vitro (8) and upon cell entry (20). A recent biochemical analysis of purified VP7 demonstrated that the dissociation of VP7 trimers to monomers is the biochemical basis for calcium chelation-induced rotavirus uncoating (10). Trypsin cleavage of the rotavirus spike protein, VP4, into the VP8* and VP5* fragments is required for efficient and productive entry of rotavirus into cells (4). Therefore, it is likely that proteolysis primes VP4 for a specific, entry-associated conformational change.
We have described in this paper a biochemical and biophysical analysis of purified recombinant VP4. The goals of this analysis have been to define a protease-triggered, entry-associated conformational change in VP4, to obtain sequence-specific structural data on VP4, and to obtain the biochemical understanding of VP4 required for successful atomic-resolution structural studies.
Gel filtration chromatography and analytical ultracentrifugation demonstrate that purified, recombinant VP4 is a moderately elongated monomer, well behaved in solution (Fig. 1A and 2A and B). Image reconstructions from electron cryomicroscopy of rotavirus particles alone and complexed with VP4-specific Fabs provide convincing evidence that VP4 forms dimers on trypsinized rotavirus particles (29, 36, 39). These findings suggest that dimeric interactions of paired VP4 molecules on the virion probably require stabilization either through a molecular rearrangement induced by trypsin cleavage or through interactions with VP6 (middle capsid layer) or VP7 (outer capsid layer).
Arias and coworkers showed that, during trypsin activation of virus, VP4 is initially cleaved C terminal to R241 and then to R231 and R247 (3). Enhancement of infectivity is specifically associated with the relatively inefficient cleavage after R247. The other potential trypsin recognition sites on virion-associated VP4 are protected from cleavage (3). Further evidence for this activation pathway was provided by a mutational analysis in which VP4 cleavage after R247 (but not after R231 or R241) was required for the induction of cell-cell fusion by virus-like particles (15).
Trypsin cleavage of purified, soluble VP4 is much more extensive (Fig. 3). Trypsin whittles 3.3 kDa from the N terminus of VP8* and approximately 30 kDa from the C terminus of VP5*. These findings suggest that the N-terminal 3.3 kDa and the C-terminal 30 kDa of virion-associated VP4 are sequestered from protease or folded into protease-resistant structures as a direct or indirect consequence of interactions between VP4 and VP6 or VP7.
As summarized in Fig. 3, the most abundant tryptic core of the VP5* region of purified VP4 lacks the entry-associated N terminus at A248 due to preferential trypsin cleavage after K258. Chymotrypsin digestion of purified VP4 protects the site after K258 and allows subsequent specific trypsin cleavage C terminal to R247. VP8CT and VP5CT have the primary structure of the VP4 activation region found on entry-competent virions. Other investigators have found that chymotrypsin cleaves in the activation region of virion-associated VP4 (3) but that this cleavage results in only a transient (4) or minimal (3) increase in infectivity. Subsequent trypsin treatment of chymotrypsin-treated rotavirus results in full enhancement of infectivity (3).
VP8CT is a homogeneous monomer, which is folded into a relatively detergent-resistant structure and is composed primarily of β-sheets (Fig. 2C, 4A, 5, and 6C and Table 1). Reducing SDS-PAGE of unboiled VP5CT indicates that it forms stable, SDS- and β-mercaptoethanol-resistant dimers (Fig. 5A). The absence of VP4 spikes from icosahedrally averaged electron cryomicroscopy-based image reconstructions of uncleaved rotavirus virions suggests that cleavage triggers a major rearrangement in VP4 to produce the dimeric spikes observed on trypsinized virions (8a). The cleavage-dependent dimerization of VP5CT may reflect the same underlying molecular property.
