

Abstract
INTRODUCTION
In Down syndrome (DS), white matter hyperintensities (WMHs) are highly prevalent, yet their topography and association with sociodemographic data and Alzheimer's disease (AD) biomarkers remain largely unexplored.
METHODS
In 261 DS adults and 131 euploid controls, fluid‐attenuated inversion recovery magnetic resonance imaging scans were segmented and WMHs were extracted in concentric white matter layers and lobar regions. We tested associations with AD clinical stages, sociodemographic data, cerebrospinal fluid (CSF) AD biomarkers, and gray matter (GM) volume.
RESULTS
In DS, total WMHs arose at age 43 and showed stronger associations with age than in controls. WMH volume increased along the AD continuum, particularly in periventricular regions, and frontal, parietal, and occipital lobes. Associations were found with CSF biomarkers and temporo‐parietal GM volumes.
DISCUSSION
WMHs increase 10 years before AD symptom onset in DS and are closely linked with AD biomarkers and neurodegeneration. This suggests a direct connection to AD pathophysiology, independent of vascular risks.
Highlights
White matter hyperintensities (WMHs) increased 10 years before Alzheimer's disease symptom onset in Down syndrome (DS).
WMHs were strongly associated in DS with the neurofilament light chain biomarker.
WMHs were more associated in DS with gray matter volume in parieto‐temporal areas.
Keywords: Alzheimer's disease, Down syndrome, magnetic resonance imaging, neuroimaging, small vessel disease, white matter hyperintensities
1. BACKGROUND
Down syndrome (DS) is a genetic condition caused by an extra copy of chromosome 21 that has been associated with an ultra‐high risk of developing Alzheimer's disease (AD), with virtually all individuals with DS showing full‐blown AD neuropathology by the age of 40. 1 , 2 A key factor driving this increased risk is the triplication of the amyloid precursor protein (APP) gene, which is necessary and sufficient to cause AD pathology. 3 As a result of the overexpression of APP, amyloid beta (Aβ) plaques, a hallmark feature of AD, accumulate early and rapidly in the brain and eventually lead to cognitive impairment and dementia. 4 Thus, DS is currently considered a genetically determined form of AD. 2
It is increasingly recognized that studying AD in individuals with DS can provide valuable insights into its pathogenesis. Alongside the predictable course of AD, 5 adults with DS present a distinct profile of vascular risk factors from aging in the general population, 6 as they are relatively protected from some conventional age‐related vascular risk factors, such as hypertension. 7 Thus, this population provides a unique opportunity to further understand the emergence of cerebrovascular diseases, specifically small vessel disease (SVD), in the context of AD without the confounds of age‐related vascular risk factors.
One of the main imaging markers of SVD is white matter hyperintensities (WMHs) of presumed vascular origin. 8 WMHs are areas of increased signal on T2‐weighted or fluid‐attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI). 9 , 10 They can reflect distinct pathological processes related to SVD, including increased interstitial fluid, demyelination, gliosis, loss of fibers and oligodendrocyte precursor cells, and failure of the glymphatic system. 8 , 11 These lesions are frequently found in healthy older adults and their prevalence and severity increase with age. 12 The occurrence of WMHs is also increased in individuals with sporadic 10 , 13 and other genetic forms of AD. 14 , 15 In adults with DS, the prevalence and severity of WMHs are increased in both asymptomatic and symptomatic DS with respect to euploid cognitively unimpaired controls 16 and WMH volume increases along the AD continuum. 17 Altogether, this suggests that these lesions may be related to AD development. However, little is known about the spatial distribution of WMHs in DS, which could provide insight into their etiology. Moreover, to our knowledge, no study has yet assessed the relationships between WMHs and fluid and neuroimaging biomarkers of AD in DS.
In the present study, we aimed to better characterize WMHs and their relationship to sociodemographic, clinical, genetic, and AD biomarkers in a large population‐based cohort of adults with DS. We particularly focused on regional WMH burden along the AD continuum and its associations with AD biomarkers. We hypothesize that the early accumulation of AD pathologies in DS will lead to WMHs years before symptoms onset, which will eventually increase as the disease progresses.
