

Abstract
Careful histopathologic examination remains the cornerstone in the diagnosis of the clinically and biologically heterogeneous group of lymphoid malignancies. However, recent advances in genomic and epigenomic characterization using high‐throughput technologies have significantly improved our understanding of these tumors. Although no single genomic alteration is completely specific for a lymphoma entity, some alterations are highly recurrent in certain entities and thus can provide complementary diagnostic information when integrated in the hematopathological diagnostic workup. Moreover, other alterations may provide important information regarding the clinical course, that is, prognostic or risk‐stratifying markers, or response to treatment, that is, predictive markers, which may allow tailoring of the patient's treatment based on (epi)genetic characteristics. In this review, we will focus on clinically relevant diagnostic, prognostic, and predictive biomarkers identified in more common types of B‐cell malignancies, and discuss how diagnostic assays designed for comprehensive molecular profiling may pave the way for the implementation of precision diagnostics/medicine approaches. We will also discuss future directions in this rapidly evolving field, including the application of single‐cell sequencing and other omics technologies, to decipher clonal dynamics and evolution in lymphoid malignancies.
Keywords: biomarkers, comprehensive molecular profiling, (epi)genetics, lymphoid malignancies, precision medicine, single‐cell sequencing
Introduction
The classification of lymphoid neoplasms has continuously undergone evolution with an increasing number of distinct diagnostic entities, and in the latest WHO classification from 2016, more than 80 entities of lymphoid malignancies were recognized [1]. Whilst the majority of lymphoid tumors derive from different stages of normal B‐cell development, they can also originate from T cells or NK cells. There is an extensive heterogeneity of morphologic, immunophenotypic, and genetic features between and within different entities of lymphoid neoplasms. Similarly, there is a corresponding great clinical heterogeneity, ranging from patients with indolent forms to very aggressive clinical courses.
In the last 10 years, with the introduction of next‐generation sequencing (NGS) technologies, we have witnessed a major leap in our understanding of the genomic landscapes of major entities of lymphoid malignancies [2, 3]. While a few entities showed a highly recurrent gene mutation [4, 5], most subtypes of lymphoid tumors demonstrated a very heterogeneous mutation landscape including hundreds of recurrently mutated genes [2, 3], probably mirroring the clinical heterogeneity observed between and among lymphoma entities. Based on a high number of publications in this field, novel biomarkers have been identified that may aid disease classification and risk stratification and guide therapy selection.
In parallel, efforts have focused on unraveling the epigenomic landscape of lymphoid malignancies [6]. From these studies, it has become apparent that many lymphoid malignancies carry a “footprint” from the cell of origin (COO), while they also gain tumor‐specific epigenetic aberrations [7]. Using DNA methylation profiling, it is possible to classify patients into clinically relevant subgroups and also to assess the proliferative history of lymphoid tumors [8, 9]. In more recent years, we have also started to understand the complex landscape of histone modifications and their involvement in gene regulation and disease pathology [10].
In this review, we will discuss the recently uncovered genomic landscapes of the most frequent mature B‐cell malignancies, and highlight important diagnostic, prognostic, and predictive biomarkers. We will also discuss how comprehensive genomic profiling may provide new tools for the implementation of precision diagnostics in everyday patient care. Furthermore, we will outline recent developments in deciphering the impact of epigenomics in the onset and evolution of lymphoid malignancies, but also discuss how single‐cell sequencing and other omics technologies may further our understanding of the disease pathobiology of these tumors.
Clinically relevant genomic markers in lymphoid malignancies
Since the REAL classification in 1994, detection of genetic aberrations is an integral part of diagnostics of lymphoid malignancies [11]. This has, until recently, mainly included detection of recurrent chromosomal aberrations using fluorescence in situ hybridization (FISH) technology that is more prevalent in certain lymphoid tumors. The most important include t(14;18) involving BCL2 and IGH in follicular lymphoma (FL), t(8;14) involving MYC and IGH in Burkitt lymphoma (BL) and high‐grade B‐cell lymphomas (HGBCL) with MYC and BCL2 and/or BCL6 translocation (so‐called double‐hit lymphoma), and t(11;14) involving CCND1 and IGH in mantle cell lymphoma (MCL) [1]. These different translocations can be captured using FISH probes designed for each translocation partner or using break‐apart probes for individual genes and are applicable both for smears/imprints and formalin‐fixed paraffin‐embedded (FFPE) tissue.
With the advent and now widespread use of NGS technologies, a more comprehensive and unbiased molecular characterization of lymphoid malignancies was possible, particularly thanks to whole‐exome (WES) or genome sequencing (WGS). In the next sections, the recently uncovered genomic landscapes of the most frequent mature B‐cell malignancies will be reviewed and clinically relevant genomic markers highlighted.
Diagnostic markers
In small B‐cell lymphomas, where morphologic or immunophenotypic overlaps sometimes pose diagnostic difficulties, several alterations have been described to be recurrent and can help in the differential diagnosis. In 2011, a point mutation in the BRAF gene (BRAF V600E), which is present in almost all hairy cell leukemia (HCL) cases, could be first identified thanks to WES of one single patient [4], highlighting the utility of using a comprehensive rather than targeted approach, since BRAF was previously not known to be involved in lymphoid malignancies. Later studies have confirmed this finding and refined the mutational landscape, identifying additional recurrent mutations in CDKN1B [12] (16% of HCL) and KMT2C [13] (15% of HCL). The HCL variant does not carry BRAF mutations but was found to have mutations in MAP2K1 (around 50% of cases), which pertains to the same signaling pathway [14]. MAP2K1 mutations can also be found in classic HCL cases, which lack the BRAF V600E mutation and use the IGHV4‐34 gene, although it remains unclear whether these cases are better classified as classic or variant HCL [14]. Similarly, the MYD88 L265P mutation was first found to be present in a very high proportion of lymphoplasmacytic lymphoma/Waldenström's macroglobulinemia (LPL/WM) by performing WGS in 30 patients [5]. Later studies confirmed this finding and found additional recurrent mutations, including CXCR4 (30%–40%), ARID1A (17%), and CD79B (8%–15%), as well as the previously known frequent deletion of chromosome 6q [15]. MYD88 L265P is not exclusive to LPL/WM and can be detected in a lower proportion of cases of chronic lymphocytic leukemia (CLL), marginal zone lymphomas (MZL) [16], and diffuse large B‐cell lymphoma (DLBCL).
Several studies have investigated the genomic landscape of CLL [17, 18, 19, 20], which shows a remarkable heterogeneity [21] when compared to the aforementioned LPL/WM or HCL, with highly recurrent alterations of single genes. The most frequent recurrent alterations in CLL are cytogenetic aberrations, which can be identified in more than 80% of cases and include deletion 13q (55%), deletion 11q (18%), trisomy 12 (16%), deletion 17p (7%), and deletion 6q (6%) [22]. Thus far, more than 60 “driver” mutations have been identified, although none of the mutations is highly prevalent nor specific to CLL. The most frequent mutations occur in NOTCH1, SF3B1, TP53, and ATM.
MCL includes two disease subtypes—conventional MCL (cMCL) and leukemic non‐nodal MCL (nnMCL)—which originate from different B‐cell maturation stages, but share the translocation t(11;14) [23]. The most frequently mutated genes include ATM, KMT2D, CCND1, and KMT2D [24]. A recent comprehensive genomic profiling of MCL [25] highlighted the differences in genetic alterations of the two subtypes—cMCL with frequent alterations of ATM, KMT2D, BIRC3, and NSD2 and nnMCL with frequent CCND1 somatic hypermutation (SHM) induced mutations and TERT alterations, whereas TP53 alterations are relatively frequent in both subtypes, although slightly enriched in the non‐nodal subtype. WES has also been performed in a series of primary and relapsed MCL samples [26] and novel mutation targets were identified, including CARD11 (5.5%) and S1PR1 (7.8%), with the latter significantly enriched in the relapsed samples [27].
FL is characterized by frequent alterations in epigenetic regulators (e.g., KMT2D, CREBBP, EZH2, MEF2B, EP300, and HIST1 genes) [28, 29], which together with the BCL2 translocation are involved in early FL pathogenesis [30]. Other involved pathways include B‐cell receptor (BcR) signaling (e.g., CARD11), mTOR signaling (e.g., RRAGC) [31], JAK‐STAT signaling (SOCS1, STAT6, and STAT3), as well as the NOTCH pathway [32]. Pediatric‐type FL, which does not carry the IGH‐BCL2 translocation (by definition), shows a different landscape of alterations with frequent mutations in TNFRSF11 (54%) and MAP2K1 (49%), as well as IRF8 (15%) [33].
The different types of MZL share common alterations and have few mutations that are prevalent in certain types [34]. Nodal marginal zone lymphoma (NMZL) has recurrent mutations in genes involved in chromatin remodeling/transcriptional regulation (71% of cases), NF‐κB (51% of cases), and NOTCH pathway (40% of cases), with KMT2D, NOTCH2, PTPRD, and KLF2 mutations as the most frequent [35], of which only PTPRD seems to be enriched in NMZL. WES studies of splenic marginal zone lymphoma (SMZL) [36, 37, 38, 39, 40, 41] have found frequent mutations in TP53, KLF2, KMT2D, MYD88, NOTCH2, and TNFAIP3, among others, in addition to the known cytogenetic aberrations of chromosomes 7 (deletion 7q) and chromosome 3 (trisomy 3 or gain of 3q). The recent large genetic characterization study of SMZL by the International Extranodal Lymphoma Study Group (IELSG; 303 spleen samples), confirmed previous results and separated the cases into four genetic subgroups based on the involved gene modules/pathways, termed NNK (involving NF‐κB, NOTCH, and KLF2), DMT (involving DNA damage response, MAPK, and TLR), CBS (involving cytokine, BcR signaling, and spliceosome), and PA (involving PI3K‐AKT), of which NNK and DMT include 90% of all studied cases [42].
HGBCL or aggressive lymphomas have been extensively studied and several mutations can have diagnostic value. In addition to the translocation involving MYC and the IGH/IGK/IGL loci, BL was found to carry mutations in transcription factor TCF3 or its negative regulator ID3 in 70% of cases [43, 44, 45, 46, 47]—mutations that are not typical of DLBCL, although they can be encountered (especially in double‐hit lymphomas). While not routinely used in diagnostics, transcriptomic analysis can be used for the diagnosis of BL, with a clear separation from DLBCL [48].
Efforts in the characterization of DLBCL through WES/WGS [49, 50, 51] have led to a refined molecular classification over the previous COO classification, which was mainly based on gene expression. Although there are slight differences in the proposed classifications between studies, there is a broad overlap among the new molecular subgroups; the major subgroups are BN2/C1 (BCL6 translocation and NOTCH2 mutation), A53/C2 (ABC subtype, TP53 and CDKN2A inactivation), EZB/C3 (GCB subtype, EZH2 mutations, and BCL2 translocation/mutations), ST2/C4 (GCB subtype, SGK1, NFKBIA, and SOCS1 mutations), MCD/C5 (ABC subtype, MYD88 and CD79B mutations), and N1 (ABC subtype and NOTCH1 mutations). Later studies [52, 53, 54] have developed classifiers that allow inferring of the DLBCL molecular subgroups when using targeted NGS gene panels. Although the prototypic cases in these subgroups carry the mentioned mutations, around 20%–35% of DLBCL cannot be assigned to a specific group. Furthermore, DLBCLs associated with infections such as hepatitis B were characterized with distinct genetic profiles and may represent either a different entity or subtype [55]. Hence, additional molecular subtypes of DLBCL are expected to be discovered in the coming years. In plasmablastic lymphomas, MYC translocations are the most frequent cytogenetic alteration [56], occurring in 50%–90% of cases, and frequently mutated pathways include MYC, MAPK, and JAK‐STAT, with recurrent mutations of STAT3, NRAS, TP53, MYC, and EP300 [57, 58].
