

Abstract
INTRODUCTION
This study investigates primary lateral sclerosis (PLS) as a rare manifestation of the presenilin 1 (PSEN1) NM_000021 c.851C > T p.Pro284Leu variant in three siblings of a Colombian family, outlining its clinical and neuropathological features and their relationship to Alzheimer's disease (AD).
METHODS
Data were gathered using clinical evaluations, next‐generation genetic sequencing, magnetic resonance imaging, biomarker analysis, and neuropathological examination.
RESULTS
Carriers of the PSEN1 Pro284Leu variant exhibited classic PLS symptoms, including unilateral onset and bulbar syndromes, along with cognitive decline. Neuropathology showed corticospinal tract degeneration without amyloid beta deposition in spinal white matter.
DISCUSSION
Our findings suggest an overlap between PLS and AD pathology in PSEN1 variant carriers. Results support considering PLS when diagnosing AD‐related motor syndromes and including PSEN1 evaluation when performing genetic testing for PLS. The study highlights the need for further research to clarify the PLS–AD relationship, informing future treatments and clinical trials.
Highlights
Pathogenic variants in presenilin 1 (PSEN1) can manifest as hereditary primary lateral sclerosis
PSEN1 Pro284Leu carriers present motor, cognitive, and behavioral alterations
Cases had corticospinal tract microgliosis and severe Aβ pathology in motor cortex
There was no evidence of amyloid deposition in the spinal cord white matter
All the neuropathology images are available for online visualization
Myelin pallor in the spinal cord is confined to the lateral corticospinal tracts
Keywords: autosomal dominant Alzheimer's disease, cotton wool plaques, neuropathology, primary lateral sclerosis, PSEN1, spastic paraparesis
1. BACKGROUND
Alzheimer's disease (AD) is the most prevalent neurodegenerative disorder and the most common cause of dementia worldwide. 1 Although the most common presentation of AD is amnestic dementia, 2 some patients develop motor syndromes such as spastic paraparesis (SP) 3 or primary lateral sclerosis (PLS), 4 which are far less well understood.
Neurodegenerative disorders such as SP and PLS are characterized by an insidious onset of upper motor neuron dysfunction without clinical signs of lower motor neuron involvement. 5 Patients experience stiffness, decreased balance and coordination, as well as progressive lower limb weakness, which are followed by difficulty speaking, swallowing, and emotional lability if the bulbar region is involved. The diagnosis is based on clinical history, typical examination findings, and diagnostic testing that is negative for other causes of upper motor neuron dysfunction. 6
In the early stages, SP and PLS are difficult to differentiate, but as the illness evolves, SP is usually restricted to the legs, whereas PLS can affect both upper and lower limbs, even compromising speaking and swallowing. The lack of vibratory sensation and foot deformities is suggestive of SP, 7 whereas asymmetrical symptoms are more predictive of PLS. In addition, clinicians often also rely on a positive family history to favor a diagnosis of SP 8 and rarely order genetic testing when suspecting PLS, given its sporadic nature. 9
SP has been documented widely as one of the atypical AD syndromes that occurs in carriers of pathogenic variants of presenilin 1 (PSEN1), 10 whereas there is only scarce evidence for PSEN1 pathogenic variants manifesting as PLS. A previous study by Vasquez‐Costa reported two families with PSEN1 Pro88Leu and PSEN1 Leu166Pro presenting with PLS. 11 However, none of the described individuals had neuropathological assessment. Here, we present the clinical characterization of three members of a family that carry the pathogenic PSEN1 variant Pro284Leu and manifests as PLS, and we include extensive neuropathologic data for two of them.
2. METHODS
This family was identified by the Neuroscience Group of Antioquia (GNA) in a previous study. 12 All the participants or their legally authorized proxies signed an informed consent approved by the institutional review board (IRB) of the Medical Research Institute, Universidad de Antioquia.
2.1. Clinical evaluation
All the individuals had an extensive neurological and neuropsychological evaluation with population‐validated batteries (see Supplementary methods). The pedigree reconstruction was done by patient and family member interviews during the medical assessment.
2.2. Laboratory tests
All the subjects were tested for vitamin B12 serum levels, syphilis (Venereal Disease Research Laboratory [VDRL]), free thyroxine (T4), thyroid‐stimulating hormone (TSH), antinuclear antibodies (ANAs), and human immunodeficiency virus (HIV).
2.3. Neuroimaging
Magnetic resonance imaging (MRI) of the brain and spinal cord were done on a 3T Siemens Skyra machine (Erlangen, Germany). Brain imaging included axial and coronal T2‐weighted spin echo (SE) images, axial fluid‐attenuated inversion recovery (FLAIR), three‐dimensional (3D) T1‐weighted magnetization‐prepared rapid gradient‐echo imaging (MPRAGE), axial susceptibility weighted imaging (SWI), and axial diffusion MR sequence. Cervical and thoracic spinal cord sequences included axial and sagittal 2D T1 and T2‐weighted fast spin echo (FSE) sequences and axial T2*‐weighted multi‐gradient echo (GRE).
