

Abstract
INTRODUCTION
We aimed to unravel the underlying pathophysiology of the neurodegeneration (N) markers neurogranin (Ng), neurofilament light (NfL), and hippocampal volume (HCV), in Alzheimer's disease (AD) using cerebrospinal fluid (CSF) proteomics.
METHODS
Individuals without dementia were classified as A+ (CSF amyloid beta [Aβ]42), T+ (CSF phosphorylated tau181), and N+ or N− based on Ng, NfL, or HCV separately. CSF proteomics were generated and compared between groups using analysis of covariance.
RESULTS
Only a few individuals were A+T+Ng−. A+T+Ng+ and A+T+NfL+ showed different proteomic profiles compared to A+T+Ng− and A+T+NfL−, respectively. Both Ng+ and NfL+ were associated with neuroplasticity, though in opposite directions. Compared to A+T+HCV−, A+T+HCV+ showed few proteomic changes, associated with oxidative stress.
DISCUSSION
Different N markers are associated with distinct neurodegenerative processes and should not be equated. N markers may differentially complement disease staging beyond amyloid and tau. Our findings suggest that Ng may not be an optimal N marker, given its low incongruency with tau pathophysiology.
Highlights
In Alzheimer's disease, neurogranin (Ng)+, neurofilament light (NfL)+, and hippocampal volume (HCV)+ showed differential protein expression in cerebrospinal fluid.
Ng+ and NfL+ were associated with neuroplasticity, although in opposite directions.
HCV+ showed few proteomic changes, related to oxidative stress.
Neurodegeneration (N) markers may differentially refine disease staging beyond amyloid and tau.
Ng might not be an optimal N marker, as it relates more closely to tau.
Keywords: Alzheimer's disease, biomarkers, cerebrospinal fluid, hippocampal volume, neurodegeneration markers, neurofilament light, neurogranin, pathophysiology, proteomics
1. BACKGROUND
The biological ATN staging scheme for Alzheimer's disease (AD) categorizes individuals based on amyloid (A), tau (T), and neurodegeneration (N) biomarkers in cerebrospinal fluid (CSF) or on imaging. 1 Proposed N biomarkers include, among others, neurogranin (Ng), neurofilament light (NfL), and hippocampal volume (HCV). 1 , 2 To date, the pathophysiological processes underlying these candidate N biomarkers within the ATN staging scheme in AD are not fully understood.
The initially proposed N markers were CSF total tau (t‐tau), glucose metabolism on fluorodeoxyglucose positron emission tomography, and atrophy on structural magnetic resonance imaging (MRI) in AD‐related brain regions, including the hippocampus. 1 More recently, other N markers in CSF have been proposed, including Ng and NfL proteins. 2 In the present study, we focus on the candidate N markers CSF Ng, CSF NfL, and HCV. Ng is a neuron‐specific postsynaptic protein, which plays a role in synaptic plasticity. Increased CSF Ng levels have been reported specifically in AD compared to other neurodegenerative diseases, and are presumably associated with synaptic dysregulation. 2 , 3 , 4 , 5 NfL is an intermediate filament, which plays a role in the assembly and maintenance of the neuronal cytoskeleton. High CSF NfL levels are associated with large‐caliber axonal degeneration in AD, but also in other neurodegenerative diseases. 2 , 6 , 7 The hippocampus is part of the limbic system and plays an important role in memory and learning. It is among the earliest regions showing atrophy in AD. Reduced HCV is an important early marker of brain atrophy in AD, as well as in other neurodegenerative diseases. 8 , 9 To date, it remains unclear to what extent different N markers reflect similar or distinct neurodegenerative processes in AD. Previous studies showed that CSF Ng reflects amyloid beta (Aβ)‐dependent neurodegeneration, while CSF NfL is associated with neurodegeneration independently of Aβ pathology. 10 CSF and neuroimaging biomarkers of neurodegeneration often show low correlation, which suggests that these markers may reflect different neurodegenerative aspects. 11 , 12
To our knowledge, no study has yet compared the proteomic signature underlying distinct N markers, that is, Ng, NfL, and HCV. Therefore, the aim of this study is to use large‐scale proteomics to assess commonalities and differences across Ng, Nfl, and HCV, and to understand whether these N markers reflect similar or distinct underlying pathophysiological processes in AD. CSF protein level alterations reflect ongoing biochemical and metabolic changes in the brain and studying a large number of proteins can provide a robust characterization of the underlying pathophysiological mechanisms in AD. 13 , 14 In the present study, we included individuals without dementia and with abnormal amyloid and tau markers (A+T+) and compared CSF proteomic profiles of those with an abnormal N marker to those with a normal N marker. Secondary analyses were conducted in A+T− individuals without dementia using the same methodology. More knowledge about the underlying biological processes of the different N markers in AD will be important for improving AD staging, which may be relevant for the design of clinical trials. 15 , 16 , 17 , 18
2. METHODS
2.1. Participants
Four hundred seven participants were enrolled from the Maastricht BioBank Alzheimer Center Limburg cohort (BB‐ACL, n = 52) memory clinic study, 19 the Washington University (WashU) Knight Alzheimer Disease Research Center (ADRC, n = 90), study 20 and the European Medical Information Framework for Alzheimer's Disease Multimodal Biomarker Discovery study (EMIF‐AD MBD, n = 265). 21 All patients provided informed consent for research. All centers approved participation in this study after local medical ethics committee approval. Participants were included in the current project if they had normal cognition/subjective cognitive decline (NC) or mild cognitive impairment (MCI), baseline CSF samples available, and baseline data of CSF Aβ42 and CSF phosphorylated tau (p‐tau)181 measures, and at least one of the following baseline measures: CSF NfL, CSF Ng, or HCV.
2.2. Neuropsychological assessment
All participants were administered a neuropsychological assessment, including the Mini‐Mental State Examination (MMSE) and tests assessing several cognitive domains, including memory. Memory tests differed between centers but most common tests were the Rey Auditory Verbal Learning Test (BB‐ACL and EMIF‐AD MBD studies) and the Free and Cued Selective Reminding Test (WashU Knight ADRC study). Detailed information about the neuropsychological tests and calculation of Z scores can be found elsewhere. 19 , 21 , 22 NC was defined as neuropsychological test performance ranged within 1.5 standard deviation (SD) of the average corrected for age, sex, and years of education. MCI was defined according to the criteria of Petersen, and based on <1.5 SD in at least one of the neuropsychological tests assessing several cognitive domains. 21 , 23
2.3. CSF protein analysis
CSF was obtained by lumbar puncture, centrifuged, and stored at −80°C in polypropylene tubes. CSF samples were shipped on dry ice to the neurochemistry lab of the University of Gothenburg in Mölndal, Sweden, where central proteomic and peptidomic analyses were performed using an untargeted tandem mass tag (TMT) technique with 10+1 plexing, using high‐pH reverse phase high‐performance liquid chromatography for peptide prefractionation 14 , 24 , 25 to quantify ≈ 500 proteins as well as endogenous peptides in the same CSF sample aliquot. More information is described elsewhere. 14 , 24 , 25 In total, 3102 proteins were quantified using TMT spectrometry. We selected proteins that had at least one third of observations per participant group. For related proteins that had identical values due to fragment non‐specificity, we randomly selected one protein for analysis. 14 All analyses were performed according to the manufacturer's instructions and using two batches (batch 1 n = 285, batch 2 n = 122) of reagents by board‐certified laboratory technicians who were blinded to clinical information.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed. In the ATN framework of Alzheimer's disease (AD), various neurodegeneration (N) markers have been suggested, such as neurogranin (Ng), neurofilament light (NfL), and hippocampal volume (HCV). It is unclear whether these N markers reflect similar or distinct underlying mechanisms.
Interpretation: Ng, NfL, and HCV were associated with distinct cerebrospinal fluid proteomic profiles, reflecting different neurodegenerative processes in AD. While Ng and NfL were both associated with neuroplasticity, they acted in opposite directions. Only a few individuals were A+T+Ng−, indicating that Ng may not be an optimal N marker. Different N markers should not be equated and may differentially complement disease staging beyond amyloid and tau.