Due to the low number of tumor cells, classic Hodgkin lymphomas (cHL) are not as easily accessible for WES or WGS as other lymphomas. Nevertheless, thanks to flow sorting [59, 60], circulating free tumor DNA [61, 62], as well as labor‐intensive laser capture microdissection [63], the genomic landscape of cHL has been revealed, with finding of recurrent mutations in JAK‐STAT, NF‐κB, and PI3K pathways. Primary mediastinal B‐cell lymphoma (PMBCL) is thought to be related to cHL, and indeed shares mutational profiles in the same pathways, such as JAK‐STAT, NF‐κB, and immune escape (e.g., SOCS1, STAT6, NFKBIE, and CIITA) [64, 65]. In addition, cytogenetic alterations involving 9p24 (which includes the genes encoding PD‐L1 and PD‐L2) are a characteristic and shared feature of cHL and PMBCL [66, 67].
As illustrated in this section and Table 1 (small cell and indolent B‐cell lymphomas and leukemias) and Table 2 (classic Hodgkin, large‐cell, and aggressive B‐cell lymphoma), there is a significant overlap between different entities, highlighting how more comprehensive genetic characterization could be helpful in the diagnostic procedure, for example by using targeted NGS panels that include frequent genetic alterations, as discussed below.
Table 1.
Overview of mutations in selected genes in small cell and indolent B‐cell lymphomas and leukemias
| Pathway | Gene | CLL (%) | MCL (%) | HCL (%) | LPL (%) | NMZL (%) | SMZL (%) | FL (%) |
|---|---|---|---|---|---|---|---|---|
| JAK‐STAT | SOCS1 | 5–15 | ||||||
| STAT3 | <5 | |||||||
| STAT6 | 5–15 | |||||||
| MAPK | BRAF | <5 | >90 | <5DMT | ||||
| MAP2K1 | <5 | <5a | b | |||||
| NRAS | <5 | |||||||
| KRAS | <5 | |||||||
| BcR | CD79B | 5–15 | <5 | |||||
| CARD11 | 5–10 | 5–10 | 5–10 | 5–15 | ||||
| Epigenetic | KMT2D | 10–20% | 20–30 | 5–15 | 40–50 | |||
| KMT2C | 5–15 | 10–20% | ||||||
| CREBBP | 5–10 | <5 | 40–50 | |||||
| EP300 | <5 | 5–10 | 5–10 | 10–20 | ||||
| EZH2 | 10–20 | |||||||
| HIST1 c | <5 | 5–10 | 5–10 | 30–40 | ||||
| MEF2B | 5–10 | 5–15 | ||||||
| ARID1A | 10–20% | 10–20% | 5–10 | 5–15 | ||||
| TERT | 10–20% | |||||||
| NSD2 | 5–15 | |||||||
| Cell cycle | CCND1 | 20–30 | ||||||
| CDKN1B | 5–15 | 10–20% | ||||||
| CDKN2A | 10–20% | |||||||
| DNA damage | TP53 | 5–10 | 20–30 | 5–10 | <5 | 10–20% DMT | 5–10 | |
| ATM | 5–10 | 40–50 | <5 | <5 DMT | ||||
| NF‐κB | NFKBIE | <5 | <5 | |||||
| NOTCH | NOTCH1 | 5–15 | 5–10 | <5 | 5–10 NNK | <5 | ||
| NOTCH2 | <5 | <5 | <5 | 10–20% | 20–30% NNK | <5 | ||
| SPEN | 5–15 | 5–10 NNK | ||||||
| TLR | MYD88 | <5 | >90 | 5–10 | 5–10 DMT | |||
| Apoptosis | BCL2 | 20–30 | ||||||
| BIRC3 | <5 | 10–20% | 5–10 | 5–10 | ||||
| Immune | TNFRSF14 | 10–20% | 20–30 | |||||
| CXCR4 | 30–40 | <5 | ||||||
| TNFAIP3 | 10–20% | 5–15 NNK | ||||||
| RNA editing | SF3B1 | 10–20% | ||||||
| RPS15 | <5 | |||||||
| Transcription | ID3 | 5–15 | ||||||
| EGR2 | <5 | |||||||
| KLF2 | 10–20% | 10–20% | 20–30NNK | |||||
| mTOR | RRAGC | 10–20% | ||||||
| Others | PTPRD | 10–20% |
| Mutation frequency ranges (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| <5 | 20–30 | |||||||
| 5–10 | 30–40 | |||||||
| 5–15 | 40–50 | |||||||
| 10–20 | >90 | |||||||
Note: Frequency ranges are approximated, in a majority of cases stemming from two or more studies.
Abbreviations: BcR, B‐cell receptor; CLL, chronic lymphocytic leukemia; DMT, DNA damage response, MAPK, and TLR; FL, follicular lymphoma; HCL, hairy cell leukemia; LPL, lymphoplasmacytic lymphoma; MCL, mantle‐cell lymphoma; NMZL, nodal marginal zone lymphoma; NNK, NF‐κB, NOTCH, and KLF2; SMZL, splenic marginal zone lymphoma; TLR, toll‐like receptor.
Frequent in BRAF unmutated HCL and in HCL variant.
40%–50% in pediatric‐type FL.
HIST1 gene family.
DMTMutations identifying DMT SMZL subtype.
NNKMutations identifying NNK SMZL subtype.
Table 2.
Overview of mutations in selected genes in classic Hodgkin, large cell, and aggressive B‐cell lymphomas
| Pathway | Gene | BL (%) | DLBCL (%) | PMBCL (%) | cHL (%) | PBL (%) |
|---|---|---|---|---|---|---|
| JAK‐STAT | SOCS1 | ST2 | 40–50 | 30–40 | 5–10 | |
| STAT3 | ST2 | 5–15 | 30–40 | |||
| STAT6 | <5 | 30–40 | 30–40 | |||
| JAK1 | 5–15 | 10–20% | ||||
| MAPK | NRAS | 10–20% | ||||
| KRAS | <5 | 5–10 | ||||
| BcR | CD79B | MCD | ||||
| CARD11 | 5–15 | |||||
| Epigenetic | KMT2D | 5–10 | EZB | 5–15 | ||
| CREBBP | 5–10 | EZB | 5–10 | |||
| EP300 | EZB | 10–20% | ||||
| EZH2 | EZB | 10–20% | 30–40 | |||
| HIST1 a | 30–40 | 5–15 | 20–30 | 10–20% | ||
| ARID1A | 5–10 | 10–20 | ||||
| TET2 | ST2 | <5 | 5–15 | |||
| ZNF217 | 20–30 | 10–20% | ||||
| Cell cycle | MYC | 40–50 | 5–15 | 10–20% | ||
| DNA damage | TP53 | 10–20% | 10–20% | 10–20% | 5–10 | 20–30 |
| ATM | <5 | <5 | 5–15 | |||
| NF‐κB | NFKBIE | <5 | 10–20% | 10–20% | ||
| NOTCH | NOTCH1 | 5–10 | N1 | <5 | ||
| NOTCH2 | BN2 | <5 | ||||
| SPEN | 5–10 | 5–15 | ||||
| TLR | MYD88 | MCD | ||||
| Apoptosis | BCL2 | EZBb | ||||
| Immune | TNFRSF14 | EZB | <5 | |||
| CXCR4 | <5 | <5 | ||||
| B2M | 5–10 | 20–30 | 20–30 | |||
| TNFAIP3 | BN2 | 20–30 | 20–30 | |||
| IL4R | 20–30 | |||||
| Transcription | ID3 | 30–40 | <5 | |||
| TCF3 | 20–30 | |||||
| KLF2 | 5–15 | <5 | ||||
| Others | PTPN1 | 20–30 | 20–30 |
| Mutation frequency ranges (%) | ||||||
|---|---|---|---|---|---|---|
| <5 | 20–30 | |||||
| 5–10 | 30–40 | |||||
| 5–15 | 40–50 | |||||
| 10–20 | >90 | |||||
Note: Frequency ranges are approximated, in a majority of cases stemming from two or more studies. In the DLBCL column, EZB, BN2, MCD, N1, and ST2 denote mutations typically present in the said molecular subtype.
Abbreviations: BcR, B‐cell receptor; BL, Burkitt lymphoma; BN2, BCL6 fusion and NOTCH2 mutation; cHL, classic Hodgkin lymphoma; DLBCL, diffuse large B‐cell lymphoma; EZB, EZH2 mutation and BCL2 translocation; MCD, MYD88 and CD79B mutations; N1, NOTCH1 mutation; PBL, plasmablastic lymphoma; PMBCL, primary mediastinal B‐cell lymphoma; ST2, SGK1 and TET2 mutations; TLR, toll‐like receptor.
HIST1 gene family.
BCL2 translocation.
Prognostic markers
In several lymphoma entities, a substantial number of genetic alterations have been demonstrated to be prognostically significant and could ideally provide information to guide treatment decisions. However, most of the described prognostic alterations have not yet entered clinical routine. CLL is the best‐studied example, where several molecular prognostic biomarkers are now included in the standard workup for all patients with CLL. The IGHV gene mutation status [68] allows to separate CLL patients with mutated IGHV genes (M‐CLL) and a favorable prognosis from patients with unmutated IGHV genes (U‐CLL) and inferior outcome, and in more recent years, studies have shown that the IGHV mutation status also has a predictive impact [69]. Based on IG gene sequencing, it is also possible to identify subsets of patients expressing quasi‐identical or stereotyped BcRs [70], which can be identified in up to 41% of cases. In particular, patients belonging to subset 2 (IGHV3‐21/IGLV3‐21) or carrying the IGLV3‐21 R110 mutation have clinically aggressive disease and do not benefit from chemoimmunotherapy [71, 72], while subset 8 patients have the highest risk of Richter transformation [73, 74].
The previously mentioned frequent cytogenetic aberrations in CLL [22] are routinely investigated with FISH and have prognostic significance, from worst to best: del(17p), del(11q), trisomy 12, no alteration, and del(13q). In addition to del(17p) (includes TP53), TP53 sequence analysis [75] has also entered routine practice, having an adverse impact even without del(17p) [76, 77, 78]. Interestingly, TP53 mutations detected at low (subclonal) frequency also has a negative impact on the outcome, at least in patients treated with chemoimmunotherapy [79, 80]. Considering the independent prognostic strength of TP53 alterations and IGHV subtype, they were included in the clinical prognostic index CLL‐IPI [33] and the iwCLL guidelines [81]. There are several other genes that are promising prognostic biomarkers, for example, NOTCH1, SF3B1 [20, 82–86], ATM [87], EGR2 [88], and BIRC3 [89] among others, although it will be important to study which of them will remain as independent prognostic biomarkers in well‐characterized larger patient series [90, 91, 92].