2.4. Cerebrospinal fluid biomarkers
Cerebrospinal fluid (CSF) was obtained from all three cases and amyloid beta (Aβ) 1‐42, tau protein (t‐tau), and phosphorylated‐tau (p‐tau)_protein levels were measured using the Lumipulse G chemiluminescent enzyme immunoassay (CLEIA) on the LUMIPULSE G600II benchtop analyzer (Fujirebio Inc).
2.5. Electrophysiology
Nerve conduction studies and electromyography were performed with a Cadwell Sierra Wave machine focusing on representative muscles of the bulbar, cervical, and lumbar segments. Nerve conduction studies were performed with the recommended standard technique, evaluating sensory and motor responses in the median, ulnar, peroneal, and tibial nerves.
2.6. Genetic sequencing
Whole genome sequences from the siblings were obtained and screened for pathogenic variants in genes associated with adult‐onset dementia or motor neuron diseases. Target genes including those in the following the Online Mendelian Inheritance in Man (OMIM) phenotypic series and phenotypes; spastic paraplegia [MIM:PS303350], frontotemporal dementia, and/or amyotrophic lateral sclerosis [MIM:PS105550, MIM:PS167320, MIM:PS105400]; Parkinson disease [MIM:PS168600], and Alzheimer's disease [MIM:104300, MIM:607822, MIM:606889] as described in Acosta‐Uribe, et al. 12 Participants were also genotyped for PSEN1 NM_000021.4 c.851C > T p.Pro284Leu (rs63750863) through Sanger sequencing.
2.7. Neuropathological study
After the deaths of two of the three patients, the family authorized the donation of their brains for autopsy studies and signed the informed consent approved by the IRB. Brains were extracted following standard protocols. Extensive brain examination and staining were done. Tissue was stained with hematoxylin and eosin (H&E), Luxol fast blue/periodic acid Schiff (LFB/PAS), and immunohistochemical (IHC) stains for Aβ (BAM‐10), p‐tau (AT8), and TAR DNA‐binding protein 43 TDP‐43 (phospho Ser409/410). Automated immunostaining was performed with a Ventana Benchmark GX system (Roche) according to the manufacturer's instructions [see Supplementary methods]. Additional automated IHC staining for phosphorylated neurofilament (SMI‐31) was performed on sections of the spinal cord using a Bond RX system (Leica).
RESEARCH IN CONTEXT
Systematic review: Authors reviewed the literature for “PSEN1,” “primary lateral sclerosis (PLS),” “spastic paraparesis (SP),” and “neuropathology.” It is worth noting that although the association between SP, cotton wool plaques (CWPs), and Alzheimer's disease (AD) is well‐established, the documentation of PLS as a manifestation of genetic AD, and its neuropathological characterization, remains scarce.
Interpretation: The evidence suggests that PLS may represent a motor phenotype of AD and recommends testing for pathogenic variants in presenilin 1 (PSEN1) in cases of hereditary PLS. Neuropathological findings highlight the association of CWPs with AD motor phenotypes, regardless of TAR DNA‐binding protein 43 (TDP‐43) accumulation or Lewy body presence.
Future directions: The mechanisms linking amyloid beta accumulation, CWP, and myelin degeneration observed in SP and PLS are yet to be clarified. Future research on brain tissues across PSEN1 variants is crucial for uncovering the pathways that contribute to the AD motor phenotype.
3. RESULTS
3.1. Case 1 (index case)
A 40‐year‐old woman experienced lower limb weakness, initially affecting her left side, which caused her to drag her foot while walking. This condition began at the age of 37 and was soon accompanied by slurred speech and impaired balance. By the time of her initial assessment, she also exhibited symptoms of depression and had significant memory complaints. Neurological examination identified mild dysarthria, spastic quadriparesis, and hyperreflexia, with greater involvement of the left lower limb. No fasciculations were observed. MRI of the brain and spine, as well as electromyography and nerve conduction studies, were reported as normal. Neuropsychological evaluation highlighted normal attention and executive functioning but revealed deficits in controlled learning, language skills, and constructional praxis, meeting the criteria for mild cognitive impairment (MCI) (Tables 1 and S1). The patient had a family history of an autosomal dominant form of neurodegeneration (Figure 1) but had never undergone genetic testing. Whole genome sequencing was performed and screened for pathogenic variants in genes associated with adult‐onset dementia or motor neuron diseases. The patient was found to be a carrier of the PSEN1 pathogenic variant Pro284Leu (rs63750863). No other pathogenic variants were identified in the candidate genes (see genetic sequencing methods).
TABLE 1.