2. METHODS
2.1. Study design and participants
This cross‐sectional study was conducted at a single center, where adults with DS aged ≥ 18 were recruited from the population‐based Down‐Alzheimer Barcelona Neuroimaging Initiative (DABNI) cohort. 2 A sample of euploid cognitively unimpaired control (HC) individuals was also included from the Sant Pau Initiative on Neurodegeneration (SPIN) cohort. 18 The study was approved by the Sant Pau Ethics Committee following the standards for medical research in humans recommended by the Declaration of Helsinki. All participants or their legally authorized representatives gave written informed consent before enrollment.
The participants with DS were screened for apolipoprotein E (APOE) haplotype and underwent a comprehensive neurological and neuropsychological evaluation, including, among other tests, the Cambridge Cognitive Examination for Older Adults with Down Syndrome (CAMCOG‐DS) Spanish version for assessing global cognition. 19 , 20 The CAMCOG‐DS is a cognitive battery that evaluates orientation, language, memory, attention, praxis, abstract thinking, and perception.
As in previous studies, 2 , 21 participants were clinically classified during a consensus meeting between neurologists and neuropsychologists, masked from biomarker data, into the following groups: asymptomatic (aDS) when there was no clinical or neuropsychological suspicion of AD‐related cognitive decline, prodromal AD (pDS) when there was evidence of cognitive decline due to AD, but no significant impact on baseline activities of daily living, and AD dementia (dDS) when the cognitive decline impaired daily activities. This functional status, differentiating pDS and dDS, was assessed using anamnesis, Dementia Questionnaire for Persons with Mental Retardation, and the Cambridge Examination for Mental Disorders of Older People with Down Syndrome and Others with Intellectual Disabilities. Additionally, the Diagnostic and Statistical Manual of Mental Disorder, Fifth Edition, was used to stratify the level of intellectual disability (ID) into mild, moderate, or severe/profound based on the individuals’ best‐ever level of functioning. 22
The HC group are volunteers, usually spouses or children of patients from the Sant Pau Memory Unit, that are informed about our studies at the outpatient clinics. They exhibited no cognitive complaints, scoring 0 on the Clinical Dementia Rating scale, and their neuropsychological evaluation was within the normal range for their respective normative groups regarding age and education. None of the participants in the HC group reported any neurological or psychiatric disorders, or other major medical illnesses. 18 We selected only the HC within the same age range as the DS group, from 18 to 63 years.
RESEARCH IN CONTEXT
Systematic review: White matter hyperintensities (WMHs) are recognized as vascular markers that contribute to cognitive decline. Previous studies have shown that WMHs are prevalent in Down syndrome (DS), a genetically determined form of Alzheimer's disease (AD) not complicated by some traditional age‐related vascular risk factors. However, the evolution and topography of WMHs across age and disease severity, as well as their relationship to AD pathophysiology, have not been thoroughly studied in DS.
Interpretation: WMHs increased with age and AD clinical stage, and were correlated with cerebrospinal fluid AD biomarkers and gray matter volume in AD vulnerable areas. Our findings suggest that WMHs might be intrinsically related to AD pathophysiological processes in DS.
Future directions: Post mortem studies could shed light on the pathological substrates of WMHs in DS and other genetically determined forms of AD.
2.2. Neuroimaging data
2.2.1. MRI acquisition
T1‐weighted and FLAIR images were acquired on a 3T Philips‐Achieva scanner at Hospital del Mar (Barcelona, Spain) or a 3T Siemens Prisma scanner at Hospital Clinic (Barcelona, Spain). The detailed acquisition parameters of the imaging protocols are provided in Table S1 in supporting information.
The quality of each T1‐weighted and FLAIR image was visually assessed. MRI scans of unsatisfactory quality in terms of low transverse resolution or low structural visibility due to movement or other sources of noise were excluded (n = 21 scans, 4.64%).