In MCL, proliferation signatures have long been described [93] to have an important prognostic impact and can now be investigated in FFPE material [94, 95, 96, 97]. TP53 is also an adverse prognostic factor in MCL [98, 99], as well as in DLBCL [100, 101, 102, 103] and LPL/WM [104]. In FL, similar to CLL, genetic alterations have been included in a risk model termed m7‐FLIPI [105], which combines clinical parameters with mutations in seven genes (EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP, and CARD11) and improves the prognostication of FL patients. MYC and BCL2 and/or BCL6 translocation confer an adverse prognosis in comparison to other DLBCL [106, 107], but these lymphomas are now classified as a separate entity (HGBCL with MYC and BCL2 and/or BCL6 translocation).
Predictive markers
Identification of predictive markers is the least developed biomarker field in B‐cell malignancies. However, there is promising mounting evidence, especially when considering signaling pathways, rather than single genes. The best example for this is BcR pathway inhibitors (BTK, STK, and PI3K inhibitors), which show high activity in lymphomas with an activated BcR pathway [108]. In CLL, the presence of TP53 alterations or unmutated IGHV genes identifies patients that benefit from treatment with BcR [109, 110] or BCL2 inhibitors [111]. NOTCH1 mutations in CLL seem to not benefit from the addition of an anti‐CD20 antibody [91]. In MCL, the presence of TP53 mutations in MCL identifies a group of young patients who do not benefit from traditional intensified chemoimmunotherapy regimens [112], highlighting the need for alternative treatment regimens for these patients. In LPL/WM, CXCR4 warts, hypogammaglobulinemia, infections and myelokathexis (WHIM)‐like mutations identify patients with higher disease activity [113] and lower response to the BTK inhibitor ibrutinib [114, 115, 116].
Another example of a potential predictive marker is the aforementioned new molecular subgroups of DLBCL, where the hope is that targeted treatments exploiting dependencies of malignant cells towards certain pathways will improve the outcome compared to the classic R‐CHOP treatment regimen [53], although this still has to be proven.
Despite the promising efficacy of these novel agents targeting signaling pathways, resistance to treatment can be observed over time, as was seen with targeted treatments in other malignancies (e.g., EGFR‐targeting tyrosine kinase inhibitors in lung cancer or BCR‐ABL1 inhibitors in chronic myeloid leukemia). In this context, predictive biomarkers of treatment resistance have been described, for example, BTK and PLCG2 mutations, which confer resistance to ibrutinib treatment [117, 118, 119]. Similarly, acquisition of the BCL2 G101V mutation has been shown to confer resistance to the BCL2 inhibitor venetoclax [120].
Comprehensive genomic profiling
Considering the increasing number of clinically relevant aberrations in lymphoid malignancies, custom NGS‐based targeted sequencing has been developed, which allows cost‐effective screening of many genes simultaneously and with a higher sensitivity than Sanger sequencing [121, 122]. For this purpose, commercial or laboratory‐developed amplicon‐based NGS assays have been designed to cover the most recurrent genes in major entities of lymphoid malignancies [3]. While these gene panels can detect single nucleotide variants (SNVs) and indels accurately, polymerase chain rection (PCR) amplification can introduce biases in amplification and sequence artifacts (in particular with FFPE tissue as input), and sequencing of GC‐rich regions can be problematic. To study the robustness of amplicon‐based assays, a multicenter study, testing three different amplicon‐based gene panels (including 11 recurrently mutated CLL‐related genes) and 48 CLL samples, was carried out at six European centers [123]. While a very high concordance was demonstrated between assays and centers above a variant allele frequency (VAF) of 5%, more diverse results were seen for variants with low VAF (<5%). A conclusion from the study was that the introduction of unique molecular identifiers is necessary to reliably detect variants with lower VAFs.
In more recent years, the focus has shifted to hybridization‐based targeted enrichment panels. Using baits that capture the regions of interest, these assays give a more uniform sequencing quality, and hence more difficult regions and difficult samples can be sequenced [124, 125]. While a larger number of genes can be included in capture‐based gene panels, different types of genomic aberrations can also be assessed, that is, SNVs/indels, copy‐number aberrations (CNAs), and structural variants (e.g., translocations). In addition, they can provide information on more complex markers, such as IG gene and T‐cell receptor rearrangements. Two such capture‐based gene panels were recently published focusing on lymphoid malignancies, that is, the LYNX panel [126] and the EuroClonality‐NGS capture panel [127]. Another advantage with broad capture‐based panels is that they may allow detection of the genomic aberrations needed to identify disease subgroups, for instance, the recently proposed new subtypes of DLBCL [53]. Moreover, capture‐based panels are not limited to the detection of genomic aberrations but can also include targeted RNA sequencing and even epigenomic alterations [124]. With this type of panel, it will be possible to combine different types of molecular tests currently performed in routine diagnostics into a single test. This will hopefully pave the way for the implementation of precision diagnostics for patients with lymphoid malignancies.
Another option for comprehensive genomic profiling would be to perform WGS or WES to capture all the relevant genomic markers needed for diagnostic purposes. Clinical WGS is tested for acute leukemias within different national/local initiatives with the goal to replace the many different genetic analyses currently performed for these patients [128, 129]. A pilot study in CLL has also compared WGS versus targeted NGS and FISH with high concordance for SNVs/indels, while the concordance was lower for CNAs. However, considering the cost for WGS is still high and that WGS is challenging for FFPE samples, this is currently not a viable option.
Epigenomics of lymphoid malignancies
The altered phenotype and function of neoplastic lymphoid cells are not only achieved through the acquisition of genetic alterations, but also a variety of epigenetic changes [6]. Generally speaking, epigenetics is defined as the discipline that studies changes in gene expression in the absence of any genetic alteration [6, 7]. This gene expression regulation is achieved through different layers that act in concert, including DNA methylation, histone modifications, chromatin accessibility, and 3D folding of the genome. DNA methylation thus far is the best‐characterized layer, for the most part consisting of addition of a methyl group to cytosines mostly in a cytosine‐guanine dinucleotide (CpG) context. Although DNA methylation is generally perceived as a repressive epigenetic mark, with gene silencing through promoter hypermethylation as a widely reported phenomenon [130], its role depends on the genomic and chromatin context where methylation takes place [131, 132]. In addition to DNA methylation, histone marks comprise a variety of chemical modifications of histones that strongly associate with particular genomic functions [133]. Looking at a combination of histone marks, different chromatin states can be defined (e.g., active, weak or poised promoters, active or weak enhancers, transcriptional elongation, as well as various modes of silent chromatin). Chromatin accessibility gives information on the open regions of the genome where transcription factors bind. Finally, the 3D structure of the genome provides epigenetic information regarding distant regulation between enhancers and promoters through DNA looping [134, 135, 136, 137]. Therefore, to gain a deep insight into epigenetic deregulation in lymphoid neoplasms, an integrative analysis of several epigenetic layers is essential. Epigenetics in precision medicine is currently an understudied aspect in comparison to genetics, but has the potential to give a plethora of information, ranging from a better understanding of pathophysiological processes, diagnostic and prognostic information, as well as potentially actionable targets for treatment.
DNA methylation changes during normal B‐cell differentiation
To understand how the DNA methylome of B‐cell neoplasms changes, it is essential to define what happens during the physiological maturation of B cells. In this context, two seminal works have described the dynamics of DNA methylation changes during the entire B‐cell differentiation program at a single base‐pair resolution [138, 139]. The first observation is that during B‐cell maturation, gradual and global demethylation of the genome can be observed, particularly at mature stages (GC B cells, memory B cells, and plasma cells). A more detailed look reveals the presence of local demethylation in B‐cell‐related enhancers and promoters, and more abundantly, changes that do not imply a direct impact on gene expressions, such as gains of methylation in CpG‐rich regions marked by the polycomb group, and methylation loss in heterochromatic regions. Different steps in the maturation of B cells are accompanied by specific changes in DNA methylation, most prominently upon entering the GC and when differentiating to memory B cells or plasma cells, and can accordingly be used to infer the differentiation stage of B cells, which is also possible with lower coverage methods, such as methylation microarrays or even just by analyzing a reduced set of CpGs [139]. Importantly, the characterization of normal‐B‐cell differentiation allows interpretation of methylation changes of B‐cell lymphomas from a COO standpoint.
Methylation‐based proliferative clock
As mentioned above, normal B‐cell maturation involves DNA methylation changes without an impact on gene expression. Based on recent studies, these changes (hypermethylation in polycomb regions and hypomethylation in heterochromatin) have been linked to cell division [140, 141, 142]. Therefore, DNA methylation in these repressed regions becomes accumulated during sequential cell divisions and can be used as a mitotic clock. In normal and neoplastic B cells, this concept has led to the development of an epigenetic mitotic clock, termed epiCMIT (epigenetically‐determined cumulative mitoses), which captures both DNA hypo‐ and hypermethylation associated with cell proliferation [8]. The epiCMIT of normal B cells at different differentiation stages reflects the past proliferative history, rather than their current proliferation state, highlighting how DNA methylation can provide information on the past processes a cell has gone through. In malignant B cells, the epiCMIT reflects the sum of proliferation during normal B‐cell development and during malignant transformation. In comparison to other epigenetic clocks like the Horvath clock [143] (which predicts the chronological age), the epiCMIT is independent of the age of the individual. Analysis of the epiCMIT not only traces the past proliferative history but has an important role in predicting the future clinical behavior of the patients. Changes in the epiCMIT have been shown to independently predict the clinical outcome in ALL, CLL, and MCL, as well as being associated with a greater number of genetic driver lesions, reflecting the accumulation of alterations with time [8].
Methylation landscape and subtypes of B‐cell lymphomas
The DNA methylation landscape of a majority of B‐cell lymphomas has already been characterized, in most cases by means of high density, single base‐pair resolution methylation microarrays (i.e., 450k or EPIC methylation arrays). A general observation is that across mature B‐cell lymphomas, there is a global DNA methylation loss when compared to normal B cells, most likely due to loss of methylation in heterochromatic regions occurring with proliferation [6].
Early work on CLL [144] showed that approximately half of the differentially methylated CpGs between CLL subtypes (U‐CLL and M‐CLL) are related to their respective resemblance to the COO. In addition, a new epigenetic subtype termed intermediate CLL (i‐CLL) could be identified, besides naïve‐like CLL (n‐CLL, corresponding to U‐CLL) and memory‐like CLL (m‐CLL, corresponding to M‐CLL). This finding was confirmed in a later study [140] using an alternative terminology (low‐programmed CLL for n‐CLL, intermediate‐programmed CLL for i‐CLL, and high‐programmed CLL for m‐CLL) and in several clinical series [145, 146, 147, 148], showing intermediate clinical behavior of i‐CLL, between the more aggressive n‐CLL and m‐CLL. Of interest, recent work has shed further light on the i‐CLL subtype, finding that a majority of cases with adverse clinical behavior harbor a point mutation in the lambda light chain variable gene (IGLV3‐21R110), which leads to constitutively active BcR signaling, and a subset of those cases also belong to stereotyped subset #2 [71, 72]. Moreover, analysis of CLL with trisomy 12 found that this subgroup has a distinct methylation profile, which, together with altered chromatin activation, explains some of the biological differences of this cytogenetically defined subtype [149].