Clinical and paraclinical findings in evaluated cases.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Age at onset of specific symptoms, years | |||
| Age at first symptom | 37 | 32 | 27 |
| Difficulty walking | 37 | 32 | 36 |
| Dysarthria | 37.5 | 34 | 36 |
| Spastic paraparesis | 40 | 36 | Not present at last evaluation |
| Cognitive symptoms | 40 | 36 | 31 |
| Behavioral symptoms | 40 | 37 | 27 |
| Dysphagia | 42 | 37 | Not present at last evaluation |
| Pseudobulbar affect | 42 | 37 | Not present at last evaluation |
| Spastic Quadriparesis | 43 | 39 | Not present at last evaluation |
| Urinary incontinence | 43 | 39 | Not present at last evaluation |
| Age at death | 46 | 40 | Alive, 37 years old |
| Clinical evaluation | |||
| Clonus | + | + | Not present at last evaluation |
| Babinski | + | + | + |
| Facial palsy | + | + | Not present at last evaluation |
| Sensitivity | Normal | Normal | Normal |
| Fasciculations | Not present | Not present | Not present at last evaluation |
| Paraclinical tests | |||
| APOE | ε3/ε3 | ε3/ε3 | ε3/ε3 |
| EMG and NCV | Normal | Normal | Not performed |
| MRI brain | Global atrophy with white matter hyperintensities | Global atrophy with white matter hyperintensities | Small non‐confluent white matter hyperintensities |
| MRI spine | Cervicodorsal atrophy | Cervicodorsal atrophy | Normal |
| NIA/AA ABC score | A3, B3, C3 | A3, B3, C3 | Not applicable |
Notes: EMG and NCV, electromyography and nerve conduction velocity in the four extremities. MRI, Magnetic resonance imaging.
NIA/AA ABC, National Institute on Aging (NIA) in collaboration with the Alzheimer's Association (AA) neuropathologic assessment score. ABC is in relation to: Thal phases of amyloid beta (Aβ) deposition (“A”), Braak staging for neurofibrillary degeneration (“B”), and Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuritic plaque score (“C”).
FIGURE 1.
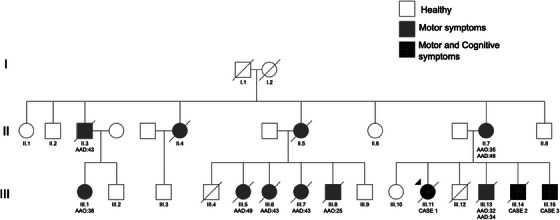
Pedigree of the family. The diagram represents the extended family using the standardized human pedigree nomenclature. Circles represent females, squares represent males. AAO, age at onset; AAD, age at death.
By age 43, the patient's condition had progressed, necessitating the use of a walker for ambulation due to severe spastic quadriparesis, now more pronounced on the right side. She also exhibited corticobulbar dysfunction, dysphagia, and pseudobulbar affect. She maintained the ability to perform tasks with her hands, such as eating with a spoon.
The patient lost the ability to walk at the age of 44, requiring wheelchair use. She developed urinary and fecal incontinence and lost the ability to perform motor tasks with her hands, becoming dependent for dressing and bathing. In addition, the patient had a compromise of facial musculature, leading to anarthria, sialorrhea, and facial paresis. There were no fasciculations or sensory function impairment. Eye movements remained normal, with adequate saccades and no compromise of upward or downward gaze. A neuropsychological examination could not be conducted, as the patient was unable to understand simple commands.
By 45, the patient became completely dependent on all basic activities of daily living (ADLs), experienced severe dysphagia, and could not track objects with her eyes. Brain MRI revealed moderate generalized atrophy, with cortical volume loss and widening of peripheral sulci. Multiple hyperintensities were observed in subcortical and periventricular white matter, classified as Fazekas scale grade 2–3 on axial FLAIR images. SWI sequence identified multiple subcortical microbleeds. In addition, significant loss of brainstem volume was noted, predominantly affecting the midbrain and pons (Figure 2A). The cervical and thoracic spinal cord showed atrophy, with annular disruptions and hyperintensity of the posterolateral columns.
FIGURE 2.
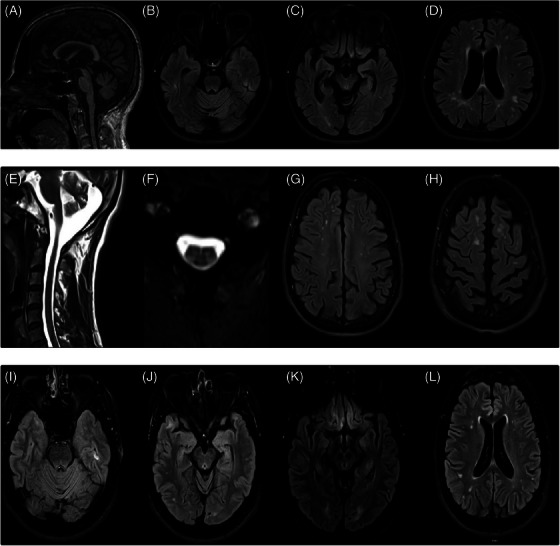
MRI of Case 1. (A) Sagittal T1 sequence showing generalized atrophy and loss of brainstem volume. (B–D) Axial flair sequence showing subcortical and periventricular white matter hyperintensities at three different levels. Magnetic resonance imaging (MRI) of Case 2. (E) Sagittal T1 showing atrophy. (F) Axial T2 images of cervical spinal cord showing posterolateral column hyperintensity. (G,H) Axial fluid‐attenuated inversion recovery (FLAIR) images showing peripheral sulci widening, and subcortical hyperintensities. MRI of Case 3. (I–L) Axial FLAIR images at different levels evidence small white matter hyperintensities and gliosis in the straight gyrus.
The patient died at the age of 46 due to sepsis secondary to broncho‐aspiration. Her brain was donated following ethical guidelines.