2.2.2. Gray matter volume
The Computational Anatomy Toolbox (CAT12) 23 for the Statistical Parametric Mapping 12 (SPM12; Welcome Center for Human Neuroimaging) toolbox was used to preprocess the structural T1‐weighted image and extract gray matter (GM) and total intracranial volumes.
2.2.3. WMH segmentation
For the segmentation of WMHs, raw FLAIR images were first coregistered onto their corresponding T1‐weighted scan using Advanced Normalization Tools (ANTs) 24 (Figure S1A in supporting information). Then, WMHs were segmented in the MRI native space using the lesion prediction algorithm (LPA) 25 implemented in the Lesion Segmentation Toolbox for SPM12. Specifically, the LPA method computed a WMH probability map for each participant based on a logistic regression algorithm to classify each voxel as either a lesion or non‐lesion. The probability maps were then binarized by applying a threshold of 0.3 (Figure S1B).
2.2.4. White matter parcellation
White matter (WM) was parcellated for each subject using an in‐house pipeline inspired by the bull's‐eye classification system previously developed by Sudre et al. 26 Specifically, two distinct sets of parcellations were created: concentric layers and lobar regions (for details, see supporting information). For the concentric layers, we followed the approach proposed by Jiménez‐Balado et al. 27 and obtained a normalized distance map reflecting the distance of each WM voxel to the lateral and third ventricles (Figure S1C). For the lobar regions, we used the anatomical Hammers atlas to label the WM according to the lobes (frontal, parietal, temporal, occipital, and basal ganglia; Figure S1D). The normalized distance map was applied to each lobar region and subsequently divided into four concentric layers of similar volume. These layers spanned from the periventricular regions to juxtacortical areas. Regional WMH volumes were then extracted and summarized as an infographic in a bull's‐eye plot (e.g., see Figure S1). Because different MRI acquisition protocols were used to acquire the data, we harmonized total and regional WMH volumes using the ComBat harmonization method 28 as implemented in the R library neuroCombat.
2.3. Fluid biomarkers
A subset of participants underwent, within 1 year of the MRI acquisition, a lumbar puncture to acquire cerebrospinal fluid (CSF) sampling, as previously described. 21 , 29 Concentration levels of Aβ40 (n = 251), Aβ42 (n = 251), and tau phosphorylated at threonine 181 (pTau181; n = 251) were quantified using a commercially available immunoassay in a fully automated platform (Lumipulse; Fujirebio‐Europe), following a previously published protocol. 18 CSF glial fibrillary acidic protein (GFAP; n = 183) concentrations were measured using the SR‐X single molecule array. CSF neurofilament light chain (NfL; n = 267) and YKL‐40 (also known as chitinase 3‐like 1; n = 236) levels were measured with enzyme‐linked immunosorbent assay (ELISA; NF‐Light Assay; UmanDiagnostics) according to the manufacturer's recommendations. CSF Aβ42/Aβ40 ratio and p‐tau181 were used to stratify individuals with DS according to the presence of amyloid (A+) and tau (T+) pathologies using previously established cutoff values (i.e., CSF Aβ42/Aβ40 < 0.062; CSF p‐tau181 > 63 pg/mL 30 ).
2.4. Statistical analyses
All statistical analyses were performed with R software, version 4.3.1. Differences in baseline sociodemographic characteristics between DS and HC participants were assessed using the compareGroups library. 31 This package tests the normality of continuous variables and subsequently performs parametric or non‐parametric statistical tests accordingly. Chi‐squared tests were used for categorical variables, such as sex and APOE ε4 status, while Mann–Whitney tests were applied for continuous variables, including age, and CSF biomarkers, because they did not follow normality. To reduce skewness, total and regional volumes of WMHs, as well as concentrations of CSF biomarkers, were log‐transformed.
To examine the association between WMH volume and age, we performed locally estimated scatterplot smoothing (LOESS) curves with a tricubic weight function and a span parameter of 0.75, providing an informative representation of the data. To assess the effect of factors such as sex, APOE ε4 status, ID, and AD clinical status on the distribution of WMH volume, we performed either a Mann–Whitney or Kruskal–Wallis test followed by the Dunn test as post hoc analysis. To investigate the relationship between total and regional WMH volume and CSF biomarkers, we performed Spearman correlation tests.
