

Abstract
RAFT aqueous emulsion polymerization of isopropylideneglycerol monomethacrylate (IPGMA) is used to prepare a series of PGEO5MA46‐PIPGMAy nanoparticles, where PGEO5MA is a hydrophilic methacrylic steric stabilizer block bearing pendent cis‐diol groups. TEM studies confirm a spherical morphology while dynamic light scattering (DLS) analysis indicated that the z‐average particle diameter can be adjusted by varying the target degree of polymerization for the core‐forming PIPGMA block. Periodate oxidation is used to convert the cis‐diol groups on PGEO5MA46‐PIPGMA500 and PGEO5MA46‐PIPGMA1000 nanoparticles into the analogous aldehyde‐functionalized nanoparticles, which are then reacted with histidine via reductive amination. In each case, the extent of functionalization is more than 99% as determined by 1H NMR spectroscopy. Aqueous electrophoresis studies indicate that such derivatization converts initially neutral nanoparticles into zwitterionic nanoparticles with an isoelectric point at pH 7. DLS studies confirm that such histidine‐derivatized nanoparticles remain colloidally stable over a wide pH range. A quartz crystal microbalance is employed at 25°C to assess the adsorption of both the cis‐diol‐ and histidine‐functionalized nanoparticles onto planar stainless steel at pH 6. The histidine‐bearing nanoparticles adsorb much more strongly than their cis‐diol counterparts. For the highest adsorbed amount of 70.5 mg m–2, SEM indicates a fractional surface coverage of 0.23 for the adsorbed nanoparticles.
Keywords: block copolymer self‐assembly, nanoparticle adsorption, polymerisation‐induced self‐assembly, quartz crystal microbalance, RAFT aqueous emulsion polymerisation
The authors examine the adsorption of a series of sterically‐stabilized diblock copolymer nanoparticles onto a model stainless steel substrate using a quartz crystal microbalance. The adsorbed amount depends on both the mean diameter and the surface functionality of the nanoparticles. Introducing histidine groups via periodate oxidation and reductive amination leads to twice the adsorbed amount compared to cis‐diol groups.
![]()
1. Introduction
Polymerization‐induced self‐assembly (PISA) is a powerful platform technology that enables the rational and efficient synthesis of a wide range of block copolymer nanoparticles of controllable size and shape.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 ] Typically, PISA involves growing an insoluble block from one end of a soluble block in a suitable selective solvent.[ 8 ] Once a critical degree of polymerization (DP) has been achieved for the growing second block, micellar nucleation occurs and the initially homogeneous polymerization becomes heterogeneous.[ 8 ] The nascent micelles continue to grow in size until the second monomer is fully consumed, leading to sterically‐stabilized diblock copolymer nanoparticles.[ 9 ]
In particular, aqueous PISA formulations have been exploited by many research groups to prepare various types of functional nanoparticles,[ 10 , 11 , 12 , 13 , 14 ] most commonly using reversible addition‐fragmentation chain transfer (RAFT) polymerization.[ 2 , 4 , 8 , 15 , 16 , 17 ] If the growth of the second block involves using a water‐miscible vinyl monomer, this is known as RAFT aqueous dispersion polymerization. Such formulations provide convenient access to spherical, worm‐like or vesicular copolymer morphologies for which the design rules are now well‐understood.[ 2 , 9 , 18 ] However, there are many more examples of water‐immiscible vinyl monomers, for which PISA can be conducted via RAFT aqueous emulsion polymerization.[ 3 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 ] The latter formulations are often associated with kinetically‐trapped spherical morphologies even when targeting highly asymmetric diblock copolymer compositions,[ 30 , 31 , 32 , 33 ] although access to either worms or vesicles can be achieved for certain formulations.[ 12 , 34 , 35 , 36 , 37 ]
Two of the most common hydrophilic steric stabilizers used in the aqueous PISA literature are poly(ethylene glycol) (PEG)[ 38 , 39 ] and poly(glycerol monomethacrylate) (PGMA).[ 12 , 34 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ] Recently, we reported a new hybrid water‐soluble polymer that combined the structural feature associated with PEG and PGMA (see Scheme S1, Supporting Information).[ 48 , 49 , 50 ] More specifically, the oligo(ethylene glycol) spacer between the methacrylic backbone and the pendent cis‐diol group enables the latter to be converted into the corresponding aldehyde without any loss in water solubility when using a selective oxidant such as sodium periodate.[ 48 , 49 , 50 , 51 ] This approach enables further chemical modification with various amino acids to be conducted in aqueous solution via Schiff base chemistry, with subsequent reduction to a more stable secondary amine linkage being achieved using NaCNBH3.[ 48 , 49 , 52 ]
Herein, we have extended this concept to include cis‐diol functionalized diblock copolymer nanoparticles, which are prepared via RAFT aqueous emulsion polymerization of isopropylideneglycerol monomethacrylate (IPGMA) using PGEO5MA as the steric stabilizer block (Scheme 1 ). Like many similar formulations,[ 13 , 42 , 53 , 54 ] this aqueous PISA protocol produces well‐defined kinetically‐trapped spheres. The pendent cis‐diol groups on such sterically‐stabilized diblock copolymer nanoparticles were then oxidized using periodate and subsequently reacted with histidine to introduce imidazole functionality into the steric stabilizer chains. Such derivatization led to a profound change in the electrophoretic footprint of the nanoparticles without any loss in colloidal stability. Finally, their adsorption onto a planar stainless steel substrate was examined using a quartz crystal microbalance (QCM), with the precursor cis‐diol‐functionalized nanoparticles serving as an appropriate reference. In principle, this model system should be relevant for the development of aqueous lubrication formulations and anti‐corrosion coatings.
