

Abstract
We report a unique case of high‐grade B‐cell lymphoma, not otherwise specified in a 5‐year‐old child. Whole‐genome sequencing revealed a DDX3X::MLLT10 fusion, usually seen in T‐cell acute lymphoblastic leukaemia (ALL). This suggests the novel idea that MLLT10 fusions are capable of driving B‐cell malignancies. An IGH deletion usually only seen in adults was also found. These unique genetic findings provide novel insights into B‐cell lymphomagenesis. The child remains in remission 7 year post chemotherapy, which demonstrates that novel complex molecular findings do not always denote high‐risk disease.
Keywords: HGBL, lymphoma, MLLT10, NOS
Abbreviations
- ALL
acute lymphoblastic leukaemia
- AML
acute myeloid leukaemia
- BL
Burkitt lymphoma
- CLL
chronic lymphocytic leukaemia
- DLBCL
diffuse large B‐cell lymphoma
- HGBL
high‐grade B‐cell lymphoma
- HGBL, NOS
high‐grade B‐cell lymphoma, not otherwise specified
- LBCL
large B‐cell lymphoma
- LBL
lymphoblastic lymphoma
- NHL
non‐Hodgkin lymphoma
- WGS
whole‐genome sequencing
1. INTRODUCTION
Non‐Hodgkin lymphoma (NHL) is a heterogenous group of malignancies that typically present as high‐grade extranodal disease, are managed with chemotherapy, and have excellent survival outcomes. 1 The WHO classification currently distinguishes over 70 different types of B‐cell NHLs, but Burkitt lymphoma (BL) and diffuse large B‐cell lymphoma (DLBCL) account for the vast majority of paediatric cases. Rarer types of NHL that can occur in children include large B‐cell lymphoma (LBCL) with IRF4 rearrangement, high‐grade B‐cell lymphoma (HGBL) with 11q aberration, EBV‐positive DLBCL, ALK‐positive LBCL and plasmablastic lymphoma. 2 In even rarer cases, NHLs are difficult to classify due to atypical presentation, histology, immunohistochemistry or genetics. This makes the optimal management and prognosis unclear. 3 We report the case of a 5‐year‐old with NHL that was difficult to classify. It possessed features of both mature and immature B‐cell NHL, and whole‐genome sequencing (WGS) revealed mutations not reported before in a childhood lymphoma. These provide novel insights into B‐cell lymphomagenesis.
2. CASE DESCRIPTION AND RESULTS
A 5‐year‐old male with no significant past medical history presented with rapidly enlarging masses over the right temple and parotid, plus lymphadenopathy. The rest of their systemic examination was normal, as were their peripheral blood counts and lactate dehydrogenase. Magnetic resonance imaging (MRI) and staging computed tomography (CT) revealed no additional lesions, including no lesions within the central nervous system, but bone marrow assessment revealed 45%–50% blasts. Histopathological and immunohistochemical assessment of the tumour revealed medium‐sized CD79a‐ and PAX5‐positive B cells with blastoid morphology, irregular rasinoid nuclei, small nucleoli or less frequently one large nucleolus, and an overall ‘starry sky’ appearance (Figure S1). Further immunohistochemical analyses identified other immature features consistent with the observed blastoid morphology, including CD10 positivity and only patchy CD20 positivity. However, mature B‐cell features were also present, including positive surface immunoglobulin, MUM1 and BCL6, strongly positive CD20 in focal areas of lymphoma, and negative terminal deoxynucleotidyl transferase (TdT) and CD34. In addition, Ki67 staining identified the proliferative fraction to be 60%, and limited genetic analyses, which were standard in the clinic at the time of diagnosis, demonstrated no rearrangements or copy number changes in MYC, BCL2 or BCL6. Overall, histopathological, immunohistochemical and genetic analysis of this child's tumour led to the rare diagnosis of stage IV HGBL, not otherwise specified (HGBL, NOS) with blastoid morphology. The child was managed with 7 months of chemotherapy for mature B‐cell NHL with bone marrow involvement following a FAB/LMB96 approach, and remains in remission 7 years post chemotherapy.