Future directions: Future studies should characterize further the dysregulated processes associated with each neurodegeneration biomarker, as well as the associated clinical outcomes, to improve AD diagnosis and prognosis, which may be relevant for the design of clinical trials.
Targeted analyses were performed for well‐established CSF markers, that is, Aβ40, Aβ42, Aβ42/40 ratio, p‐tau181, t‐tau, NfL, and Ng. For the BB‐ACL and the EMIF‐AD MBD cohorts, the neurochemistry lab of University of Gothenburg in Sweden analyzed centrally the levels of Aβ40, Aβ42 (using V‐PLEX Plus Aβ Peptide Panel 1 [6E10] Kit from Meso Scale Discovery [MSD]), NfL (using NF‐light enzyme‐linked immunosorbent assay [ELISA], UmanDiagnostics), and Ng (using an in‐house immunoassay). Moreover, Aβ42, t‐tau and p‐tau levels were measured locally with INNOTEST ELISAs (Fujirebio) and for a subset with Alzbio3 xMAP Luminex (n = 29). For the WashU Knight ADRC cohort, levels of Aβ40, Aβ42, t‐tau, and p‐tau were measured by chemiluminescent enzyme immunoassay with a LUMIPULSE G1200 (Fujirebio), NfL was measured with the NF‐light ELISA (UmanDiagnostics), and Ng was measured by quantitative fluorescent two‐site immunoassays using single‐molecule counting (SMC) technology on the Singulex Erenna platform. 26 , 27
2.4. Genetic analysis
Protocols for apolipoprotein E (APOE) genotyping are described elsewhere. 21 , 28 , 29 In brief, APOE genotype was assessed using polymerase chain reaction (PCR) techniques for two single nucleotide polymorphisms (SNPs; rs429358 for the “ε4 allele” and rs7412 the “ε2 allele”). Participants were classified as APOE ε4 carriers or non‐carriers, determined by the presence of at least one APOE ε4 allele.
In a subset of participants of the EMIF‐AD MBD study (n = 234), polygenic risk score (PGRS) analyses were performed on imputed genome‐wide SNP genotyping data generated with the Global Screening Array (Illumina, Inc.) using PRSice (v2.3). 30 PGRSs were calculated by adding the sum of each allele weighted by the strength of its association with AD risk as calculated previously by a genome‐wide association study (GWAS) on AD. 31 Prior to calculating PGRS, clumping was performed to remove SNPs that are in linkage disequilibrium (r 2 > 0.1) within a slicing 1 M bp window. After clumping, PGRS were computed using various SNP inclusion thresholds. 14 PGRS results were validated in the WashU Knight ADRC cohort (n = 91). 32 , 33
2.5. Image analysis
At each site, MRI scans were acquired using local protocols. For all studies, a thorough quality check was performed. Images were segmented using FreeSurfer (version 5.3.0 for EMIF‐AD MB and BB‐ACL studies and version 5.0 for the WashU Knight ADRC study, https://surfer.nmr.mgh.harvard.edu). 34 Subcortical volumes (including HCV) were normalized by total intracranial volume (TIV). 35 , 36
2.6. Participant classification
Persons were classified using the ATN scheme. Local CSF Aβ42 was used as a measure of amyloid (A) and local CSF p‐tau as a measure of tau (T). We used cohort‐specific cut‐offs to define abnormal biomarker levels. The Aβ42 cut‐offs were redefined for each cohort using unbiased Gaussian mixture modeling, as different methodologies had been used by the centers to define Aβ42 cut‐offs (Table S1 in supporting information). CSF NfL, CSF Ng, or HCV were used as a measure of neurodegeneration (N). As those parameters were measured centrally for EMIF‐AD MBD and BB‐ACL studies, common cut‐offs were calculated for both studies and separately for the WashU Knight ADRC cohort, using unbiased Gaussian mixture modeling. 37 , 38 If the Gaussian mixture modeling showed only one distribution, the Youden index was used. 39 The cut‐offs for N markers are presented in Figure S1 in supporting information. For primary analysis, we included individuals with A+T+N+ and A+T+N−. For secondary analysis, we included individuals with A+T−N+ and A+T−N−. Individuals with NC A−T− were included as control group.
2.7. Pathway enrichment analysis
Gene Ontology (GO) enrichment analysis was performed using Protein ANalysis THrough Evolutionary Relationships (PANTHER, version 15.0) 40 to identify the biological processes, cellular components, and molecular functions related to the increased or decreased proteins of each group comparison. This tool used the Fisher exact test with false discovery rate (FDR; using the Benjamini–Hochberg procedure 41 ) and we only reported pathways with an FDR–corrected P value < 0.05. 42 , 43 Associated GO terms were clustered in broader categories to reduce redundancy and facilitate interpretation. We validated these pathways and categories using the online database STRING version 11.0 44 and ClueGO, a Cytoscape plug‐in. 45 We further annotated proteins as indicative of increased blood–brain barrier (BBB) permeability from Neumeier et al., 46 Dayon et al., 47 and Rapoport and Pettigrew 48 or as highly expressed by the choroid plexus (ChP) of the lateral ventricles according to the Allen Brain Map 49 through Harmonizome 50 and ABAEnrichment analysis. 51
2.8. Statistical analysis
To investigate the associations among the different ATN biomarkers, we performed Spearman rank correlation tests. For parametric analyses, we transformed the biomarker values to Z scores as absolute values for biomarkers varied across assays. To characterize the N groups, clinical, CSF, and imaging measures were compared between groups using analyses of covariance (ANCOVA) corrected for age and sex for continuous variables and chi‐square for categorical variables. CSF protein levels were normalized according to the mean and SD of the control group. PGRS were compared between groups using linear models.
Individuals with NC and MCI were combined for analyses. For the main analysis, we compared proteomic profiles of individuals without dementia and with A+T+N+ to those with A+T+N−. In secondary analysis, we compared proteomic profiles of individuals without dementia and with A+T−N+ to those with A+T−N−. Groups were also compared to controls (NC A−T−). In post hoc analyses, proteomic profiles were studied separately in individuals with NC and MCI. In post hoc analyses, our main analyses were corrected for potential batch effects, and as there is an age‐dependent increase of CSF NfL, we also determined post hoc an age‐adjusted cut‐off of ≥ 2 SD, using previously reported age‐adjusted Z score formulas. 52 For EMID‐AD MBD and Maastricht BB‐ACL, the formula for age‐adjusted Z scores was: Z score = (log2[NfL value] − [5.957 + (age × 0.053)])/396.549. For WashU Knight ADRC, the formula for age‐adjusted Z scores was: Z score = (log2[NfL value] – [6.911 + (age × 0.038)])/507.565.
Statistical analyses were performed using R 4.1.3. and IBM SPSS Statistics version 26. Two‐sided statistical significance was used and set at P < 0.05.
3. RESULTS
3.1. CSF proteomic profiles for neurodegeneration biomarkers
We compared CSF protein levels in persons with AD (A+T+) with an abnormal N marker to those with a normal N marker. Comparisons to controls are presented in Tables S2–S4 in supporting information. A similar secondary analysis in A+T− individuals is presented in Supplementary Results and in Tables S2–S5 in supporting information.
3.1.1. CSF neurogranin as a neurodegeneration marker
Using Ng as N marker, 12 participants were classified as A+T+N− and 143 as A+T+N+ (Figure 1A). A+T+N+ individuals showed higher levels of Aβ40, Aβ42, p‐tau, and t‐tau compared to A+T+N−. No significant differences in the Aβ42/40 ratio were found between A+T+N+ and A+T+N−. There were no A+T+N− individuals with NC (Table 1). No significant AD PGRS differences were found between N− and N+ (Figure S2A in supporting information).
FIGURE 1.