Scheme 1.
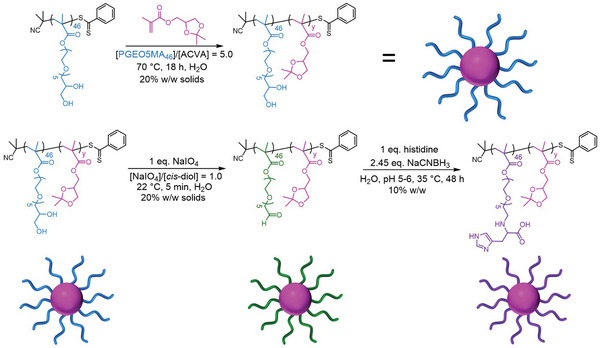
Synthesis route used to prepare the PGEO5MA46‐PIPGMAy spherical nanoparticles employed in this study. The first step involves the RAFT aqueous emulsion polymerization of IPGMA using a water‐soluble PGEO5MA46 precursor to form a series of sterically‐stabilized PGEO5MA46‐PIPGMAy spherical nanoparticles (where y = 500 to 2000). The second step involves the selective periodate oxidation of the pendent cis‐diol groups on the PGEO5MA46 chains to form the corresponding aldehyde‐functional spherical nanoparticles, followed by reductive amination to produce histidine‐functionalized nanoparticles.
2. Results and Discussion
A PGEO5MA46 precursor was synthesized via RAFT solution polymerization of GEO5MA in ethanol (Scheme S2 and Figure S1, Supporting Information). DMF GPC analysis (using a series of poly(methyl methacrylate) calibration standards) indicated that this homopolymer had an M n of 20.5 kg mol–1 and a dispersity, Đ of 1.30. This PGEO5MA46 precursor was then chain‐extended via RAFT aqueous emulsion polymerization of IPGMA at 20% w/w solids (Scheme 1). A series of PGEO5MA46‐PIPGMAy diblock copolymer nanoparticles were prepared. All polymerizations had high IPGMA conversions (>99% as judged by 1H NMR spectroscopy; Figure S2, Supporting Information). DMF GPC analysis indicated relatively high copolymer dispersities (Figure S3, Supporting Information), which are not uncommon for PISA formulations when targeting a relatively high DP for the core‐forming block.[ 40 , 56 , 57 , 58 ] Nevertheless, DLS analysis of the resulting series of PGEO5MA46‐PIPGMAy nanoparticles indicated relatively narrow particle size distributions in each case (DLS polydispersity ≤ 0.06), see Figure 1 . TEM analysis confirmed a spherical morphology for such nanoparticles (Figure 2a,b). Moreover, DLS studies indicated a linear increase in particle size with increasing PIPGMA DP (Figure S4, Supporting Information). Comparable results were obtained by Jesson et al. for similar spherical nanoparticles prepared by RAFT aqueous emulsion polymerization of IPGMA.[ 59 ]
Figure 1.
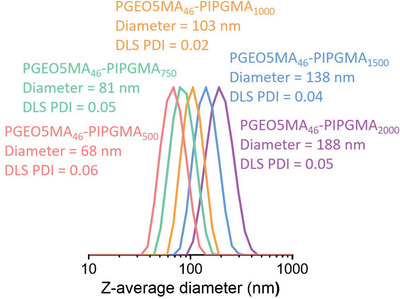
Dynamic light scattering (DLS) particle size distributions obtained for a series of PGEO5MA46‐PIPGMA500‐2000 spherical nanoparticles prepared by RAFT aqueous emulsion polymerization of IPGMA at 70 °C. Increasing the target DP of the hydrophobic core‐forming PIPGMA block enables the z‐average particle diameter to be systematically varied from 68 nm to 188 nm while retaining good control over the particle size distribution.
Figure 2.

Representative TEM images recorded for (a) PGEO5MA46‐PIPGMA500 and (b) PGEO5MA46‐PIPGMA1000 diblock copolymer nanoparticles.
PGEO5MA46‐PIPGMA500 and PGEO5MA46‐PIPGMA1000 nanoparticles were oxidized using aqueous NaIO4 (Scheme 1). A NaIO4/cis‐diol molar ratio of unity was employed to ensure full oxidation of the PGEO5MA chains. The extent of oxidation was determined to be more than 99% by 1H NMR spectroscopy in each case (Figure S5a, Supporting Information). DLS analysis indicated minimal change in the particle size distribution after oxidation (Figure 3a) and TEM studies showed that the original spherical morphology was retained after oxidation (Figure S6a,b, Supporting Information). Unfortunately, DMF GPC analysis could not be performed on these PAGEO5MA46‐PIPGMAy nanoparticles as the corresponding diblock copolymer solutions could not be passed through a 0.50 µm filter. This suggests some degree of crosslinking between the copolymer chains, possibly via hemiacetal formation. This is not surprising: Brotherton et al.[ 48 ] reported a large increase in copolymer dispersity for a series of PAGEO5MA26‐PHPMAy nano‐objects when subjected to periodate oxidation at 10% w/w solids. In the present study, the PGEO5MA46‐PIPGMAy nanoparticles were oxidized at 20% w/w solids, which suggests that the copolymer concentration has a significant effect on the degree of inter‐chain crosslinking that can occur during periodate oxidation. In principle, the formation of hemiacetal bonds could also occur between neighboring nanoparticles, as well as within nanoparticles. If so, this would be expected to lead to inter‐particle crosslinking. However, DLS studies provide no evidence for such a side‐reaction (see Figure 3a).