Tumour and matched germline WGS were performed for the 100,000 Genomes Project. This revealed no pertinent germline findings and a relatively low somatic mutation burden. 4 Mutational signature analysis identified the ‘clock‐like’ signatures 1 and 5 contributing to most of the mutation burden (Figure 1A and Figure S2). Given the low overall mutational frequency, these signatures likely represent random mutation acquisition during normal ageing/growing. 5 In total, four somatic events were deemed pertinent (Figure 1B,C). Of these, the DDX3X::MLLT10 translocation and IGH deletion have not been reported in a paediatric B‐cell lymphoma before.
FIGURE 1.
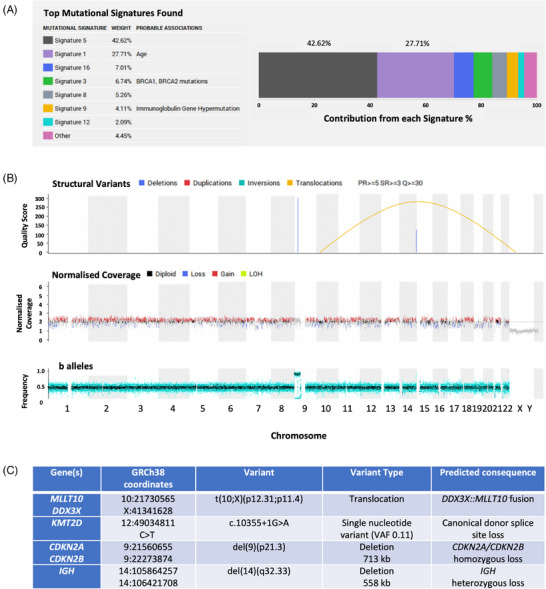
Whole‐genome sequencing results. (A) Somatic mutational signature analysis. Mathematical methods (decomposition by non‐negative least squares) were used to assess the top mutation signatures contributing to the overall mutation burden observed in the tumour. These mutation signatures have been derived using the analysis of large sequencing datasets (10,952 exomes and 1048 whole‐genomes from 40 distinct tumour types) in order to group patterns of relative contextual frequencies of different single nucleotide variants. (B and C) Somatic events. (B) Linear genome plot of global somatic structural variants (top track) and copy number aberrations (middle track). (C) Tabular summary of the four identified clinically pertinent somatic variants. VAF: variant allele frequency.
MLLT10 fusions, including with DDX3X, are found in T‐cell acute lymphoblastic leukaemia (ALL), 6 , 7 T‐cell lymphoblastic lymphoma (LBL) 8 and acute myeloid leukaemia (AML). 9 In this case, the translocation, t(10;X)(p12.31;p11.4), results in an in‐frame fusion of DDX3X exons 1–4 and MLLT10 exons 18–24 (Figure 2 and Figure S3). 4 Multiple breakpoints exist in T‐cell malignancies for DDX3X::MLLT10, including one similar to this case: exons 1−3 of DDX3X and exons 17−24 of MLLT10. 7 In all, the MLLT10 octapeptide motif‐leucine‐zipper domain is maintained, which is crucial for leukaemogenesis. 8 , 10 Other maintained features include the DDX3X nuclear export signal and MLLT10 nuclear localisation signal. 7 Moreover, we suggest the novel idea that MLLT10 rearrangements drive B‐cell malignancies too.
FIGURE 2.