In MCL, two epigenetic subtypes were identified [150], which correspond to the clinicopathological subtypes of cMCL and leukemic nnMCL (C1 and C2 MCL, respectively). By integrating the knowledge on methylation changes in normal B cells, these two subtypes could again be related to their COO (naïve‐like for C1 MCL and memory‐like for C2 MCL), confirming earlier results (e.g., through IGHV sequencing) [151]. In plasma cell myeloma/multiple myeloma (PCM/MM), apart from the aforementioned global loss of methylation, specific hypermethylation of enhancer regions could be observed, as opposed to the widely reported hypermethylation of CpG islands (CpG‐island methylator phenotype) reported in some solid tumors [152]. In BL and FL, despite both originating from the GC, significant differences could be observed [153]. Interestingly, as BL and FL are thought to originate from the dark zone (DZ) and light zone (LZ), respectively; several genes associated with the DZ were shown to be hypomethylated and upregulated in BL and hypermethylated and downregulated in FL, whereas other genes associated with the LZ exhibited the opposite pattern. Analysis of primary central nervous system lymphomas (PCNSL) [154] showed a distinct methylation pattern from DLBCL, underlining the previously postulated separation of PCNSL as a distinct subtype of DLBCL, rather than merely representing DLBCL with isolated CNS manifestation. Recently, DNA methylation analysis of LPL/WM identified two disease subtypes, which resemble memory B cells and plasma cells (PC‐like) [155]. The two subtypes show several different clinical, morphological (plasma cell differentiation in PC‐like WM), immunophenotypical, and genetic characteristics, highlighting the clinicopathological significance of these previously unknown subtypes.
To sum up, although the classical role of DNA methylation is to regulate gene expression, recent epigenetic studies in B‐cell neoplasms have revealed that DNA methylation is strongly related to cellular memory, without a regulatory role, in terms of identifying the cellular origin and measuring the past proliferative history. These two components of cellular memory are variables independently associated with the clinical behavior of the patients and they could represent an important complement to the detection of genetic changes in clinical diagnostics.
Towards a DNA methylation–based classification of B‐cell lymphomas
DNA methylation has been successfully used to generate tumor classifiers, first for central nervous system tumors [156] and more recently for sarcomas [157]. As the characterization of B‐cell malignancies (which include more than 40 different entities) is still not complete, a globally applicable classifier is not available yet. Duran‐Ferrer et al. recently designed a two‐step methylation classifier that accurately classified an unknown B‐cell malignancy into general categories first, such as ALL, CLL, MCL, PCM/MM, and DLBCL, and their subtypes in a second step (e.g., n‐CLL, i‐CLL, and m‐CLL) [8]. Importantly, this classifier is based on a limited number of CpG sites and therefore is amenable to be used in targeted gene panels or locus‐specific assays. Looking forward, with further characterization of rare B‐cell lymphomas, a pan B‐cell lymphoma methylation classifier can be achieved in the coming years, offering a powerful tool to complement the traditional classification of lymphomas. Figure 1a summarizes DNA methylation changes in B‐cell lymphomas and their prognostic implications.
Fig. 1.
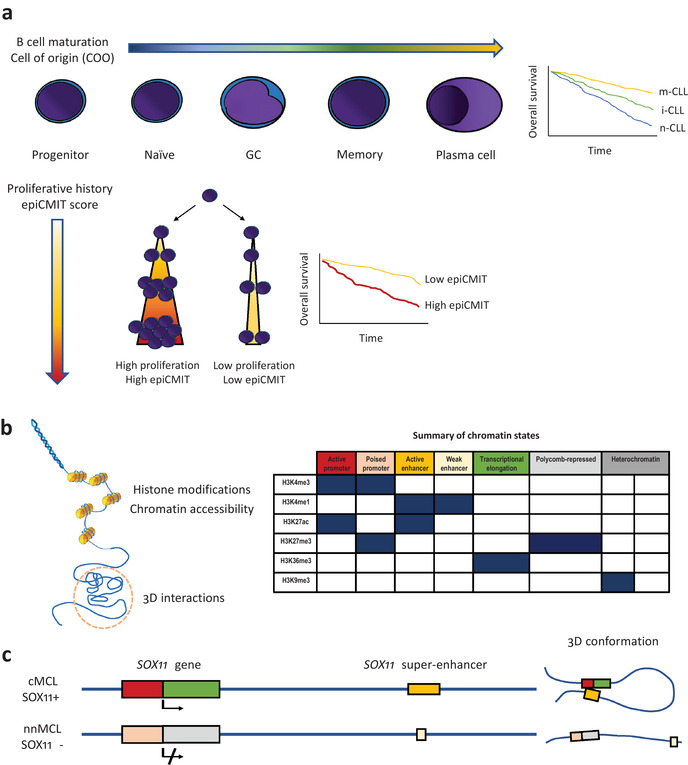
Epigenetics in B‐cell malignancies. (a) DNA methylation changes during B‐cell maturation (upper panel); in B‐cell neoplasms, DNA methylation imprints of normal B‐cell maturation allow classification of them into different clinicobiological subtypes (the Kaplan–Meier curve on the right shows chronic lymphocytic leukemia [CLL] as an example). Cell proliferation is also imprinted into the DNA methylome, which allows to determine the past proliferative history (epigenetically determined cumulative mitoses score), whose magnitude also has important prognostic value in B‐cell malignancies (lower panel). (b) Histone modifications, chromatin accessibility, and 3D genome interactions are important epigenetic marks (left). The combination of different histone marks allows identification of segments of the genome with different functions, the so‐called chromatin states (right). (c) Example of multilayered epigenetic analysis for SOX11 in mantle cell lymphoma (MCL). Chromatin state analysis in SOX11 positive MCL shows active promoter/distal super‐enhancer regions, which are in close contact in the 3D space (chromatin loop). SOX11 negative MCL has a poised promoter and a small weak enhancer, which are maintained distant in the 3D space (no chromatin loop).
Chromatin landscape of B‐cell lymphomas
Overall, the chromatin profile of B‐cell lymphomas is less explored than their DNA methylome, but CLL has been the subject of several integrative chromatin studies, which have revealed important insights into deregulated genes, pathways, and transcription factors networks [10, 158–160]. Chromatin accessibility studies of CLL [161] can reproduce the subclassification of the major disease subtypes (U‐CLL and M‐CLL), and like in the aforementioned methylation studies, an intermediate group could also be identified, highlighting the added value of epigenetics in refining the subclassification of lymphoid malignancies. The reference epigenome of CLL [10], which includes six histone marks and chromatin accessibility besides DNA methylation and gene expression, revealed the chromatin state changes CLL cells undergo in malignant transformation, as well as subtype (U‐CLL and M‐CLL) specific differences. Other findings include more active and open chromatin in the clinically more aggressive U‐CLL subgroup, de novo activation of regions enriched for several transcription factor binding sites (NFAT, FOX, and TCF/LEF), and some associations of chromatin configurations with genetic alterations (e.g., MYD88 mutated CLL and cases with trisomy 12). Interestingly, differential methylation in trisomy 12 CLLs is linked to differential chromatin activation in these cases [149]. Figure 1 (b) illustrates the different aforementioned epigenetic layers and a summary of chromatin states based on histone marks.
In MCL, through analysis of chromatin states and chromatin conformation [150], a distant SOX11 enhancer could be identified, which is active only in SOX11 positive MCL, shedding light on the mechanisms that lead to the expression of this important oncogene (Fig. 1c). These results have been recently confirmed, including demonstration of physical proximity by FISH analysis and identification, within the enhancer element, of a small accessible region that is exclusively active in SOX11 expressing MCL [162]. Recent work on the genome‐wide 3D genome architecture of CLL and MCL [163] highlighted changes in active and inactive compartments during B‐cell development and lymphomas, as well as identified an intermediate compartment that is enriched in poised and polycomb repressed chromatin. In PCM/MM, the chromatin landscape has been characterized and revealed the presence of a widespread activation of regulatory elements leading to upregulation of members of the NOTCH, NF‐κB, MTOR signaling, and TP53 pathways as well as other processes such as osteoblast differentiation and response to oxidative stress [164].
New technologies for precision medicine in lymphoma
As elaborated in the previous sections, bulk genomic, epigenomic, and transcriptomic studies have provided a comprehensive and unbiased view of lymphoid malignancies. Integration of the unbiased functional screen by CRISPR‐Cas9 based methods has also been useful for the identification of druggable vulnerabilities in lymphoma cell lines [49, 165]. However, bulk sequencing or screen of cell lines offers limited insight into the clonal composition of tumors, a key factor in treatment resistance and tumor progression [166]. Another missing element of bulk analysis is the tumor microenvironment (TME), which closely interacts with malignant cells [167] and is especially relevant with the advent of immunotherapies, such as immune checkpoint inhibitors. The rise of single‐cell technologies has opened new possibilities, allowing capture of both tumoral heterogeneity and the microenvironment in one single experiment [168], even combining different information layers (multi‐omics) [169] and spatial characterization of cell populations in tissue samples [170]. The Human Cell Atlas (HCA) project leverages these new technologies and aims to provide a comprehensive catalog of cells in human tissues and organs [171], including transcriptomic, epigenetic, and spatial characteristics. Thus far, HCA projects have already investigated several tissues and organs, some of them with prominent immune cell populations. For instance, the thymus cell atlas [172] provides a comprehensive map of the cellular composition of this lymphoid organ and especially of T‐cell development. Several studies have investigated the single‐cell composition of the liver [173, 174, 175], including the hepatic immune microenvironment and fetal hepatic hematopoiesis. As expected, the normal human heart contains only a relatively modest amount of immune cells, mainly myeloid and T cells [176]. The lung on the other hand is constantly challenged by potential infectious agents and therefore relies on well‐functioning mucosal immunity, especially rich in alveolar macrophages [177]. Upon completion, the HCA will provide a comprehensive map of the normal cell populations of different organs, allowing to better interpret changes observed in the disease. Other single‐cell studies have looked specifically at the B‐cell lineage, elucidating the maturation process and different cell subtypes/states [178, 179, 180, 181, 182]. In the study by King et al. [178], tonsillar B cells were sequenced, characterizing the maturation process from naïve B cell to plasmablast. Interestingly, a previous unappreciated heterogeneity of memory B cells could be shown, as well as comparable SHM rates between memory B cells and plasmablasts. In mice [179], two different subsets of memory B cells were identified, which were shown to arise from activated B cells and GC B cells. Detailed analysis of GC B cells [180] expanded the traditional DZ and LZ subdivision of GC B cells, defining four DZ states, six intermediate states, an LZ state, as well as prememory and plasmablast states. Using mass cytometry, surface molecules of B cells from various sources (blood, bone marrow, lymph node, and tonsil) were characterized [181], proposing classification into 12 different B‐cell subsets (immature/transitional, two naïve, GC, six memory, plasma cell, and tonsil‐specific subsets). Similarly, using single‐cell transcriptomics [182] of fluorescence‐activated cell sorting (FACS)‐sorted peripheral blood mononuclear cells, 10 different B‐cell clusters were defined, expanding classic B‐cell characterization by traditional flow sorting. These studies provide a framework for the interpretation of changes in malignant transformation of B cells and allow a novel transcriptomic‐based refined COO classification.