Figure 3.
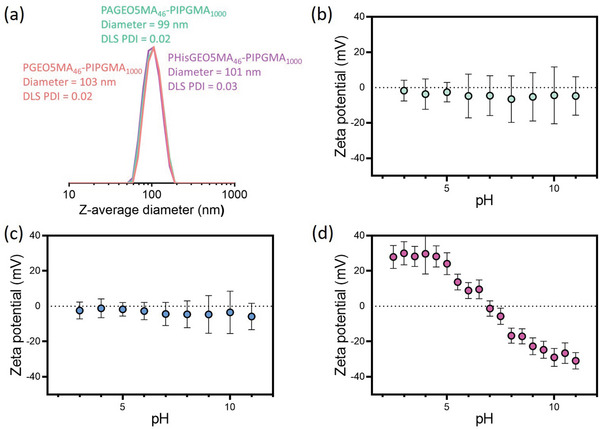
a) DLS particle size distributions obtained for PGEO5MA46‐PIPGMA1000, PAGEO5MA46‐PIPGMA1000 and PHisGEO5MA46‐PIPGMA1000 spherical nanoparticles. Clearly, chemical derivatization of the steric stabilizer chains leads to no significant change in either the z‐average diameter or the particle size distribution. Zeta potential versus pH curves obtained for b) PGEO5MA46‐PIPGMA1000, c) PAGEO5MA46‐PIPGMA1000 and d) PHisGEO5MA46‐PIPGMA1000.
According to Zhang and co‐workers, imidazole groups (such as those found in histidine) can bind strongly to stainless steel.[ 60 ] Similarly, there is good literature precedent to suggest that cis‐diol groups can act as a bidentate ligand to promote strong binding to iron atoms present within a surface oxide layer.[ 61 , 62 , 63 ]
Thus we sought to investigate whether introducing histidine functionality into the steric stabilizer chains could promote additional nanoparticle adsorption onto stainless steel.
Accordingly, PAGEO5MA46‐PIPGMAy nanoparticles were functionalized with histidine via reductive amination using excess NaCNBH3 as the reducing agent (Scheme 1). The extent of functionalization was confirmed to be more than 99% in all cases as determined by 1H NMR spectroscopy (Figure S5b, Supporting Information). DLS analysis of the resulting PHisGEO5MA46‐PIPGMAy nanoparticles indicated that such derivatization did not affect the particle size distribution (Figure 3a) while TEM studies confirmed that the original spherical morphology was retained (Figure S6c,d, Supporting Information).
However, aqueous electrophoresis studies revealed a substantial change in the electrophoretic footprint of the nanoparticles (Figure 3b–d).[ 48 ] As expected, the non‐ionic PGEO5MA46‐PIPGMAy and PAGEO5MA46‐PIPGMAy nanoparticles exhibit zeta potentials close to zero regardless of the solution pH. In contrast, the PHisGEO5MA46‐PIPGMAy nanoparticles exhibited positive zeta potentials at low pH, an isoelectric point at around pH 7, and negative zeta potentials above pH 7. Similar results were reported by Brotherton et al. for the analogous PHisGEO5MA46‐functionalized diblock copolymer vesicles.[ 48 ]
A series of QCM experiments were conducted to examine the adsorption of PGEO5MA46‐PIPGMA500/1000 and PHisGEO5MA46‐PIPGMA500/1000 nanoparticles onto a model planar substrate (stainless steel; Figure 4 ). According to the literature, QCM has been employed to determine the adsorption of various types of nanoparticles, including gold, silica, clay, and diblock copolymer nanoparticles.[ 64 , 65 , 66 , 67 , 68 ] In the present study, a substantial reduction in frequency was observed within 200 s for all four types of nanoparticles, indicating relatively fast adsorption kinetics onto the stainless steel surface.
Figure 4.
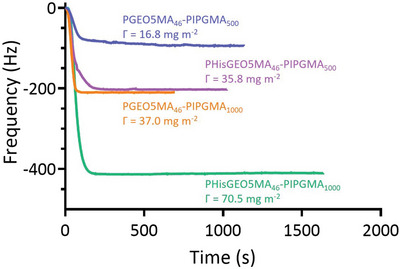
Change in frequency observed over time during the adsorption of PGEO5MA46‐PIPGMA500, PHisGEO5MA46‐PIPGMA500, PGEO5MA46‐PIPGMA1000 or PHisGEO5MA46‐PIPGMA1000 nanoparticles onto a planar stainless steel substrate using a quartz crystal microbalance at 25 °C.