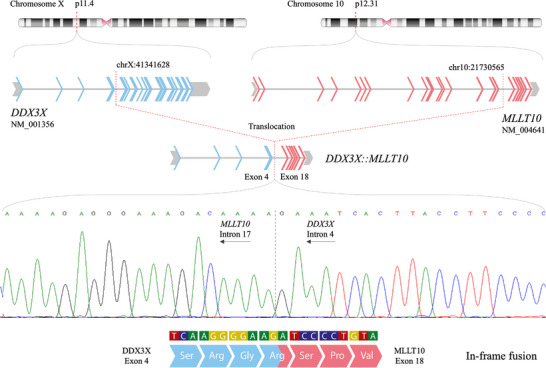
DDX3X::MLLT10 gene fusion. Fusion‐specific PCR primers were designed to confirm the presence of the DDX3X::MLLT10 gene fusion detected in the patient's tumour sample by whole‐genome sequencing. An amplicon at the expected size of 780 bp was detected in the patient's tumour sample only. This amplicon was subsequently sequenced to confirm the genomic breakpoints of the fusion, which are demonstrated by the schematic. Sequencing demonstrated a somatically acquired translocation between the short arm of chromosome X (cytogenetic band p11.4) and the short arm of chromosome 10 (cytogenetic band p12.31). The genomic coordinates of the breakpoints (genome reference build 38) map to intron 4 of the DDX3X gene (transcript NM_001356) and intron 17 of the MLLT10 gene (transcript NM_004641). This translocation opposes exon 4 of DDX3X to exon 18 of MLLT10, leading to a DDX3X::MLLT10 gene fusion. Fluorescent sequencing analysis (sequence data show the negative strand in the opposite direction) confirms this fusion. Upon splicing and transcription, this fusion is predicted to maintain the amino acid read frame and result in a DDX3X::MLLT10 chimeric protein. Block arrows: exons; grey: untranslated region; blue: DDX3X coding exons; red: MLLT10 coding exons; grey connecting lines: introns; amino acid Ser: serine; arg: arginine; Gly: glycine; Pro: proline; Val: valine.
The second somatic change that has not been identified in paediatric lymphomas before is an IGH deletion. These occur in mature adult B‐cell malignancies, including chronic lymphocytic leukaemia (CLL) 11 and DLBCL. 12
The other somatic events (Figure 1C) are not unexpected. KMT2D is a tumour suppressor that regulates homeobox genes and is important in B‐cell receptor signalling and apoptosis. 13 CDKN2A/CDKN2B are tumour suppressors with roles in the cell cycle, apoptosis and immune responses. 14 Inactivation of these genes is common in leukaemias and lymphomas, 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 including HGBL, NOS. 15 , 20 Indeed, concurrent CDKN2A/CDKN2B deletions were seen in three of four ALL cases with DDX3X::MLLT10 fusions. 7
In summary, we report a HGBL, NOS with unique molecular findings. The tumour possessed a DDX3X::MLLT10 fusion usually only seen in T‐cell disease and an IGH deletion usually only seen in adults.
3. DISCUSSION
We present a case of paediatric HGBL, NOS with unusual histology and immunohistochemistry and previously undescribed genetic changes. It is highly unusual to see a paediatric B‐cell lymphoma with genetic events usually found in malignancies with other origins, such as T‐cell ALL and CLL. This tumour appears to have been driven by a DDX3X::MLLT10 fusion and an accompanying IGH deletion, both of which have not been seen before in paediatric B‐cell NHLs. When DDX3X::MLLT10 fusions are observed in ALL, they increase MLLT10 expression, leading to increased homeobox gene activity. This promotes cell cycle progression and drives leukaemogenesis. 8 , 9 Although DDX3X::MLLT10 translocations have not been reported in B‐cell malignancies before, DDX3X loss of function mutations have been in BL, 21 , 22 DLBCL, 21 HGBL, NOS 15 and CLL. 23 In BL, these buffer MYC‐induced proteotoxic stress during early lymphomagenesis. 21 As DDX3X::MLLT10 translocations decrease DDX3X expression in T‐cell LBL, 8 a similar effect may have facilitated early lymphomagenesis in this case, although independently of MYC. However, the oncogenic and tumour suppressor functions of DDX3X require further characterisation. 7 IGH deletions are usually seen in CLL 11 and DLBCL, 12 and it is thought that they may activate an unknown oncogene at 14q. 11 Together, these changes, seen individually in either immature or mature B‐cell malignancies, challenge our understanding of the genesis of the lymphoma seen in this case.