3.2. Case 2
A male sibling of the index case, presented with progressive asymmetric lower limb spasticity, dysarthria, and hypophonia starting at age 32. At 34, during his first neurological evaluation, he exhibited hypometric saccades, left facial palsy, an exaggerated palmomental reflex, and emotional incontinence. Muscle strength was normal across all limbs, but presented generalized hyperreflexia and spasticity. Babinski's sign was positive on both sides. There were no fasciculations, atrophy, or sensory impairments. He was a PSEN1 Pro284Leu carrier as well and did not have any other pathogenic variant in a gene associated with the phenotype.
By 36, the patient required a walker for ambulation but remained independent in all ADLs. He reported mild forgetfulness and occasional anomia. Neurological examination revealed sialorrhea, pseudobulbar affect, scissor gait, bilateral patellar clonus, and an inexhaustible glabellar reflex, without sensory impairment or fasciculations. Brain MRI indicated mild involutional changes, peripheral sulci widening, and subcortical hyperintensities, especially in the superior frontal lobe (Fazekas scale grade 2) (Figure 2G,H). Cervical spinal cord T2 images showed mild volume loss and subtle posterolateral column hyperintensity (Figure 2F).
At 37, he lost the capability to walk and developed spastic quadriparesis, leading to complete dependence for ADLs. Significant mood alterations and aggressive behaviors were noted. A comprehensive neuropsychological assessment identified anterograde amnesia, attentional deficits, executive dysfunction, constructional apraxia, and lexical access difficulties. These cognitive and functional impairments led to a diagnosis of mild dementia (Tables 1 and S1).
He died at 40 from pulmonary infection complications. His brain was donated following ethical guidelines.
3.3. Case 3
A male sibling of Cases 1 and 2 was included in the current study. He had no motor symptoms but was identified as a carrier of the PSEN1 Pro284Leu variant. At his initial evaluation at 31, he reported a decline in episodic memory over the previous year. Following a traumatic brain injury, the family observed personality changes, including disruptive behavior, childish speech, inappropriate laughter, and an inability to manage his finances. Although these behavioral symptoms remained stable, his memory continued to decline. His medical and neurological examinations were normal.
Neuropsychological assessment revealed normal attention, executive function, and lexical access capabilities. However, his performance was below the age and schooling expectations in episodic memory, controlled learning, and constructional praxis. Despite retaining full independence in ADLs the patient met the criteria for MCI (Tables 1 and S1). At the age of 35, brain MRI identified a few small, non‐confluent signal hyperintensities in the white matter (Fazekas grade 1). The brainstem was morphologically normal with proper signal intensity, but showed signal hyperintensity and gliosis in the straight gyrus, likely a result of his traumatic brain injury history (Figure 2K). By the age of 36, the participant began experiencing asymmetric gait difficulties, pronounced weakness in the lower right limb, and a wide‐based gait. At the same time, his healthy relatives noticed a worsening of his episodic memory and slurred speech.
3.4. Neuropathological findings
3.4.1. Case 1 (index case)
The brain weighed 1039 g and exhibited generalized gyral atrophy, particularly in the frontoparietal regions. Sections of the cerebral hemispheres showed slight dilation of the lateral ventricles and a moderate reduction in the size of the hippocampi. There was moderate thinning of the cerebral cortex throughout. The substantia nigra displayed slight depigmentation, whereas the locus coeruleus had normal pigmentation. There was a reduction in cerebellar white matter, although the folia and vermis remained normal. The spinal cord, especially in the cervical region, showed a slight reduction in caliber.
Light microscopy demonstrated abundant p‐tau (AT8) immunoreactivity in the form of neurofibrillary tangles (NFTs), neuritic plaques (NPs), and neuropil threads (NTs) in the hippocampus, neocortex (including primary motor cortex) (Figures 3 and 4), and a portion of the calcarine cortex, corresponding to Braak stage VI and the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) plaque grade “frequent.” This pathology was also present in the striatum. Aβ cotton wool plaques (CWPs) were abundant in the hippocampus, neocortex (including the primary motor cortex) (Figures 3 and 4), and striatum, mirroring the distribution of p‐tau immunoreactive NP, as well as amyloid angiopathy. Aβ plaques and angiopathy were also observed in the cerebellar gray matter (Thal stage 5) and the gray matter of the spinal cord (Figures S1 and S2). The density of tangles, plaques, and dystrophic neurites was similar between the primary motor cortex and the non‐motor cortical areas examined. The ABC score was A3 B3 C3, corresponding to “high‐level AD neuropathologic change” according to the 2012 National Institute on Aging/Alzheimer's Association (NIA/AA) guidelines. There was no tau immunoreactivity in sections of the spinal cord.
FIGURE 3.