As expected, increasing the mean nanoparticle diameter led to significantly greater adsorption for both the PGEO5MA46‐PIPGMA500/1000 and the PHisGEO5MA46‐PIPGMA500/1000 nanoparticles (Figure 4). For example, the adsorbed amount, Γ, obtained for the PGEO5MA46‐PIPGMA500 and PGEO5MA46‐PIPGMA1000 nanoparticles was 16.8 and 37.0 mg m–2 respectively at pH 6 (i.e., near the IEP). Similarly, PHisGEO5MA46‐PIPGMA500 nanoparticles exhibited a Γ of 35.8 mg m–2 while the corresponding PHisGEO5MA46‐PIPGMA1000 nanoparticles had a Γ of 70.5 mg m–2. These observations are physically reasonable: if relatively large and relatively small nanoparticles adsorb onto stainless steel at approximately the same surface number density, then the more massive nanoparticles will inevitably produce a higher adsorbed mass. Moreover, introducing histidine groups into the steric stabilizer chains led to an approximate two‐fold increase in the adsorbed amount. In view of this latter observation, we examined whether the histidine‐functionalized nanoparticles became colloidally unstable around their isoelectric point of pH 7. This control experiment was considered prudent because the QCM experiments were conducted at around pH 6. However, DLS analysis confirmed that there was essentially no change in the nanoparticle diameter for the PHisGEO5MA46‐PIPGMA1000 nanoparticles over a wide range of solution pH. Thus, it seems that the enhanced adsorbed amount is not related to colloidal instability. It is perhaps also worth emphasizing that these QCM experiments were conducted at a fixed nanoparticle concentration of 1.0% w/w. This means that the number concentration of the larger nanoparticles is lower than that of the smaller nanoparticles. Nevertheless, the adsorbed amount obtained for the former nanoparticles is twice as high as that observed for the latter. Moreover, the curvature of the QCM adsorption data shown in Figure 4 differs significantly when comparing the large and small nanoparticles at short run times (< 200 s). For both the cis‐diol‐ and the histidine‐functionalized nanoparticles, the smaller nanoparticles take longer to reach the equilibrium adsorbed amount compared to the larger nanoparticles. This suggests that the latter species adsorb more efficiently than the former.
An SEM image recorded for the PHisGEO5MA46‐PIPGMA1000 nanoparticles adsorbed onto the stainless steel substrate after a typical QCM experiment (Γ = 70.5 mg m–2) is shown in Figure 5 . These nanoparticles were selected because their relatively large size (mean particle diameter ≈100 nm) aids their visualization. At first sight, the adsorbed nanoparticles appear to be weakly aggregated. However, DLS studies indicate no change in the apparent nanoparticle diameter regardless of the solution pH, so such incipient aggregation appears to be an experimental artifact. Alternatively, surface‐confined aggregation may occur after nanoparticle adsorption. The extent of adsorption of these nanoparticles on the stainless steel substrate was assessed using ImageJ software Figure S7 (Supporting Information). The fractional surface coverage, θ, was estimated to be 0.23, which is comparable to that reported by Hayes and co‐workers for the electrostatic adsorption of anionic nanoparticles onto a cationic planar substrate.[ 69 ] In summary, appropriate chemical derivatization of sterically‐stabilized diblock copolymer nanoparticles can significantly increase their propensity to adsorb onto stainless steel.
Figure 5.

Representative SEM image recorded for PHisGEO5MA46‐PIPGMA1000 nanoparticles adsorbed onto planar stainless steel after a QCM experiment.
3. Conclusion
A series of PGEO5MA46‐PIPGMAy nanoparticles has been synthesized via RAFT aqueous emulsion polymerization of IPGMA. TEM studies confirmed that kinetically‐trapped spherical nanoparticles were obtained while DLS analysis indicated that the z‐average particle diameter could be adjusted from 68 to 188 nm by systematically varying the target DP for the core‐forming PIPGMA block. The pendent cis‐diol groups on PGEO5MA46‐PIPGMA500 and PGEO5MA46‐PIPGMA1000 nanoparticles were oxidized using aqueous NaIO4 to produce the analogous aldehyde‐functionalized nanoparticles, which were subsequently derivatized with histidine via reductive amination. 1H NMR spectroscopy studies confirmed that the extent of functionalization was more than 99% in each case. Moreover, histidine modification led to a dramatic change in the nanoparticle electrophoretic footprint, with an isoelectric point being observed at around pH 7. Interestingly, DLS studies confirmed that such histidine‐derivatized nanoparticles remained colloidally stable over a wide pH range. Adsorption of both the cis‐diol‐ and histidine‐functionalized nanoparticles onto a planar stainless steel substrate at pH 6 was assessed using a quartz crystal microbalance at 25 °C. Larger nanoparticles led to higher adsorbed amounts, as expected. More importantly, the adsorbed amount obtained for PHisGEO5MA45‐PIPGMA1000 nanoparticles was 70.5 mg m–2, which is significantlyhigher than that observed for the corresponding cis‐diol‐functionalized precursor nanoparticles. SEM analysis suggested a fractional surface coverage of 0.23 for the former nanoparticles.
4. Experimental Section
Materials
All reagents were used as received unless otherwise stated. GEO5MA monomer was synthesized as previously described.[ 49 ] Isopropylideneglycerol monomethacrylate (IPGMA) was kindly provided by GEO Specialty Chemicals (Hythe, UK). 4,4’‐Azobis(4‐cyanopentanoic acid) (ACVA; >98%), glycine (≥98%), histidine (≥98%), arginine (≥99.5%), sodium periodate (NaIO4, ≥99.8%) and sodium cyanoborohydride (NaCNBH3, 95%), were purchased from Sigma‐Aldrich (UK). 2‐Cyano‐2‐propyl dithiobenzoate (CPDB, >97%) was purchased from Strem Chemicals Ltd (Cambridge, UK). Dimethylformamide (DMF), tetrahydrofuran (THF), methanol, ethanol, and diethyl ether were purchased from Fisher Scientific (UK). d 4‐Methanol and d 7‐DMF were purchased from Goss Scientific Instruments Ltd (Cheshire, UK). Deionized water was used for all experiments involving aqueous solutions.