We used univariate linear regression models to assess the predictive capacity of individual CSF biomarkers on total WMH volume in the whole DS cohort, but also in aDS and symptomatic DS (sDS; comprising pDS and dDS) separately. Subsequently, we conducted a multivariate analysis incorporating all the CSF biomarkers to assess their relative contribution to the prediction of WMH volume. Additionally, we repeated both univariate and multivariate models including age as a predictor to evaluate its effect in relation to the CSF biomarkers. The standardized regression coefficients were used to compare the strength of the association between the predictors and total WMH volume. The threshold for significance was set at p = 0.05 and Bonferroni corrections were applied in regional volume analyses to account for multiple comparisons.
Finally, we used voxelwise linear models to compare the association of GM volume and WMHs in the DS cohort. Analyses were performed in a mask that excluded non‐GM voxels, and the statistical models were corrected for total intracranial volume and the MRI acquisition protocol. Voxelwise results are presented at a corrected threshold p < 0.05 (family‐wise error [pFWE], cluster size k > 100 mm3).
3. RESULTS
3.1. Population
The study included 261 adults with DS from the DABNI cohort and 131 HC from the SPIN study (see Table 1 for demographic data). Individuals in the DS group spanned the whole AD continuum; 158 were aDS, 41 pDS, and 62 dDS. Regarding the level of ID, 76 DS subjects had mild ID, 149 moderate ID, and 34 severe/profound ID. The HC group was significantly older than the DS group (W = 24666, p < 0.05). There was also a significant difference in the sex distribution, with fewer male participants in the HC group (29.01%) than in the DS group (58.62%, p < 0.001). No significant between‐group differences were found for APOE ε4 status. Only 1.5% of individuals with DS presented hypertension, a significantly lower proportion compared to the HC group (21.9%, p < 0.001). Additionally, 18% had dyslipidemia and 4.7% had diabetes mellitus, versus 27% (p = 0.18) and 3.12% (p = 0.74), respectively. As expected, individuals with DS demonstrated lower values in CSF for the Aβ42/Aβ40 ratio and higher concentrations of pTau181, NfL, and GFAP than HC (all p < 0.001). CSF YKL‐40 levels were not significantly different across groups (p = 0.50).
TABLE 1.
Study participants.
| HC | DS | p‐value | |
|---|---|---|---|
| N = 131 | N = 261 | ||
| Age, y, median [IQR] | 53.65 [48.70;57.81] | 46.96 [37.77;51.81] | <0.001 |
| Sex, no. (%) | <0.001 | ||
| Female | 93 (70.99%) | 108 (41.38%) | |
| APOE ε4 status, no. (%) | 0.15 | ||
| ε4+ | 36 (27.69%) | 51 (20.56%) | |
| Intellectual disability, no. (%) | |||
| Mild | 76 (29.46%) | ||
| Moderate | 148 (57.36%) | ||
| Severe/profound | 34 (13.18%) | ||
| AD diagnostic group, no. (%) | |||
| aDS | 158 (60.54%) | ||
| pDS | 41 (15.71%) | ||
| dDS | 62 (23.75%) | ||
| Vascular risk factors, no (%) | |||
| Hypertension (N = 323) | 14 (21.88%) | 4 (1.54%) | <0.001 |
| Dyslipidemia (N = 323) | 17 (26.56%) | 47 (18.15%) | 0.18 |
| Diabetes mellitus (N = 322) | 2 (3.12%) | 12 (4.65%) | 0.74 |
| CAMCOG‐DS, median [IQR] | 69.00 [51.00;82.00] | ||
| CSF biomarkers, pg/mL, median [IQR] | |||
| Aβ42/Aβ40 (N = 251) | 0.11 [0.10;0.11] | 0.06 [0.04;0.08] | <0.001 |
| pTau181 (N = 251) | 32.30 [24.90;41.40] | 55.05 [24.52;133.55] | <0.001 |
| NfL (N = 267) | 349.80 [298.92;418.61] | 542.30 [304.32;871.20] | <0.001 |
| GFAP (N = 183) | 3.35 [3.14;3.50] | 3.61 [3.40;3.78] | <0.001 |
| YKL‐40 (N = 236) | 2.20 [2.11;2.29] | 2.26 [2.03;2.37] | 0.50 |
Notes: Data are n (%) or median (IQR).