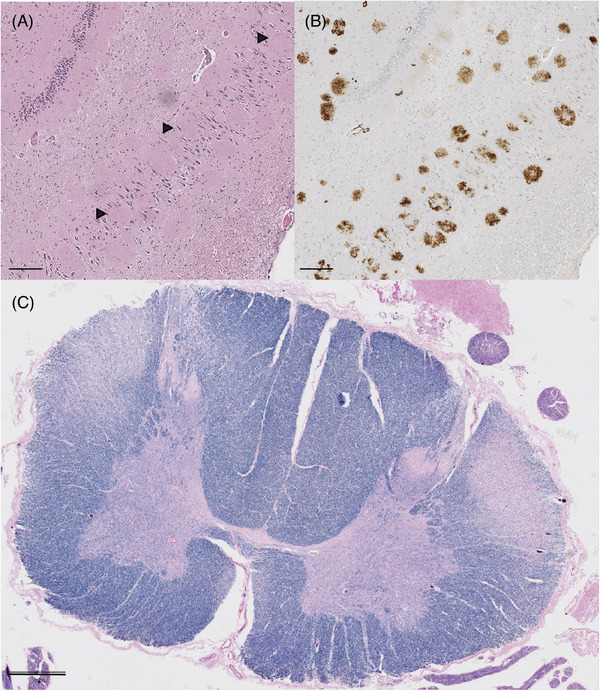
(A) Hippocampus stained with hematoxylin and eosin (H&E). Black arrows show the cotton wool plaques displacing neurons in the pyramidal cell layer; scale bar 200 µm. (B) Amyloid beta (Aβ) immunohistochemical staining of hippocampus section displayed in A; scale bar 200 µm. (C) Cross section of spinal cord of Case 2 demonstrating decreased Luxol fast blue staining for myelin in the lateral corticospinal tracts; scale bar 1 mm.
FIGURE 4.
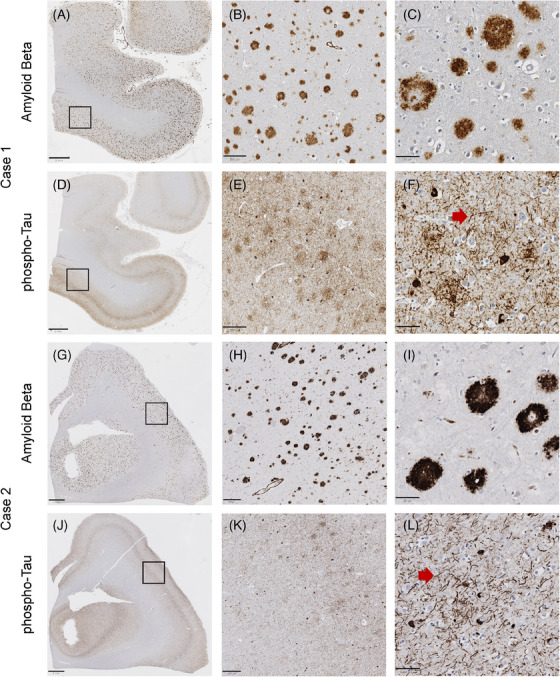
Amyloid beta (Aβ) and tau deposition in primary motor cortex of Cases 1 and 2. Low‐ (A), medium‐ (B), and high‐ (C) power views of Aβ immunohistochemical staining demonstrating frequent neocortical amyloid plaques from Case 1. Low‐ (G), medium‐ (H), and high‐ (I) power views of Aβ immunohistochemical staining demonstrating frequent neocortical amyloid plaques from Case 2. Low‐ (D), medium‐ (E), and high‐ (F) power views of ‐tau immunohistochemical staining demonstrating frequent neocortical neuritic plaques, neurofibrillary tangles, and dystrophic neurites from Case 1. Low‐ (J), medium‐ (K), and high‐ (L) power views of p‐tau immunohistochemical staining demonstrating frequent neocortical neuritic plaques, neurofibrillary tangles, and dystrophic neurites from Case 2. (A, D, G, J) Scale bar 2 mm. (B, E, H, K) Scale bar 200 µm. (C, F, I, L) Scale bar 50 µm.
LFB/PAS myelin staining demonstrated myelin pallor in the lateral corticospinal tracts at cervical, thoracic, and lumbar levels of the spinal cord (Figure 3); there was also slight myelin pallor in the midportion of the cerebral peduncle, corresponding to the location of the corticospinal tract. Phosphorylated neurofilament staining of the spinal cord demonstrated a slight decrease in the density of staining in the lateral corticospinal tracts (Figures S1 and S2).
No Lewy bodies, α‐synuclein, or TDP‐43 immunoreactivity was observed in the motor cortex, medulla, or spinal anterior horn. Bunina bodies were not observed in spinal cord sections. 13 , 14 , 15
3.4.2. Case 2 (sibling of index case)
The brain weighed 1143 g and displayed moderate generalized atrophy, most notably in the fronto‐parietotemporal regions. Sections of the cerebral hemispheres showed moderate dilation of the lateral ventricles and moderate hippocampal atrophy. The cerebral cortex exhibited slight thinning across its extent. The caudate nucleus was slightly diminished in size. Both the substantia nigra and locus coeruleus had moderate depigmentation. The cerebellar folia, dentate nucleus, and white matter were reduced in size. The spinal cord was grossly normal.
