

Abstract
Electrochemical CO2 reduction reaction (CO2RR) to produce value‐added multi‐carbon chemicals has been an appealing approach to achieving environmentally friendly carbon neutrality in recent years. Despite extensive research focusing on the use of CO2 to produce high‐value chemicals like high‐energy‐density hydrocarbons, there have been few reports on the production of propane (C3H8), which requires carbon chain elongation and protonation. A rationally designed 0D/2D hybrid Cu2O anchored‐Ti3C2Tx MXene catalyst (Cu2O/MXene) is demonstrated with efficient CO2RR activity in an aqueous electrolyte to produce C3H8. As a result, a significantly high Faradaic efficiency (FE) of 3.3% is achieved for the synthesis of C3H8 via the CO2RR with Cu2O/MXene, which is ≈26 times higher than that of Cu/MXene prepared by the same hydrothermal process without NH4OH solution. Based on in‐situ attenuated total reflection‐Fourier transform infrared spectroscopy (ATR‐FTIR) and density functional theory (DFT) calculations, it is proposed that the significant electrocatalytic conversion originated from the synergistic behavior of the Cu2O nanoparticles, which bound the *C2 intermediates, and the MXene that bound the *CO coupling to the C3 intermediate. The results disclose that the rationally designed MXene‐based hybrid catalyst facilitates multi‐carbon coupling as well as protonation, thereby manipulating the CO2RR pathway.
Keywords: C2‐C1 coupling, electrochemical CO2 reduction, in‐situ ATR‐FTIR, propane production, proton‐coupled electron transfer
A rational design of Cu2O/Ti3C2Tx MXene catalyst presents an excellent activity for saturated hydrocarbon C3H8 from electrochemical CO2 reduction, providing *C2‐*C1 coupling sites and facilitating further protonation. Based on in‐situ attenuated total reflection‐Fourier transform infrared spectroscopy and density functional theory calculations, results offer insights into green electrochemical carbon utilization for achieving carbon neutrality as well as lowering global warming potential.
1. Introduction
With the continuously increasing demand to reduce CO2 emissions, the conversion of CO2 into value‐added hydrocarbon products has gained significant attention.[ 1 ] From an energy perspective, utilization of CO2 to produce high‐value chemicals, particularly those with high energy density, such as hydrocarbons with long carbon chains or saturated (i.e., hydrogen‐rich) hydrocarbons, is desirable. In particular, propane (C3H8) has a high specific energy (50.4 MJ kg−1) with a remarkably low global warming potential (< 1) compared to other CO2‐driven products including CO, CH4, and C2H4, thereby considered a desirable renewable energy source to reduce the impact on carbon footprint with versatile applications.[ 2 ] Among the various technologies to utilize CO2 to produce C3H8, the electrochemical conversion of CO2 can be effective since the electrochemical CO2 reduction reaction (CO2RR) operates under environmentally benign mild conditions, using renewable electricity. However, the electrochemical CO2RR to C3H8 in an aqueous electrolyte is challenging because the reaction involves several steps, including carbon chain elongation and protonation with 20 electrons. Thus, modulating the electrochemical catalysts to promote the protonation of longer‐chain carbons is critical to produce C3H8. To the best of our knowledge, few studies report a notable Faradaic efficiency for C3H8 of the electrochemical CO2RR in an aqueous electrolyte, but most of the research achieved a modest Faraday efficiency (FE) of ≈1%, without the aid of other ionomers or carbon sources.[ 3 ]
A recent study with imidazolium‐functionalized Mo3P coated with the ionomer (ImF‐Mo3P) showed FE of 91% C3H8, marking a significant advance over previous reports that typically showed FE less than 1% or even trace amounts.[ 4 ] However, the carbon sources, i.e., carbon functional groups, namely imidazolium moieties on the catalyst, as well as the coated ionomers, are participating in the CO2RR.[ 5 ] These carbon sources inhibit the identification of the reaction mechanism to find the crucial *C2 and *C3 intermediates for C3H8. Especially, in‐situ Raman data pinpointing appropriate CO2RR intermediates other than *CO to support the proposed reaction mechanism from CO2 to C3H8 is required. Given that the *CO formation is an initial step of CO2RR, peaks for *CO alone cannot directly substantiate the C3H8 production. Thus, a rational strategy by facilitating multi‐carbon coupling and protonation supported by substantial experimental evidence to represent the reaction mechanism is required.
Cu‐based catalyst is generally considered to produce multiple hydrocarbons of various chain lengths from CO2.[ 6 ] Some studies on Cu‐based catalysts reported a wide selectivity for C3 products including n‐propanol, acetone, and hydroxyacetone,[ 7 ] albeit the FE of C3H8 is generally less than 0.3%, or even a traceable amount, indicating a lack of protonation.[ 8 ] To increase the selectivity toward multi‐carbon, Zhang et al. reported that not only the electron transfer for CO dimerization at catalytic active sites but also several protonation steps are necessary.[ 9 ]
Ti3C2Tx MXene with 2D layered structure has received considerable attention due to its catalytic performance in the electrochemical CO2RR with excellent chemical durability and abundant adsorption sites with tunable functional groups on the surface.[ 10 ] Especially, in an aqueous electrolyte, the naturally modulated O‐ and OH‐ functional group of MXene can share H with CO2RR intermediates, enabling proton‐coupled electron transfer (PCET) for the protonation of hydrocarbon products, thus increasing the energy density of CO2 reduction products.[ 11 ] Besides, modulating the MXene functional groups with O‐ or OH‐ can result in high chemical reactivity and an increase in the number of active sites for catalytic reactions in an aqueous electrolyte.[ 12 ] Thus, the composite of Cu2O and MXene can be useful for hydrocarbon generation with long‐term stability, given that the modulated MXene surfaces and oxygen‐derived Cu offer stable adsorption sites for CO2 molecules, further promoting C‐C coupling and effective protonation of longer‐chain hydrocarbons.[ 11 , 13 ]
In this study, we designed a rational hybrid structure of Cu2O incorporated into MXene (Cu2O/MXene) for the efficient conversion of CO2 into multi‐carbon products, especially C3H8 which requires C3 coupling and sufficient protonation. To understand the synergistic effect of the Cu2O and MXene with the mechanism of CO2 conversion to C3H8, Cu2O/MXene was characterized by X‐ray photoelectron spectroscopy (XPS), Fourier‐transform infrared spectroscopy (FT‐IR), Auger electron spectroscopy (AES) and X‐ray absorption spectroscopy (XAS). Notably, attenuated total reflection‐Fourier transform infrared spectroscopy (ATR‐FTIR) revealed that the interfacial effect between Cu2O and MXene was significant for *C2–*C1 coupling. Specifically, Cu2O strongly bound and preserved *C2 intermediates. In contrast, the MXene bound to the sole *C1 site and provided sufficient protons to the CO2RR intermediates. As a result, Cu2O/MXene reveals an efficient C3H8 production with a FE of 3.3% at −1.3 V versus reversible hydrogen electrode (RHE) in CO2 saturated 0.1 M KHCO3, without the aid of carbon sources. We envision that our strategy of catalyst design combined with Cu2O and MXene, which could tune the selectivity toward multi‐carbon products and proton‐coupled electron transfer, respectively, is able to produce saturated hydrocarbon products with high energy density, such as C3H8.
2. Results
2.1. Characterizations for Cu2O/MXene
Cu2O/MXene was fabricated via a facile hydrothermal process. First, Ti3C2Tx MXene nanosheets were prepared by a selective Al etching method, starting from Ti3AlC2 MAX powder with further delamination, following the previous study with some modifications.[ 14 ] Cu nanoparticles (CuNPs) were prepared separately by the hot‐injection method. After individually fabricating MXene and CuNPs, the mixture solution of MXene and CuNPs was transferred to a Teflon liner, with 4 mmol NH4OH solution added to adjust pH ≈10, and autoclaved for 30 min at 70 °C. During the synthesis, some of the functional groups of the MXene nanosheets were modulated to hydroxyl (‐OH) moieties in the presence of an NH4OH solution. In addition, the NH4OH solution induced partial oxidation of CuNPs to form Cu2O and further consolidated Cu2O on the MXene. The as‐prepared samples were then freeze‐dried to prevent further oxidation and restacking. To compare the CO2RR activity, Cu/MXene was synthesized in the same manner as Cu2O/MXene without the use of an NH4OH solution, so the CuNPs were not significantly oxidized when anchored to MXene during the synthesis.
Transmission electron microscopy (TEM) was used to examine the structural morphology of Cu2O/MXene (Figure 1 ). As shown in Figure 1a‐b, the Cu NPs adhered to the MXene nanosheets. As shown in Figure 1b, the CuNPs were distributed on the surface of MXene with 2.6 nm of average diameter, showing a typical log‐normal distribution. The high‐resolution TEM (HRTEM) image in Figure 1c shows that the CuNPs in Cu2O/MXene were crystalline structures with lattice fringes of 0.24 nm for Cu2O (111). This indicates the partial oxidation of CuNPs during the hydrothermal process in the presence of NH4OH solution to form Cu2O. However, no morphological variance was observed between MXene and NH4OH‐treated MXene (AT‐MXene), as shown in Figure S1 (Supporting Information). In addition, a clear interface was observed between the CuNPs and MXene. EDS line profile scanning was conducted to assign Ti (black), Cu (red), and O (blue) (Figure 1d,g), where a clear interface between the CuNPs and MXene was observed. The elemental O distribution was higher in the presence of Cu, indicating the incorporation of CuNPs as Cu2O phase into MXene.
Figure 1.