Difficult to classify B‐cell NHLs are currently allocated HGBL, NOS or double/triple hit lymphoma within the WHO classification. 2 These are rare and biological courses are difficult to predict, but they are typically thought to have worse outcomes than other lymphomas. 3 , 15 Of note here, poor prognoses are also seen with: MLLT10 rearrangements in T‐cell ALL 6 , 7 and AML 9 ; IGH changes in CLL and prolymphocytic leukaemia 11 ; KMT2D mutations in DLBCL 24 and mantle cell lymphoma 16 ; and CDKN2A/CDKN2B deletions in DLBCL, 14 follicular lymphoma, 25 cutaneous T‐cell lymphoma 18 and ALL. 19 , 26 Moreover, there are few sufficiently comparable cases of paediatric HGBL, NOS in the literature, as well as limited available understanding of the genetic changes seen in the current case. In situations such as this, clinicians may consider whether intensified therapy is required and/or whether there is a place for the use of novel therapeutics, such as MAPK 27 or KDM5 28 inhibitors for KMT2D mutations; CDK4/6 inhibitors for CDKN2A/CDKN2B deletions 19 ; and DOT1L inhibitors for MLLT10 changes. 7 However, the child remains disease‐free 7 years after chemotherapy alone. This cautions against overtreating patients with novel complex molecular findings due to assumptions that these confer high risk. Indeed, as WGS is not ubiquitous in clinical practice, we may have more sequencing data for unusual and/or aggressive cases. Therefore, we should presume neither that all findings are responsible for this phenotype, nor that these findings would not be found in typical disease.
In summary, paediatric HGBL, NOS is a rare entity that is difficult to diagnose, select treatment, and predict prognosis for. The child in this case had novel genetic changes, but remains disease‐free following chemotherapy alone. The case provides novel insights into paediatric B‐cell lymphomagenesis and highlights that NHLs are a more heterogenous group than the WHO classification currently accounts for. Indeed it is most likely that lymphoma is a continuous spectrum of disease, and the classification boundaries imposed are artificial. Therefore, a multi‐national effort to sequence these rare tumours is required to improve understanding of B‐cell lymphomagenesis, and to optimally categorise and manage these cases.
CONFLICT OF INTEREST STATEMENT
Lucy Hare, Jamie Trotman, Patrick Tarpey and Elizabeth Hook declare they have no conflicts of interest. G. A. Amos Burke has received institutional consultancy fees from Roche, Takeda, Novartis and Janssen.
FUNDING INFORMATION
Lucy Hare is supported with funding from a Cancer Research UK Cambridge Centre clinical research fellowship (grant number: C9685/A25117).
Supporting information
SUPPORTING INFORMATION FIGURE S1 Bone marrow smear histology. Histological appearances of the bone marrow smear at 100× (A) and 400× (B) magnification. Images were obtained using an Olympus BX51 microscope and Infinity Capture (Infinity 2‐1C) camera.
{kind=link}
SUPPORTING INFORMATION FIGURE S2 Somatic mutational signature analysis. The counts of each mutation type (i.e., base substitution) at each mutation context (i.e., base situated immediately 3′ and 5′ to the mutated nucleotide) are normalised by the total number of tri‐nucleotide counts in the reference genome. All substitutions are referred to by the pyrimidine context of the mutated base pair. Mutation types are given on the horizontal, while the percentage of mutations attributed to a specific mutation type are on the vertical axis.
{kind=link}
SUPPORTING INFORMATION FIGURE S3 PCR confirmation of the DDX3X::MLLT10 gene fusion detected in the patient's tumour sample. Fusion‐specific PCR primers were designed to confirm the presence of the DDX3X::MLLT10 gene fusion detected in the patient's tumour sample by whole‐genome sequencing. An amplicon at the expected size of 780 bp was detected in the patient's tumour sample only.