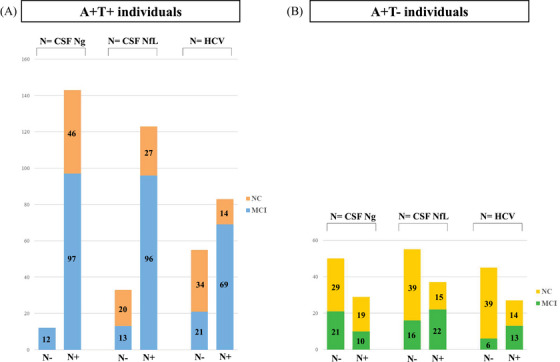
A, Stacked bar graph showing the numbers of N+ and N− participants within the A+T+ group for the three neurodegeneration markers (i.e., CSF neurogranin, CSF neurofilament light, and hippocampal volume). The number of individuals with normal cognition (NC; orange) and mild cognitive impairment (MCI; blue) is also shown. B, Stacked bar graph showing the numbers of N+ and N− participants within the A+T− group for the three neurodegeneration markers (i.e., CSF neurogranin, CSF neurofilament light, and hippocampal volume). The number of individuals with NC (yellow) and MCI (green) is also shown. A−, absence of amyloid pathology determined by CSF Aβ42 level above cut point; A+, presence of amyloid pathology determined by CSF Aβ42 level below cut point; T−, normal CSF p‐tau level below cut point; T+, abnormal CSF p‐tau level above cut point; N−, absence of neurodegeneration or neuronal injury determined by CSF neurogranin, CSF neurofilament light, or hippocampal volume; N+, presence of neurodegeneration or neuronal injury determined by CSF neurogranin, CSF neurofilament light, or hippocampal volume. Aβ, amyloid beta; CSF, cerebrospinal fluid; HCV, hippocampal volume; MCI, mild cognitive impairment; NC, normal cognition; NfL, neurofilament light; Ng, neurogranin; p‐tau, phosphorylated tau.
TABLE 1.
Sample characteristics of A+T+ individuals classified using different neurodegeneration (N) biomarker.
| A. Neurodegeneration biomarker (N) = neurogranin (Ng) | ||||
|---|---|---|---|---|
| Controls n = 145 | A+T+N− n = 12 | A+T+N+ n = 143 | P value N+ vs. N− | |
| Age, years | 64.6 (8.6) | 71.6 (5.9) | 71.1 (6.8) | 0.798 |
| Female (%) | 83 (57.2) | 7 (59.3) | 80 (55.9) | 0.873 |
| Education, years | 13.2 (3.6) | 10.9 (3.1) | 12.4 (3.9) | 0.226 |
| APOE ε4 carriers (%) | 28 (24.1) | 6 (54.5) | 94 (67.1) | 0.395 |
| MCI diagnosis | / | 12 (100.0) | 97 (67.8) | 0.019 |
| MMSE | 28.9 (1.2) | 26.3 (3.1) | 26.6 (2.8) | 0.749 |
| Memory, Z score | 0.2 (0.9) | −1.0 (1.5) | −1.9 (1.4) | 0.798 |
| CSF Aβ40, Z score | 0.3 (1.2) | −1.0 (1.1) | 0.1 (1.8) | 0.025 |
| CSF Aβ42, Z score | 0.5 (1.3) | −1.6 (0.4) | −0.9 (1.2) | 0.031 |
| CSF Aβ42/40 ratio, Z score | 0.9 (1.0) | −1.7 (0.6) | −1.7 (0.8) | 0.808 |
| CSF p‐tau, Z score | −0.4 (0.5) | 0.99 (0.43) | 1.8 (1.3) | 0.049 |
| CSF t‐tau, Z score | −0.3 (0.5) | 0.8 (0.7) | 1.6 (1.3) | 0.050 |
| CSF Ng, Z score | −0.3 (0.7) | −0.9 (0.3) | 0.6 (1.1) | <0.001 |
| CSF NfL, Z score | 0.0 (1.5) | 0.6 (1.8) | 1.3 (4.0) | 0.530 |
| HCV, Z score | 0.1 (1.3) | −0.7 (1.5) | −1.7 (0.7) | 0.151 |
| B. Neurodegeneration biomarker (N) = neurofilament light (NfL) | ||||
|---|---|---|---|---|
| Controls n = 145 | A+T+N− n = 33 | A+T+N+ n = 123 | p‐value N+ vs. N− | |
| Age, years | 68.1 (6.9) | 72.0 (6.5) | 0.003 | |
| Female (%) | 26 (78.8) | 61 (49.6) | 0.003 | |
| Education, years | 13.2 (4.0) | 12.1 (3.8) | 0.136 | |
| APOE ε4 carriers (%) | 19 (59.4) | 83 (69.2) | 0.295 | |
| MCI diagnosis | 13 (39.4) | 96 (78.0) | <0.001 | |
| MMSE | 27.1 (3.1) | 26.4 (2.8) | 0.212 | |
| Memory, Z score | −0.6 (1.4) | −1.0 (1.4) | 0.488 | |
| CSF Aβ40, Z score | −0.7 (0.8) | 0.2 (1.8) | <0.001 | |
| CSF Aβ42, Z score | −0.7 (1.0) | −1.0 (1.2) | 0.373 | |
| CSF Aβ42/40 ratio, Z score | −1.5 (0.7) | −1.8 (0.8) | 0.007 | |
| CSF p‐tau, Z score | 1.3 (0.9) | 1.8 (1.4) | 0.008 | |
| CSF t‐tau, Z score | 1.0 (0.9) | 1.7 (1.3) | 0.001 | |
| CSF Ng, Z score | 0.2 (0.8) | 0.6 (1.2) | 0.032 | |
| CSF NfL, Z score | −0.4 (0.7) | 1.7 (4.3) | 0.013 | |
| HCV, Z score | −1.0 (1.6) | −1.8 (1.7) | 0.018 | |
| C. Neurodegeneration biomarker (N) = hippocampal volume (HCV) | ||||
|---|---|---|---|---|
| Controls n = 145 | A+T+N− n = 55 | A+T+N+ n = 83 | p‐value N+ vs. N− | |
| Age, years | 68.8 (6.6) | 72.0 (5.9) | 0.004 | |
| Female (%) | 26 (47.3) | 49 (59.0) | 0.174 | |
| Education, years | 13.6 (3.8) | 12.0 (3.5) | 0.018 | |
| APOE ε4 carriers (%) | 32 (59.3) | 60 (74.1) | 0.070 | |
| MCI diagnosis | 21 (38.2) | 69 (83.1) | <0.001 | |
| MMSE | 27.8 (2.2) | 26.2 (2.9) | 0.001 | |
| Memory, Z score | −0.1 (1.2) | −1.4 (1.4) | <0.001 | |
| CSF Aβ40, Z score | 0.0 (1.6) | 0.3 (1.8) | 0.161 | |
| CSF Aβ42, Z score | −0.5 (1.3) | −1.0 (1.0) | 0.026 | |
| CSF Aβ42/40 ratio, Z score | −1.6 (0.9) | −1.8 (0.7) | 0.029 | |
| CSF p‐tau, Z score | 1.3 (0.9) | 2.0 (1.5) | 0.001 | |
| CSF t‐tau, Z score | 1.3 (1.0) | 1.9 (1.5) | 0.008 | |
| CSF Ng, Z score | 0.5 (1.0) | 0.6 (1.2) | 0.465 | |
| CSF NfL, Z score | 1.3 (5.4) | 1.1 (1.9) | 0.656 | |
| HCV, Z score | 0.1 (0.9) | −2.6 (1.2) | <0.001 | |
Notes: Controls were individuals with normal cognition and normal levels of Aβ42 and p‐tau. A+T+N− individuals were non‐demented (NC+MCI) individuals with abnormal levels of Aβ42 (A) and p‐tau (T) and normal levels of neurodegeneration marker (N, either Ng, NfL, or HCV). A+T+N+ individuals were non‐demented (NC+MCI) individuals with abnormal levels of Aβ42 (A), p‐tau (T), and neurodegeneration marker (N, either Ng, NfL, or HCV). Values represent mean (standard deviation) or number (percentages). Significant p‐values (< 0.05) are bold. The sample size was smaller for some variables: A total of 13 values are missing for education, 36 for APOE ε4, and 54 for memory. Twenty‐three values are missing for Aβ40, Aβ42, and HCV; 37 for NfL; 45 for Ng; and 74 for HCV. Aβ40, Aβ42, t‐tau, p‐tau, Ng, and NfL values are presented as Z scores with controls as a reference.