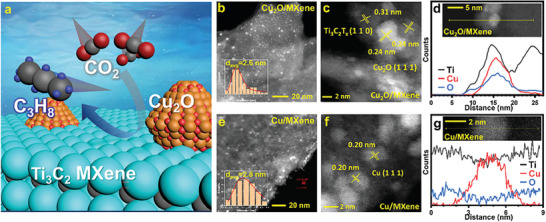
a) Schematic illustration of CO2RR with Cu2O/MXene. TEM, HRTEM, and EDS line profile mapping for b–d) Cu2O/MXene and e–g) Cu/MXene. b) and e) TEM images of Cu2O/MXene and Cu/MXene. c) and f) HRTEM for Cu2O/MXene and Cu/MXene. d) and g) EDS line profile mapping Ti, Cu, and O for Cu2O/MXene and Cu/MXene.
As for the Cu/MXene, which was synthesized without NH4OH treatment, the average diameter of the CuNPs on Cu/MXene was 2.8 nm with a narrower size distribution compared to Cu2O/MXene (Figure 1e). In addition, the CuNPs in Cu/MXene, which predominantly presents a lattice fringe with 0.20 nm for Cu (111), are metallic Cu (Figure 1f). In Figure 1g, metallic CuNPs are observed with no change in the elemental O distribution, whereas an increase in the Ti content is observed in Cu/MXene. Thus, the presence of NH4OH during the synthesis induced the incorporation of CuNPs being transformed to Cu2O on the surface of the MXene nanosheets.
Following up on the TEM results, electron energy‐loss spectrometry (EELS) was performed to determine the valence states of the Cu species in Cu2O/MXene and Cu/MXene (Figure 2a). From the EELS spectra, the slope for Cu2O/MXene in the range of 930–940 eV was between those of Cu and Cu2O, but close to that of Cu2O. Therefore, it was difficult to distinguish the valence state of Cu in Cu/MXene from that in Cu2O/MXene. These results may imply that the valence state of Cu in both Cu2O/MXene and Cu/MXene was a combination of metallic Cu and Cu+. Additionally, by comparing the EELS spectra of Cu2O/MXene and Cu/MXene in the range of 400–750 eV, representing MXene, we found a low residual F intensity with a higher O intensity in Cu2O/MXene (Figure S2, Supporting Information). This indicates that the NH4OH treatment induced not only the partial oxidation of CuNPs but also the successful functional group modulation of MXene in Cu2O/MXene during the hydrothermal process.
Figure 2.
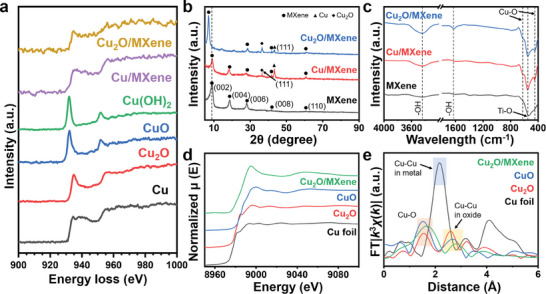
a) EELS spectra of Cu/MXene and Cu2O/MXene. b) XRD and c) FT‐IR spectra of MXene, Cu/MXene and Cu2O/MXene. d) XANES and e) FT‐EXAFS data for Cu2O/MXene.
The crystal and phase structures of Cu2O/MXene were characterized by XRD (Figure 2b). The two distinct main peaks (i.e., (002) and (004)) represent MXene, which is consistent with the results of previous studies.[ 15 ] The (002) peak at 8.87° is a typical peak for the Ti3C2Tx MXene, indicating a c‐lattice parameter (c‐LP, ≈ 19.9 Å).[ 16 ] This (002) peak did not shift in Cu/MXene. However, the (002) peak shifted toward a lower value (7.06°) in Cu2O/MXene with c‐LP to 25.0 Å when Cu2O was anchored to the MXene. This result may imply that the CuNPs were anchored to the functional group of MXene as Cu2O phase, affecting the lattice structure and eventually increasing the interlayer distance of MXene.[ 17 ] In Cu2O/MXene, the composition of Cu2O (111) was higher than the Cu (111) (JCPDS 77–0199 and JCPDS 04–0836, respectively). The ratio from the quantitative phase analysis by Rietveld refinement for Cu:Cu2O was 0.17:0.83, indicating a high fraction of Cu2O. A sharp peak with low intensity for Cu2O (111) was also observed for Cu/MXene. Unlike Cu2O/MXene, the ratio of Cu:Cu2O in Cu/MXene was 0.91:0.09, indicating that most of the CuNPs were composed of Cu0. Thus, NH4OH treatment affected the oxidation state of Cu in Cu2O/MXene.
FT‐IR was conducted to determine the chemical bonds and functional groups on the surface of Cu2O/MXene. As shown in Figure 2c, the peak at 540 cm−1 is attributed to the vibration of the Ti‐O bond from the MXene.[ 18 ] Cu‐O stretching vibration peaks are also observed at 425 cm−1 and 620 cm−1,[ 19 ] presumably the CuNPs were hybridized onto the ‐O or ‐OH functional group of MXene forming Cu2O. The broad absorption band with a peak at 3445 cm−1 corresponds to the ‐OH stretching vibration of the water molecules in the MXene interlayer.[ 20 ] For the Cu2O/MXene where NH4OH treatment was applied, the FT‐IR spectra showed a slight peak at 1625 cm−1 which is attributed to the OH groups.[ 21 ] Thus, this result suggests that the NH4OH treatment modulated the functional group of the MXene to the ‐OH.
The coordination environment of the CuNPs in Cu2O/MXene was further characterized by synchrotron‐based X‐ray absorption near‐edge structure (XANES) and extended X‐ray absorption fine structure (EXAFS). As shown in Figure 2d, the Cu absorption edge of Cu2O/MXene resided between those of commercial Cu and Cu2O. Especially, the Cu k‐edge for Cu2O/MXene was in between those of Cu2O and CuO but close to that of Cu2O, suggesting that the average oxidation state of Cu is likely to be higher than that of Cu+ but lower than that of Cu2+. This valence state also reflected the successful hybridization of Cu2O and MXene, where Cu2O was anchored to MXene. The Fourier transform (FT)‐EXAFS spectra of Cu2O/MXene and the references (Figure 2e) showed the intensity of the Cu‐O peak at ≈1.6 Å, which was intermediate between the intensities found in Cu2O and CuO. The increase in Cu‐O distance on Cu2O/MXene also shows evidence of Cu‐O‐Ti bonding by hybridization, as the length of the Cu‐O bond from the Cu‐O‐Cu is shorter than the Cu‐O‐Ti bond length.[ 22 ] The Cu‐Cu peak from the oxide (≈2.8 Å) is distinct in Cu2O/MXene which is associated with the scattering path of the second Ti shell (i.e., Ti L‐shell).[ 22 ]
XPS was further performed to elucidate the chemical structure of Cu2O anchored to MXene from Cu2O/MXene, compared with MXene and Cu/MXene (Figure 3 ). In the XPS high‐resolution spectra of C 1s (Figure 3a), the binding energy peak at 284.8 eV was assigned to the adventitious carbon of C‐C.[ 20 ] Furthermore, the binding energy of C‐Ti in Cu2O/MXene shifted to a lower value (282.3 eV) than that of bare MXene (282.6 eV), indicating that Cu2O transferred electrons to MXene. The oxygen contents with carbon (C‐O, C = O, and O‐C = O) peaks may result from MXene oxidation and carbon networks.[ 23 ] In the XPS Ti 2p spectra (Figure 3b), the binding energies of Ti‐C, Ti2+, Ti3+, and Ti4+ for MXene are 455.4, 456.1, 457.3, and 458.8 eV, respectively. Specifically, the Ti2+, Ti3+, and Ti4+ peaks are attributed to Ti‐X, Ti‐O, and Ti‐Ox (including Ti‐(OH)x), respectively.[ 24 ] When CuNPs are hybridized onto the MXene (Cu/MXene and Cu2O/MXene), the CuNPs are consolidated at the oxygen‐rich functional group of MXene. The evidence of anchoring CuNPs on MXene is further characterized by the O 1s XPS. The O 1s region of the MXene was deconvoluted by components corresponding to C‐Ti‐Ox, Ti‐O‐Ti, C‐Ti‐(OH)x, Oads at BE = 530.5, 531.2, 532.0, and 533.2 eV, respectively (Figure 3c).[ 25 ] Oads is the surface adsorbed oxygen species in MXene, and the peak at BE = 533.2 eV corresponds to adsorbed water.[ 26 ] The peak for the adsorbed water was significantly increased when Cu2O was applied to the MXene, affecting the hydrophilicity. In addition, compared to the MXene, Cu/MXene and Cu2O/MXene display a positive shift of C‐Ti‐Ox and Ti‐O‐Ti, demonstrating that the CuNPs were anchored onto the MXene terminals. Notably, the N 1s peak in AT‐MXene or Cu2O/MXene was not observed, indicating that the NH4OH treatment affected the functional group modulation of MXene to the ‐OH group and did not participate in the N‐doped or amine groups (Figure S7, Supporting Information). Besides, as MXene was fabricated and stored as a solution, the MXene already has some ‐OH functional groups, even without NH4OH treatment. Thus, the NH4OH treatment slightly increased the number of ‐OH functional groups. The result is in accordance with the previous research by Gogotsi's group, which modulated the MXene functional group by the NH4OH solution to the ‐OH.[ 27 ]
Figure 3.