Methods
1H NMR spectroscopy: Spectra were recorded in either CD3OD or d 7‐dimethylformamide using a 400 MHz Bruker Avance‐400 spectrometer at 298 K with 16 scans being averaged per spectrum.
Aqueous electrophoresis: Zeta potentials for diblock copolymer nanoparticles were analyzed using a Malvern Zetasizer Nano ZS instrument equipped with a 4 mW He‐Ne laser (λ = 633 nm) operating at a fixed scattering angle of 173°. Samples were diluted to 0.1% w/w using 1 mM KCl, with either dilute NaOH or HCl being used for pH adjustment as required. Zeta potentials were calculated from the Henry equation using the Smoluchowski approximation.
DMF Gel Permeation Chromatography (GPC): DMF GPC was used to determine the number‐average molecular weights (M n) and dispersities (Đ) for all (co)polymers. The instrument set‐up comprised two Agilent PL gel 5 µm Mixed‐C columns and a guard column connected in series to an Agilent 1260 Infinity GPC system operating at 60 °C. The GPC eluent was HPLC‐grade DMF containing 10 mmol LiBr at a flow rate of 1.0 mL min–1, the copolymer concentration was typically 1.0% w/w and calibration was achieved using a series of ten near‐monodisperse poly(methyl methacrylate) standards ranging from 1 080 to 905 000 g mol–1. Chromatograms were analyzed using Agilent GPC/SEC software.
Dynamic Light Scattering (DLS): DLS studies were performed using a Malvern Zetasizer Nano‐ZS instrument equipped with a 4 mW He–Ne laser (λ = 633 nm) operating at a fixed scattering angle of 173°. Copolymer dispersions were diluted to 0.1% w/w using deionized water prior to light scattering studies at 25 °C, with 2 min being allowed for thermal equilibrium prior to each measurement. The hydrodynamic z‐average particle diameter was calculated via the Stokes–Einstein equation.
Scanning Electron Microscopy (SEM): Images were obtained using a field emission Inspect‐F instrument operating at an accelerating voltage of 10 kV. Each sample was dispersed and dried onto a stainless steel substrate following a QCM experiment, before being sputter‐coated with a 5 nm overlayer of gold to prevent sample charging.
Transmission Electron Microscopy (TEM): Copper/palladium TEM grids (Agar Scientific, UK) were coated in‐house to yield a thin film of amorphous carbon and were subjected to a glow discharge for 30 s. Aqueous droplets of copolymer dispersions (5.0 µl, 0.1% w/w) were placed on freshly‐treated grids for 1 min and then carefully blotted with filter paper to remove excess solution. An aqueous droplet of uranyl formate solution (5 µL, 0.75% w/w) was placed on each sample‐loaded grid for 20 s and then blotted with filter paper to remove excess stain. This negative staining protocol was required to ensure sufficient electron contrast. Each grid was then carefully dried using a vacuum hose. Imaging was performed at 100 kV using a Phillips CM100 microscope fitted with a Gatan 1k CCD camera.
Quartz crystal microbalance (QCM) studies of nanoparticle adsorption onto stainless steel: QCM measurements were performed using an openQCM NEXT instrument (Novaetech S.r.l., Pompeii, Italy) equipped with a temperature‐controlled cell connected to a Masterflex Digital Miniflex peristaltic pump (Cole‐Parmer Instrument Company, UK). Stainless steel substrates (SS2343, 5 MHz) were supplied by Q‐Sense AB (Sweden). Prior to adsorption, these substrates were thoroughly cleaned by sonication in DMF, ethanol and acetone for 10 min in each case, followed by exposure to UV/ozone for 10 min, and finally dried under a flow of compressed air. The cleaned substrates were initially equilibrated with deionized water, followed by the introduction of an aqueous dispersion of 1.0% w/w nanoparticles at pH 6.0 prior to rinsing with deionized water to remove any weakly adhered nanoparticles. Measurements were performed at 25 °C using a constant flow rate of 0.50 mL min−1. The change in resonance frequency of the crystal (Δf) is measured in a QCM experiment, which corresponds to a change in mass attached to the oscillating crystal surface (Δm). The Sauerbrey equation (see Equation 1) was used to convert Δf into the corresponding adsorbed mass per unit area, Γ.[ 55 ]
| (1) |
Here n is the overtone number and C is a sensitivity constant, which is equal to –0.177 (mg (m2 Hz)−1) for the 5 MHz crystal employed in this study. The fundamental frequency (n = 1) was used to calculate Γ, which is expressed in mg m–2.
Synthesis of a PGEO5MA46 Precursor via RAFT Solution Polymerization of GEO5MA
GEO5MA (30 g, 0.079 mol), CPDB (0.349 g, 1.58 mmol), and ACVA (88.4 mg, 0.315 mmol; CPDB/ACVA molar ratio = 5.0) were weighed into a 250 mL round‐bottom flask. The reaction mixture was degassed for 40 min using a N2 purge before being placed into an oil bath set at 70 °C for 180 min. The polymerization was quenched by removing the flask from the oil bath and subsequent exposure of the reaction mixture to air. The GEO5MA conversion was determined to be 80% by 1H NMR spectroscopy. The crude PGEO5MA homopolymer was purified by precipitation into diethyl ether (to remove any unreacted monomer and other impurities), before being filtered and redissolved in methanol. This precipitation step was repeated and the purified homopolymer was dried in a vacuum oven set at 35 °C overnight to produce a pink/red viscous liquid. The mean DP of this PGEO5MA46 precursor was determined by end‐group analysis using 1H NMR spectroscopy. More specifically, the integrated signals between 7.34 and 8.03 ppm assigned to the five aromatic protons of the dithiobenzoate chain‐end were compared to that of the five proton signals assigned to the methacrylate backbone at 0.78–2.71 ppm.