Light microscopy demonstrated abundant p‐tau immunoreactive NFTs, NPs, and NTs in the hippocampus and neocortex (including primary motor cortex), corresponding to Braak stage VI and CERAD plaque grade “frequent.” This pathology extended to the striatum and midbrain tegmentum. In addition, there were abundant CWPs in the hippocampus, neocortex (including the primary motor cortex) (Figure 4), and striatum, mirroring the distribution of p‐tau immunoreactive NPs, alongside amyloid angiopathy. Aβ plaques and amyloid angiopathy were also present in the midbrain tegmentum, in the gray matter of the cerebellum (Thal stage 5), and in the gray matter of the spinal cord. The density of tangles, plaques, and dystrophic neurites was similar between the primary motor cortex and the non‐motor cortical areas examined. The ABC score was A3 B3 C3, corresponding to “high‐level AD neuropathologic change” by 2012 NIA/AA guidelines. There was no tau immunoreactivity in sections of the spinal cord.
LFB/PAS staining showed myelin pallor in the lateral corticospinal tracts at the cervical, thoracic, and lumbar levels of the spinal cord; slight pallor was also noted in the medullary pyramid. Phosphorylated neurofilament staining of the spinal cord revealed only a faint decrease in the density of staining in the lateral corticospinal tracts (Figures S1 and S2).
No Lewy bodies, α‐synuclein, or TDP‐43 immunoreactivity was observed in the motor cortex, medulla, or spinal anterior horn. Bunina bodies were not observed in spinal cord sections
4. DISCUSSION
In this study, we explored the clinical and neuropathological characteristics of a Colombian family exhibiting a PLS phenotype linked to the PSEN1 Pro284Leu variant. Our findings provide a compelling link between AD, CWP, and upper motor neuron syndrome in these subjects.
The association of AD, CWP, and SP has been discussed widely following the initial report by Crook et al. 16 Subsequent documentation by Tabira et al. 17 detailed five Japanese patients with SP and CWP, including an individual carrying the PSEN1 Pro284Leu variant. More recently, other PSEN1 variants (Pro88Leu and Leu166Pro) were identified in association with PLS. 11
The clinical manifestations in this family more closely resemble those of PLS rather than SP. Their symptoms initially present unilaterally and involve bulbar and pseudobulbar syndromes from the outset. They experience loss of strength in both lower and upper extremities, cognitive decline, and behavioral alterations. However, there are no gaze abnormalities, signs of parkinsonism, or lower motor neuron involvement to suggest progressive supranuclear palsy (PSP) or amyotrophic lateral sclerosis (ALS). Thus our patients meet the criteria for defined PLS (age ≥25 years, symptoms of progressive upper motor neuron (UMN) disease for at least 2 years in at least two of the three regions: lower extremity, upper extremity, and bulbar; and absence of significant active lower motor neuron degeneration 4 or more years from symptom onset).
Memory impairment was a common finding across all cases. Cases 1 and 3, diagnosed with MCI, experienced difficulties in memory recall during controlled learning exercises, indicating potential disruptions in information storage mechanisms. Case 2, diagnosed with dementia, exhibited extensive deficits in verbal and visual memory along with other cognitive impairments. All three cases also had trouble with visuoconstructional tasks.
Although some studies, such as those of Piquard, 18 Caselli, 19 and De Vries, 20 highlight the influence of executive functions on memory tasks in non‐dementia patients, including those with ALS, our findings suggest that memory deficits might not stem solely from executive dysfunction but could also be indicative of direct information storage issues. 21
CSF levels of amyloid markers (Aβ42 and Aβ42‐40 ratio) in the three cases were consistent with AD (Table 2). Specifically, Case 2 exhibited an abnormal increase in CSF p‐tau levels, indicating a more extensive degree of neurodegeneration.
TABLE 2.
Biomarkers in cerebrospinal fluid from each of the assessed siblings.
| Case 1 | Case 2 | Case 3 | Reference values | |
|---|---|---|---|---|
| Age at testing (years) | 45 | 36 | 35 | NA |
| Aβ 1‐42 (pg/mL) | 55 | 81 | 72 | >600 pg/mL |
| Aβ 1‐42/1‐40 ratio | 0.02 | 0.02 | 0.02 | >0.07 |
| Total tau (pg/mL) | 269 | 1058 | 259 | <385 pg/mL |
| Phosphorylated tau (pg/mL) | 54 | 221 | 50 | <65 pg/mL |
Abbreviations: Aβ, amyloid beta; NA, not applicable.
Neuropathological assessment of Cases 1 and 2 revealed significant similarities with other PSEN1 SP cases in the existing literature, including the presence of CWPs, cerebral amyloid angiopathy, and degeneration of the corticospinal tracts and motor cortex, features also associated with PLS. 10 , 22 , 23 , 24 , 25 Although PLS has been observed in concomitance with PSP, the extensive presence of Aβ is against it. 26 , 27
The neuropathological findings in the motor cortex and the lateral column pallor in these patients point to death or dysfunction of primary motor cortex neurons resulting in Wallerian degeneration. We do not believe that the observed condition is due to spinal cord white matter toxicity from myelin deposits for the following reasons. First, there is no evidence of amyloid deposition in the spinal cord white matter. Second, the observed pallor in myelin stains, and in at least one case, the neurofilament stain as well, is confined to the lateral corticospinal tracts. This indicates an isomorphic pattern, where changes align precisely with a specific neuroanatomic tract. Furthermore, there is pronounced involvement of the brain across multiple structures beyond the corticospinal tract, which contrasts with the lesser involvement observed in various structures of the spinal cord. Given that the burden of tangles in the neocortex is not extreme, whereas the amount of amyloid deposition (especially in the form of CWPs) is very high, we believe that the cerebral cortical amyloid burden more than the p‐tau burden is the more likely basis of upper motor neuron dysfunction. A plausible explanation is that there may be an element of amyloid toxicity causing neuronal dysfunction without a requirement for significant tangle formation.