{kind=link}
ACKNOWLEDGEMENTS
This report was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. We also gratefully acknowledge the members of the Genomics England Research Consortium, listed below, for providing the detailed data and infrastructures to enable clinical cancer WGS: J. C. Ambrose1; P. Arumugam1; R. Bevers1; M. Bleda1; F. Boardman‐Pretty1,2; C. R. Boustred1; H. Brittain1; M. A. Brown; M. J. Caulfield1,2; G. C. Chan; A. Giess1; J. N. Griffin; A. Hamblin1; S. Henderson1,2; T. J. P. Hubbard; R. Jackson1; L. J. Jones1,2; D. Kasperaviciute1,2; M. Kayikci1; A. Kousathanas1; L. Lahnstein1; A. Lakey; S. E. A. Leigh1; Leong, I. U. S.1; Lopez, F. J.1; Maleady‐Crowe, F.1; McEntagart, M.1; Minneci F.1; Mitchell, J.1; Moutsianas, L.1,2; M. Mueller1,2; N. Murugaesu1; A. C. Need1,2; P. O‘Donovan1; C. A. Odhams1; C. Patch1,2; D. Perez‐Gil1; M. B. Pereira1; J. Pullinger1; T. Rahim1; A. Rendon1; T. Rogers1; K. Savage1; K. Sawant1; R. H. Scott1; A. Siddiq1; A. Sieghart1; S. C. Smith1; A. Sosinsky1,2; A. Stuckey1; M. Tanguy1; A. L. Taylor Tavares1; E. R. A. Thomas1,2; S. R. Thompson1; A. Tucci1,2; M. J. Welland1; E. Williams1; K. Witkowska1,2; S. M. Wood1,2; M. Zarowiecki1 (1. Genomics England, London, UK; 2. William Harvey Research Institute, Queen Mary University of London, London, UK).
Hare L, Trotman J, Tarpey P, Hook E, Burke GAA. Challenging our understanding of B‐cell lymphomagenesis and risk: Paediatric high‐grade B‐cell lymphoma, not otherwise specified with a DDX3X::MLLT10 fusion and an IGH deletion. Pediatr Blood Cancer. 2024;71:e30810. 10.1002/pbc.30810
REFERENCES
- 1. Uzunova L, Burke A. Update on non‐Hodgkin lymphoma in children. Paediatrics and Child Health. 2016;26(2):57‐62. [Google Scholar]
- 2. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720‐1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li J, Liu X, Yao Z, Zhang M. High‐grade B‐cell lymphomas, not otherwise specified: a study of 41 cases. Cancer Manag Res. 2020;12:1903‐1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. The National Genomics Research and Healthcare Knowledgebase v5. Genomics England; 2019. doi: 10.6084/m9.figshare.4530893.v5 [DOI] [Google Scholar]
- 5. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941‐D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandimarte L, Pierini V, Di Giacomo D, et al. New MLLT10 gene recombinations in pediatric T‐acute lymphoblastic leukemia. Blood. 2013;121(25):5064‐5067. [DOI] [PubMed] [Google Scholar]
- 7. Brandimarte L, La Starza R, Gianfelici V, et al. DDX3X‐MLLT10 fusion in adults with NOTCH1 positive T‐cell acute lymphoblastic leukemia. Haematologica. 2014;99(5):64‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salmerón‐Villalobos J, Ramis‐Zaldivar JE, Balagué O, et al. Diverse mutations and structural variations contribute to Notch signaling deregulation in paediatric T‐cell lymphoblastic lymphoma. Pediatr Blood Cancer. 2022;69(11):e29926. [DOI] [PubMed] [Google Scholar]
- 9. Ries RE, Leonti AR, Triche TJ, et al. Structural variants involving MLLT10/AF10 are associated with adverse outcome in AML regardless of the partner gene—a COG/Tpaml study. Blood. 2019;134:461. [Google Scholar]
- 10. Deshpande AJ, Rouhi A, Lin Y, et al. The clathrin‐binding domain of CALM and the OM‐LZ domain of AF10 are sufficient to induce acute myeloid leukemia in mice. Leukemia. 2011;25(11):1718‐1727. [DOI] [PubMed] [Google Scholar]
- 11. Reindl L, Bacher U, Dicker F, et al. Biological and clinical characterization of recurrent 14q deletions in CLL and other mature B‐cell neoplasms. Br J Haematol. 2010;151(1):25‐36. [DOI] [PubMed] [Google Scholar]
- 12. Tirado CA, Chen W, García R, Kohlman KA, Rao N. Genomic profiling using array comparative genomic hybridization define distinct subtypes of diffuse large B‐cell lymphoma: a review of the literature. J Hematol Oncol. 2012;5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ortega‐Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jardin F, Jais J‐P, Molina T‐J, et al. Diffuse large B‐cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R‐CHOP treatment: a GELA study. Blood. 2010;116(7):1092‐1104. [DOI] [PubMed] [Google Scholar]