Synthesis of PGEO5MA46‐PIPGMAy Diblock Copolymer Nanoparticles via RAFT Aqueous Emulsion Polymerization of IPGMA
The synthesis of PGEO5MA46‐PIPGMA500 is representative of the general protocol. IPGMA monomer (2.00 g, 10.0 mmol), PGEO5MA46 precursor (0.384 g, 18.8 µmol; target PIPGMA DP = 500), ACVA initiator (1.10 mg, 3.93 µmol; PGEO5MA46/ACVA molar ratio = 4.0) and water (9.54 g; targeting 20% w/w solids) were weighed into a 30 mL glass vial. The reaction mixture was purged using N2 gas for 20 min and then the sealed vial was placed in an oil bath set at 70 °C. After 18 h, the polymerization was quenched by removing the vial from the oil bath and exposing its contents to air. The final IPGMA conversion was determined to be 99% by 1H NMR spectroscopy by comparing the integrated vinyl IPGMA monomer signals at 5.67 and 6.16 ppm with that assigned to the methacrylate backbone signals at 0.81–2.30 ppm arising from the monomer and polymer. [N.B. A shorthand notation is used to describe two or more diblock copolymers in this manuscript. Thus, a pair of PGEO5MA46‐PIPGMAy diblock copolymers for which the mean DP of the PIPGMA block (y) was either 500 or 1000, respectively, is denoted as PGEO5MA46‐PIPGMA500/1000 for brevity. Similarly, a series of PGEO5MA46‐PIPGMAy diblock copolymers for which the PIPGMA block DP ranges from 500 to 2000 is denoted as PGEO5MA46‐PIPGMA500–2000].
Selective Oxidation of PGEO5MA46‐PIPGMAx Nanoparticles using NaIO4
The oxidation of PGEO5MA46‐PIPGMA500 nanoparticles is representative of the general protocol. NaIO4 (0.041 g, 0.19 mmol) was dissolved in a 20% w/w aqueous dispersion of PGEO5MA46‐PIPGMA500 nanoparticles (2.50 g, 21.0 µmol). A NaIO4/cis‐diol molar ratio of unity was used to target full oxidation of the PGEO5MA chains. The reaction solution was stirred in the dark for 30 min at 22 °C. The mean degree of oxidation was determined by 1H NMR spectroscopy. Finally, the reaction mixture was dialyzed for 24 h against deionized water with three changes of water to remove any impurities.
Histidine Conjugation to PAGEO5MA46‐PIPGMAx Nanoparticles via Reductive Amination
The synthesis of PHisGEO5MA46‐PIPGMA500 is representative of the general protocol. A 20% w/w aqueous dispersion of PAGEO5MA46‐PIPGMA500 nanoparticles (1.00 g, 1.70 µmol) was diluted to 10% w/w with deionized water. Histidine (12.3 mg, 0.0793 mmol) was added and the solution pH was adjusted to pH 5 using 0.1 M HCl. Excess NaCNBH3 (6.9 mg, 79 µmol; 2.45 mol excess) was carefully added to the reaction mixture, which was then stirred at 35 °C for 48 h. The degree of functionalization was determined to be 99% by 1H NMR spectroscopy by comparing the residual geminal diol signal at 6.06 ppm with the methacrylate backbone signals and the acetal signals from the PIPGMA at 0.83–2.61 ppm. Finally, the reaction mixture was dialyzed for 24 h against deionized water with three changes of water to remove any impurities.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
EPSRC is thanked for providing a CDT PhD studentship for E.E.B. (EP/L016281/1) and a Programme grant to support E.C.J. (EP/T012455/1). In addition, S.P.A. acknowledges EPSRC for a four‐year Established Career Particle Technology Fellowship (EP/R003009). GEO Specialty Chemicals (Hythe, UK) is thanked for partial funding of E.E.B.’s PhD project and Lubrizol Ltd. (Hazelwood, UK) is acknowledged for funding a PhD studentship for C.G. Finally, Novaetech S.r.l. (Pompeii, Italy) is thanked for their excellent technical support regarding the QCM studies.