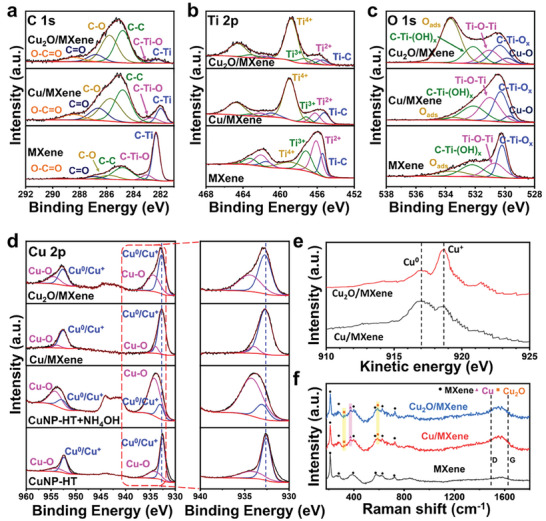
XPS spectra of a) C 1s, b) O 1s, and c) Ti 2p of MXene, Cu/MXene and Cu2O/MXene. d) Cu 2p of Cu/MXene and Cu2O/MXene displayed with a magnified view of XPS Cu 2p3/2 comparing with CuNP prepared with hydrothermal process in the presence and absence of NH4OH treatment. e) Cu LMM AES spectra of Cu/MXene and Cu2O/MXene. f) Raman spectra of MXene, Cu/MXene, and Cu2O/MXene.
To determine the valence state of Cu, the Cu 2p XPS for Cu2O/MXene was compared with that of Cu/MXene, as well as CuNPs with the hydrothermal process (CuNP‐HT) in the absence and presence of NH4OH treatment (Figure 3d). The Cu0/Cu+ and Cu2+ peaks of Cu 2p3/2 were observed at 933.0 and 934.2 eV. Without MXene, the NH4OH solution easily oxidized the surface of CuNP to form Cu2O or even further oxidized to CuO. Thus, a significant increase in the Cu2+ peak was observed compared with CuNP‐HT and Cu2O (CuNP‐HT+NH4OH). The typical Cu2+ satellites at 940–945 eV and 960–965 eV also confirmed the further oxidation of Cu to CuO with NH4OH treatment in the absence of MXene, in accordance with the XRD data (Figure S3, Supporting Information).[ 28 ] In the presence of MXene, although the Cu0/Cu+ peak was shifted to a higher binding energy, NH4OH treatment cannot fully oxidize Cu to Cu2+ since NH4OH also affects the partial substitution of the functional group of MXene to ‐OH. Cu LMM Auger electron spectra (AES) were tested to further carefully distinguish the oxidation state of the CuNPs decorated on MXene (Figure 3e). Comparing the intensities of Cu0 and Cu+, Cu+ was dominant in Cu2O/MXene, whereas Cu0 was dominant in Cu/MXene.
The chemical structure of Cu2O/MXene was revealed using Raman spectroscopy (Figure 3f). Signals representing the vibrations of Ti and C atoms were observed in MXene, Cu/MXene, and Cu2O/MXene (i.e., peaks at 201, 274, 386, 582, 626, and 726 cm−1).[ 10 ] The peak located at 201 cm−1 is attributed to the out‐of‐plane vibrations of Ti atoms, whereas the peaks at 274 and 386 cm−1 are ascribed to the in‐plane vibrations of the surface groups attached to the Ti atoms.[ 29 ] The two peaks at 1373 cm−1 and 1576 cm−1 represent the D band and G band, respectively, where the former D‐band is generally related to the disordered graphite formed by defects in carbon‐based materials, whereas the latter G‐band is ascribed to the stacking of the graphite hexagonal network plane.[ 10 , 15 ] For Cu/MXene and Cu2O/MXene, both Cu and Cu2O were observed (376 cm−1 for Cu, and 322 and 590 cm−1 for Cu2O, respectively).[ 30 ] Thus, Cu could be slightly oxidized when anchored to MXene, even when the NH4OH treatment was not applied to Cu/MXene.
2.2. Electrochemical Analysis for CO2 Reduction
Linear sweep voltammetry (LSV) curves in Ar and CO2 saturated electrolyte were used to evaluate the CO2RR performance of Cu2O/MXene, and then the results were compared with those of MXene and Cu/MXene (Figure 4a). MXene, Cu/MXene, and Cu2O/MXene showed activity on CO2RR. Anchoring Cu2O on MXene further increased the activities of both CO2RR and hydrogen evolution reaction (HER), according to the results obtained with CO2 and Ar saturation. The current density of Cu2O/MXene in CO2 purged electrolyte was higher than that in the Ar, demonstrating its good electrocatalytic activity for CO2 reduction. In addition, the CO2 saturated LSV curves for all samples exhibited more positive onset potentials than the Ar‐saturated ones, suggesting that their activities on CO2 participated in the reaction. Note that these current density data can be enhanced with different cell configuration such as a membrane electrode assembly (MEA) cell. The electrochemical impedance spectroscopy (EIS) was conducted to investigate the kinetic behaviors of the electrocatalyst for CO2RR in CO2‐saturated 0.1 M KHCO3 (Figure S9, Supporting Information). For the Cu NPs showed low charge transfer resistance reflecting efficient CO2RR activity in high frequency region but extremely large diffusion resistance in low frequency region. In cases of MXene and AT‐MXene, both exhibited large semi‐circles which indicate insufficient CO2RR, which are coherent to the product analysis. The Cu/MXene showed smaller semi‐circle than those of MXene and AT‐MXene by enhanced charge transfer due to the Cu anchoring. Especially, the Cu2O/MXene displayed the lowest charge transfer resistance among the MXene‐based electrocatalysts reflecting superior activity for CO2RR. From the CO2RR, products with CO, CH4, C2H4, C2H6, and C3H8 were observed. The FEs of the CO2RR products at the applied potentials for MXene, AT‐MXene, Cu/MXene, and Cu2O/MXene are shown in Figure S13 (Supporting Information). As shown in Figure S14 (Supporting Information), the GC‐FID chromatograms clearly confirm that the gaseous product is comprised with CH4, C2H4, and C3H8, not including C3H6. The FE of the CO2RR products on Cu2O/MXene at −1.3 V versus RHE was compared with that of Cu/MXene in Figure 4b, where the highest FEC3H8 was observed. Cu2O/MXene was the most active electrocatalyst, affording 3.3% of FEC3H8 at −1.3 V versus RHE as the optimum potential, suggesting effective electron and proton transfer during CO2 reduction. These results were comparable to those obtained for Cu/MXene with ≈0.1% FEC3H8. Although MXene also exhibited activity in CO2, the products of the CO2RR were C1 gases composed of CO and CH4. Especially, as the major products of Cu or Cu2O are C1 and C2 products,[ 31 ] the presence of MXene in the composite may affect the C3 intermediate production toward C3H8. We also compared the experimental C2 & C3 to C1 ratio with −0.9 to −1.5 V versus RHE, as shown in Figure 4c. When the applied potential increased, the (C2+C3)/C1 ratio decreased, indicating that the time for carbon coupling became insufficient as the HER is significantly affected by the higher applied potential, which is the competitive reaction of CO2RR. Additionally, the FE of both CO and CH4 decreased with increasing applied potential (Figure S13, Supporting Information), indicating that the HER favored over the CO2RR.[ 7a ] An interesting result on the ratio of C3 to H2 was found at −1.3 V versus RHE, showing the highest value. In general, the C3/H2 ratio is low as *H adsorption onto the catalyst during the HER requires less energy than the protonation of the hydrocarbon products.[ 3b ] However, as the MXene could provide protons to the C3 intermediate, the highest C3H8 FE at −1.3 V versus RHE could be achieved by a proton‐coupled electron‐transfer step, while concurrently reducing the HER.[ 11 ]
Figure 4.
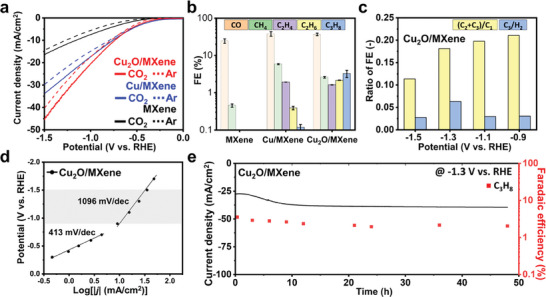
Electrochemical tests of Cu2O/MXene. a) LSV curves of Cu/MXene and Cu2O/MXene saturated with CO2 (straight lines) and Ar (dashed lines). Applied potential is corrected with iR compensation, and ECSA is considered for current density. b) Faradaic efficiency on each carbonaceous CO2RR product at −1.3 V versus RHE. Error bars represent the standard deviation in FE calculated after three tests for repeatability. The other non‐carbon product is listed in Table S2 (Supporting Information). c) Relative ratio of the FE with Cu2O/MXene. d) Tafel slope with Cu2O/MXene. The intermediate overpotential region where significant C3 production was detected is highlighted in grey. e) Stability tests of Cu2O/MXene at −1.3 V versus RHE.