In this study, we examined a Colombian family with PLS and identified the pathogenic PSEN1 Pro284Leu variant through a combination of clinical assessments, genetic sequencing, neuroimaging, biomarker analysis, and neuropathological examination. The findings provide insights into the genetic basis and pathological mechanisms of PLS associated with PSEN1 variants, underscoring the potential genotypic and neuropathological overlap among various neurodegenerative diseases. We conclude that PLS should be considered as a motor syndrome linked to AD, which reinforces the importance of genetic testing in diagnosing hereditary motor neuron syndromes. 25 Future research will help understand the nature of the relationship between PLS and AD, which will be of great relevance, both for identifying therapeutic targets and for selecting patients for clinical trials that target these neurodegenerative diseases.
CONFLICT OF INTEREST STATEMENT
J.A.‐U. has grants from the Tau Consortium. Y.T.Q. consults for Biogen. F.L. consults for Biogen and Viewmind and has grants from the National Institutes of Health (NIH), Multi‐Partner Consortium to Expand Dementia Research in Latin America (ReD‐Lat), Alzheimer's Association, Biogen, Dominantly Inherited Alzheimer Network Trials Unit (DIAN‐TU), Dominantly Inherited Alzheimer Network Observational Study (DIAN‐Obs), The Latin American Research consortium on the GEnetics of Parkinson's Disease (LARGE‐PD), and Enroll‐HD. K.S.K. consults for ADRx and Expansion Therapeutics and is a member of the Tau Consortium board of directors. The remaining authors have no interest to declare. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Each patient or their designated proxy provided informed consent for all evaluations and assessments conducted. This consent included explicit permission to publish the findings. Ethics committee/institutional review board (IRB) of the Medical Research Institute, Universidad de Antioquia gave ethical approval for this work (National Institutes of Health [NIH] Office for Human Research Protections IRB identification code: IRB00012257).
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We would like to thank our patients and their families who consented to participate in this study and allowed us to publish their information in a scientific journal. In addition, we thank Carlos M. Cardona for assisting with staining and tissue processing, Dr. Russell O. Kosik for the neuroimaging assessment, and Dr. Mark H. Ellisman, Willy W. Wong, and Steven T. Peltier at the National Center for Microscopy and Imaging Research (NCMIR), University of California San Diego, for the support of histology images visualization. This study was funded by the Tau Consortium (F.L.), National Institutes of Health (NIH) grant 1RF1AG062479‐01*** (F.L., K.S.K., T.G.B., and C.LW.) and R01AG054671 (Y.T.Q. and F.L.). Y.T.Q. is supported by the NIH (R01AG054671, RF1AG077627, and RM1NS132996), the Alzheimer's Association, and the Massachusetts General Hospital Executive Committee on Research.
Acosta‐Uribe J, Villegas‐Lanau A, Vallejo D, et al. Primary lateral sclerosis associated with PSEN1 Pro284Leu variant in a Colombian family: Clinical and neuropathological features. Alzheimer's Dement. 2024;20:6384–6394. 10.1002/alz.14133
Charles L. White and Margarita Giraldo‐Chica contributed equally
DATA AVAILABILITY STATEMENT
Histological images from the comprehensive neuropathological analyses of Cases 1 and 2 are available at the CIL Cell Image Library (DOI: https://doi.org/10.7295/W9P20489). These materials may be used and shared by anyone, provided that they are not modified, used for non‐commercial purposes only, and that the original authors are duly credited and cited.