Data obtained under nondissociating conditions indicate that VP5CT dimers weakly self-associate. Specifically, by size exclusion chromatography, VP5CT migrated in two relatively broad peaks at apparent molecular masses of 90.1 and 231 kDa (Fig. 4A); by native gel electrophoresis, VP5CT migrated in a broad band (Fig. 5B); and by IEF, VP5CT formed a smear between two main states (Fig. 5C). The broad main peak of VP5CT observed by size exclusion chromatography and the broad band observed by native gel electrophoresis may contain mixtures of self-associated states of VP5CT dimers in rapid equilibrium. Cross-linking VP5CT with BS3 under nondenaturing conditions yielded VP5CT dimers and tetramers as the major stabilized products (Fig. 6). The equilibrium distribution of VP5CT upon centrifugation is consistent with a weak pairing of dimers to form tetramers (Fig. 2D). The biochemical specificity and biological significance of the SDS-sensitive association between dimers is unclear.
Gel filtration chromatography of VP5C showed that it, too, had oligomerized (not shown). It is likely that dimerization of VP5C sequesters its potential trypsin cleavage site after K258 (Fig. 3B). This site must also be protected, but by a different mechanism, on virion-associated VP4, which does not require chymotrypsin predigestion for efficient trypsin activation. Possibly, the enforced proximity between paired VP4 monomers on the virion leads to rapid dimerization after trypsin cleavage at the more trypsin-sensitive sites C terminal to R241 and R231. This rapid dimerization would prevent cleavage after K258 and permit activation by subsequent specific trypsin cleavage C terminal to R247.
Activated forms of viral fusion proteins and viral particles are often insoluble. For example, both isolated influenza virus HA and intact influenza virions aggregate at the pH of membrane fusion (35). Poliovirus aggregates after binding to a soluble form of its receptor (16). The solubility of proteolytically produced VP5CT (Fig. 2D and 4A) and trypsinized rotavirus virions suggests that they may not be fully activated for membrane attack. Indeed, trypsinized rotavirus particles do not permeabilize liposomes until after they are uncoated by calcium chelation (25). Therefore, the conformational change in VP4 triggered by trypsin cleavage probably primes the molecule for a subsequent rearrangement that actually accomplishes membrane penetration.
In contrast to VP5CT produced proteolytically, VP5CT expressed directly as a glutathione S-transferase-tagged fusion protein in bacteria or as a histidine-tagged fusion protein in insect cells or bacteria is insoluble (data not shown). Proteolytic removal of the glutathione S-transferase tag did not yield soluble VP5CT (not shown). Other investigators have found that directly expressed VP5* constructs are mainly insoluble (personal communication, Erich Mackow) but that they selectively permeabilize liposomes (9). When expressed directly, influenza HA2 (the fusion domain of HA) folds in its activated, low-pH form (6). Thus, proteolytically produced, soluble VP5CT may be a metastable precursor of the directly expressed, insoluble form. Future studies will compare the membrane interactions of directly expressed and proteolytically produced VP5CT and will examine the possibility of a triggered transition between the two forms.
The protease analysis of purified VP4 defines structural domains that match previously defined functional regions. VP8CT contains the mapped VP8*-specific neutralizing-antibody escape mutations (14, 22, 27, 41), the minimal VP8* antigenic peptide (23), and the hemagglutination region (13). VP5CT contains the hydrophobic region associated with membrane interaction (9), the mapped VP5*-specific neutralization escape mutations (22, 27, 37), the minimal VP5* antigenic peptide (23), and a short heptad repeat region.
The protease mapping of VP4 provides sequence-specific data that can be correlated with the shape of the protein as seen by electron cryomicroscopy. For example, the N-terminal 5.0 kDa and the C-terminal 27.4 kDa of purified VP4 were digested by the combination of chymotrypsin and trypsin (Fig. 3B), but they are protected on virion-associated VP4 (3). Image reconstructions from electron cryomicroscopy of virion-associated VP4 show that approximately 30 kDa of each VP4 monomer is buried beneath the VP7 shell (36, 39). Thus, the protease-sensitive C-terminal 27.4 kDa of VP4 probably makes up a substantial proportion of this buried domain.