Generally, selectivity is highly dependent on the competitive adsorption of intermediates such as *CO and *OCCO, as well as the reaction kinetic energy barrier of the rate‐determining step in the electrochemical CO2RR for C3H8 production.[ 32 ] The Tafel plot was tested for Cu2O/MXene and Cu/MXene, highlighting the CO2RR intermediate overpotential region (−0.9 to −1.5 V versus RHE) where significant C3 products were observed (Figure 4d; Figure S15, Supporting Information). The Tafel slope of the Cu/MXene at −0.9 to −1.5 V versus RHE was 957 mV dec−1, lower than that of Cu2O/MXene (1096 mV dec−1). However, the C2 and C3 production for Cu/MXene was much lower than that for Cu2O/MXene (Figure 4b). These results indicate sluggish reaction kinetics for the C2 and C3 hydrocarbon production which require complex multistep reactions including carbon coupling and protonation.[ 33 ] Thus, Cu2O/MXene could effectively enforce the reaction pathway toward multi‐carbon production when sufficient time for carbon coupling and protonation was provided during electrochemical CO2 reduction. In addition, the long‐term stability of Cu2O/MXene at −1.3 V versus RHE up to 48 h was investigated as shown in Figure 4e. After the stability test, Cu2O NPs stably anchored onto the MXene were observed by HAADF‐STEM, and the catalyst was characterized by XRD and XPS, which showed no significant changes (Figure S17, Supporting Information).
To determine the mechanism of how the Cu2O/MXene affected the selectivity toward C3 product in CO2RR, in‐situ ATR‐FTIR spectroscopy was performed and compared with the Cu2O to reveal the coupling effect of the intermediates such as *CO and *OCCO at the interface. As shown in Figure 5a, the peak at 1560–1640 cm−1 corresponds to the asymmetric O‐C‐O stretching peak associated with Cu2O (Figure 5b) when CO2 is adsorbed and the C atom is bound to the metallic atom of the catalyst.[ 34 ] This O‐C‐O stretching peak remained after 20 min of CO2 reduction, indicating that the Cu2O anchored on the MXene could continuously and strongly bind the CO2 intermediate.[ 35 ] *CHO peak at 1660–1740 cm−1 was expected to produce the C1 pathway toward the formation of CH4.[ 36 ] *COOH peak was found at 1210–1280 cm−1 which ultimately led to CO.[ 37 ] Notably, *COOH favors the production of CO, which is not a desirable intermediate for HCOOH formation, as has already been proven by several studies.[ 37 , 38 ] C = C peaks were found at 820–850 cm−1 and 940−1030 cm−1, significant evidence of alkene products including C2H4.[ 39 ] Moreover, peaks for CO dimerization were found at 1470−1530 cm−1 and 1390–1460 cm−1 which belong to *OCCO and *OCCOH onto the Cu2O, respectively.[ 36 , 40 ] These CO dimerization intermediates triggered the C2+ and C3+ pathways, as reported in previous studies for the CO2RR mechanism to produce C3 through the C1‐C2 coupling step.[ 6 , 41 ] The key feature of this mechanism is the high coverage of the C2 intermediate, which can be stabilized by a well‐designed catalyst morphology and electronic structure.[ 6d ] Here, as the *OCCO and *OCCOH peaks increased with CO2 electrolysis time, Cu2O (111) strongly bound CO2 to C2 intermediates and provided catalytic sites for a longer time for additional carbon chain elongation and protonation toward C3H8, as discussed in the Tafel slopes (Figure 4d; Figure S15, Supporting Information).[ 41 , 42 ] To determine the weak C3 intermediate peak masked by stronger noise, the first derivative of the ATR‐FTIR was processed, which is a common method used for spectral data processing and interpretation (Figure 5c‐d).[ 43 ] Compared to Cu2O (CuNP‐HT + NH4OH), an additional peak at ≈690 cm−1 for C‐CH2‐C peak was found for Cu2O/MXene, confirming that the C3 intermediate could generate C3H8 (propane), n‐C3H7OH (n‐propanol) or C2H3CHO (acrolein) during CO2 reduction.[ 44 ] With Cu2O, although there are peaks for *CO, *OCCO and *OCCOH, no C‐CH2‐C peak at ≈690 cm−1 is found, indicating that the C3 intermediate is not solely found at the Cu2O catalytic site, as well as additional *CO intermediate adsorption site from the MXene is necessary for C3 intermediate coupling. Considering that MXene and AT‐MXene could not produce C2 products (Figure S13, Supporting Information), the interface between Cu2O and MXene in Cu2O/MXene was the key site for coupling *C2 intermediates and *CO toward C3 products, where Cu2O provided adsorption sites for *C2 intermediates and MXene provided adsorption sites for *CO.
Figure 5.
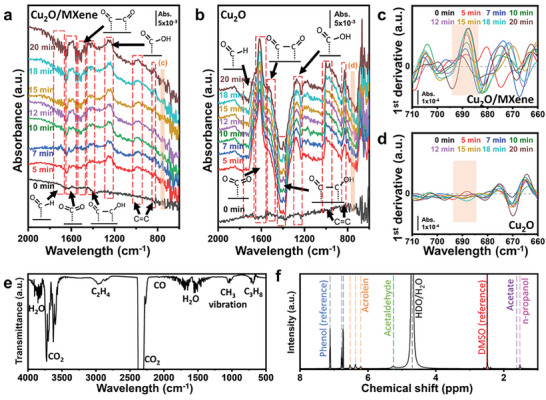
a) and b) ATR‐FTIR spectra for Cu2O/MXene and Cu2O at varying CO2RR time. c) and d) 1st derivative of ATR‐FTIR spectra for Cu2O/MXene and Cu2O in ranges of 660–710 cm−1. e) FT‐IR spectra of gas species from Cu2O/MXene CO2RR. f) 1H NMR spectra of Cu2O/MXene. All the tests were conducted at −1.3 V versus RHE.
As *CO and *C2 bound to couple the carbon chain to C3+ products,[ 3 , 7 , 45 ] the gas and liquid products were analyzed by IR and 1H NMR to determine the hydrocarbon products (Figure 5e–f). Several peaks representing hydrocarbons from the CO2RR were observed. Some unreacted gases, such as CO2 and H2O, were also found.[ 46 ] Notably, C‐C‐C skeletal vibration was found at ≈720 cm−1, confirming C3H8 production.[ 47 ] Through 1H NMR, we found additional liquid hydrocarbon products, including C2 and C3 products such as acetaldehyde, acetate, acrolein, and n‐propanol.[ 48 ] However, only traceable amounts of liquid products were generated during CO2RR.
2.3. Proposed Mechanism for Electrochemical CO2 Reduction to C3H8
Based on the above mechanistic analysis, we highlighted the key steps and crucial intermediates in C3H8 production (Figure S19, Supporting Information). CO2 was initially adsorbed onto Cu2O/MXene at both Cu2O and MXene sites to form *COOH, which underwent further reduction to *CO with H2O removal.[ 49 ] The *CO intermediates on Cu2O underwent CO dimerization to *OCCO and were further hydrogenated to form *OCCOH.[ 50 ] After protonation, *OCCO formed *OCCOH, which is the core step for the formation of C2 and C3 hydrocarbons, including C2H4, C2H6, and C3H7OH, following sufficient protonation with electron transfer. Furthermore, the stabilized and protonated *C2 intermediates on Cu2O can be coupled with *CO adsorbed on MXene by NH4OH treatment to form the C3 intermediate. Additional protonation/electron‐transfer steps, including the hydrogenation of the carbon from the intermediate, can transfer this C3 intermediate to the saturated hydrocarbon, i.e., C3H8.[ 51 ]
The production of C3H8 on Cu2O/MXene benefits from its nanostructure. The catalyst is composed of Cu2O with a prevalent Cu2O (111) facet, which is in favor of binding *C2 intermediates.[ 52 ] MXene also favors CO2 adsorption because the Gibbs free energy of CO2 adsorption on MXene is negative, indicating that CO2 adsorption on MXene is thermodynamically favorable.[ 53 ] This hybrid of Cu2O and MXene couple *C2‐*C1 intermediates into C3 products at the interface between Cu2O and MXene. The NH4OH treatment modulated the surface group of MXene to ‐O and ‐OH, which favors the transfer of protons with low H adsorption energy for the protonation of CO2RR intermediates.[ 54 ] Thus, these synergistic effects on the Cu2O/MXene structure could increase the C3H8 production.
We performed density functional theory (DFT) calculations to gain further insight into the role of MXene on the improved CO2RR activity for C3H8(g) production in the 0D/2D heterostructure composed of Cu2O (or Cu)‐anchored MXene compared to pure Cu2O and Cu under aqueous condition (Figure 6 ). Preferentially, we investigated the CO2RR activity of pure Cu2O(111) and Cu(111) structures through their limiting potential (UL) evaluated by the free energy diagram (FED) constructed for experimentally observed intermediates in the CO2RR mechanism depicted in Figure 6a and Figure S19 (Supporting Information) (Details of calculated thermodynamic values for FED are listed in Tables S6 and S7, Supporting Information).[ 55 ] The calculated FED in Figure 6a shows that the potential determining steps (PDS) correspond to C1‐C1 coupling (*CO+*CO → *C2O2H) and additional *CO supply (*CO → *CO+*CO) steps with UL values of −1.45 V and −1.20 V at Cu(111) and Cu2O(111), respectively. The difference in PDS steps for pure Cu2O(111) and Cu(111) structures can be interpreted by the binding energy of the intermediate. Cu(111) surface has a higher energy barrier in C1‐C1 coupling for *C2O2H formation due to the relatively weak binding energy of intermediates, especially *C2O2H and *CO. In contrast, Cu2O(111) provides a very strong *CO binding at a well‐known coordinatively unsaturated Cu site (Cucus), and additional *CO supply for *C2O2H formation cannot be easily achieved because neighboring *CO binding sites are coordinatively saturated Cu site (Cucss) to which *CO cannot stably bind, and instead Cucus sites are located far apart, as shown in Figure S21 (Supporting Information).[ 56 ] Our results imply that *C2O2H formation is a thermodynamically unfavorable process in pure Cu2O(111) and Cu(111) structures, resulting in low *C3 intermediate selectivity. Therefore, MXene can be expected to play a positive role in this *C2O2H formation process.