Abbreviations: APOE, apolipoprotein E; Aβ, amyloid beta; CSF, cerebrospinal fluid; HCV, hippocampal volume; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; NC, normal cognition; NfL, neurofilament light; Ng, neurogranin; p‐tau, phosphorylated tau; t‐tau, total tau.
CSF proteomic profiling showed that 177 proteins were increased and 14 were decreased in A+T+N+ compared to A+T+N− (Figure 2A, Table S2A). Increased proteins were associated with nervous system development, cell adhesion and migration, protein secretion and modification, and blood vessel development. No pathways were related to the decreased proteins (Figure 2B,C, Table S2B). Twenty‐nine percent (four proteins) of those decreased proteins were associated with increased BBB permeability.
FIGURE 2.
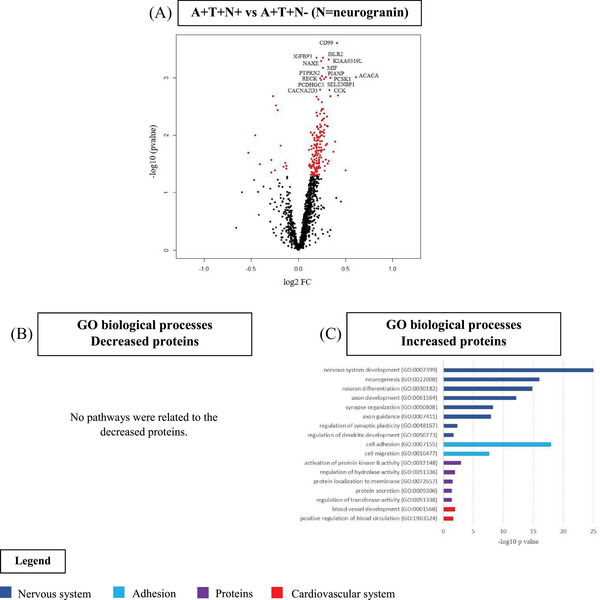
Cerebrospinal fluid (CSF) proteomics in A+T+ individuals without dementia by neurogranin status. A, Volcano plot displaying the log2 fold‐change against the −log10 statistical p‐value for the comparison of A+T+N+ versus A+T+N− (N = neurogranin). Significantly different proteins are red. The top 15 proteins are named. B, C, Selected biological processes Gene Ontology (GO) terms for decreased (B) and increased (C) proteins in the comparison of A+T+N+ versus A+T+N−.
In A+T− individuals, similar proteomic results were found when comparing N+ to N− (Figure 1B, Supplementary Results, Figure S3 in supporting information, Table S2C,D).
3.1.2. CSF neurofilament light as a neurodegeneration marker
Using NfL as N marker, 33 participants were classified as A+T+N− and 123 as A+T+N+ (Figure 1A). A+T+N+ individuals , were older, more often males, and more often diagnosed with MCI than A+T+N−. A+T+N+ individuals had higher levels of Aβ40, p‐tau, t‐tau, and Ng; a lower Aβ42/40 ratio; and smaller HCV compared to A+T+N− (Table 1). No significant AD PGRS differences were found between A+T+N− and A+T+N+ (Figure S2A).
CSF proteomic analysis showed that 13 proteins were increased and 144 were decreased in A+T+N+ compared to A+T+N− (Figure 3A, Table S3A). No pathways were associated with the increased proteins. Decreased proteins were related to biological pathways linked with cell adhesion and migration, nervous system development, extracellular matrix (ECM), lipids, protein processing, hemostasis and blood vessel development, transforming growth factor β signaling, and lysosome organization (Figure 3B,C, Table S3B). Fifty‐eight decreased proteins were enriched for expression in the ChP (40%, ABAenrichment P = 0.034).
FIGURE 3.
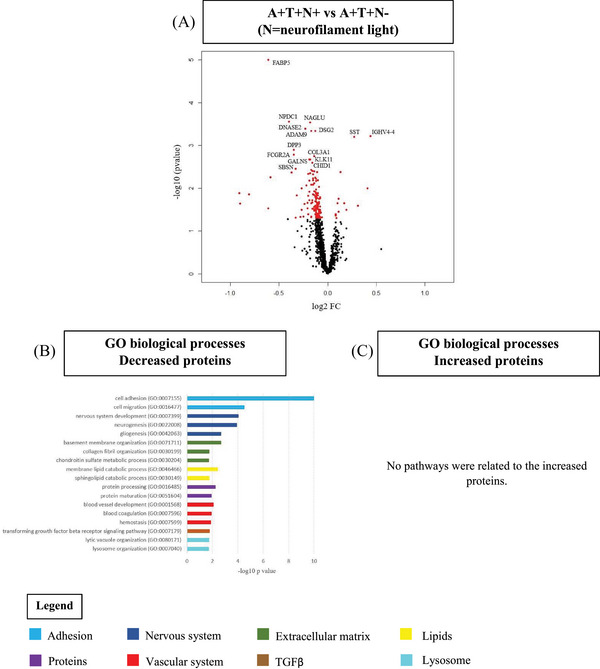
CSF proteomics in A+T+ individuals without dementia by neurofilament light status. A, Volcano plot displaying the log2 fold‐change against the −log10 statistical p‐value for the comparison of A+T+N+ versus A+T+N− (N = neurofilament light). Significantly different proteins are red. The top 15 proteins are named. B, C, Selected biological processes Gene Ontology (GO) terms for decreased (B) and increased (C) proteins in the comparison A+T−N+ versus A+T−N−. CSF, cerebrospinal fluid; TGFβ, transforming growth factor beta.
In A+T− individuals (Figure 1B), similar proteomic pattern was found as in A+T+ individuals, with mainly decreased proteins in N+ compared to N− (Supplementary Results, Figure S4A in supporting information, Table S3C). Nonetheless, biological pathways were distinct from those in A+T+. In A+T−, increased proteins were enriched for biological pathways related to the immune system, while decreased proteins were enriched for ion homeostasis, cell migration, immune system and inflammation, protein degradation, and lipids and epithelial cells (Figure S4B,C, Table S3D). Decreased proteins were also enriched for expression in the ChP (41%, ABAenrichment P = 0.001).
3.1.3. Hippocampal volume as a neurodegeneration marker
Using HCV as N marker, 55 participants were classified as A+T+N− and 83 as A+T+N+ (Figure 1A). A+T+N+ individuals were older and more often diagnosed with MCI, and had lower levels of Aβ42, a lower Aβ42/40 ratio, and higher p‐tau and t‐tau levels than A+T+N− (Table 1). No significant PGRS differences were found between A+T+N− and A+T+N+ (Figure 2A).
CSF proteomic profiling showed that 7 proteins were increased and 22 were decreased in A+T+N+ compared to A+T+N− (Figure 4A, Table S4A). No biological pathways were associated with the increased proteins. Decreased proteins were associated with oxidative stress (Figure 4B,C, Table S4B).
FIGURE 4.
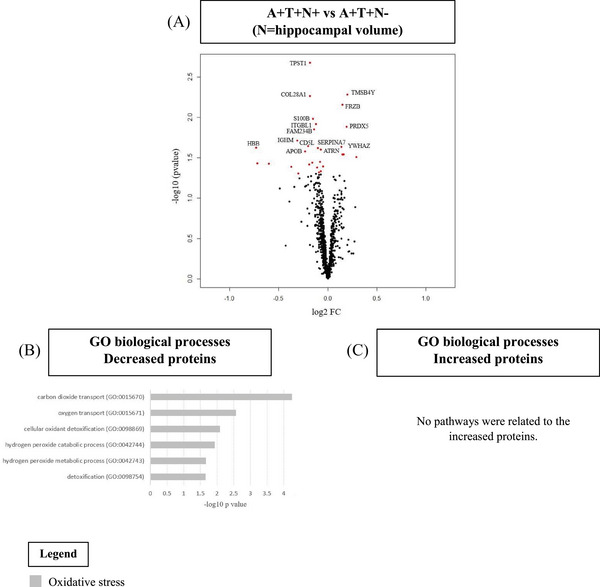
Cerebrospinal fluid (CSF) proteomics in A+T+ individuals without dementia by hippocampal volume status. A, Volcano plot displaying the log2 fold‐change against the −log10 statistical p‐value for the comparison A+T+N+ versus A+T+N− (N = hippocampal volume). Significantly different proteins are red. The top 15 proteins are named. B, C, Selected biological processes Gene Ontology (GO) terms for decreased (B) and increased (C) proteins in the comparison of A+T−N+ versus A+T−N−.