The dimerization of VP5CT suggests that it forms much of the dimeric stalk of the trypsinized VP4 spike. As VP8CT is monomeric (Fig. 2C), it probably makes up the heads of the VP4 spike, which make no dimeric contacts. VP8CT dissociates from VP5CT (Fig. 4A), while authentic VP8* remains associated with virions following trypsin activation. It is likely, therefore, that the 45 N-terminal residues of VP8*, which are missing from VP8CT, mediate its tight association with VP5*, even after trypsin cleavage. This basic arrangement is supported by images from electron cryomicroscopy of rotavirus particles complexed with Fabs recognizing VP8* and VP5* (K. A. Dryden, M. Tihova, A. R. Bellamy, H. Greenberg, and M. Yeager, Abstr. Seventh Int. Symp. Double-Stranded RNA Viruses, abstr. P2-15, 2000) (29).
Rotavirus provides an unusual opportunity to study cell entry by nonenveloped viruses. All of the known viral components involved in cell entry (cleaved and uncleaved triple-layered particles, double-layered particles, VP4, VP5*, VP7, and VP8*) have now been purified (intact or as protease-resistant cores) in substantial quantity as soluble and biochemically tractable entities. The purified outer capsid proteins undergo triggered, entry-associated conformational changes in simple in vitro systems: purified VP7 trimers dissociate upon calcium chelation (10) and purified VP4 monomers are proteolytically cleaved to yield VP5* dimers. Functional studies of the interactions of these proteins with each other and with membranes will help to define the mechanisms of virus activation and membrane penetration.
Finally, these experiments have defined tractable targets for atomic-resolution structural analysis of the rotavirus entry apparatus. In particular, the characterization of purified VP4 has provided the biochemical basis for nuclear magnetic resonance and X-ray crystallographic studies, which have recently yielded atomic-resolution structures of the VP8* core with and without bound sialic acid (unpublished data).
ACKNOWLEDGMENTS
We thank Marina Babyonyshev for her skillful technical assistance; Michael Berne of the Tufts Protein Chemistry Facility for N-terminal sequencing and mass spectrometry; Walter McMurray and Kathy Stone of the W. M. Keck Foundation Biotechnology Resource Laboratory for mass spectrometry; and Raymond Brown, Susanne Liemann, Andrea Carfi, and Mykol Larvie for helpful discussions.
This work was supported by National Institutes of Health grants K08 AI 001496 to P.R.D and CA-13202 to S.C.H and by a VA Merit Review grant to H.B.G. S.C.H. is an investigator in the Howard Hughes Medical Institute.
REFERENCES
- 1.Allison S L, Schalich J, Stiasny K, Mandl C W, Kunz C, Heinz F X. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol. 1995;69:695–700. doi: 10.1128/jvi.69.2.695-700.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anonymous. Rotavirus vaccines. Wkly Epidemiol Rec. 1999;74:33–38. [PubMed] [Google Scholar]
- 3.Arias C F, Romero P, Alvarez V, Lopez S. Trypsin activation pathway of rotavirus infectivity. J Virol. 1996;70:5832–5839. doi: 10.1128/jvi.70.9.5832-5839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnett B B, Spendlove R S, Clark M L. Effect of enzymes on rotavirus infectivity. J Clin Microbiol. 1979;10:111–113. doi: 10.1128/jcm.10.1.111-113.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bullough P A, Hughson F M, Skehel J J, Wiley D C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Wharton S A, Weissenhorn W, Calder L J, Hughson F M, Skehel J J, Wiley D C. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc Natl Acad Sci USA. 1995;92:12205–12209. doi: 10.1073/pnas.92.26.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark S M, Roth J R, Clark M L, Barnett B B, Spendlove R S. Trypsin enhancement of rotavirus infectivity: mechanism of enhancement. J Virol. 1981;39:816–822. doi: 10.1128/jvi.39.3.816-822.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen J, Laporte J, Charpilienne A, Scherrer R. Activation of rotavirus RNA polymerase by calcium chelation. Arch Virol. 1979;60:177–186. doi: 10.1007/BF01317489. [DOI] [PubMed] [Google Scholar]