Figure 6.
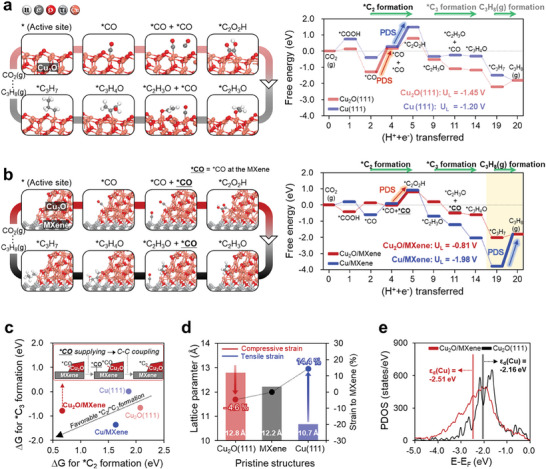
CO2RR mechanism and calculated free energy diagram (FED) for C3H8(g) production in (a) pure Cu2O(111) structure and b) 0D/2D heterostructure of Cu2O/MXene. c) Correlation between calculated reaction free energies (DG) for *C2 and *C3 formation through sequential processes of additional *CO supply/C‐C coupling in heterostructures of Cu2O/MXene and Cu/MXene and pure Cu2O(111) and Cu(111) surfaces. d) Comparison of lattice parameters of pure Cu2O(111) and Cu(111) compared to MXene structures with respect to compressive or tensile strain. e) Partial density of state (PDOS) of Cu2O/MXene heterostructure and pure Cu2O(111) structure for the d‐band center (εd) of Cu active sites.
To verify the role of MXene considering the experimentally characterized 0D/2D heterostructures (Figure 1) of Cu2O/MXene, we first constructed thermodynamically stable 2D/2D heterostructures of Cu2O and MXene, and then manipulated the Cu2O interface to expose the MXene surface to become a 0D/2D heterostructure, as shown in Figure 6b.[ 57 ] Note that 2D/2D and 0D/2D heterostructure construction processes and their formation energies are described in Figure S21 and Table S8 (Supporting Information). Using the well‐established 0D/2D heterostructures, we constructed FED to clarify the effect of MXene incorporation on the PDS and UL values (Figure 6b). In the case of Cu2O/MXene heterostructure, the FED shows improved *C2 intermediate selectivity compared to Cu2O(111) by lowering UL value from −1.45 V to −0.81 V. It is also seen that the overall binding energy of intermediates on Cu2O/MXene is weakened by MXene incorporation, ultimately overcoming the strong *CO binding problem that caused the high energy barrier on the pure Cu2O(111) surface.[ 58 ] In addition, MXene can act as an additional *CO supplier for *C2O2H formation through a thermodynamically spontaneous process due to MXene's preference for *CO, as shown in Figure S23 and Table S9 (Supporting Information). In contrast, for the FED of Cu/MXene heterostructure, the higher energy barrier for *C2O2H formation at the C1‐C1 coupling step can be reduced by the overall enhanced binding energy of the intermediate. Subsequently, both Cu2O/MXene and Cu/MXene heterostructures facilitate the formation of *C2O2H and enable further reduction processes as a spontaneous downhill reaction to produce C3 compounds such as C3H8 through *C2‐*C1 coupling. Moreover, the correlation analysis between calculated reaction free energies for *C2O2H and *C3H4O formation through sequential processes of additional *CO supply and C‐C coupling in Figure 6c and Table S10 (Supporting Information) clearly shows that the MXene incorporation plays an important role in promoting the formation reactions of *C2O2H and *C3H4O.[ 59 ] Therefore, Cu2O/MXene can proceed CO2RR to C3H8(g) production without specifically high energy barrier. However, Cu/MXene cannot produce the final C3H8(g) compound due to the enhanced intermediate binding energy. Especially, the strong binding energy of Cu/MXene for *C3H7 results in a very high energy barrier for C3H8(g) formation, thus increasing the UL value from −1.20 V to −1.98 V compared to pure Cu(111).
The conflicting behavior of intermediate binding energy can be comprehended by lattice parameter mismatch in the Cu2O/MXene and Cu/MXene heterostructures, as analyzed in Figure 6d. Cu2O and Cu in the heterostructure with MXene undergo compressive (−4.6%) and tensile (+14.4%) strain, respectively. Cu in the Cu/MXene heterostructure suffers severe lattice strain, and the extremely large tensile strain on Cu induces strong binding energy of intermediate to stabilize the strained structure through intermediate binding. For Cu2O in the Cu2O/MXene heterostructure, the weakened binding energy of the intermediate can be explained by electronic structure modulation, i.e., d‐band center shift. The binding strength of adsorbents is closely related to the filling of the anti‐bonding state near the Fermi level, which is estimated by the d‐band center (εd) theory of Hammer and Norskov.[ 60 ] The increased filling of the anti‐bonding state leads to weak binding energy of adsorbents. The partial density of states (PDOS) analysis of the d‐orbital of Cu (εd(Cu)) in Figure 6e reveals that εd(Cu) of each Cu2O(111) and Cu2O/MXene are located at −2.16 eV and −2.51 eV, respectively. Here, the downshifted εd(Cu) of Cu2O/MXene position compared to εd(Cu) of Cu2O(111) proves that the binding energy of intermediates in Cu2O/MXene is relatively weak. Consequently, our theoretical calculations clearly show that the incorporation of MXene, especially into the Cu2O/MXene heterostructure plays a crucial role in the improvement of CO2RR catalytic activity up to C3H8(g) production by regulating the electronic structure and promoting the sequential processes of *CO supplying/C‐C coupling.
The intermediates involved in this mechanism are more complicated than those suggested in our study as 3 CO2 molecules with 20 electrons are required for C3H8 production. Nevertheless, the mechanism described in our study offers opportunities for the design of advanced catalysts for the efficient production of C3H8, with a pathway for electrochemical CO2 reduction and protonation of the hydrocarbon intermediates.
3. Conclusion
In summary, we designed Cu2O/Ti3C2Tx MXene, hybridized oxide‐derived Cu (Cu2O) on OH‐modulated MXene by NH4OH treatment and achieved highly active and selective production of C3H8 via electrochemical CO2 reduction reaction in aqueous electrolyte. ATR‐FTIR showed that Cu2O can strongly bind and stably preserve the reaction intermediates on the surface, but additional active sites are required for *C2‐*C1 coupling toward C3 hydrocarbons. Thus, owing to the synergistic effects of Cu2O and MXene, the interface between Cu2O and MXene is a key factor for the CO2 reduction to C3H8, where Cu2O binds *C2 intermediates and MXene binds *CO for C3 coupling with further efficient protonation for C3H8 production. In detailed characterizations, Cu2O/MXene exhibited a remarkable FE enhancement toward C3H8 (≈3.3%), representing a 26‐fold increase compared to Cu/MXene without the aid of carbon sources neither on the surface on the catalyst nor the ionomer. DFT calculations also highlight the effective pathway for electrochemical CO2 reduction and protonation of hydrocarbon intermediates to produce C3H8 in the presence of the interface between Cu2O and MXene. This study not only provides evidence of the reaction process in the selective electrochemical reduction of CO2 to C3H8 but also suggests a rational electrocatalyst design strategy for CO2RR. This strategy involves multi‐carbon coupling as well as protonation to produce high‐energy‐density products, including hydrogen‐rich hydrocarbons.
4. Experimental Section
Synthesis of Ti3C2Tx MXene
2D Ti3C2Tx MXene nanosheets were prepared by a typical etching method previously reported with some modifications.[ 14 ] Initially, 2.0 g of the MAX powder (Ti3AlC2) was added to 20 mL of an aqueous HF solution in a Teflon liner and stirred for 36 h at 50 °C to selectively etch the Al layer from the MAX powder. The obtained suspension was washed several times with deionized water (DI water) via centrifugation to remove the acidic solvent. The precipitated samples were then collected and freeze‐dried. After freeze‐drying, the 1.0 g of MXene powder was mixed with 12 mL of DMSO and stirred for 18 h at room temperature for intercalation. After intercalation, a tip sonicator was used in an ice‐water bath for 4 h for delamination. The resulting solution was centrifuged several times with DI water to remove the remained DMSO. Finally, the obtained MXene was dispersed in DI water and stored in the fridge until use.
Synthesis of Cu Nanoparticles
First, 30 mmol of 1‐octadecene, 4 mmol of oleic acid, and 8 mmol of 1,5‐pentanediol were mixed and heated at 130 °C for 30 min under an N2 atmosphere in a two‐necked round bottom flask to form solution A. Another bottle of 1 mmol Cu(acac)2 into 5 mmol oleylamine was heated at 70 °C for 30 min with stirring to form solution B. Subsequently, solution B was injected into solution A and heated at 200 °C for 2 h under stirring. After heating at 200 °C, the obtained solution was then rapidly cooled. To remove organic impurities, the sample was washed and centrifuged several times with hexane and IPA, then freeze‐dried. Finally, the products obtained as a powder were denoted as CuNPs.