Similar proteomic results were found in A+T− (Figure 1B, Supplementary Results, Figure S5 in supporting information, Table S4C,D).
3.2. Comparison of proteomic profiles of distinct N markers
Next, we compared the proteomic results of the N+ versus N− groups for the three N markers in A+T+ individuals. NfL+ was mainly associated with decreased proteins, while we found mainly increased proteins for Ng+. There was limited overlap in these proteins (4% to 5%; Figure 5A). Yet, top biological pathways were overlapping and associated with neuronal plasticity (Figure 5B), suggesting hyperplasticity in Ng+ and hypoplasticity in NfL+. HCV+ showed limited overlap with protein changes in Ng+ and NfL+ groups (between 2% and 35% of overlap; Figure 5A). Nonetheless, A+T+HCV+ showed overlap in the top 10 dysregulated GO biological pathways with A+T−Ng+, related to oxidative stress (Figure 5B). Comparison results for A+T− individuals can be found in the Supplementary Results.
FIGURE 5.
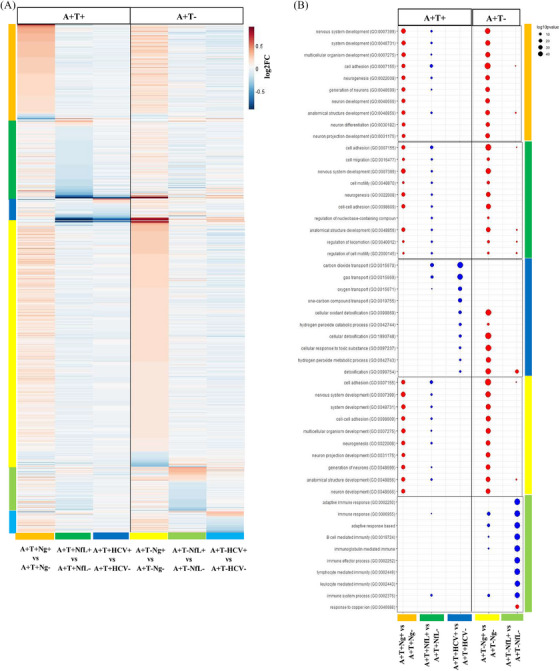
Comparison of the proteomic profiles of the groups classified using distinct N markers. A, Heatmap representing the log2 fold‐change values of the proteins with significant level changes in each comparison using different neurodegeneration markers. B, Gene Ontology biological pathway enrichment analysis with a dot plot representing the top 10 biological pathways enriched for the different comparisons.
3.3. Correlations between ATN markers
Figure 6A displays the correlation between ATN biomarkers. In a subset of 128 A+T+ individuals with availability of the three N markers, Ng was positively correlated with p‐tau, t‐tau, NfL, Aβ40, and Aβ42 and negatively correlated with the ratio Aβ42/40. NfL was positively correlated with t‐tau, p‐tau, Ng, and Aβ40, and negatively correlated to HCV. HCV was positively correlated with the ratio Aβ42/40 and Aβ42 and negatively with p‐tau, t‐tau, and NfL. In a subset of 58 A+T− individuals with availability of the three N markers, we found that Ng was positively correlated with p‐tau and t‐tau, and negatively correlated with the ratio Aβ42/40, while NfL was positively correlated with t‐tau.
FIGURE 6.
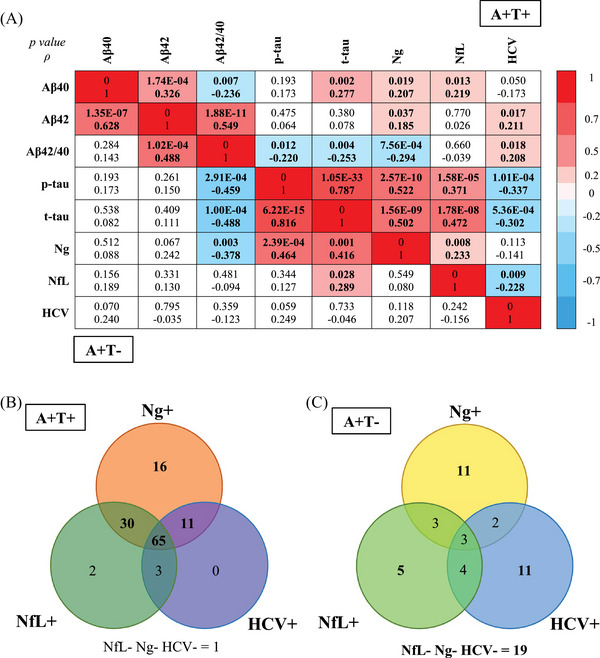
Association between the different neurodegeneration biomarkers. A, Correlation matrix for AD biomarkers using the Spearman rank correlation test in A+T+ individuals with no dementia (upper part of the graph) and in A+T− individuals with no dementia (lower part of the graph). The figure presents the p‐value and correlation coefficient (ρ). The color scale depicts the strength of the Spearman correlation coefficient in the significant correlations. B, Venn diagram depicting the distribution of the three neurodegeneration biomarkers in a subset of 128 A+T+ individuals and with availability of the three N markers. C, Venn diagram depicting the distribution of the three neurodegeneration biomarkers in a subset of 58 A+T− individuals and with availability of the three N markers. Aβ, amyloid beta; AD, Alzheimer's disease; HCV, hippocampal volume; NC, normal cognition; NfL, neurofilament light; Ng, neurogranin; p‐tau, phosphorylated tau; t‐tau, total tau.
Most of the A+T+ participants had more than one abnormal N marker, including most often Ng (Figure 6B). In A+T−, most individuals presented only one abnormal N marker (Figure 6C).
3.4. Post hoc CSF proteomic analyses
In post hoc analyses, proteomic profiles were studied separately in individuals with NC and MCI (Table S6A–F in supporting information). In A+T+, results remained similar between individuals with NC and MCI for all N markers (Table S6A–C). In A+T−, results remained similar when Ng was used as the N marker, but were somewhat different in MCI A+T− when NfL or HCV were used as N markers (Supplementary Results and Table S6D–F).
As a sensitivity analysis, we corrected our main analysis for batch effects, which resulted in similar findings. We also compared the key demographic and clinical characteristics between participants with all three N markers to those with at least one N marker, and found no statistically significant differences, indicating similar sample characteristics between the groups.
Finally, as there is an age‐dependent increase of CSF NfL, we determined post hoc an age‐adjusted cut‐off of ≥ 2 SD (see Materials and Methods). The use of this age‐adjusted cut‐off resulted in similar results.
4. DISCUSSION
Comparing neurodegeneration markers in AD individuals without dementia, we found that CSF Ng, CSF NfL, and HCV were each associated with distinct CSF proteomic profiles. Yet, Ng+ and NfL+ protein changes showed overlap in top biological pathways, which were associated with neuroplasticity, though in opposite directions. HCV+ was associated with relatively few proteomic changes that were related to oxidative stress. Overall, our results show that different N markers represent distinct pathophysiological mechanisms in AD and cannot be used interchangeably.