Brotherton E. E., Josland D., György C., Johnson E. C., Chan D. H., Smallridge M. J., Armes S. P., Histidine‐Functionalized Diblock Copolymer Nanoparticles Exhibit Enhanced Adsorption onto Planar Stainless Steel. Macromol. Rapid Commun. 2023, 44, 2200903. 10.1002/marc.202200903
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Charleux B., Delaittre G., Rieger J., D'Agosto F., Macromolecules 2012, 45, 6753. [Google Scholar]
- 2. Warren N. J., Armes S. P., J. Am. Chem. Soc. 2014, 136, 10174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zetterlund P. B., Thickett S. C., Perrier S., Bourgeat‐Lami E., Lansalot M., Chem. Rev. 2015, 115, 9745. [DOI] [PubMed] [Google Scholar]
- 4. Rieger J., Macromol. Rapid Commun. 2015, 36, 1458. [DOI] [PubMed] [Google Scholar]
- 5. Karagoz B., Esser L., Duong H. T., Basuki J. S., Boyer C., Davis T. P., Polym. Chem. 2014, 5, 350. [Google Scholar]
- 6. D'Agosto F., Rieger J., Lansalot M., Angew. Chem., Int. Ed. 2019, 59, 2. [DOI] [PubMed] [Google Scholar]
- 7. Sugihara S., Blanazs A., Armes S. P., Ryan A. J., Lewis A. L., J. Am. Chem. Soc. 2011, 133, 15707. [DOI] [PubMed] [Google Scholar]
- 8. Canning S. L., Smith G. N., Armes S. P., Macromolecules 2016, 49, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blanazs A., Madsen J., Battaglia G., Ryan A. J., Armes S. P., J. Am. Chem. Soc. 2011, 133, 16581. [DOI] [PubMed] [Google Scholar]
- 10. Simon K. A., Warren N. J., Mosadegh B., Mohammady M. R., Whitesides G. M., Armes S. P., Biomacromolecules 2015, 16, 3952. [DOI] [PubMed] [Google Scholar]
- 11. Rosselgong J., Blanazs A., Chambon P., Williams M., Semsarilar M., Madsen J., Battaglia G., Armes S. P., ACS Macro Lett. 2012, 1, 1041. [DOI] [PubMed] [Google Scholar]
- 12. Hatton F. L., Derry M. J., Armes S. P., Polym. Chem. 2020, 11, 6343. [Google Scholar]
- 13. Hatton F. L., Lovett J. R., Armes S. P., Polym. Chem. 2017, 8, 4856. [Google Scholar]
- 14. Byard S. J., Williams M., McKenzie B. E., Blanazs A., Armes S. P., Macromolecules 2017, 50, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He W.‐D., Sun X.‐L.i, Wan W.‐M., Pan C.‐Y., Macromolecules 2011, 44, 3358. [Google Scholar]
- 16. Qiu L., Xu C.‐R., Zhong F., Hong C.‐Y., Pan C.‐Y., ACS Appl. Mater. Interfaces 2016, 8, 18347. [DOI] [PubMed] [Google Scholar]
- 17. Khor S. Y., Truong N. P., Quinn J. F., Whittaker M. R., Davis T. P., ACS Macro Lett. 2017, 6, 1013. [DOI] [PubMed] [Google Scholar]
- 18. Foster J. C., Varlas S., Couturaud B., Jones J. R., Keogh R., Mathers R. T., O'reilly R. K., Angew. Chem., Int. Ed. 2018, 57, 15733. [DOI] [PubMed] [Google Scholar]
- 19. Ferguson C. J., Hughes R. J., Nguyen D., Pham B. T. T., Gilbert R. G., Serelis A. K., Such C. H., Hawkett B. S., Macromolecules 2005, 38, 2191. [Google Scholar]
- 20. Lovell P. A., Schork F. J., Biomacromolecules 2020, 21, 4396. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J., Yao H., Ma J., Polym. Chem. 2018, 9, 2532. [Google Scholar]
- 22. Guimarães T. R., Khan M., Kuchel R. P., Morrow I. C., Minami H., Moad G., Perrier S., Zetterlund P. B., Macromolecules 2019, 52, 2965. [Google Scholar]
- 23. Khan M., Guimarães T. R., Kuchel R. P., Moad G., Perrier S., Zetterlund P. B., Angew. Chem., Int. Ed. 2021, 60, 23281. [DOI] [PubMed] [Google Scholar]
- 24. Ferguson C. J., Hughes R. J., Pham B. T. T., Hawkett B. S., Gilbert R. G., Serelis A. K., Such C. H., Macromolecules 2002, 35, 9243. [Google Scholar]
- 25. Truong N. P., Dussert M. V., Whittaker M. R., Quinn J. F., Davis T. P., Polym. Chem. 2015, 6, 3865. [Google Scholar]
- 26. Sugihara S., Sudo M., Hirogaki K., Irie S., Maeda Y., Macromolecules 2018, 51, 1260. [Google Scholar]
- 27. Zhang X., Boissé S., Zhang W., Beaunier P., D'Agosto F., Rieger J., Charleux B., Macromolecules 2011, 44, 4149. [Google Scholar]
- 28. Chaduc I., Crepet A., Boyron O., Charleux B., D'Agosto F., Lansalot M., Macromolecules 2013, 46, 6013. [Google Scholar]
- 29. Clothier G. K. K., Guimarães T. R., Khan M., Moad G., Perrier S., Zetterlund P. B., ACS Macro Lett. 2019, 8, 989. [DOI] [PubMed] [Google Scholar]
- 30. Cunningham V. J., Ratcliffe L. P. D., Blanazs A., Warren N. J., Smith A. J., Mykhaylyk O. O., Armes S. P., Polym. Chem. 2014, 5, 6307. [Google Scholar]
- 31. Rieger J., Osterwinter G., Bui C., Stoffelbach F.‐O., Charleux B., Macromolecules 2009, 42, 5518. [Google Scholar]
- 32. Rieger J., Stoffelbach F.‐O., Bui C., Alaimo D., Jérôme C., Charleux B., Macromolecules 2008, 41, 4065. [Google Scholar]
- 33. Zhang W., D'Agosto F., Boyron O., Rieger J., Charleux B., Macromolecules 2011, 44, 7584. [Google Scholar]