REFERENCES
- 1. Alzheimer's Association . 2022 Alzheimer's Disease Facts and Figures. n.d. [DOI] [PubMed]
- 2. Lippa CF, Saunders AM, Smith TW, et al. Familial and sporadic Alzheimer's disease: neuropathology cannot exclude a final common pathway. Neurology. 1996;46:406‐412. doi: 10.1212/wnl.46.2.406 [DOI] [PubMed] [Google Scholar]
- 3. Ryan NS, Rossor MN. Correlating familial Alzheimer's disease gene mutations with clinical phenotype. Biomark Med. 2010;4:99‐112. doi: 10.2217/bmm.09.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ryan NS, Nicholas JM, Weston PSJ, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer's disease: a case series. Lancet Neurol. 2016;15:1326‐1335. doi: 10.1016/S1474-4422(16)30193-4 [DOI] [PubMed] [Google Scholar]
- 5. Statland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mitsumoto H. Primary lateral sclerosis. Neurol Clin. 2015;33:749‐760. doi: 10.1016/j.ncl.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turner MR, Barohn RJ, Corcia P, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020;91:373‐377. doi: 10.1136/jnnp-2019-322541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol. 2014;34:293‐305. doi: 10.1055/s-0034-1386767 [DOI] [PubMed] [Google Scholar]
- 8. Brugman F, Veldink JH, Franssen H, et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult‐onset upper motor neuron syndromes. Arch Neurol. 2009;66:509‐514. [DOI] [PubMed] [Google Scholar]
- 9. Fullam T, Statland J. Upper motor neuron disorders: primary lateral sclerosis, upper motor neuron dominant amyotrophic lateral sclerosis, and hereditary spastic paraplegia. Brain Sci. 2021;11:611. doi: 10.3390/brainsci11050611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ringman JM, Dorrani N, Fernández SG, et al. Characterization of spastic paraplegia in a family with a novel PSEN1 mutation. Brain Commun. 2023;5:fcad030. doi: 10.1093/braincomms/fcad030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vázquez‐Costa JF, Payá‐Montes M, Martínez‐Molina M, et al. Presenilin‐1 mutations are a cause of primary lateral sclerosis‐like syndrome. Front Mol Neurosci. 2021;14:721047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Acosta‐Uribe J, Aguillón D, Cochran JN, et al. A neurodegenerative disease landscape of rare mutations in Colombia due to founder effects. Genome Med. 2022;14. doi: 10.1186/s13073-022-01035-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479‐486. doi: 10.1212/wnl.41.4.479 [DOI] [PubMed] [Google Scholar]
- 14. Love S, Chalmers K, Ince P, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post‐mortem brain tissue. Am J Neurodegener Dis. 2014;3:19‐32. [PMC free article] [PubMed] [Google Scholar]
- 15. Braak H, Braak E. Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol (Berl). 1991;82:239‐259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 16. Crook R, Verkkoniemi A, Perez‐Tur J, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4:452‐455. doi: 10.1038/nm0498-452 [DOI] [PubMed] [Google Scholar]
- 17. Tabira T, Chui DH, Nakayama H, Kuroda S, Shibuya M. Alzheimer's disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70:367‐372. doi: 10.1002/jnr.10392 [DOI] [PubMed] [Google Scholar]
- 18. Piquard A, Le Forestier N, Baudoin‐Madec V, et al. Neuropsychological changes in patients with primary lateral sclerosis. Amyotroph Lateral Scler Off Publ World Fed Neurol Res Group Mot Neuron Dis. 2006;7:150‐160. doi: 10.1080/17482960600680371 [DOI] [PubMed] [Google Scholar]
- 19. Caselli RJ, Smith BE, Osborne D. Primary lateral sclerosis: a neuropsychological study. Neurology. 1995;45:2005‐2009. doi: 10.1212/wnl.45.11.2005 [DOI] [PubMed] [Google Scholar]
- 20. De Vries BS, Rustemeijer LMM, Bakker LA, et al. Cognitive and behavioural changes in PLS and PMA:challenging the concept of restricted phenotypes. J Neurol Neurosurg Psychiatry. 2019;90:141‐147. doi: 10.1136/jnnp-2018-318788 [DOI] [PubMed] [Google Scholar]
- 21. Machts J, Bittner V, Kasper E, et al. Memory deficits in amyotrophic lateral sclerosis are not exclusively caused by executive dysfunction: a comparative neuropsychological study of amnestic mild cognitive impairment. BMC Neurosci. 2014;15:83. doi: 10.1186/1471-2202-15-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolind S, Sharma R, Knight S, Johansen‐Berg H, Talbot K, Turner MR. Myelin imaging in amyotrophic and primary lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2013;14:562‐573. doi: 10.3109/21678421.2013.794843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lyoo CH, Cho H, Choi JY, et al. Tau accumulation in primary motor cortex of variant Alzheimer's disease with spastic paraparesis. J Alzheimers Dis JAD. 2016;51:671‐675. doi: 10.3233/JAD-151052 [DOI] [PubMed] [Google Scholar]
- 24. Mackenzie IRA. Neuropathology of primary lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2020;21:47‐51. doi: 10.1080/21678421.2020.1837173 [DOI] [PubMed] [Google Scholar]
- 25. Chelban V, Breza M, Szaruga M, et al. Spastic paraplegia preceding PSEN1‐related familial Alzheimer's disease. Alzheimers Dement Diagn Assess Dis Monit. 2021;13:e12186. doi: 10.1002/dad2.12186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagao S, Yokota O, Nanba R, et al. Progressive supranuclear palsy presenting as primary lateral sclerosis but lacking parkinsonism, gaze palsy, aphasia, or dementia. J Neurol Sci. 2012;323:147‐153. doi: 10.1016/j.jns.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 27. King A, Curran O, Al‐Sarraj S. Atypical progressive supranuclear palsy presenting as primary lateral sclerosis. J Neurol Sci. 2013;329:69. doi: 10.1016/j.jns.2013.03.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
Histological images from the comprehensive neuropathological analyses of Cases 1 and 2 are available at the CIL Cell Image Library (DOI: https://doi.org/10.7295/W9P20489). These materials may be used and shared by anyone, provided that they are not modified, used for non‐commercial purposes only, and that the original authors are duly credited and cited.