CLL has been more extensively studied with single‐cell technologies [183, 184, 185, 186, 187, 188, 189], focusing on tumoral heterogeneity, clonal evolution, and changes after treatment. Integration of single‐cell genomics and transcriptomics identified two new disease drivers (LCP1 and WNK1), thanks to the precise determination of tumor subclones [183]. One study focused on the effects of SF3B1 mutations on signaling pathways and alternative splicing, providing insight into the mechanistic effects of this important gene in CLL [186]. A thorough case study of one CLL patient looked at cytogenetic, transcriptomic, and genomic changes over a period of 29 years, highlighting the clonal changes in different disease phases [189]. Gaiti et al. applied a combination of single‐cell transcriptomics and methylation analysis to explore intratumoral heterogeneity in the evolution of CLL (including after treatment with ibrutinib), finding considerable heterogeneity after initial malignant transformation and clones that preferentially exited the lymph node upon ibrutinib exposure [187]. Another study also analyzed the changes CLL cells undergo when treated with ibrutinib, this time considering single‐cell transcriptomics and chromatin accessibility, uncovering NF‐κB signaling downregulation, a quiescence‐like gene signature, and changes in chromatin accessibility (e.g., decreased accessibility for CLL‐specific enhancers) upon exposure [184]. Recently, Penter et al. leveraged (naturally in vivo occurring) mitochondrial DNA mutations to obtain a precise tracking of CLL clones, which was then used to study differences in chromatin states and transcription [188], showing how mitochondrial DNA mutations are powerful in vivo markers of (sub)clones.
In the first study that used single‐cell technology in FL [190], the transcriptional heterogeneity was determined and compared to different GC B‐cell states. First, characterization of normal GC B cells allowed definition of three main cell states (DZ, intermediate zone, and LZ), which lie in a continuum and show a nuanced synchronous expression of gene clusters. When comparing FL cases with these normal GC B‐cell states, these synchronized gene clusters could not be observed and FL B cells were not found to be blocked in a specific GC stage, pointing to a desynchronization of the normal GC specific expression program. In a later study on FL [191], it was found that patient samples include multiple malignant subclones at the transcriptional level, which seem to be at least in part caused by mutational heterogeneity, although it is apparent that other (unknown) drivers could also be responsible for transcriptional heterogeneity. Furthermore, tumor‐infiltrating CD4+ Treg cells with coexpression of immune checkpoint genes were identified, highlighting the utility of the simultaneous profiling of the TME in single‐cell studies. Another interesting FL study [192] characterized tumor samples from different anatomical sites of the same patient using a multi‐omics approach. Subclones were identified (leveraging BcR sequencing) and, interestingly, were shown to sometimes coexist in both locations and other times to be location specific, suggesting divergent evolution, which might be responsible for treatment resistance. In the aforementioned study by Holmes et al. that characterized the GC into novel cell subpopulations [180], DLBCL samples were studied using a single‐cell RNA sequencing (scRNA‐seq) defined COO classification (based on normal GC data) and compared to the classical GCB versus ABC COO model. Around 80% of DLBCL cases could be assigned to a specific B‐cell subpopulation; it was found that a majority of GCB DLBCL fall between an intermediate LZ‐like and early LZ stage, most ABC DLBCL are related to late GC and prememory B cells (rather than plasmablasts), and interestingly several DLBCL cases that displayed a DZ signature were enriched for MYC and BCL2 translocations (double hit lymphomas), reinforcing the view that these lymphomas represent a separate entity (HGBCL with MYC and BCL2 and/or BCL6 translocation). Roider et al. exploited single‐cell sequencing to dissect both the tumor and microenvironment heterogeneity of four FL, two transformed FL, and three DLBCL clinical samples in an interesting proof‐of‐concept study for the use of single‐cell technology in precision oncology [193]. Different proportions of T‐cell subtypes were shown to populate the TME of different lymphomas (e.g., expected higher proportion of Tfh cells in FL). All examined samples had at least two transcriptional distinct populations of malignant cells and three cases were studied in depth, including in vitro drug‐response assays, which revealed different sensitivity of subclones to chemotherapeutic and targeted agents. In a more recent study, 17 DLBCL samples were analyzed by scRNA‐seq in depth, and 74 gene expression programs were identified from the malignant B cells, illustrating high degrees of inter‐ and intratumor heterogeneity (Ye and Pan‐Hammarström et al., manuscript submitted). Furthermore, eight nonmalignant B‐cell subclusters, 16 T‐cell subclusters, and six myeloid subpopulations were characterized, and more than 2700 pairs of cell–cell interactions were predicted, indicating a complex and highly dynamic TME in DLBCL.
cHL is another ideal candidate for single‐cell sequencing, due to the low tumor cell content and a prominent and heterogeneous inflammatory TME. Indeed, scRNA‐seq of 22 cHL [194] cases revealed a novel subset of LAG3 expressing T cells, which locate closer to the tumoral cells, are associated with loss of MHC‐II expression, and might represent a novel treatment target. In MM/PCM, malignant clones are readily identified at the single‐cell transcriptional level, including small residual clones after treatment, and transcriptionally different tumor subclones could be identified in a third of cases in one study [195] and all cases in another [196]. Zavidij et al. [197] concentrated on the TME in PCM/MM and found an increase of NK, T, and CD16+ cells and a decrease of plasmacytoid DC and CD14+ monocytes in comparison to normal samples. A detailed look at T‐cell subsets specifically highlighted an increase in Tregs and a switch from memory to effector cytotoxic cells during disease progression. Analyzing PCM/MM patients enrolled in a clinical trial [198], several genes (implicated in mitochondrial, endoplasmic reticulum, and oxidative stress) associated with therapy resistance were identified, which, combined in a signature, were shown to predict outcome better than traditional cytogenetic models. One particular identified overexpressed gene was demonstrated to confer resistance to proteasome inhibitors and shown to be targetable with cyclosporin A in vitro. Leveraging single‐cell resolution and multiple samplings during treatment, different clonal dynamics were observed in patients, with some being primarily sensitive or resistant and others showing the emergence of new resistant subclones during treatment. PCM/MM studies highlight the potential of single‐cell technologies since it is still an incurable disease in which treatment resistance develops, at least in part due to tumor heterogeneity.
Thus far, two studies have tackled MCL using single‐cell technologies. In the first [199], bone marrow cells from a single patient were sequenced, describing four transcriptional malignant subclones as well as the TME, but conclusions from this study are limited due to only analyzing one case. The second study sequenced samples from five patients at different timepoints, of which three were ibrutinib responsive and two were nonresponsive [200]. Using this approach, a 17q gain (which, among other genes, includes BIRC5) was identified in one ibrutinib‐resistant case. The overexpression of survivin (encoded by BIRC5) was then validated in a patient‐derived xenograft model and an independent series of ibrutinib‐treated MCLs, and was shown to be an actionable target to overcome ibrutinib resistance in in vitro models (cell lines).
In summary, single‐cell studies of lymphomas highlight both tumor cell heterogeneity (which might explain treatment resistance), potential new actionable targets, as well as the cellular composition of the TME.
Concluding remarks
By applying high‐throughput genomic, transcriptomic, and epigenomic technologies in the last 10 years, new biomarkers with clinical impact have been identified in most B‐cell malignancies. While the number of diagnostic and prognostic markers is steadily increasing, there are still relatively few markers that can be used to predict therapy response at the individual patient level. Hopefully, with comprehensive molecular profiling, as part of clinical trials, we will discover new predictive markers that can be applied to tailor the patient's treatment and follow‐up. Although an increasing number of targeted drugs are available for patients with lymphoid malignancies, at least for selected entities, very few precision medicine trials exist worldwide that include patients with lymphoid tumors. In the coming years, the implementation of broad gene panel sequencing in the diagnostic workup of lymphoid tumors will result in improved patient subcategorization and this will hopefully prompt the initiation of new precision medicine studies. Finally, even though new technologies, such as single‐cell sequencing and spatial transcriptomics, have not yet entered clinical practice, they are very promising and could be a key factor in unlocking the potential of precision medicine in the treatment of lymphoid malignancies. Tumor heterogeneity and the TME have long been recognized as crucial factors of tumor biology, but only with the advent of single‐cell sequencing can they be studied with sufficient depth and efficiency, particularly in patients treated with targeted therapies. This may lead to the development of new strategies and approaches for personalized treatment and care for patients with lymphoid malignancies.
Author contributions
Marco M. Bühler: conceptualization (equal); writing – original draft (equal); writing – review and editing (equal). José I. Martin‐Subero: conceptualization (equal); writing – original draft (equal); writing – review and editing (equal). Qiang Pan‐Hammarström: conceptualization (supporting); writing – original draft (supporting); writing – review and editing (supporting). Elias Campo: conceptualization (equal); writing – original draft (equal); writing – review and editing (equal). Richard Rosenquist : conceptualization (lead); writing – original draft (lead); writing – review & editing (lead).
Conflict of interest
Richard Rosenquist has received honoraria from Abbvie, AstraZeneca, Janssen, Illumina, and Roche. Elias Campo has received honoraria from Roche, NanoString, Takeda, Eusapharma, Illumina, AbbVie, and AstraZeneca.
Acknowledgments
Marco Matteo Bühler is supported by grants from the Nuovo‐Soldati Foundation for Cancer Research and the Swiss Cancer League (BIL KLS‐5130‐08‐2020). Elias Campo is funded by the Ministerio de Ciencia, Innovación y Universidades (MICIU) (Grant No. RTI2018‐094274‐B‐I00), La Caixa Foundation (CLLEvolution‐LCF/PR/HR17/52150017), Health Research 2017 Program (HR17‐00221), and Generalitat de Catalunya Suport Grups de Recerca (AGAUR 2017‐SGR‐1142). Elias Campo is an Academia Researcher at the “Institució Catalana de Recerca i Estudis Avançats” (ICREA) of the Generalitat de Catalunya, CERCA Programme/Generalitat de Catalunya. José Ignacio Martin‐Subero is funded by Ministerio de Ciencia, Innovación y Universidades (MICIU) (Grant No. PID2020‐118167RB‐I00), the Cancer Research UK Accelerator award CRUK/AIRC/AECC joint funder partnership, Fundació La Marató de TV3, and Generalitat de Catalunya Suport Grups de Recerca (AGAUR 2017‐SGR‐736). Elias Campo and José Ignacio Martin‐Subero are also funded by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Project BCLLATLAS, grant agreement 810287). Richard Rosenquist and Qiang Pan‐Hammarström are supported by grants from the Swedish Cancer Society, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, Karolinska Institutet, Karolinska University Hospital, CIMED, and Radiumhemmets Forskningsfonder, Stockholm.
Bühler MM, Martin‐Subero JI, Pan‐Hammarström Q, Campo E, Rosenquist R. Towards precision medicine in lymphoid malignancies. J Intern Med. 2022;292:221–242.
Content List – This is an article from the symposium: “Precision medicine in hematology”.