Abbreviations: Aβ42/Aβ40, concentration ratio between amyloid beta peptide 1‐42 and amyloid beta peptide 1‐40 (pg/mL); AD, Alzheimer's disease; aDS, asymptomatic Down syndrome; APOE, apolipoprotein E; CAMCOG‐DS, Cambridge Cognitive Examination for Older Adults with Down Syndrome; CSF, cerebrospinal fluid; dDS, Down syndrome with Alzheimer's disease dementia; DS, Down syndrome; GFAP, glial fibrillary acidic protein concentration (pg/mL); HC, euploid cognitively unimpaired controls; IQR, interquartile range; NfL, neurofilament light chain concentration (pg/mL); pDS, prodromal Down syndrome; pTau181, tau phosphorylated at threonine 181 concentration (pg/mL); YKL‐40, chitinase 3‐like 1.
3.2. Associations between WMH volume and sociodemographic, clinical, and genetic data
Total WMH volume was positively associated with age in both HC (rho = 0.36, p < 0.001) and adults with DS (rho = 0.67, p < 0.001). Looking at the LOESS curves (Figure 1A), we found an earlier increase in total WMH volume in DS with age. Specifically, the regression lines between DS and HC started diverging at age 40, and the confidence interval (CI) ceased to overlap at the age of 43.8. Regional associations revealed that, in HC, a significant relationship with age was mainly observed in the first layer (Figure 1B and Table S2 in supporting information for more details). In contrast, regional associations with age were more widespread and stronger in DS, including all lobes. The first and second layers (i.e., the region closest to the ventricles) showed the highest correlation strength and earliest changes (Figures 1B, S2 in supporting information, and Table S2 for more details).
FIGURE 1.
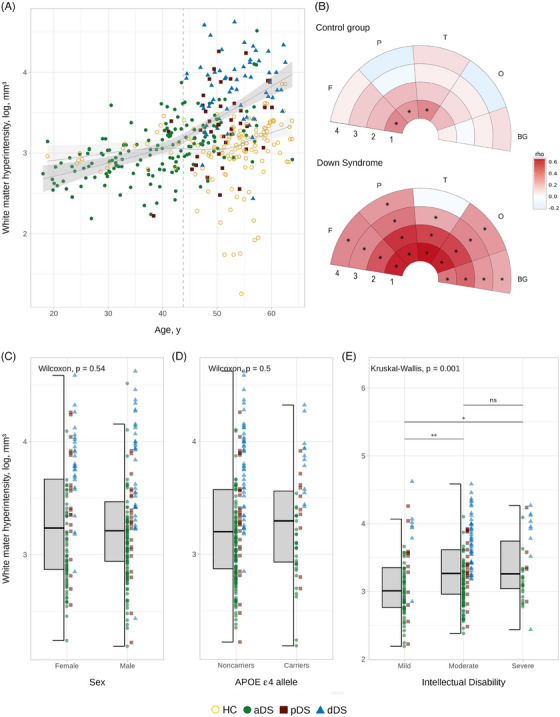
Associations between white matter hyperintensity volume and demographic, clinical, and genetic factors. (A) Association between total WMH volume and age. The points represent individual participants, and the color indicates their clinical diagnosis. Shaded areas represent 95% CI: dark gray represents the age‐related change in DS and light gray in the HC group for visual reference. The dashed line indicates when the CI stops overlapping between DS and HC groups (43.8 years). (B) Spearman correlation coefficients between age and regional WMH volumes. The color scale represents the strength (rho) of Spearman correlation for significant results (p < 0.05). The star (*) indicates results surviving to the Bonferroni correction (α = 0.05, p < 0.0025). (C), (D) and (E) Boxplots showing the effect of sex, APOE haplotype, and intellectual disability, respectively, on the distribution of the total WMH volume in DS. Abbreviations: 1, first layer; 2, second layer; 3, third layer; 4, fourth layer; aDS, asymptomatic Down syndrome; APOE, apolipoprotein E; BG, basal ganglia; CI, confidence interval; DS, Down syndrome; F, frontal; HC, euploid cognitively unimpaired controls; dDS, Down syndrome with Alzheimer's disease dementia; O, occipital; P, parietal; pDS, prodromal Down syndrome; rho, strength of the correlation; T, temporal; WMH, white matter hyperintensity.