Synthesis of Cu2O/MXene
The as‐prepared MXene solution was diluted to 1 mg mL−1 in DI water and sonicated for 30 min to prevent restacking. The synthesis of Cu2O/MXene adopted a hydrothermal method. 6 mg of Cu NPs and 4 mmol of ammonia solution (NH4OH) were added into 25 mL of the MXene solution and kept at 70 °C for 30 min under N2 to inhibit further oxidation of MXene. For comparison, different mass ratios of CuNPs to MXene were prepared: 2:25, 6:25, and 10:25 wt./wt. (denoted as Cu2O/MXene 2:25, 6:25, and 10:25, respectively). The mixture was then ultrasonicated at room temperature for 1 h. After sonication, the obtained Cu2O/MXene sample was washed several times with DI water, centrifuged, and vacuum dried to prevent further oxidation. Cu/MXene was prepared following the same procedure as for Cu2O/MXene, except for adding 4 mmol of NH4OH. Additionally, MXene subjected to a hydrothermal process with NH4OH treatment (AT‐MXene) was prepared following the same procedure as for Cu2O/MXene, except for adding CuNPs.
Electrochemical Measurements
The electrochemical CO2 reduction reaction (CO2RR) measurements were conducted by a three‐electrode configuration using an electrochemical working station (Gamry Reference 600+). To prepare the working electrode, 2 mg of catalyst was dispersed in 0.5 mL isopropyl alcohol (IPA) with a sonicator. After the sonication, 5 µL of Nafion (5 wt.%) was additionally mixed with the catalyst solution and further ultrasonicated for 30 min to obtain a catalyst ink. The 40 µl of ink was then dropped onto the carbon cloth with a geometric area of 1 cm2 and dried in the vacuum oven. The carbon cloth was pretreated by soaking it in 3 M HCl for 15 min and then washed several times with DI water and ethanol, followed by N2 blowing until dry. The Pt plate and Ag/AgCl (3 M NaCl) electrode served as the counter and reference electrodes, respectively. As an electrolyte, CO2 saturated with 0.1 M KHCO3 (pH = 6.8) was prepared and kept at a CO2 flow rate of 20 sccm using a mass flow controller during electrochemical CO2RR measurements. LSV was performed at a scan rate of 5 mV s−1. All potentials were reported with respect to the RHE with the iR correction. Electrochemical impedance spectroscopy (EIS) was conducted in the frequency range of 105–10−1 Hz and amplitude of 5 mV at −1.0 V versus RHE. The electrochemical double‐layer capacitance method was used for the electrochemical active surface area (ECSA) measurements, which were extracted from the cyclic voltammograms (CV) at different scan rates (2, 5, 10, 20, and 25 mV s−1) in the non‐Faradaic region. Note that all electrochemical data (except stability testing) were repeated more than three times, with error bars representing the standard deviation of the data.
Computational Details
All density functional theory (DFT) calculations were performed with the Vienna Ab initio Simulation Package (VASP 5.4.4).[ 61 ] The Projector Augmented Wave (PAW) method[ 61 , 62 ] was employed and exchange‐correlation interactions were treated through Perdew‐Burke‐Ernzerohf (PBE)[ 63 ] functional under the generalized gradient approximation (GGA). Monkhorst‐Pack k‐point meshes of 2 × 4 × 1 and 1 × 4 × 1 were used in each primitive lattice vector of the reciprocal space for geometry optimization of pure Cu2O(111) and Cu(111) and Cu2O (or Cu)/MXene heterostructures, respectively with Brillouin zone sampling.[ 64 ] And DFT‐D3 dispersion correction method was used to reflect the non‐bonding interactions correlation in the Cu2O/MXene system.[ 63 , 65 ] Lattice constants and internal atomic positions were fully optimized using a plane‐wave cutoff energy of 500 eV and spin‐polarized calculations until the residual forces were less than 0.04 eV Å−1. Additional description of the structural information and CO2RR catalytic activity evaluation is described in the Supporting Information.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
J.Y.K. and W.T.H. contributed equally to this work. J.Y.K. designed the experiment and methodology and conducted the formal analysis and investigations. W.T.H. performed the data curation, material synthesis, characterization, and investigation. T.K.C.P. performed the methodology, assisted with investigations, and contributed to the initiation of this work. S.C.C. conducted the DFT calculation. B.K. and U.B. assisted with the investigation and methodology. H.‐S.O., J.H.K., X.Y., C.‐H.C., and J.P. conducted the validations, assisted with the formal analysis, and conducted funding acquisition. S.U.L. supervised the DFT calculation, provided the resources and revised the manuscript. C.‐H.C. provided the resources, conducted the investigation and funding acquisition, and assisted with the formal analysis. J.K.K. conceptualized this work, supervised this project, performed project administration, and conducted funding acquisition. All authors contributed to the writing, revising, and editing of this manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF‐2022R1A2C1011559, and 2020R1A6A1A03048004). This work was partly supported by the GRRC program of Gyeonggi province [(GRRCKYUNGHEE2023‐B01), Development of ultra‐fine process materials based on the sub‐nanometer class for the next‐generation semiconductors]. This work was supported by the Technology Innovation Program (RS‐2024‐00419747, Development of Materials and Devices Based on Tandem Device to Achieve High Efficiency and Long Lifetime Blue OLEDs for IT Display Applications) funded By the Ministry of Trade, Industry & Energy (MOTIE, Korea). This work is funded by Korea Institute of Science and Technology (KIST) Institutional Program (2E33251). Experiments at PLS‐II were supported in part by MSIT and POSTECH.
Kim J. Y., Hong W. T., Phu T. K. C., Cho S. C., Kim B., Baeck U., Oh H.‐S., Koh J. H., Yu X., Choi C. H., Park J., Lee S. U., Chung C.‐H., Kim J. K., Proton‐Coupled Electron Transfer on Cu2O/Ti3C2Tx MXene for Propane (C3H8) Synthesis from Electrochemical CO2 Reduction. Adv. Sci. 2024, 11, 2405154. 10.1002/advs.202405154
Contributor Information
Jongwook Park, Email: jongpark@khu.ac.kr.
Sang Uck Lee, Email: suleechem@skku.edu.
Jung Kyu Kim, Email: legkim@skku.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. De Luna P., Hahn C., Higgins D., Jaffer S. A., Jaramillo T. F., Sargent E. H., Science 2019, 364, eeav3506. [DOI] [PubMed] [Google Scholar]
- 2.a) Climate Change 2022 – Mitigation of Climate Change, Cambridge University Press, Cambridge, 2023; [Google Scholar]; b) Kim J. Y., Kim D., Li Z. J., Dariva C., Cao Y. K., Ellis N., Energy 2023, 263, 125900. [Google Scholar]
- 3.a) Du J. H., Cheng B. G., Yuan H. Q., Tao Y., Chen Y., Ming M., Han Z. J., Eisenberg R., Angew. Chem. Int. Ed. 2023, 62, e202211804; [DOI] [PubMed] [Google Scholar]; b) Liu Y. Q., Guo Z. Y., Qiu Z. Y., Wang W. W., Lin H. P., Zhao X., Dang J. S., ACS Appl. Mater. Interfaces 2022, 14, 46657. [DOI] [PubMed] [Google Scholar]
- 4. Esmaeilirad M., Jiang Z., Harzandi A. M., Kondori A., Tamadoni Saray M., Segre C. U., Shahbazian‐Yassar R., Rappe A. M., Asadi M., Nat. Energy 2023, 8, 891. [Google Scholar]