References
- 1. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenquist R, Beà S, Du M‐Q, Nadel B, Pan‐Hammarström Q. Genetic landscape and deregulated pathways in B‐cell lymphoid malignancies. J Intern Med. 2017;282:371–94. [DOI] [PubMed] [Google Scholar]
- 3. Rosenquist R, Rosenwald A, Du M‐Q, Gaidano G, Groenen P, Wotherspoon A, et al. Clinical impact of recurrently mutated genes on lymphoma diagnostics: state‐of‐the‐art and beyond. Haematologica. 2016;101:1002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy‐cell leukemia. N Engl J Med. 2011;364:2305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med. 2012;367:826–33. [DOI] [PubMed] [Google Scholar]
- 6. Oakes CC, Martin‐Subero JI. Insight into origins, mechanisms, and utility of DNA methylation in B‐cell malignancies. Blood. 2018;132:999–1006. [DOI] [PubMed] [Google Scholar]
- 7. Martin‐Subero JI, Oakes CC. Charting the dynamic epigenome during B‐cell development. Semin Cancer Biol. 2018;51:139–48. [DOI] [PubMed] [Google Scholar]
- 8. Duran‐Ferrer M, Clot G, Nadeu F, Beekman R, Baumann T, Nordlund J, et al. The proliferative history shapes the DNA methylome of B‐cell tumors and predicts clinical outcome. Nat Cancer. 2020;1:1066–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mansouri L, Wierzbinska JA, Plass C, Rosenquist R. Epigenetic deregulation in chronic lymphocytic leukemia: clinical and biological impact. Semin Cancer Biol. 2018;51:1–11. [DOI] [PubMed] [Google Scholar]
- 10. Beekman R, Chapaprieta V, Russiñol N, Vilarrasa‐Blasi R, Verdaguer‐Dot N, Martens JHA, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat Med. 2018;24:868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris N, Jaffe E, Stein H, Banks P, Chan J, Cleary M, et al. A revised European–American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361–92. [PubMed] [Google Scholar]
- 12. Dietrich S, Hüllein J, Lee SC‐W, Hutter B, Gonzalez D, Jayne S, et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood. 2015;126:1005–8. [DOI] [PubMed] [Google Scholar]
- 13. Durham BH, Getta B, Dietrich S, Taylor J, Won H, Bogenberger JM, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130:1644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, et al. High prevalence of MAP2K1 mutations in variant and IGHV4‐34–expressing hairy‐cell leukemias. Nat Genet. 2014;46:8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Treon SP, Xu L, Guerrera ML, Jimenez C, Hunter ZR, Liu X, et al. Genomic landscape of Waldenström macroglobulinemia and its impact on treatment strategies. J Clin Oncol. 2020;38:1198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swerdlow SH, Kuzu I, Dogan A, Dirnhofer S, Chan JKC, Sander B, et al. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016;468:259–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burns A, Alsolami R, Becq J, Stamatopoulos B, Timbs A, Bruce D, et al. Whole‐genome sequencing of chronic lymphocytic leukaemia reveals distinct differences in the mutational landscape between IgHVmut and IgHVunmut subgroups. Leukemia. 2018;32:332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landau DA, Tausch E, Taylor‐Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puente XS, Beà S, Valdés‐Mas R, Villamor N, Gutiérrez‐Abril J, Martín‐Subero JI, et al. Non‐coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–24. [DOI] [PubMed] [Google Scholar]
- 20. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole‐genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nadeu F, Diaz‐Navarro A, Delgado J, Puente XS, Campo E. Genomic and epigenomic alterations in chronic lymphocytic leukemia. Annu Rev Pathol Mech Dis. 2020;15:149–77. [DOI] [PubMed] [Google Scholar]
- 22. Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. [DOI] [PubMed] [Google Scholar]
- 23. Puente XS, Jares P, Campo E. Chronic lymphocytic leukemia and mantle cell lymphoma: crossroads of genetic and microenvironment interactions. Blood. 2018;131:2283–96. [DOI] [PubMed] [Google Scholar]
- 24. Pararajalingam P, Coyle KM, Arthur SE, Thomas N, Alcaide M, Meissner B, et al. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood. 2020;136:572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nadeu F, Martin‐Garcia D, Clot G, Díaz‐Navarro A, Duran‐Ferrer M, Navarro A, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136:1419–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu C, De Miranda NF, Chen L, Wasik AM, Mansouri L, Jurczak W, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: impact of recurrent CARD11 mutations. Oncotarget. 2016;7:38180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wasik AM, Wu C, Mansouri L, Rosenquist R, Pan‐Hammarström Q, Sander B. Clinical and functional impact of recurrent S1PR1 mutations in mantle cell lymphoma. Blood Adv. 2018;2:621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okosun J, Bödör C, Wang J, Araf S, Yang C‐Y, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Küppers R, Stevenson FK. Critical influences on the pathogenesis of follicular lymphoma. Blood. 2018;131:2297–306. [DOI] [PubMed] [Google Scholar]
- 31. Ortega‐Molina A, Deleyto‐Seldas N, Carreras J, Sanz A, Lebrero‐Fernández C, Menéndez C, et al. Oncogenic Rag GTPase signalling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat Metab. 2019;1:775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huet S, Sujobert P, Salles G. From genetics to the clinic: a translational perspective on follicular lymphoma. Nat Rev Cancer. 2018;18:224–39. [DOI] [PubMed] [Google Scholar]
- 33. International CLL‐IPI Working Group . An international prognostic index for patients with chronic lymphocytic leukaemia (CLL‐IPI): a meta‐analysis of individual patient data. Lancet Oncol. 2016;17:779–90. [DOI] [PubMed] [Google Scholar]
- 34. Vela V, Juskevicius D, Dirnhofer S, Menter T, Tzankov A. Mutational landscape of marginal zone B‐cell lymphomas of various origin: organotypic alterations and diagnostic potential for assignment of organ origin. Virchows Arch. 2021. 10.1007/s00428-021-03186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spina V, Khiabanian H, Messina M, Monti S, Cascione L, Bruscaggin A, et al. The genetics of nodal marginal zone lymphoma. Blood. 2016;128:1362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clipson A, Wang M, De Leval L, Ashton‐Key M, Wotherspoon A, Vassiliou G, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2015;29:1177–85. [DOI] [PubMed] [Google Scholar]
- 37. Parry M, Rose‐Zerilli MJJ, Ljungström V, Gibson J, Wang J, Walewska R, et al. Genetics and prognostication in splenic marginal zone lymphoma: revelations from deep sequencing. Clin Cancer Res. 2015;21:4174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi D, Deaglio S, Dominguez‐Sola D, Rasi S, Vaisitti T, Agostinelli C, et al. Alteration of BIRC3 and multiple other NF‐κB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118:4930–4. [DOI] [PubMed] [Google Scholar]
- 39. Piva R, Deaglio S, Famà R, Buonincontri R, Scarfò I, Bruscaggin A, et al. The Krüppel‐like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia. 2015;29:503–7. [DOI] [PubMed] [Google Scholar]
- 40. Martínez N, Almaraz C, Vaqué JP, Varela I, Derdak S, Beltran S, et al. Whole‐exome sequencing in splenic marginal zone lymphoma reveals mutations in genes involved in marginal zone differentiation. Leukemia. 2014;28:1334–40. [DOI] [PubMed] [Google Scholar]
- 41. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bonfiglio F, Bruscaggin A, Guidetti F, Terzi Di Bergamo L, Faderl MR, Spina V, et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood. 2021. 10.1182/blood.2021012386 [DOI] [PubMed] [Google Scholar]
- 43. Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316–20. [DOI] [PubMed] [Google Scholar]
- 44. López C, Kleinheinz K, Aukema SM, Rohde M, Bernhart SH, Hübschmann D, et al. Genomic and transcriptomic changes complement each other in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun. 2019;10:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Panea RI, Love CL, Shingleton JR, Reddy A, Bailey JA, Moormann AM, et al. The whole‐genome landscape of Burkitt lymphoma subtypes. Blood. 2019;134:1598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma E‐J, et al. Molecular diagnosis of Burkitt's lymphoma. N Engl J Med. 2006;354:2431–42. [DOI] [PubMed] [Google Scholar]
- 49. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171:481–94.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmitz R, Wright GW, Huang DaW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N Engl J Med. 2018;378:1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pedrosa L, Fernández‐Miranda I, Pérez‐Callejo D, Quero C, Rodríguez M, Martín‐Acosta P, et al. Proposal and validation of a method to classify genetic subtypes of diffuse large B cell lymphoma. Sci Rep. 2021;11:1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood. 2020;135:1759–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ren W, Ye X, Su H, Li W, Liu D, Pirmoradian M, et al. Genetic landscape of hepatitis B virus–associated diffuse large B‐cell lymphoma. Blood. 2018;131:2670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Valera A, Balagué O, Colomo L, Martínez A, Delabie J, Taddesse‐Heath L, et al. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol. 2010;34:1686–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Z, Filip I, Gomez K, Engelbrecht D, Meer S, Lalloo PN, et al. Genomic characterization of HIV‐associated plasmablastic lymphoma identifies pervasive mutations in the JAK–STAT pathway. Blood Cancer Discov. 2020;1:112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramis‐Zaldivar JE, Gonzalez‐Farré B, Balagué O, Celis V, Nadeu F, Salmerón‐Villalobos J, et al. Distinct molecular profile of IRF4‐rearranged large B‐cell lymphoma. Blood. 2020;135:274–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reichel J, Chadburn A, Rubinstein PG, Giulino‐Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed–Sternberg cells. Blood. 2015;125:1061–72. [DOI] [PubMed] [Google Scholar]
- 60. Wienand K, Chapuy B, Stewart C, Dunford AJ, Wu D, Kim J, et al. Genomic analyses of flow‐sorted Hodgkin Reed–Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019;3:4065–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Desch A‐K, Hartung K, Botzen A, Brobeil A, Rummel M, Kurch L, et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia. 2020;34:151–66. [DOI] [PubMed] [Google Scholar]
- 62. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131:2413–25. [DOI] [PubMed] [Google Scholar]
- 63. Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive mutations of JAK‐STAT pathway genes in classical Hodgkin lymphoma. Blood. 2018;131:2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B‐cell lymphoma. Blood. 2016;128:2666–70. [DOI] [PubMed] [Google Scholar]
- 65. Mottok A, Hung SS, Chavez EA, Woolcock B, Telenius A, Chong LC, et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B‐cell lymphoma. Blood. 2019;134:802–13. [DOI] [PubMed] [Google Scholar]
- 66. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD‐1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B‐cell lymphoma. Blood. 2010;116:3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Roemer MGM, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD‐L1 and PD‐L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34:2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rosenquist R, Ghia P, Hadzidimitriou A, Sutton L‐A, Agathangelidis A, Baliakas P, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017;31:1477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tausch E, Schneider C, Robrecht S, Zhang C, Dolnik A, Bloehdorn J, et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood. 2020;135:2402–12. [DOI] [PubMed] [Google Scholar]