In DS, sex and APOE ε4 status did not significantly influence total WMH volume (Figure 1C,D) or the association between total WMH volume and age (Figure S3 in supporting information). However, we found a significant difference according to the degree of ID, with the moderate and severe ID levels showing greater WMH volume than the mild level (Figure 1E). Given that this result was unexpected, we assessed whether it may be explained by differences in clinical status across the ID groups. Results showed a higher proportion of moderate ID in dDS (77.97%) than aDS (50.64%; X 2 [4, N = 259] = 14.462, p < 0.05). Moreover, the effect of ID on WMH volume was no longer significant when groups were stratified by AD clinical diagnosis (p > 0.05; Figure S4 in supporting information) and associations with age appeared similar across ID groups (Figure S3).
3.3. Spatial distribution of WMHs in DS
To assess the spatial distribution of WMHs in DS, we computed a frequency map of WMHs for all adults with DS. Results showed that WMHs had a symmetrical distribution, and were most frequently located in periventricular and deep regions (Figure 2). Specifically, WMHs were predominantly found in the corona radiata (mainly the posterior part), the posterior thalamic radiation, and the corpus callosum, especially in the splenium.
FIGURE 2.
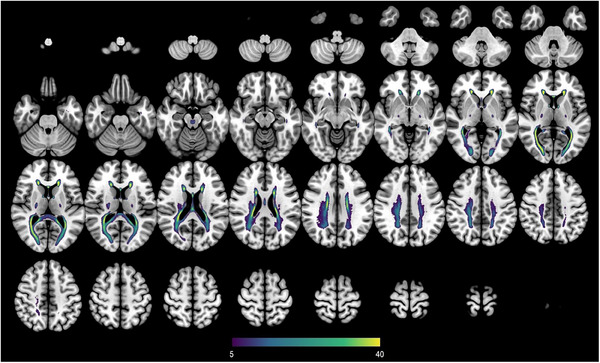
Lesion prevalence map of white matter hyperintensities in Down syndrome. Lesion frequency map of white matter hyperintensities is shown in the Montreal Neurological Institute 152 T1 template. The color of each voxel indicates the percentage of subjects that had white matter hyperintensities in this voxel, ranging from at least 5% (purple) to 40% or more (yellow).
3.4. Effect of AD clinical status on WMH volume
Total WMH volume progressively increased with AD clinical status in DS: dDS and pDS groups demonstrated significantly greater WMH volumes than aDS and HC, and dDS greater volumes compared to pDS. No significant differences were observed between HC and aDS (Figure 3A). Regarding regional WMH volumes, aDS showed significantly greater volumes in the temporal lobe with respect to the HC, while HC showed greater WMH in the occipital lobe and basal ganglia (Figure 3C). pDS presented significantly more WMH volume than aDS in the first and second WM layers, and in all lobes (Figure 3B,C). In the other regions (i.e., layers 3–4, and basal ganglia), there was a visible trend toward higher volumes in pDS compared to aDS, although statistical significance was not reached (Figure 3B‐D). dDS consistently had greater WMH volumes compared to pDS in all regions of interest, except in the fourth WM layer (Figure 3B‐D). Analyses considering both the concentric layers and lobar regions showed similar results, where the first WM layer and the occipital lobes showed the greatest WMH load in all the diagnostic groups. The dDS groups demonstrated both higher and more widespread WMH load compared to other disease stages (Figure 3E).
FIGURE 3.