A high number of increased proteins were associated with Ng+ compared to Ng−. Those increased proteins were related to aberrant neuroplasticity. 30 This is consistent with the global function of Ng in the central nervous system and with its role as a biomarker of synaptic dysregulation in AD. Findings could be also partially linked to tau, as CSF t‐tau is linked in AD with changes in proteins associated with neuronal plasticity. 30 Other dysregulated pathways associated with Ng were linked to angiogenesis. Cerebral hypoperfusion is a key hallmark of AD and a previous study reported a high positive correlation between CSF Ng levels and CSF levels of vascular endothelial growth factor, a protein involved in modulation of vascular remodeling, permeability, and angiogenesis. Angiogenesis could also be part of the neuronal plasticity response, as previously reported. 30 , 53 A high percentage of the decreased proteins related to Ng+, in both A+T− and A+T+, showed an association with BBB functioning (39% to 41%). This is in line with a study on traumatic brain injury reporting increased CSF Ng levels as a result of damage to the BBB. 54 The proteomic profile of Ng+ was similar across AD pathology stages (A+T+ and A+T−), as well as across clinical stages (NC or MCI). This suggests that Ng reflects Aβ‐dependent degeneration 10 independently of the clinical stage and tau status. Still, it should be noted that the levels of Ng were higher in A+T+N+ compared to A+T−N+. T‐tau, p‐tau, and Ng incongruency was uncommon, which is in line with the high correlation between Ng and tau (both p‐ and t‐tau) in our study, as also previously reported. 55 Along with the shared associations of Ng and tau with aberrant neuroplasticity, this suggests that Ng may serve more as a tau‐related marker, rather than a neurodegeneration marker.
A high number of decreased proteins were associated with NfL+ compared to NfL−. The proteomic profile of NfL+ was different across AD pathology stages (A+T− and A+T+), and for the A+T− also across clinical stages (NC or MCI). This could be explained by the fact that NfL reflects neurodegeneration independently of Aβ pathology. 10 Hypoplasticity pathways in A+T+, including downregulated pathways related to neurogenesis and nervous system development, might be related to the degeneration of neurons associated with the increased levels of NfL in the CSF. 2 , 6 , 7 The opposite profile of Ng and NfL, in which Ng is associated with hyperplasticity and NfL with hypoplasticity, may reflect different temporal dynamics in AD pathology. Changes in Ng levels may occur earlier in the disease progression, reflecting synaptic dysfunction 2 , 3 , 4 , 5 and hyperplasticity, and potentially precede neurodegenerative processes. Conversely, alterations in NfL levels may occur later, reflecting axonal degeneration and hypoplasticity. A high percentage of the decreased proteins in NfL+ were highly expressed by the ChP (40% to 41%). The ChP is located inside the brain ventricles and is responsible for the production of CSF; transport of ions, proteins, nutrients, lipids, and metabolic precursors across the epithelium to the CSF; and clearance of proteins from the CSF. 56 Hence, pathways dysregulated in NfL+ could be linked with ChP dysfunction, that is, pathways associated with ion homeostasis, immune system, lipids, and ECM. 57 , 58 The ChP seems to also play an important role in neuroplasticity and synaptic functions. 59 Yet, the implication of ChP functioning in AD in relation to CSF NfL changes warrants further investigation. Pathways associated with protein degradation and processing were also downregulated in NfL+, which could partially explain the downregulation of proteins.
Contrary to our expectations, HCV+ in AD was not associated with many protein changes in CSF. In A+T+, we found that decreased proteins were associated with oxidative stress, while in A+T− we found no pathways associated with the significant proteins. Oxidative stress is one of the earliest events occurring in AD and the hippocampus is a brain structure highly sensitive to oxidative stress. 60 , 61 Oxidative stress in the hippocampus alters neurogenesis, dendritic complexity, and learning and could lead to atrophy. 62 As we only found few proteomic changes in HCV, it might be that the pathophysiological mechanisms associated with hippocampal atrophy are not directly reflected in the CSF, but maybe more in the tissue itself. 63 Alternatively, as hippocampal atrophy is a relatively late marker in AD, it might be difficult to identify HCV changes using dichotomization in early AD stages, especially in preclinical AD. Yet, several previous studies have successfully used dichotomized HCV measures in early AD. 64 , 65 Another hypothesis could propose that, because lower HCV are not exclusive to AD but also occur in other neurodegenerative diseases, 66 , 67 each of these conditions might exhibit its own distinct proteomic profile. In cases in which multiple pathologies coexist, the proteomic signatures associated with each pathology could potentially be diluted or obscured by the presence of others.
The non‐interchangeability between N markers is in line with conclusions from previous studies. 12 , 68 Nonetheless, it should be noted that most of the A+T+ participants showed abnormalities in more than one N marker, and almost all had abnormal levels of Ng. This is in line with the strong correlation that we observed between tau and Ng levels, consistent with previous findings. 10 Moreover, NfL showed a positive correlation with Ng and negative correlation with HCV. In A+T−, the levels of the three N markers were not significantly correlated and most of the A+T− individuals had only one abnormal N marker. This indicates that, whenever only abnormal levels of Aβ42 are found, distinct neurodegeneration mechanisms can occur in different persons with AD. The N markers can represent AD but also non‐AD pathologies, as N markers are not AD specific. 1
Our study has several strengths and limitations. To the best of our knowledge, this is the first study reporting CSF proteomic profiling of distinct neurodegeneration biomarkers in AD. Our results are a first step toward a better pathophysiological characterization of the distinct neurodegeneration processes happening in AD. Another main strength of this study is that our overall sample size for proteomic analyses was large, encompassing 407 individuals. Yet, for some subgroups, the sample size was rather small. While this reflects lower frequency of subgroups, this could have reduced our statistical power. Furthermore, to define biomarker abnormality, we had to calculate cut‐offs for the neurodegeneration markers, as no reference values were available. Future studies should validate our cut‐points in independent or larger datasets. Furthermore, the methodology to quantify ATN biomarkers varied between centers, potentially introducing variability into the results. However, we defined center‐ or study‐specific cutoffs to account for this. Further research is also needed to unravel the causes and consequences of the dysregulated processes associated with the distinct N markers. Longitudinal studies are needed to investigate the cognitive outcomes of each neurodegeneration marker in relation to proteomic profiles.
Together, our findings suggest that N markers cannot be used interchangeably, as Ng, NfL, and HCV markers measure largely different neurodegenerative processes in AD. Yet, Ng+ and NfL+ showed some overlap in biological pathways related to neuroplasticity. Ng might not be a good N marker given its close associations with tau pathophysiology. HCV+ was associated with fewer proteomic differences. The use of different N markers may refine disease staging beyond amyloid and tau by providing complementary information, which has implications for clinical trial design.