- 70. Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher‐order connections between stereotyped subsets: implications for improved patient classification in CLL. Blood. 2021;137:1365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Maity PC, Bilal M, Koning MT, Young M, Van Bergen CAM, Renna V, et al. IGLV3‐21 * 01 is an inherited risk factor for CLL through the acquisition of a single‐point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci. 2020;117:4320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nadeu F, Royo R, Clot G, Duran‐Ferrer M, Navarro A, Martín S, et al. IGLV3‐21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood. 2021;137:2935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, De Paoli L, et al. Stereotyped B‐cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15:4415–22. [DOI] [PubMed] [Google Scholar]
- 74. Rossi D, Spina V, Bomben R, Rasi S, Dal‐Bo M, Bruscaggin A, et al. Association between molecular lesions and specific B‐cell receptor subsets in chronic lymphocytic leukemia. Blood. 2013;121:4902–5. [DOI] [PubMed] [Google Scholar]
- 75. Malcikova J, Tausch E, Rossi D, Sutton LA, Soussi T, Zenz T, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia—update on methodological approaches and results interpretation. Leukemia. 2018;32:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28:4473–9. [DOI] [PubMed] [Google Scholar]
- 77. Campo E, Cymbalista F, Ghia P, Jäger U, Pospisilova S, Rosenquist R, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103:1956–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009;23:117–24. [DOI] [PubMed] [Google Scholar]
- 79. Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123:2139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127:2122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hallek M, Cheson BD, Catovsky D, Caligaris‐Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60. [DOI] [PubMed] [Google Scholar]
- 82. Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine‐refractoriness. Blood. 2011;118:6904–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. [DOI] [PubMed] [Google Scholar]
- 85. Mansouri L, Cahill N, Gunnarsson R, Smedby KE, Tjönnfjord E, Hjalgrim H, et al. NOTCH1 and SF3B1 mutations can be added to the hierarchical prognostic classification in chronic lymphocytic leukemia. Leukemia. 2013;27:512–4. [DOI] [PubMed] [Google Scholar]
- 86. Sportoletti P, Baldoni S, Cavalli L, Del Papa B, Bonifacio E, Ciurnelli R, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B‐CLL. Br J Haematol. 2010;151:404–6. [DOI] [PubMed] [Google Scholar]
- 87. Guarini A, Marinelli M, Tavolaro S, Bellacchio E, Magliozzi M, Chiaretti S, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2012;97:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Young E, Noerenberg D, Mansouri L, Ljungström V, Frick M, Sutton L‐A, et al. EGR2 mutations define a new clinically aggressive subgroup of chronic lymphocytic leukemia. Leukemia. 2017;31:1547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild‐type chronic lymphocytic leukemia. Blood. 2012;119:2854–62. [DOI] [PubMed] [Google Scholar]
- 90. Baliakas P, Hadzidimitriou A, Sutton L‐A, Rossi D, Minga E, Villamor N, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29:329–36. [DOI] [PubMed] [Google Scholar]
- 91. Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Döhner K, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123:3247–54. [DOI] [PubMed] [Google Scholar]
- 92. Tausch E, Beck P, Schlenk RF, Jebaraj BJ, Dolnik A, Yosifov DY, et al. Prognostic and predictive role of gene mutations in chronic lymphocytic leukemia: results from the pivotal phase III study COMPLEMENT1. Haematologica. 2020;105:2440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185–97. [DOI] [PubMed] [Google Scholar]
- 94. Ramsower CA, Maguire A, Robetorye RS, Feldman AL, Syrbu SI, Rosenthal AC, et al. Clinical laboratory validation of the MCL35 assay for molecular risk stratification of mantle cell lymphoma. J Hematop. 2020;13:231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rauert‐Wunderlich H, Mottok A, Scott DW, Rimsza LM, Ott G, Klapper W, et al. Validation of the MCL35 gene expression proliferation assay in randomized trials of the European Mantle Cell Lymphoma Network. Br J Haematol. 2019;184:616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Holte H, Beiske K, Boyle M, Trøen G, Blaker YN, Myklebust J, et al. The MCL35 gene expression proliferation assay predicts high‐risk MCL patients in a Norwegian cohort of younger patients given intensive first line therapy. Br J Haematol. 2018;183:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Scott DW, Abrisqueta P, Wright GW, Slack GW, Mottok A, Villa D, et al. New molecular assay for the proliferation signature in mantle cell lymphoma applicable to formalin‐fixed paraffin‐embedded biopsies. J Clin Oncol. 2017;35:1668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ferrero S, Rossi D, Rinaldi A, Bruscaggin A, Spina V, Eskelund CW, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high‐dose therapy: a FIL study. Haematologica. 2020;105:1604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Clot G, Jares P, Giné E, Navarro A, Royo C, Pinyol M, et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood. 2018;132:413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J Cancer. 2017;141:1381–8. [DOI] [PubMed] [Google Scholar]
- 101. Xu‐Monette ZY, Wu L, Visco C, Tai YuC, Tzankov A, Liu W‐M, et al. Mutational profile and prognostic significance of TP53 in diffuse large B‐cell lymphoma patients treated with R‐CHOP: report from an International DLBCL Rituximab‐CHOP Consortium Program Study. Blood. 2012;120:3986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Young KH, Leroy K, Møller MB, Colleoni GWB, Sánchez‐Beato M, Kerbauy FR, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B‐cell lymphoma: an international collaborative study. Blood. 2008;112:3088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, et al. Mutations in the DNA‐binding codons of TP53, which are associated with decreased expression of TRAILreceptor‐2, predict for poor survival in diffuse large B‐cell lymphoma. Blood. 2007;110:4396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Poulain S, Roumier C, Bertrand E, Renneville A, Caillault‐Venet A, Doye E, et al. TP53 mutation and its prognostic significance in Waldenstrom's macroglobulinemia. Clin Cancer Res. 2017;23:6325–35. [DOI] [PubMed] [Google Scholar]
- 105. Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first‐line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population‐based registry. Lancet Oncol. 2015;16:1111–22. [DOI] [PubMed] [Google Scholar]
- 106. Johnson NA, Savage KJ, Ludkovski O, Ben‐Neriah S, Woods R, Steidl C, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114:2273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben‐Neriah S, Rogic S, et al. Concurrent expression of MYC and BCL2 in diffuse large B‐cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012;30:3452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wiestner A. Targeting B‐cell receptor signaling for anticancer therapy: The Bruton's tyrosine kinase inhibitor ibrutinib induces impressive responses in B‐Cell malignancies. J Clin Oncol. 2013;31:128–30. [DOI] [PubMed] [Google Scholar]
- 109. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Byrd JC, Harrington B, O'brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP‐196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130:1903–10. [DOI] [PubMed] [Google Scholar]
- 113. Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169:795–803. [DOI] [PubMed] [Google Scholar]
- 114. Castillo JJ, Xu L, Gustine JN, Keezer A, Meid K, Dubeau TE, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187:356–63. [DOI] [PubMed] [Google Scholar]
- 115. Cao Y, Hunter ZR, Liu X, Xu L, Yang G, Chen J, et al. The WHIM‐like CXCR4S338X somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom's macroglobulinemia. Leukemia. 2015;29:169–76. [DOI] [PubMed] [Google Scholar]
- 116. Gustine JN, Xu L, Tsakmaklis N, Demos MG, Kofides A, Chen JG, et al. CXCR4 S338X clonality is an important determinant of ibrutinib outcomes in patients with Waldenström macroglobulinemia. Blood Adv. 2019;3:2800–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Jones D, Woyach JA, Zhao W, Caruthers S, Tu H, Coleman J, et al. PLCG2 C2 domain mutations co‐occur with BTK and PLCG2 resistance mutations in chronic lymphocytic leukemia undergoing ibrutinib treatment. Leukemia. 2017;31:1645–7. [DOI] [PubMed] [Google Scholar]
- 118. Woyach JA, Furman RR, Liu Ta‐M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ahn IE, Underbayev C, Albitar A, Herman SEM, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129:1469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Blombery P, Anderson MA, Gong J‐N, Thijssen R, Birkinshaw RW, Thompson ER, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9:342–53. [DOI] [PubMed] [Google Scholar]
- 121. Kohlmann A, Klein H‐U, Weissmann S, Bresolin S, Chaplin T, Cuppens H, et al. The Interlaboratory RObustness of Next‐generation sequencing (IRON) study: a deep sequencing investigation of TET2, CBL and KRAS mutations by an international consortium involving 10 laboratories. Leukemia. 2011;25:1840–8. [DOI] [PubMed] [Google Scholar]
- 122. Sutton L‐A, Ljungstrom V, Mansouri L, Young E, Cortese D, Navrkalova V, et al. Targeted next‐generation sequencing in chronic lymphocytic leukemia: a high‐throughput yet tailored approach will facilitate implementation in a clinical setting. Haematologica. 2015;100:370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sutton L‐A, Ljungström V, Enjuanes A, Cortese D, Skaftason A, Tausch E, et al. European Research Initiative On CLL (ERIC) comparative analysis of targeted next‐generation sequencing panels for the detection of gene mutations in chronic lymphocytic leukemia: an ERIC multi‐center study. Haematologica. 2021;106:682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. He J, Abdel‐Wahab O, Nahas MK, Wang K, Rampal RK, Intlekofer AM, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hung SS, Meissner B, Chavez EA, Ben‐Neriah S, Ennishi D, Jones MR, et al. Assessment of capture and amplicon‐based approaches for the development of a targeted next‐generation sequencing pipeline to personalize lymphoma management. J Mol Diagnostics. 2018;20:203–14. [DOI] [PubMed] [Google Scholar]
- 126. Navrkalova V, Plevova K, Hynst J, Pal K, Mareckova A, Reigl T, et al. LYmphoid NeXt‐generation sequencing (LYNX) panel. J Mol Diagnostics. 2021;23:959–74. [DOI] [PubMed] [Google Scholar]
- 127. Stewart JP, Gazdova J, Darzentas N, Wren D, Proszek P, Fazio G, et al. Validation of the EuroClonality‐NGS DNA capture panel as an integrated genomic tool for lymphoproliferative disorders. Blood Adv. 2021;5:3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Duncavage EJ, Schroeder MC, O'laughlin M, Wilson R, Macmillan S, Bohannon A, et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N Engl J Med. 2021;384:924–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rosenquist R ISRCTN ‐ ISRCTN66987142: Evaluation of genome sequencing as a diagnostic test in acute leukemia. 10.1186/ISRCTN66987142 [DOI]
- 130. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. [DOI] [PubMed] [Google Scholar]
- 131. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. [DOI] [PubMed] [Google Scholar]
- 132. Kulis M, Queirós AC, Beekman R, Martín‐Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta. 2013;1829:1161–74. [DOI] [PubMed] [Google Scholar]
- 133. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Robson MI, Ringel AR, Mundlos S. Regulatory landscaping: how enhancer‐promoter communication is sculpted in 3D. Mol Cell. 2019;74:1110–22. [DOI] [PubMed] [Google Scholar]
- 135. Schoenfelder S, Fraser P. Long‐range enhancer–promoter contacts in gene expression control. Nat Rev Genet. 2019;20:437–55. [DOI] [PubMed] [Google Scholar]
- 136. Kim S, Shendure J. Mechanisms of interplay between transcription factors and the 3D genome. Mol Cell. 2019;76:306–19. [DOI] [PubMed] [Google Scholar]
- 137. Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kulis M, Merkel A, Heath S, Queirós AC, Schuyler RP, Castellano G, et al. Whole‐genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. 2015;47:746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48:253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, et al. Proliferation‐dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–25. [DOI] [PubMed] [Google Scholar]