- 34. Brotherton E. E., Hatton F. L., Cockram A. A., Derry M. J., Czajka A., Cornel E. J., Topham P. D., Mykhaylyk O. O., Armes S. P., J. Am. Chem. Soc. 2019, 141, 13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hatton F. L., Park A. M., Zhang Y., Fuchs G. D., Ober C. K., Armes S. P., Polym. Chem. 2019, 10, 194. [Google Scholar]
- 36. Byard S. J., O'Brien C. T., Derry M. J., Williams M., Mykhaylyk O. O., Blanazs A., Armes S. P., Chem. Sci. 2020, 11, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hunter S. J., Penfold N. J. W., Jones E. R., Zinn T., Mykhaylyk O. O., Armes S. P., Macromolecules 2022, 55, 3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Penfold N. J. W., Whatley J. R., Armes S. P., Macromolecules 2019, 52, 1653. [Google Scholar]
- 39. Warren N. J., Mykhaylyk O. O., Mahmood D., Ryan A. J., Armes S. P., J. Am. Chem. Soc. 2014, 136, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunningham V. J., Derry M. J., Fielding L. A., Musa O. M., Armes S. P., Macromolecules 2016, 49, 4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sponchioni M., O'Brien C. T., Borchers C., Wang E., Rivolta M. N., Penfold N. J. W., Canton I., Armes S. P., Chem. Sci. 2020, 11, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cunningham V. J., Alswieleh A. M., Thompson K. L., Williams M., Leggett G. J., Armes S. P., Musa O. M., Macromolecules 2014, 47, 5613. [Google Scholar]
- 43. Madsen J., Madden G., Themistou E., Warren N. J., Armes S. P., Polym. Chem. 2018, 9, 2964. [Google Scholar]
- 44. Kocik M. K., Mykhaylyk O. O., Armes S. P., Soft Matter 2014, 10, 3984. [DOI] [PubMed] [Google Scholar]
- 45. Canton I., Warren N. J., Chahal A., Amps K., Wood A., Weightman R., Wang E., Moore H., Armes S. P., ACS Cent. Sci. 2016, 2, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verber R., Blanazs A., Armes S. P., Soft Matter 2012, 8, 9915. [Google Scholar]
- 47. Lovett J. R., Warren N. J., Armes S. P., Smallridge M. J., Cracknell R. B., Macromolecules 2016, 49, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brotherton E. E., Smallridge M. J., Armes S. P., Biomacromolecules 2021, 22, 5382. [DOI] [PubMed] [Google Scholar]
- 49. Brotherton E. E., Jesson C. P., Warren N. J., Smallridge M. J., Armes S. P., Angew. Chem., Int. Ed. 2021, 60, 12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brotherton E. E., Neal T. J., Kaldybekov D. B., Smallridge M. J., Khutoryanskiy V. V., Armes S. P., Chem. Sci. 2022, 13, 6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Smith M. B., March J., March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Inc., Hoboken, NJ, 2007. [Google Scholar]
- 52. Borch R. F., Bernstein M. D., Durst H. D., J. Am. Chem. Soc. 1971, 93, 2897. [Google Scholar]
- 53. Akpinar B., Fielding L. A., Cunningham V. J., Ning Y., Mykhaylyk O. O., Fowler P. W., Armes S. P., Macromolecules 2016, 49, 5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jesson C. P., Pearce C. M., Simon H., Werner A., Cunningham V. J., Lovett J. R., Smallridge M. J., Warren N. J., Armes S. P., Macromolecules 2017, 50, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sauerbrey G., Z. Med. Phys. 1959, 155, 206. [Google Scholar]
- 56. Deane O. J., Musa O. M., Fernyhough A., Armes S. P., Macromolecules 2020, 53, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parker B. R., Derry M. J., Ning Y., Armes S. P., Langmuir 2020, 36, 3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mcbride R. J., Miller J. F., Blanazs A., Hähnle H. ‐J., Armes S. P., Macromolecules 2022, 55, 7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jesson C. P., Cunningham V. J., Smallridge M. J., Armes S. P., Macromolecules 2018, 51, 3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Z., Int. J. Electrochem. Sci. 2016, 11, 9175. [Google Scholar]
- 61. Yuan J. ‐J., Armes S. P., Takabayashi Y., Prassides K., Leite C. A. P., Galembeck F., Lewis A. L., Langmuir 2006, 22, 10989. [DOI] [PubMed] [Google Scholar]
- 62. Wan S., Zheng Y., Liu Y., Yan H., Liu K., J. Mater. Chem. 2005, 15, 3424. [Google Scholar]
- 63. Wan S., Huang J., Yan H., Liu K., J. Mater. Chem. 2006, 16, 298. [Google Scholar]
- 64. Strøm M. B., Rekdal Ø., Svendsen J. S., J. Pept. Sci. 2002, 8, 431. [DOI] [PubMed] [Google Scholar]
- 65. Jin Y., Huang Y., Liu G., Zhao R., Analyst 2013, 138, 5479. [DOI] [PubMed] [Google Scholar]
- 66. Krozer A., Nordin S.‐A., Kasemo B., J. Colloid Interface Sci. 1995, 176, 479. [Google Scholar]
- 67. Xu D., Hodges C., Ding Y., Biggs S., Brooker A., York D., Langmuir 2010, 26, 8366. [DOI] [PubMed] [Google Scholar]
- 68. Sakai K., Smith E. G., Webber G. B., Schatz C., Wanless E. J., Bütün V., Armes S. P., Biggs S., Langmuir 2006, 22, 5328. [DOI] [PubMed] [Google Scholar]
- 69. Hayes R. A., Böhmer M. R., Fokkink L. G. J., Langmuir 1999, 15, 2865. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.