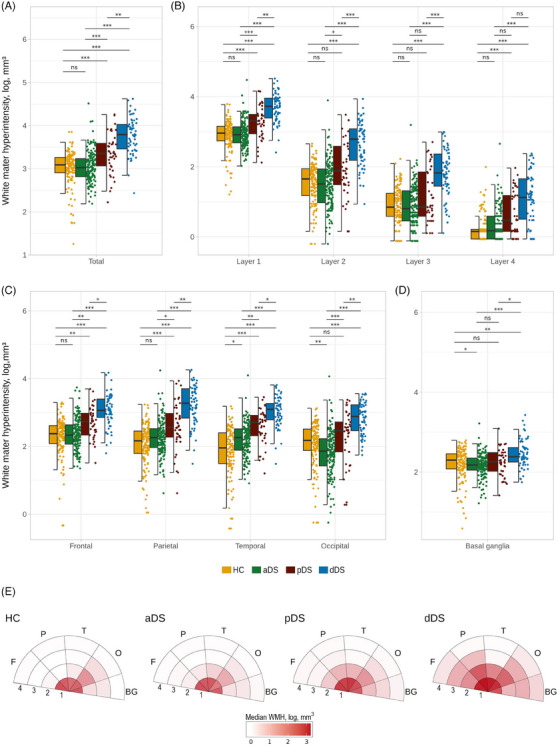
Between‐group differences for total and regional white matter hyperintensity volumes. (A), (B), (C) and (D) The boxplots illustrate WMH volumes across the brain regions. Significant results at p < 0.05 using the Bonferroni correction. (E) Median of WMH volume per region represented in bull's‐eye plot. Colors represent the burden of WMH volume in log mm3, darker colors indicate higher WMH load. Abbreviations: ns, no significance; *p < 0.05; **p < 0.01; ***p < 0.001; 1, first layer; 2, second layer; 3, third layer; 4, fourth layer; aDS, asymptomatic Down syndrome; BG, basal ganglia; dDS, Down syndrome with Alzheimer's disease dementia; DS, Down syndrome; F, frontal; HC, euploid cognitively unimpaired controls; O, occipital; P, parietal; pDS, prodromal Down syndrome; T, temporal; WMH, white matter hyperintensity.
3.5. Associations between WMH volume and CSF biomarkers
In DS, total WMH volume was negatively related to CSF Aβ42/Aβ 40 ratio (rho = −0.58, p < 0.001) and positively associated with CSF pTau181 (rho = 0.63, p < 0.001), CSF NfL (rho = 0.72, p < 0.001), CSF GFAP (rho = 0.61, p < 0.001), and CSF YKL‐40 (rho = 0.63, p < 0.001) concentrations (Figure 4A). Additionally, A+ and T+ individuals with DS presented greater WMH volume compared to negative participants (p < 0.001; Figure 4A). When assessed regionally, the strongest associations between CSF biomarkers of AD and WMHs were found in the frontal, parietal, and occipital lobes and in the first and second layers (Figure 4B). The univariate models revealed that all five CSF biomarkers were significant predictor variables in the whole DS cohort (Aβ42/Aβ 40: p < 0.001, estimate: −0.59, 95% CI: [−0.70, −0.47]; pTau181: p < 0.001, estimate: 0.60 [0.48, 0.71]; NfL: p < 0.001, estimate: 0.68 [0.57, 0.79]; GFAP: p < 0.001, estimate: 0.57 [0.42, 0.69]; YKL‐40: p < 0.001, estimate: 0.58 [0.47, 0.70]). Considering the groups separately, we found significant relationships between the five biomarkers and WMHs in aDS (Aβ42/Aβ 40: p < 0.001, estimate: −0.40, [−0.60, −0.21]; pTau181: p < 0.001, estimate: 0.37 [0.18, 0.56]; NfL: p < 0.001, estimate: 0.44, [0.27, 0.62]; GFAP: p < 0.001, estimate: 0.32 [0.12, 0.52]; YKL‐40: p < 0.001, estimate: 0.46 [0.29, 0.63]). In sDS, only the association with NfL concentrations reached significance (Aβ42/Aβ 40: p = 0.81, estimate: 0.03 [−0.21, 0.27]; pTau181: p = 0.06, estimate: 0.21 [−0.01, 0.44]; NfL: p < 0.001, estimate: 0.44 [0.22, 0.66]; GFAP: p = 0.08, estimate: 0.21 [−0.03, 0.45]; YKL‐40: p = 0.20, estimate: 0.16 [−0.09, 0.40]). When performing a multivariate analysis incorporating all five CSF AD biomarkers, only NfL significantly predicted total WMH volume in the whole DS cohort (p < 0.001, estimate: 0.37 [0.13, 0.61]) and in sDS (p < 0.001, estimate: 0.54 [0.18, 0.90]) group (Figure 4C). When age was included as a covariate, several results lost significance, especially in the aDS group, but the associations with NfL remained significant or showed a trend toward significance in the whole DS and sDS cohorts (Figure 5S in supporting information).