- 141. Aran D, Toperoff G, Rosenberg M, Hellman A. Replication timing‐related and gene body‐specific methylation of active human genes. Hum Mol Genet. 2011;20:670–80. [DOI] [PubMed] [Google Scholar]
- 142. Zhou W, Dinh HQ, Ramjan Z, Weisenberger DJ, Nicolet CM, Shen H, et al. DNA methylation loss in late‐replicating domains is linked to mitotic cell division. Nat Genet. 2018;50:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kulis M, Heath S, Bibikova M, Queirós AC, Navarro A, Clot G, et al. Epigenomic analysis detects widespread gene‐body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. [DOI] [PubMed] [Google Scholar]
- 145. Giacopelli B, Zhao Q, Ruppert AS, Agyeman A, Weigel C, Wu Y‐Z, et al. Developmental subtypes assessed by DNA methylation‐iPLEX forecast the natural history of chronic lymphocytic leukemia. Blood. 2019;134:688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bhoi S, Ljungström V, Baliakas P, Mattsson M, Smedby KE, Juliusson G, et al. Prognostic impact of epigenetic classification in chronic lymphocytic leukemia: the case of subset #2. Epigenetics. 2016;11:449–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wojdacz TK, Amarasinghe HE, Kadalayil L, Beattie A, Forster J, Blakemore SJ, et al. Clinical significance of DNA methylation in chronic lymphocytic leukemia patients: results from 3 UK clinical trials. Blood Adv. 2019;3:2474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Queirós AC, Villamor N, Clot G, Martinez‐Trillos A, Kulis M, Navarro A, et al. A B‐cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2015;29:598–605. [DOI] [PubMed] [Google Scholar]
- 149. Tsagiopoulou M, Chapaprieta V, Duran‐Ferrer M, Moysiadis T, Psomopoulos F, Kollia P, et al. Chronic lymphocytic leukemias with trisomy 12 show a distinct DNA methylation profile linked to altered chromatin activation. Haematologica. 2020;105:2864–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Queirós AC, Beekman R, Vilarrasa‐Blasi R, Duran‐Ferrer M, Clot G, Merkel A, et al. Decoding the DNA methylome of mantle cell lymphoma in the light of the entire B cell lineage. Cancer Cell. 2016;30:806–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72:5307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Agirre X, Castellano G, Pascual M, Heath S, Kulis M, Segura V, et al. Whole‐epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell‐specific enhancers. Genome Res. 2015;25:478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kretzmer H, Bernhart SH, Wang W, Haake A, Weniger MA, Bergmann AK, et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat Genet. 2015;47:1316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Nakamura T, Yamashita S, Fukumura K, Nakabayashi J, Tanaka K, Tamura K, et al. Genome‐wide DNA methylation profiling identifies primary central nervous system lymphoma as a distinct entity different from systemic diffuse large B‐cell lymphoma. Acta Neuropathol. 2017;133:321–4. [DOI] [PubMed] [Google Scholar]
- 155. Roos‐Weil D, Giacopelli B, Armand M, Della‐Valle V, Ghamlouch H, Decaudin C, et al. Identification of 2 DNA methylation subtypes of Waldenström macroglobulinemia with plasma and memory B‐cell features. Blood. 2020;136:585–95. [DOI] [PubMed] [Google Scholar]
- 156. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ott CJ, Federation AJ, Schwartz LS, Kasar S, Klitgaard JL, Lenci R, et al. Enhancer architecture and essential core regulatory circuitry of chronic lymphocytic leukemia. Cancer Cell. 2018;34:982–95.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Pastore A, Gaiti F, Lu SX, Brand RM, Kulm S, Chaligne R, et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat Commun. 2019;10:1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Mallm J‐P, Iskar M, Ishaque N, Klett LC, Kugler SJ, Muino JM, et al. Linking aberrant chromatin features in chronic lymphocytic leukemia to transcription factor networks. Mol Syst Biol. 2019;15:e8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, et al. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype‐specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vilarrasa‐Blasi R, Verdaguer‐Dot N, Belver L, Soler‐Vila P, Beekman R, Chapaprieta V, et al. Insights into the mechanisms underlying aberrant SOX11 oncogene expression in mantle cell lymphoma. Leukemia. 2021. 10.1038/s41375-021-01389-w [DOI] [PubMed] [Google Scholar]
- 163. Vilarrasa‐Blasi R, Soler‐Vila P, Verdaguer‐Dot N, Russiñol N, Di Stefano M, Chapaprieta V, et al. Dynamics of genome architecture and chromatin function during human B cell differentiation and neoplastic transformation. Nat Commun. 2021;12:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ordoñez R, Kulis M, Russiñol N, Chapaprieta V, Carrasco‐Leon A, García‐Torre B, et al. Chromatin activation as a unifying principle underlying pathogenic mechanisms in multiple myeloma. Genome Res. 2020;30:1217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Nie M, Du L, Ren W, Joung J, Ye X, Shi Xi, et al. Genome‐wide CRISPR screens reveal synthetic lethal interaction between CREBBP and EP300 in diffuse large B‐cell lymphoma. Cell Death Dis. 2021;12:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Mcgranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28. [DOI] [PubMed] [Google Scholar]
- 167. Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, et al. A pan‐cancer blueprint of the heterogeneous tumor microenvironment revealed by single‐cell profiling. Cell Res. 2020;30:745–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. González‐Silva L, Quevedo L, Varela I. Tumor functional heterogeneity unraveled by scRNA‐seq technologies. Trends in Cancer. 2020;6:13–9. [DOI] [PubMed] [Google Scholar]
- 169. Lee J, Hyeon DY, Hwang D. Single‐cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52:1428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. [DOI] [PubMed] [Google Scholar]
- 171. Rozenblatt‐Rosen O, Stubbington MJT, Regev A, Teichmann SA. The Human Cell Atlas: from vision to reality. Nature. 2017;550:451–3. [DOI] [PubMed] [Google Scholar]
- 172. Park J‐E, Botting RA, Domínguez Conde C, Popescu D‐M, Lavaert M, Kunz DJ, et al. A cell atlas of human thymic development defines T cell repertoire formation. Science. 2020;367:eaay3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Popescu D‐M, Botting RA, Stephenson E, Green K, Webb S, Jardine L, et al. Decoding human fetal liver haematopoiesis. Nature. 2019;574:365–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Aizarani N, Saviano A, Sagar, Mailly L, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Macparland SA, Liu JC, Ma X‐Z, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Litviňuková M, Talavera‐López C, Maatz H, Reichart D, Worth CL, Lindberg EL, et al. Cells of the adult human heart. Nature. 2020;588:466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019;25:1153–63. [DOI] [PubMed] [Google Scholar]
- 178. King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, et al. Single‐cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol. 2021;6:eabe6291. [DOI] [PubMed] [Google Scholar]
- 179. Viant C, Wirthmiller T, Eltanbouly MA, Chen ST, Cipolla M, Ramos V, et al. Germinal center–dependent and –independent memory B cells produced throughout the immune response. J Exp Med. 2021;218:e20202489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single‐cell analysis of germinal‐center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020;217:e20200483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Glass DR, Tsai AG, Oliveria JP, Hartmann FJ, Kimmey SC, Calderon AA, et al. An integrated multi‐omic single‐cell atlas of human B cell identity. Immunity. 2020;53:217–32.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Stewart A, Ng JC‐F, Wallis G, Tsioligka V, Fraternali F, Dunn‐Walters DK. Single‐cell transcriptomic analyses define distinct peripheral B cell subsets and discrete development pathways. Front Immunol. 2021;12:602539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Wang L, Fan J, Francis JM, Georghiou G, Hergert S, Li S, et al. Integrated single‐cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. 2017;27:1300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Rendeiro AF, Krausgruber T, Fortelny N, Zhao F, Penz T, Farlik M, et al. Chromatin mapping and single‐cell immune profiling define the temporal dynamics of ibrutinib response in CLL. Nat Commun. 2020;11:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Vickovic S, Ståhl PL, Salmén F, Giatrellis S, Westholm JO, Mollbrink A, et al. Massive and parallel expression profiling using microarrayed single‐cell sequencing. Nat Commun. 2016;7:13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell. 2016;30:750–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Gaiti F, Chaligne R, Gu H, Brand RM, Kothen‐Hill S, Schulman RC, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature. 2019;569:576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Penter L, Gohil SH, Lareau C, Ludwig LS, Parry EM, Huang T, et al. Longitudinal single‐cell dynamics of chromatin accessibility and mitochondrial mutations in chronic lymphocytic leukemia mirror disease history. Cancer Discov. 2021. 10.1158/2159-8290.CD-21-0276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zhao Z, Goldin L, Liu S, Wu L, Zhou W, Lou H, et al. Evolution of multiple cell clones over a 29‐year period of a CLL patient. Nat Commun. 2016;7:13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Milpied P, Cervera‐Marzal I, Mollichella M‐L, Tesson B, Brisou G, Traverse‐Glehen A, et al. Human germinal center transcriptional programs are de‐synchronized in B cell lymphoma. Nat Immunol. 2018;19:1013–24. [DOI] [PubMed] [Google Scholar]
- 191. Andor N, Simonds EF, Czerwinski DK, Chen J, Grimes SM, Wood‐Bouwens C, et al. Single‐cell RNA‐Seq of follicular lymphoma reveals malignant B‐cell types and coexpression of T‐cell immune checkpoints. Blood. 2019;133:1119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Haebe S, Shree T, Sathe A, Day G, Czerwinski DK, Grimes SM, et al. Single‐cell analysis can define distinct evolution of tumor sites in follicular lymphoma. Blood. 2021;137:2869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Roider T, Seufert J, Uvarovskii A, Frauhammer F, Bordas M, Abedpour N, et al. Dissecting intratumour heterogeneity of nodal B‐cell lymphomas at the transcriptional, genetic and drug‐response levels. Nat Cell Biol. 2020;22:896–906. [DOI] [PubMed] [Google Scholar]
- 194. Aoki T, Chong LC, Takata K, Milne K, Hav M, Colombo A, et al. Single‐cell transcriptome analysis reveals disease‐defining T‐cell subsets in the tumor microenvironment of classic Hodgkin lymphoma. Cancer Discov. 2020;10:406–21. [DOI] [PubMed] [Google Scholar]
- 195. Ledergor G, Weiner A, Zada M, Wang S‐Y, Cohen YC, Gatt ME, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. 2018;24:1867–76. [DOI] [PubMed] [Google Scholar]
- 196. Jang JS, Li Y, Mitra AK, Bi L, Abyzov A, Van Wijnen AJ, et al. Molecular signatures of multiple myeloma progression through single cell RNA‐Seq. Blood Cancer J. 2019;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zavidij O, Haradhvala NJ, Mouhieddine TH, Sklavenitis‐Pistofidis R, Cai S, Reidy M, et al. Single‐cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer. 2020;1:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Cohen YC, Zada M, Wang S‐Y, Bornstein C, David E, Moshe A, et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single‐cell sequencing. Nat Med. 2021;27:491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Wang L, Mo S, Li X, He Y, Yang J. Single‐cell RNA‐seq reveals the immune escape and drug resistance mechanisms of mantle cell lymphoma. Cancer Biol Med. 2020;17:726–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Zhang S, Jiang VC, Han G, Hao D, Lian J, Liu Y, et al. Longitudinal single‐cell profiling reveals molecular heterogeneity and tumor‐immune evolution in refractory mantle cell lymphoma. Nat Commun. 2021;12:2877. [DOI] [PMC free article] [PubMed] [Google Scholar]