- 15. Ramis‐Zaldivar JE, Gonzalez‐Farré B, Balagué O, et al. Distinct molecular profile of IRF4‐rearranged large B‐cell lymphoma. Blood. 2020;135(4):274‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrero S, Rossi D, Rinaldi A, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high‐dose therapy: a FIL study. Haematologica. 2020;105(6):1604‐1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li JF, Dai YT, Lilljebjörn H, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. 2018;115(50):E11711‐E11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laharanne E, Chevret E, Idrissi Y, et al. CDKN2A‐CDKN2B deletion defines an aggressive subset of cutaneous T‐cell lymphoma. Mod Pathol. 2010;23(4):547‐558. [DOI] [PubMed] [Google Scholar]
- 19. Zhang W, Kuang P, Liu T. Prognostic significance of CDKN2A/B deletions in acute lymphoblastic leukaemia: a meta‐analysis. Ann Med. 2019;51(1):28‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Olszewski A, Kurt H, Evens AM. Defining and treating high‐grade B‐cell lymphoma, NOS. Blood. 2022;140(9):943‐954. [DOI] [PubMed] [Google Scholar]
- 21. Gong C, Krupka JA, Gao J, et al. Sequential inverse dysregulation of the RNA helicases DDX3X and DDX3Y facilitates MYC‐driven lymphomagenesis. Mol Cell. 2021;81(19):4059‐4075.e11. [DOI] [PubMed] [Google Scholar]
- 22. Burkhardt B, Michgehl U, Rohde J, et al. Clinical relevance of molecular characteristics in Burkitt lymphoma differs according to age. Nat Commun. 2022;13(1):3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ojha J, Secreto CR, Rabe KG, et al. Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br J Haematol. 2015;169(3):445‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Juskevicius D, Lorber T, Gsponer J, et al. Distinct genetic evolution patterns of relapsing diffuse large B‐cell lymphoma revealed by genome‐wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30(12):2385‐2395. [DOI] [PubMed] [Google Scholar]
- 25. Alhejaily A, Day AG, Feilotter HE, Baetz T, Lebrun DP. Inactivation of the CDKN2A tumor‐suppressor gene by deletion or methylation is common at diagnosis in follicular lymphoma and associated with poor clinical outcome. Clin Cancer Res. 2014;20(6):1676‐1686. [DOI] [PubMed] [Google Scholar]
- 26. Braun M, Pastorczak A, Fendler W, et al. Biallelic loss of CDKN2A is associated with poor response to treatment in pediatric acute lymphoblastic leukemia. Leuk Lymphoma. 2017;58(5):1162‐1171. [DOI] [PubMed] [Google Scholar]
- 27. Fagan RJ, Dingwall AK. COMPASS ascending: emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019;458:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heward J, Konali L, D'Avola A, et al. KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas. Blood. 2021;138(5):370‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION FIGURE S1 Bone marrow smear histology. Histological appearances of the bone marrow smear at 100× (A) and 400× (B) magnification. Images were obtained using an Olympus BX51 microscope and Infinity Capture (Infinity 2‐1C) camera.
SUPPORTING INFORMATION FIGURE S2 Somatic mutational signature analysis. The counts of each mutation type (i.e., base substitution) at each mutation context (i.e., base situated immediately 3′ and 5′ to the mutated nucleotide) are normalised by the total number of tri‐nucleotide counts in the reference genome. All substitutions are referred to by the pyrimidine context of the mutated base pair. Mutation types are given on the horizontal, while the percentage of mutations attributed to a specific mutation type are on the vertical axis.
SUPPORTING INFORMATION FIGURE S3 PCR confirmation of the DDX3X::MLLT10 gene fusion detected in the patient's tumour sample. Fusion‐specific PCR primers were designed to confirm the presence of the DDX3X::MLLT10 gene fusion detected in the patient's tumour sample by whole‐genome sequencing. An amplicon at the expected size of 780 bp was detected in the patient's tumour sample only.