- 8a.Crawford S E, Mukherjee S K, Estes M K, Lawton J A, Shaw A L, Ramig R F, Prasad B V. Trypsin cleavage stabilizes the rotavirus VP4 spike. J Virol. 2001;75:6052–6061. doi: 10.1128/JVI.75.13.6052-6061.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denisova E, Dowling W, LaMonica R, Shaw R, Scarlata S, Ruggeri F, Mackow E R. Rotavirus capsid protein VP5* permeabilizes membranes. J Virol. 1999;73:3147–3153. doi: 10.1128/jvi.73.4.3147-3153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dormitzer P R, Greenberg H B, Harrison S C. Purified recombinant rotavirus VP7 forms soluble, calcium-dependent trimers. Virology. 2000;277:420–428. doi: 10.1006/viro.2000.0625. [DOI] [PubMed] [Google Scholar]
- 11.Dowling W, Denisova E, LaMonica R, Mackow E R. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J Virol. 2000;74:6368–6376. doi: 10.1128/jvi.74.14.6368-6376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estes M K, Graham D Y, Mason B B. Proteolytic enhancement of rotavirus infectivity: molecular mechanisms. J Virol. 1981;39:879–888. doi: 10.1128/jvi.39.3.879-888.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuentes-Panana E M, Lopez S, Gorziglia M, Arias C F. Mapping the hemagglutination domain of rotaviruses. J Virol. 1995;69:2629–2632. doi: 10.1128/jvi.69.4.2629-2632.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giammarioli A M, Mackow E R, Fiore L, Greenberg H B, Ruggeri F M. Production and characterization of murine IgA monoclonal antibodies to the surface antigens of rhesus rotavirus. Virology. 1996;225:97–110. doi: 10.1006/viro.1996.0578. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert J M, Greenberg H B. Cleavage of rhesus rotavirus VP4 after arginine 247 is essential for rotavirus-like particle-induced fusion from without. J Virol. 1998;72:5323–5327. doi: 10.1128/jvi.72.6.5323-5327.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez Yafal A, Kaplan G, Racaniello V R, Hogle J M. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology. 1993;197:501–505. doi: 10.1006/viro.1993.1621. [DOI] [PubMed] [Google Scholar]
- 17.Kyte J. Structure in protein chemistry. New York, N.Y: Garland Publishing, Inc.; 1995. [Google Scholar]
- 18.Lopez S, Arias C F, Bell J R, Strauss J H, Espejo R T. Primary structure of the cleavage site associated with trypsin enhancement of rotavirus SA11 infectivity. Virology. 1985;144:11–19. doi: 10.1016/0042-6822(85)90300-9. [DOI] [PubMed] [Google Scholar]
- 19.Lopez S, Lopez I, Romero P, Mendez E, Soberon X, Arias C F. Rotavirus YM gene 4: analysis of its deduced amino acid sequence and prediction of the secondary structure of the VP4 protein. J Virol. 1991;65:3738–3745. doi: 10.1128/jvi.65.7.3738-3745.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludert J E, Michelangeli F, Gil F, Liprandi F, Esparza J. Penetration and uncoating of rotaviruses in cultured cells. Intervirology. 1987;27:95–101. doi: 10.1159/000149726. [DOI] [PubMed] [Google Scholar]
- 21.Mackow E R, Barnett J W, Chan H, Greenberg H B. The rhesus rotavirus outer capsid protein VP4 functions as a hemagglutinin and is antigenically conserved when expressed by a baculovirus recombinant. J Virol. 1989;63:1661–1668. doi: 10.1128/jvi.63.4.1661-1668.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackow E R, Shaw R D, Matsui S M, Vo P T, Dang M N, Greenberg H B. The rhesus rotavirus gene encoding protein VP3: location of amino acids involved in homologous and heterologous rotavirus neutralization and identification of a putative fusion region. Proc Natl Acad Sci USA. 1988;85:645–649. doi: 10.1073/pnas.85.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackow E R, Yamanaka M Y, Dang M N, Greenberg H B. DNA amplification-restricted transcription-translation: rapid analysis of rhesus rotavirus neutralization sites. Proc Natl Acad Sci USA. 1990;87:518–522. doi: 10.1073/pnas.87.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McRorie D K, Voelker P J. Self-associating systems in the analytical ultracentrifuge. Fullerton, Calif: Beckman Instruments; 1993. [Google Scholar]