CONFLICT OF INTEREST STATEMENT
A.D. received funding from Alzheimer Nederland (grant No. WE.15‐2022‐01). J.G. has nothing to disclose. S.E.S. has analyzed data provided by C2N Diagnostics to Washington University. She has served on scientific advisory boards for Eisai. M.K. has nothing to disclose. L.M.R. has nothing to disclose. V.D. has nothing to disclose. B.M.T. has nothing to disclose. T.L.S.B. has investigator‐initiated research funding from the NIH, the Alzheimer's Association, the Barnes‐Jewish Hospital Foundation, and Siemens. She participates as a site investigator in clinical trials sponsored by Avid Radiopharmaceuticals, Eli Lilly, Biogen, Eisai, Jaansen, and Roche. She serves as a consultant to Biogen, Lilly, Eisai, and Siemens. C.C. has nothing to disclose. C.E.T. has nothing to disclose. I.R. has nothing to disclose. P.M.L. has nothing to disclose. M.T. has nothing to disclose. R.V.’s institution has clinical trial agreements (R.V. as P.I.) with Alector, Biogen, Denali, EliLilly, J&J, UCB. R.V.’s institution has consultancy agreements (R.V. as DSMB member) with AC Immune. J.S. is a senior postdoctoral fellow (12Y1623N) of FWO. J.S. receives funding from Stichting Alzheimer Onderzoek (SAO‐FRA 2021/0022). S.E. has nothing to disclose. E.D.R. has nothing to disclose. J.P. served as a consultant and on advisory boards for the Nestlé Institute of Health Sciences, Ono Pharma, OM Pharma, Schwabe Pharma, Lilly, Roche, and Fujirebio Europe. All his disclosures are unrelated to the present work. The VD cohort was supported by grants from the Swiss National Research Foundation (SNF 320030_204886), Synapsis Foundation – Dementia Research Switzerland (Grant No. 2017‐PI01). G.P. has nothing to disclose. M.T. has nothing to disclose. Y.F.L. has nothing to disclose. S.L. has nothing to disclose. J.S. has nothing to disclose. F.B. is a steering committee or Data Safety Monitoring Board member for Biogen, Merck, Eisai, and Prothena; an advisory board member for Combinostics, Scottish Brain Sciences; a consultant for Roche, Celltrion, Rewind Therapeutics, Merck, Bracco. F.B. has research agreements with ADDI, Merck, Biogen, GE Healthcare, Roche. F.B. is co‐founder and shareholder of Queen Square Analytics LTD. L.B. has nothing to disclose. K.B. has served as a consultant and on advisory boards for AC Immune, Acumen, ALZPath, AriBio, BioArctic, Biogen, Eisai, Lilly, Moleac Pte. Ltd., Novartis, Ono Pharma, Prothena, Roche Diagnostics, and Siemens Healthineers; has served on data monitoring committees for Julius Clinical and Novartis; has given lectures, produced educational materials, and participated in educational programs for AC Immune, Biogen, Celdara Medical, Eisai, and Roche Diagnostics; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper. H.Z. has served on scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Amylyx, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Merry Life, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Alzecure, Biogen, Cellectricon, Fujirebio, Lilly, Novo Nordisk, and Roche, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). P.J.V. received funding from the European Commission, IMI 2 Joint Undertaking (JU), AMYPAD, grant No. 115952; European Commission, IMI 2 JU, RADAR‐AD, grant No. 806999; European Commission, IMI 2 JU, EPND, grant No. 101034344. The IMI JU receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA. P.J.V. received also funding from Zon‐MW, Redefining Alzheimer's disease, grant No. 733050824736; and Biogen (Amyloid biomarker study group). Grants were paid to the university. S.J.B.V. received funding from ZonMW (SNAP VIMP grant No. 7330505021), Stichting Adriana van Rinsum‐Ponssen, and the EPND project, which received funding from the European Commision, IMI 2 Joint Undertaking (JU) under grant agreement No. 101034344. The IMI JU receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects provided informed consent for research.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The present study was supported by the Memorabel program of ZonMw (the Netherlands Organization for Health Research and Development) grant No. 733050502 and 7330505021, an anonymous foundation, and EMIF‐AD. The EMIF‐AD project has received support from the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement No. 115372, resources of which are composed of financial contribution from the European Union's Seventh Framework Program (FP7/2007‐2013) and EFPIA companies’ in‐kind contribution. The DESCRIPA study was funded by the European Commission within the 5th Framework Program (QLRT‐2001‐2455). The EDAR study was funded by the European Commission within the 5th Framework Program (contract # 37670). San Sebastian GAP study is partially funded by the Department of Health of the Basque Government (allocation 17.0.1.08.12.0000.2.454.01.41142.001.H), Provincial Government of Gipuzkoa (124/16), Kutxa Fundazioa, and by the Carlos III Institute of Health (PI15/00919, PN de I+D+I 2013‐2016). The Lausanne study was funded by a grant from the Swiss National Research Foundation (SNF 320030_141179). Collection of data from the Knight ADRC was supported by the National Institute on Aging grants K23AG053426, P30AG066444, P01AG003991, and P01AG026276. F.B. is supported by the NIHR Biomedical Research Centre at UCLH. H.Z. is a Wallenberg Scholar and a Distinguished Professor at the Swedish Research Council supported by grants from the Swedish Research Council (#2023‐00356; #2022‐01018, and #2019‐02397), the European Union's Horizon Europe Research and Innovation Programme under grant agreement No. 101053962, Swedish State Support for Clinical Research (#ALFGBG‐71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C, #ADSF‐21‐831377‐C, and #ADSF‐24‐1284328‐C), the Bluefield Project, Cure Alzheimer's Fund, the Olav Thon Foundation, the Erling‐Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022‐0270), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021‐00694), the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and the UK Dementia Research Institute at UCL (UKDRI‐1003).
Delvenne A, Gobom J, Schindler SE, et al. CSF proteomic profiles of neurodegeneration biomarkers in Alzheimer's disease. Alzheimer's Dement. 2024;20:6205–6220. 10.1002/alz.14103
REFERENCES
- 1. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mielke MM, Syrjanen JA, Blennow K, et al. Comparison of variables associated with cerebrospinal fluid neurofilament, total‐tau, and neurogranin. Alzheimers Dement. 2019;15(11):1437‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Z, Yang J, Zhu W, Tang Y, Jia J. The synaptic marker neurogranin as a disease state biomarker in Alzheimer's disease: a systematic review and meta‐analysis. Int J Neurosci. 2021:1‐9. [DOI] [PubMed] [Google Scholar]
- 4. Nilsson J, Cousins KAQ, Gobom J, et al. Cerebrospinal fluid biomarker panel of synaptic dysfunction in Alzheimer's disease and other neurodegenerative disorders. Alzheimer Dement. 2023;19(5):1775‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Portelius E, Olsson B, Höglund K, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136(3):363‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giacomucci G, Mazzeo S, Bagnoli S, et al. Plasma neurofilament light chain as a biomarker of Alzheimer's disease in Subjective Cognitive Decline and Mild Cognitive Impairment. J Neurol. 2022;269(8):4270‐4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiong YL, Meng T, Luo J, Zhang H. The potential of neurofilament light as a biomarker in Alzheimer's disease. Eur Neurol. 2021;84(1):6‐15. [DOI] [PubMed] [Google Scholar]
- 8. Schuff N, Woerner N, Boreta L, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(Pt 4):1067‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jahn H. Memory loss in Alzheimer's disease. Dialogues Clin Neurosci. 2013;15(4):445‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mattsson N, Insel PS, Palmqvist S, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer's disease. EMBO Mol Med. 2016;8(10):1184‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bucci M, Chiotis K, Nordberg A. Alzheimer's Disease Neuroimaging I. Alzheimer's disease profiled by fluid and imaging markers: tau PET best predicts cognitive decline. Mol Psychiatry. 2021;26(10):5888‐5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boerwinkle AH, Wisch JK, Chen CD, et al. Temporal correlation of CSF and neuroimaging in the amyloid‐tau‐neurodegeneration model of Alzheimer disease. Neurology. 2021;97(1):e76‐e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noor Z, Ahn SB, Baker MS, Ranganathan S, Mohamedali A. Mass spectrometry‐based protein identification in proteomics‐a review. Brief Bioinform. 2021;22(2):1620‐1638. [DOI] [PubMed] [Google Scholar]
- 14. Delvenne A, Gobom J, Tijms B, et al. Cerebrospinal fluid proteomic profiling of individuals with mild cognitive impairment and suspected non‐Alzheimer's disease pathophysiology. Alzheimers Dement. 2023;19:807‐820. [DOI] [PubMed] [Google Scholar]