- 5. Fortunati A., Risplendi F., Re Fiorentin M., Cicero G., Parisi E., Castellino M., Simone E., Iliev B., Schubert T. J. S., Russo N., Hernandez S., Commun. Chem. 2023, 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Zhu Q., Sun X., Yang D., Ma J., Kang X., Zheng L., Zhang J., Wu Z., Han B., Nat. Commun. 2019, 10, 3851; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Y. H., Liu J. L., Zheng G. F., Adv. Mater. 2021, 33, 2005798; [Google Scholar]; c) Woldu A. R., Huang Z. L., Zhao P. X., Hu L. S., Astruc D., Coord. Chem. Rev. 2022, 454, 214340; [Google Scholar]; d) Zheng Y., Vasileff A., Zhou X. L., Jiao Y., Jaroniec M., Qiao S. Z., J. Am. Chem. Soc. 2019, 141, 7646. [DOI] [PubMed] [Google Scholar]
- 7.a) Zhi W. Y., Liu Y. T., Shan S. L., Jiang C. J., Wang H., Lu J. X., J CO2 Util 2021, 50, 101594; [Google Scholar]; b) Abdelnaby M. M., Liu K. L., Hassanein K., Yin Z. Y., Chem. Nano. Mat. 2021, 7, 969; [Google Scholar]; c) Ren H. J., Kovalev M., Weng Z. Y., Muhamad M. Z., Ma H. Y., Sheng Y., Sun L. B., Wang J. J., Rihm S., Yang W. F., Lapkin A. A., Ager J. W., Nat. Catal. 2022, 5, 1169. [Google Scholar]
- 8.a) Ting L. R. L., García‐Muelas R., Martín A. J., Veenstra F. L. P., Chen S. T. J., Peng Y. J., Per E. Y. X., Pablo‐García S., López N., Pérez‐Ramírez J., Yeo B. S., Angew. Chem. Int. Ed. 2020, 59, 21072; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Z. D., Attanayake N. H., Blackburn J. L., Miller E. M., Energy Environ. Sci. 2021, 14, 1696; [Google Scholar]; c) Kortlever R., Kortlever R., Peters I., Peters I., Balemans C., Balemans C., Kas R., Kas R., Kwon Y., Kwon Y., Mul G., Mul G., Koper M. T. M., Koper M. T. M., Chem. Commun. 2016, 52, 10229. [DOI] [PubMed] [Google Scholar]
- 9. Zhang J. C., Cai W. Z., Hu F. X., Yang H. B., Liu B., Chem. Sci. 2021, 12, 6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu C. J., Yang L., Yan B. Z., Sun L. B., Zhang P. G., Zhang W., Sun Z. M., Adv. Funct. Mater. 2020, 30, 2000852. [Google Scholar]
- 11. Chen H. T., Handoko A. D., Xiao J. W., Feng X., Fan Y. C., Wang T. S., Legut D., Seh Z. W., Zhang Q. F., ACS Appl. Mater. Interfaces 2019, 11, 36571. [DOI] [PubMed] [Google Scholar]
- 12. Zhao D. Y., Zhao R. Z., Dong S. H., Miao X. G., Zhang Z. W., Wang C. X., Yin L. W., Energy Environ. Sci. 2019, 12, 2422. [Google Scholar]
- 13. Fu W. L., Liu Z., Wang T. Y., Liang J. S., Duan S., Xie L. F., Han J. T., Li Q., ACS Sustain. Chem. Eng. 2020, 8, 15223. [Google Scholar]
- 14. Nguyen D. N., Gund G. S., Jung M. G., Roh S. H., Park J., Kim J. K., Park H. S., ACS Nano 2020, 14, 17615. [DOI] [PubMed] [Google Scholar]
- 15.a) Le T. A., Bui Q. V., Tran N. Q., Cho Y., Hong Y., Kawazoe Y., Lee H., ACS Sustain. Chem. Eng. 2019, 7, 16879; [Google Scholar]; b) Kong X. L., Peng Z. B., Jiang R., Jia P. P., Feng J., Yang P. P., Chi Q. Q., Ye W., Xu F. C., Gao P., ACS Appl. Nano Mater. 2020, 3, 1373; [Google Scholar]; c) Zhao Q., Zhang C., Hu R. M., Du Z. G., Gu J. N., Cui Y. L. S., Chen X., Xu W. J., Cheng Z. J., Li S. M., Li B., Liu Y. F., Chen W. H., Liu C. T., Shang J. X., Song L., Yang S. B., ACS Nano 2021, 15, 4927. [DOI] [PubMed] [Google Scholar]
- 16. Naguib M., Mashtalir O., Carle J., Presser V., Lu J., Hultman L., Gogotsi Y., Barsoum M. W., ACS Nano 2012, 6, 1322. [DOI] [PubMed] [Google Scholar]
- 17.a) Raj K. A. S., Barman N., Radhakrishnan S., Thapa R., Rout C. S., J. Mater. Chem. A 2022, 10, 23590; [Google Scholar]; b) Wang X., Li H., Li H., Lin S., Ding W., Zhu X. G., Sheng Z. G., Wang H., Zhu X. B., Sun Y. P., Adv. Funct. Mater. 2020, 30, 1910302. [Google Scholar]
- 18. Xue Q., Zhang H. J., Zhu M. S., Pei Z. X., Li H. F., Wang Z. F., Huang Y., Huang Y., Deng Q. H., Zhou J., Du S. Y., Huang Q., Zhi C. Y., Adv. Mater. 2017, 29, 1604847. [DOI] [PubMed] [Google Scholar]
- 19. Raza M. A., Li F., Que M. D., Zhu L. L., Chen X., Adv. Funct. Mater. 2021, 2, 7187. [Google Scholar]
- 20. Xiao Y., Men C., Chu B. X., Qin Z. Z., Ji H. B., Chen J. H., Su T. M., Chem. Eng. J. 2022, 446, 137028. [Google Scholar]
- 21. Wang Y., Dou H., Wang J., Ding B., Xu Y. L., Chang Z., Hao X. D., J. Power Sources 2016, 327, 221. [Google Scholar]
- 22. Bao H. H., Qiu Y., Peng X. Y., Wang J. A., Mi Y. Y., Zhao S. Z., Liu X. J., Liu Y. F., Cao R., Zhuo L. C., Ren J. Q., Sun J. Q., Luo J., Sun X. P., Nat. Commun. 2021, 12, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao Y., Deng Q. H., Liu Z. D., Shen D. Y., Wang T., Huang Q., Du S. Y., Jiang N., Lin C. T., Yu J. H., RSC Adv. 2017, 7, 20494. [Google Scholar]
- 24.a) Nguyen D. N., Phu T. K. C., Kim J., Hong W. T., Kim J. S., Roh S. H., Park H. S., Chung C. H., Choe W. S., Shin H., Lee J. Y., Kim J. K., Small 2022, 18, 2204797; [DOI] [PubMed] [Google Scholar]; b) Zhang Y. K., Jiang H. L., Lin Y. X., Liu H. J., He Q., Wu C. Q., Duan T., Song L., Adv. Mater. Interfaces 2018, 5, 1800392. [Google Scholar]
- 25.a) Hou T. T., Luo Q. Q., Li Q., Zu H. L., Cui P. X., Chen S. W., Lin Y., Chen J. J., Zheng X. S., Zhu W. K., Liang S. Q., Yang J. L., Wang L. B., Nat. Commun. 2020, 11, 4251; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou H. Y., Han S. J., Lee H. D., Zhang D. Z., Anayee M., Jo S. H., Gogotsi Y., Lee T. W., Adv. Mater. 2022, 34, 2206377; [DOI] [PubMed] [Google Scholar]; c) Schultz T., Frey N. C., Hantanasirisakul K., Park S., May S. J., Shenoy V. B., Gogotsi Y., Koch N., Chem. Mater. 2019, 31, 6590; [Google Scholar]; d) Benchakar M., Loupias L., Garnero C., Bilyk T., Morais C., Canaff C., Guignard N., Morisset S., Pazniak H., Hurand S., Chartier P., Pacaud J., Mauchamp V., Barsoum M. W., Habrioux A., Célérier S., Appl. Surf. Sci. 2020, 530, 147209. [Google Scholar]
- 26.a) Kobayashi T., Sun Y. Y. L., Prenger K., Jiang D. E., Naguib M., Pruski M., J. Phys. Chem. C 2020, 124, 13649; [Google Scholar]; b) Zhou Y., Wang Y. H., Wang Y. J., Li X., Anal. Chem. 2020, 92, 16033; [DOI] [PubMed] [Google Scholar]; c) Chen W. Y., Jiang X. F., Lai S. N., Peroulis D., Stanciu L., Nat. Commun. 2020, 11, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H. W., Naguib M., Page K., Wesolowski D. J., Gogotsi Y., Chem. Mater. 2016, 28, 349. [Google Scholar]
- 28. Chu S. L., Kang C., Park W., Han Y., Hong S., Hao L. D., Zhang H., Lo T. W. B., Robertson A. W., Jung Y. S., Han B. X., Sun Z. Y., SmartMat 2022, 3, 194. [Google Scholar]
- 29. Sarycheva A., Gogotsi Y., Chem. Mater. 2020, 32, 3480. [Google Scholar]
- 30.a) Maaoui H., Singh S. K., Teodorescu F., Coffinier Y., Barras A., Chtourou R., Kurungot S., Szunerits S., Boukherroub R., Electrochim. Acta 2017, 224, 346; [Google Scholar]; b) Jiang Y. W., Wang X. Y., Duan D. L., He C. H., Ma J., Zhang W. Q., Liu H. J., Long R., Li Z. B., Kong T. T., Loh X. J., Song L., Ye E. N., Xiong Y. J., Adv. Sci. 2022, 9, 2105292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Yang Y., He A. B., Li H., Zou Q., Liu Z. H., Tao C. Y., Du J., ACS Catal. 2022, 12, 12942. [Google Scholar]; b) Li J. H., Xu K. Q., Liu F. M., Li Y. Z., Hu Y. F., Chen X. J., Wang H., Xu W. C., Ni Y. X., Ding G. Y., Zhao T. T., Yu M., Xie W., Cheng F. Y., Adv. Mater. 2023, 35, 2301127; [Google Scholar]; c) Timoshenko J., Bergmann A., Rettenmaier C., Herzog A., Arán‐Ais R. M., Jeon H. S., Haase F. T., Hejral U., Grosse P., Kühl S., Davis E. M., Tian J., Magnussen O., Cuenya B. R., Nat. Catal. 2022, 5, 259. [Google Scholar]
- 32. Yan C. L., Luo W., Yuan H. M., Liu G. Y., Hao R., Qin N., Wang Z. Q., Liu K., Wang Z. Y., Cui D. H., Hu Z. F., Lan Y. C., Lu Z. G., Appl. Catal. B 2022, 308, 121191. [Google Scholar]
- 33. Kim T., Kargar A., Luo Y. Q., Mohammed R., Martinez‐Loran E., Ganapathi A., Shah P., Fenning D. P., ACS Appl. Energy Mater. 2018, 1, 1965. [Google Scholar]