- 25.Nandi P, Charpilienne A, Cohen J. Interaction of rotavirus particles with liposomes. J Virol. 1992;66:3363–3367. doi: 10.1128/jvi.66.6.3363-3367.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Offit P A, Blavat G, Greenberg H B, Clark H F. Molecular basis of rotavirus virulence: role of gene segment 4. J Virol. 1986;57:46–49. doi: 10.1128/jvi.57.1.46-49.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padilla-Noriega L, Dunn S J, Lopez S, Greenberg H B, Arias C F. Identification of two independent neutralization domains on the VP4 trypsin cleavage products VP5* and VP8* of human rotavirus ST3. Virology. 1995;206:148–154. doi: 10.1016/s0042-6822(95)80029-8. [DOI] [PubMed] [Google Scholar]
- 28.Padilla-Noriega L, Werner-Eckert R, Mackow E R, Gorziglia M, Larralde G, Taniguchi K, Greenberg H B. Serologic analysis of human rotavirus serotypes P1A and P2 by using monoclonal antibodies. J Clin Microbiol. 1993;31:622–628. doi: 10.1128/jcm.31.3.622-628.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prasad B V, Burns J W, Marietta E, Estes M K, Chiu W. Localization of VP4 neutralization sites in rotavirus by three-dimensional cryo-electron microscopy. Nature. 1990;343:476–479. doi: 10.1038/343476a0. [DOI] [PubMed] [Google Scholar]
- 30.Provencher S W, Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 31.Ralston G. Introduction to analytical ultracentrifugation. Fullerton, Calif: Beckman Instruments; 1993. [Google Scholar]
- 32.Righetti P G, Drysdale J W. Isoelectric focusing in gels. J Chromatogr. 1974;98:271–321. doi: 10.1016/s0021-9673(00)92076-4. [DOI] [PubMed] [Google Scholar]
- 33.Robinson I K, Harrison S C. Structure of the expanded state of tomato bushy stunt virus. Nature. 1982;297:563–568. [Google Scholar]
- 34.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 35.Sato S B, Kawasaki K, Ohnishi S. Hemolytic activity of influenza virus hemagglutinin glycoproteins activated in mildly acidic environments. Proc Natl Acad Sci USA. 1983;80:3153–3157. doi: 10.1073/pnas.80.11.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw A L, Rothnagel R, Chen D, Ramig R F, Chiu W, Prasad B V. Three-dimensional visualization of the rotavirus hemagglutinin structure. Cell. 1993;74:693–701. doi: 10.1016/0092-8674(93)90516-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taniguchi K, Maloy W L, Nishikawa K, Green K Y, Hoshino Y, Urasawa S, Kapikian A Z, Chanock R M, Gorziglia M. Identification of cross-reactive and serotype 2-specific neutralization epitopes on VP3 of human rotavirus. J Virol. 1988;62:2421–2426. doi: 10.1128/jvi.62.7.2421-2426.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willis S H, Peng C, Ponce de Leon M, Nicola A V, Rux A H, Cohen G H, Eisenberg R J. Expression and purification of secreted forms of HSV glycoproteins from baculovirus-infected insect cells. In: Brown S M, McLean A R, editors. Methods in molecular medicine: herpes simplex virus protocols. Vol. 10. Totowa, N.J: Humana Press, Inc.; 1998. [DOI] [PubMed] [Google Scholar]
- 39.Yeager M, Berriman J A, Baker T S, Bellamy A R. Three-dimensional structure of the rotavirus haemagglutinin VP4 by cryo-electron microscopy and difference map analysis. EMBO J. 1994;13:1011–1018. doi: 10.1002/j.1460-2075.1994.tb06349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zarate S, Espinosa R, Romero P, Mendez E, Arias C F, Lopez S. The VP5 domain of VP4 can mediate attachment of rotaviruses to cells. J Virol. 2000;74:593–599. doi: 10.1128/jvi.74.2.593-599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Y J, Burns J W, Morita Y, Tanaka T, Estes M K. Localization of rotavirus VP4 neutralization epitopes involved in antibody-induced conformational changes of virus structure. J Virol. 1994;68:3955–3964. doi: 10.1128/jvi.68.6.3955-3964.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]