- 15. Delmotte K, Schaeverbeke J, Poesen K, Vandenberghe R. Prognostic value of amyloid/tau/neurodegeneration (ATN) classification based on diagnostic cerebrospinal fluid samples for Alzheimer's disease. Alzheimers Res Ther. 2021;13(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cummings J. The Role of Biomarkers in Alzheimer's Disease Drug Development. Adv Exp Med Biol. 2019;1118:29‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cummings J, Kinney J. Biomarkers for Alzheimer's disease: context of use, qualification, and roadmap for clinical implementation. Medicina (Kaunas). 2022;58(7):952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cummings J, Ritter A, Zhong K. Clinical trials for disease‐modifying therapies in Alzheimer's disease: a primer, lessons learned, and a blueprint for the future. J Alzheimers Dis. 2018;64(s1):S3‐S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bos I, Verhey FR, Ramakers I, et al. Cerebrovascular and amyloid pathology in predementia stages: the relationship with neurodegeneration and cognitive decline. Alzheimers Res Ther. 2017;9(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morris JC, Schindler SE, McCue LM, et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 2019;76(3):264‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bos I, Vos S, Vandenberghe R, et al. The EMIF‐AD Multimodal Biomarker Discovery study: design, methods and cohort characteristics. Alzheimers Res Ther. 2018;10(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grober E, Petersen KK, Lipton RB, et al. Association of stages of objective memory impairment with incident symptomatic cognitive impairment in cognitively normal individuals. Neurology. 2023;100(22):e2279‐e2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256(3):183‐194. [DOI] [PubMed] [Google Scholar]
- 24. Magdalinou NK, Noyce AJ, Pinto R, et al. Identification of candidate cerebrospinal fluid biomarkers in parkinsonism using quantitative proteomics. Parkinsonism Relat Disord. 2017;37:65‐71. [DOI] [PubMed] [Google Scholar]
- 25. Batth TS, Francavilla C, Olsen JV. Off‐line high‐pH reversed‐phase fractionation for in‐depth phosphoproteomics. J Proteome Res. 2014;13(12):6176‐6186. [DOI] [PubMed] [Google Scholar]
- 26. Herries EMBN, Sutphen CL, Fagan AM, Ladenson JH. Brain biomarkers: follow‐up of RNA expression discovery approach: CSF assays for neurogranin, SNAP‐25, and VILIP‐1. Neuromethods. 2021;168. [Google Scholar]
- 27. Schindler SE, Li Y, Todd KW, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer's disease. Alzheimers Dement. 2019;15(5):655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aalten P, Ramakers IH, Biessels GJ, et al. The Dutch Parelsnoer Institute – Neurodegenerative diseases; methods, design and baseline results. BMC Neurol. 2014;14:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Butt OH, Long JM, Henson RL, et al. Cognitively normal APOE epsilon4 carriers have specific elevation of CSF SNAP‐25. Neurobiol Aging. 2021;102:64‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Visser PJ, Reus LM, Gobom J, et al. Cerebrospinal fluid tau levels are associated with abnormal neuronal plasticity markers in Alzheimer's disease. Mol Neurodegener. 2022;17(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Rojas I, Moreno‐Grau S, Tesi N, et al. Common variants in Alzheimer's disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12(1):3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deming Y, Li Z, Kapoor M, et al. Genome‐wide association study identifies four novel loci associated with Alzheimer's endophenotypes and disease modifiers. Acta Neuropathol. 2017;133(5):839‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Del‐Aguila JL, Fernandez MV, Schindler S, et al. Assessment of the genetic architecture of Alzheimer's disease risk in rate of memory decline. J Alzheimers Dis. 2018;62(2):745‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fischl B. FreeSurfer. Neuroimage. 2012;62(2):774‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ten Kate M, Redolfi A, Peira E, et al. MRI predictors of amyloid pathology: results from the EMIF‐AD Multimodal Biomarker Discovery study. Alzheimers Res Ther. 2018;10(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Millar PR, Gordon BA, Luckett PH, et al. Multimodal brain age estimates relate to Alzheimer disease biomarkers and cognition in early stages: a cross‐sectional observational study. eLife. 2023;12:e81869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duits FH, Wesenhagen KEJ, Ekblad L, et al. Four subgroups based on tau levels in Alzheimer's disease observed in two independent cohorts. Alzheimers Res Ther. 2021;13(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu H, Bi YL, Shen XN, et al. Application of the amyloid/tau/neurodegeneration framework in cognitively intact adults: the CABLE study. Ann Neurol. 2022;92(3):439‐450. [DOI] [PubMed] [Google Scholar]
- 39. Unal I. Defining an optimal cut‐point value in ROC analysis: an alternative approach. Comput Math Methods Med. 2017;2017:3762651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mi H, Muruganujan A, Huang X, et al. Protocol Update for large‐scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc. 2019;14(3):703‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol). 1995;57:289‐300. [Google Scholar]
- 42. Lewin A, Grieve IC. Grouping Gene Ontology terms to improve the assessment of gene set enrichment in microarray data. BMC Bioinf. 2006;7:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419‐D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607‐D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neumeier M, Weigert J, Buettner R, et al. Detection of adiponectin in cerebrospinal fluid in humans. Am J Physiol Endocrinol Metab. 2007;293(4):E965‐969. [DOI] [PubMed] [Google Scholar]
- 47. Dayon L, Cominetti O, Wojcik J, et al. Proteomes of paired human cerebrospinal fluid and plasma: relation to blood‐brain barrier permeability in older adults. J Proteome Res. 2019;18(3):1162‐1174. [DOI] [PubMed] [Google Scholar]
- 48. Rapoport SI, Pettigrew KD. A heterogenous, pore‐vesicle membrane model for protein transfer from blood to cerebrospinal fluid at the choroid plexus. Microvasc Res. 1979;18(1):105‐119. [DOI] [PubMed] [Google Scholar]
- 49. Hawrylycz MJ, Lein ES, Guillozet‐Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford). 2016;2016:baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Grote S, Prufer K, Kelso J, Dannemann M. ABAEnrichment: an R package to test for gene set expression enrichment in the adult and developing human brain. Bioinformatics. 2016;32(20):3201‐3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vermunt L, Otte M, Verberk IMW, et al. Age‐ and disease‐specific reference values for neurofilament light presented in an online interactive support interface. Ann Clin Transl Neurol. 2022;9(11):1832‐1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hill NL, Kolanowski AM, Gill DJ. Plasticity in early Alzheimer's disease: an opportunity for intervention. Top Geriatr Rehabil. 2011;27(4):257‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li L, Li Y, Ji X, Zhang B, Wei H, Luo Y. The effects of retinoic acid on the expression of neurogranin after experimental cerebral ischemia. Brain Res. 2008;1226:234‐240. [DOI] [PubMed] [Google Scholar]
- 55. Agnello L, Lo Sasso B, Vidali M, et al. Neurogranin as a reliable biomarker for synaptic dysfunction in Alzheimer's disease. Diagnostics (Basel). 2021;11:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus‐cerebrospinal fluid system. Nat Rev Neurosci. 2015;16(8):445‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kratzer I, Ek J, Stolp H. The molecular anatomy and functions of the choroid plexus in healthy and diseased brain. Biochim Biophys Acta Biomembr. 2020;1862(11):183430. [DOI] [PubMed] [Google Scholar]
- 58. Balusu S, Brkic M, Libert C, Vandenbroucke RE. The choroid plexus‐cerebrospinal fluid interface in Alzheimer's disease: more than just a barrier. Neural Regen Res. 2016;11(4):534‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Snodgrass R, Johanson CE. Choroid Plexus: source of cerebrospinal fluid and regulator of brain development and function. In: Cinalli G, Ozek MM, Sainte‐Rose C, eds. Pediatric Hydrocephalus. Springer International Publishing; 2018:1‐36. [Google Scholar]
- 60. Huang TT, Leu D, Zou Y. Oxidative stress and redox regulation on hippocampal‐dependent cognitive functions. Arch Biochem Biophys. 2015;576:2‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759‐767. [DOI] [PubMed] [Google Scholar]
- 62. Huang Y, Wang J, Zhu B, Fu P. CSF VEGF was positively associated with neurogranin independent of beta‐amyloid pathology. Neuropsychiatr Dis Treat. 2020;16:1737‐1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gao Y, Liu J, Wang J, et al. Proteomic analysis of human hippocampal subfields provides new insights into the pathogenesis of Alzheimer's disease and the role of glial cells. Brain Pathol. 2022;32(4):e13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vos SJB, Gordon BA, Su Y, et al. NIA‐AA staging of preclinical Alzheimer disease: discordance and concordance of CSF and imaging biomarkers. Neurobiol Aging. 2016;44:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ingala S, De Boer C, Masselink LA, et al. Application of the ATN classification scheme in a population without dementia: findings from the EPAD cohort. Alzheimers Dement. 2021;17(7):1189‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vijayakumar A, Vijayakumar A. Comparison of hippocampal volume in dementia subtypes. ISRN Radiol. 2013;2013:174524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Riha P, Brabenec L, Marecek R, Rektor I, Rektorova I. The reduction of hippocampal volume in Parkinson's disease. J Neural Transm (Vienna). 2022;129(5‐6):575‐580. [DOI] [PubMed] [Google Scholar]
- 68. Ebenau JL, Pelkmans W, Verberk IMW, et al. Association of CSF, plasma, and imaging markers of neurodegeneration with clinical progression in people with subjective cognitive decline. Neurology. 2022;98(13):e1315‐e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information