- 34.a) Gao D. F., Zhou H., Cai F., Wang D. N., Hu Y. F., Jiang B., Cai W. B., Chen X. Q., Si R., Yang F., Miao S., Wang J. G., Wang G. X., Bao X. H., Nano Res. 2017, 10, 2181; [Google Scholar]; b) Ogura K., J CO2 Util 2013, 1, 43; [Google Scholar]; c) Lee J. E., Yamaguchi A., Ooka H., Kazami T., Miyauchi M., Kitadai N., Nakamura R., Chem. Comm. 2021, 57, 3267. [DOI] [PubMed] [Google Scholar]
- 35.a) Holmberg V. C., Korgel B. A., Chem. Mater. 2010, 22, 3698; [Google Scholar]; b) Wood J., Alldrick M. J., Winterbottom J. M., Stitt E. H., Bailey S., Catal. Today 2007, 128, 52. [Google Scholar]
- 36. Kim Y., Park S., Shin S. J., Choi W., Min B. K., Kim H., Kim W., Hwang Y. J., Energ Environ. Sci. 2020, 13, 4301. [Google Scholar]
- 37.a) Podrojková N., Sans V., Orinak A., Orinaková R., ChemCatChem 2020, 12, 1802; [Google Scholar]; b) Van den Bossche M., Rose‐Petruck C., Jónsson H., J. Phys. Chem. C 2021, 125, 13802; [Google Scholar]; c) Cheng T., Xiao H., Goddard W. A., J. Am. Chem. Soc. 2016, 138, 13802. [DOI] [PubMed] [Google Scholar]
- 38.a) Katayama Y., Nattino F., Giordano L., Hwang J., Rao R. R., Andreussi O., Marzari N., Shao‐Horn Y., J. Phys. Chem. C 2019, 123, 5951; [Google Scholar]; b) Nguyen D. L. T., Kim Y., Hwang Y. J., Won D. H., Carbon Energy 2020, 2, 72. [Google Scholar]
- 39. Choi B. N., Seo J. Y., An Z., Yoo P. J., Chung C. H., Chem. Eng. J. 2022, 430, 132807. [Google Scholar]
- 40. Zhi X., Jiao Y., Zheng Y., Vasileff A., Qiao S. Z., Nano Energy 2020, 71, 104601. [Google Scholar]
- 41. Gao J., Bahmanpour A., Kröcher O., Zakeeruddin S. M., Ren D., Grätzel M., Nat. Chem. 2023, 15, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ternero‐Hidalgo J. J., Daturi M., Clet G., Bazin P., Bañares M. A., Portela R., Guerrero‐Pérez M. O., Rodríguez‐Mirasol J., Cordero T., Catal. Today 2022, 387, 197. [DOI] [PubMed] [Google Scholar]
- 43. Chen C., Chen F. F., Yang B., Zhang K., Lv X. Y., Chen C., Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2022, 269, 120684. [DOI] [PubMed] [Google Scholar]
- 44. Socrates G., Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed., John Wiley & Sons, New York, 2004. [Google Scholar]
- 45. Rahaman M., Kiran K., Montiel I. Z., Grozovski V., Dutta A., Broekmann P., Green Chem. 2020, 22, 6497. [Google Scholar]
- 46.a) Winkler M. E. G., Gonçalves R. H., Rubira A. F., ACS Omega 2022, 7, 45067; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schwan R., Kaufmann M., Leicht D., Schwaab G., Havenith M., Phys. Chem. Chem. Phys. 2016, 18, 24063. [DOI] [PubMed] [Google Scholar]
- 47.a) Moosakhani A., Parvin P., Reyhani A., Mortazavi S. Z., Phys. Plasmas 2017, 24, 13505; [Google Scholar]; b) Harrison J. J., Bernath P. F., J. Quant. Spectrosc. Radiat. Transf. 2010, 111, 1282. [Google Scholar]
- 48.a) Kuhl K. P., Cave E. R., Abram D. N., Jaramillo T. F., Energ. Environ. Sci. 2012, 5, 7050; [Google Scholar]; b) Rihm S. D., Kovalev M. K., Lapkin A. A., Ager J. W., Kraft M., Energ. Environ. Sci. 2023, 16, 1697. [Google Scholar]
- 49. Vasileff A., Zhu Y. P., Zhi X., Zhao Y. Q., Ge L., Chen H. M., Zheng Y., Qiao S. Z., Angew. Chem. Int. Ed. 2020, 59, 19649. [DOI] [PubMed] [Google Scholar]
- 50. Nitopi S., Bertheussen E., Scott S. B., Liu X. Y., Engstfeld A. K., Horch S., Seger B., Stephens I. E. L., Chan K., Hahn C., Norskov J. K., Jaramillo T. F., Chorkendorff I., Chem. Rev. 2019, 119, 7610. [DOI] [PubMed] [Google Scholar]
- 51.a) Handoko A. D., Wei F. X., Jenndy, B. S. Y. , Seh Z. W., Nat. Catal. 2018, 1, 922; [Google Scholar]; b) Fan L., Xia C., Yang F. Q., Wang J., Wang H. T., Lu Y. Y., Sci. Adv. 2020, 6, eaay3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gao Y. G., Wu Q., Liang X. Z., Wang Z. Y., Zheng Z. K., Wang P., Liu Y. Y., Dai Y., Whangbo M. H., Huang B. B., Adv. Sci. 2020, 7, 1902820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo Z. L., Li Y., Sa B. S., Fang Y., Lin J., Huang Y., Tang C. C., Zhou J., Miao N. H., Sun Z. M., Appl. Surf. Sci. 2020, 521, 146. [Google Scholar]
- 54. Nguyen T. P., Nguyen D. M. T., Tran D. L., Le H. K., Vo D. V. N., Lam S. S., Varma R. S., Shokouhimehr M., Nguyen C. C., Le Q. V., Mol. Catal. 2020, 486, 110850. [Google Scholar]
- 55.a) Norskov J. K., Rossmeisl J., Logadottir A., Lindqvist L., Kitchin J. R., Bligaard T., Jónsson H., J. Phys. Chem. B 2004, 108, 17886; [Google Scholar]; b) Man I. C., Su H. Y., Calle‐Vallejo F., Hansen H. A., Martínez J. I., Inoglu N. G., Kitchin J., Jaramillo T. F., Norskov J. K., Rossmeisl J., ChemCatChem 2011, 3, 1159. [Google Scholar]
- 56. Wu L. N., Tian Z. Y., Qin W., Molecules 2022, 27, 6748.36235282 [Google Scholar]
- 57.a) Shinde S. S., Jung J. Y., Wagh N. K., Lee C. H., Kim D. H., Kim S. H., Lee S. U., Lee J. H., Nat. Energy 2021, 6, 592; [Google Scholar]; b) Ahmed A. A., Lee C. H., Ansari A., Pawar S. M., Han J., Park S., Shin G., Yeon S., Cho S., Seol J., Lee S. U., Kim H., Im H., Appl. Surf. Sci. 2022, 592, 153196; [Google Scholar]; c) Wagh N. K., Kim D. H., Lee C. H., Kim S. H., Um H. D., Kwon J. S. I., Shinde S. S., Lee S. U., Lee J. H., Nanoscale Horiz. 2023, 8, 921; [DOI] [PubMed] [Google Scholar]; d) Shinde S. S., Wagh N. K., Lee C. H., Kim D. H., Kim S. H., Um H. D., Lee S. U., Lee J. H., Adv. Mater. 2023, 35, 2303509; [DOI] [PubMed] [Google Scholar]; e) Yang J., Cho S. C., Lee S., Yoon J. W., Jeong W. H., Song H. C., Oh J. T., Lim S. G., Bae S. Y., Lee B. R., Ahmadi M., Sargent E. H., Yi W., Lee S. U., Choi H., ACS Nano 2022, 16, 1649. [DOI] [PubMed] [Google Scholar]
- 58.a) Wagh N. K., Shinde S. S., Lee C. H., Kim S.‐H., Kim D.‐H., Um H.‐D., Lee S. U., Lee J.‐H., Nanomicro Lett. 2022, 14, 190; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jin H., Kim H. S., Lee C. H., Hong Y., Choi J., Baik H., Lee S. U., Yoo S. J., Lee K., Park H. S., ACS Catal. 2022, 12, 13638. [Google Scholar]
- 59.a) Kuo T. C., Chou J. W., Shen M. H., Hong Z. S., Chao T. H., Lu Q., Cheng M. J., J. Phys. Chem. C 2021, 125, 2464; [Google Scholar]; b) Pablo‐García S., Veenstra F. L. P., Ting L. R. L., García‐Muelas R., Dattila F., Martín A. J., Yeo B. S., Pérez‐Ramírez J., López N., Catal. Sci. Technol. 2022, 12, 409; [Google Scholar]; c) Wang X., Wang Z. Y., Zhuang T. T., Dinh C. T., Li J., Nam D. H., Li F. W., Huang C. W., Tan C. S., Chen Z. T., Chi M. F., Gabardo C. M., Seifitokaldani A., Todorovic P., Proppe A., Pang Y. J., Kirmani A. R., Wang Y. H., Ip A. H., Richter L. J., Scheffel B., Xu A. N., Lo S. C., Kelley S. O., Sinton D., Sargent E. H., Nat. Commun. 2019, 10, 5186; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang X., Ou P. F., Ozden A., Hung S. F., Tam J., Gabardo C. M., Howe J. Y., Sisler J., Bertens K., de Arquer F. P. G., Miao R. K., O'Brien C. P., Wang Z. Y., Abed J., Rasouli A. S., Sun M. J., Ip A. H., Sinton D., Sargent E. H., Nat. Energy 2022, 7, 170. [Google Scholar]
- 60. Hammer B., Norskov J. K., Nature 1995, 376, 238. [Google Scholar]
- 61.a) Kresse G., Hafner J., Phys. Rev. B 1993, 48, 13115; [DOI] [PubMed] [Google Scholar]; b) Kresse G., Hafner J., Phys. Rev. B 1994, 49, 14251; [DOI] [PubMed] [Google Scholar]; c) Kresse G., Furthmuller J., Comput. Mater. Sci. 1996, 6, 15; [Google Scholar]; d) Kresse G., Furthmüller J., Phys. Rev. B 1996, 54, 11169. [DOI] [PubMed] [Google Scholar]
- 62.a) Blochl P. E., Phys. Rev. B 1994, 50, 17953; [DOI] [PubMed] [Google Scholar]; b) Kresse G., Joubert D., Phys. Rev. B 1999, 59, 1758. [Google Scholar]
- 63. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- 64. Monkhorst H., Pack J., Phys. Rev. B 1976, 13, 5188. [Google Scholar]
- 65. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.