FIGURE 4.
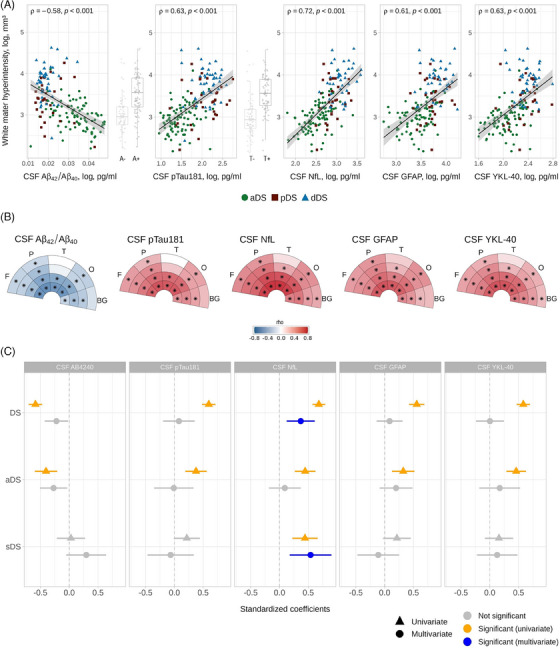
Associations between white matter hyperintensities and cerebrospinal fluid biomarkers in DS. (A) Correlation between total WMH volume and each corresponding CSF biomarker. Boxplots illustrate WMH volumes across the amyloid and tau status. The points represent individual participants, and the color indicates the clinical diagnosis. Shaded areas represent 95% CI. (B) Spearman correlation coefficients between each corresponding CSF biomarker and regional WMH volume in DS. The color scale represents the strength (rho) of Spearman correlation for significant results (p < 0.05). The star (*) indicates results surviving the Bonferroni correction (α = 0.05, p < 0.0025). (C) Standardized coefficient of univariate and multivariate linear regression models for the WMH volume. The triangles and circles represent the univariate and multivariate models, respectively. The lines represent the 95% CI. Gray color indicates non‐significant values, while orange color indicates significant values (p < 0.01) and blue color after applying Bonferroni correction (α = 0.05, p < 0.01). Abbreviations: 1, first layer; 2, second layer; 3, third layer; 4, fourth layer; A−, amyloid negative; A+, amyloid positive; Aβ42/Aβ40, concentration ratio between amyloid beta peptide 1‐42 and amyloid beta peptide 1‐40 (pg/mL); aDS, asymptomatic Down syndrome; BG, basal ganglia; CI, confidence interval; CSF, cerebrospinal fluid; dDS, Down syndrome with Alzheimer's disease dementia; DS, Down syndrome; F, frontal; GFAP, glial fibrillary acidic protein concentration (pg/mL); NfL, neurofilament light chain concentration (pg/mL); O, occipital; P, parietal; pDS, prodromal Down syndrome; pTau181, tau phosphorylated at threonine 181 concentration (pg/mL); sDS, symptomatic Down syndrome; T, temporal; T−, tau negative; T+, tau positive; WMH, white matter hyperintensity.
FIGURE 5.
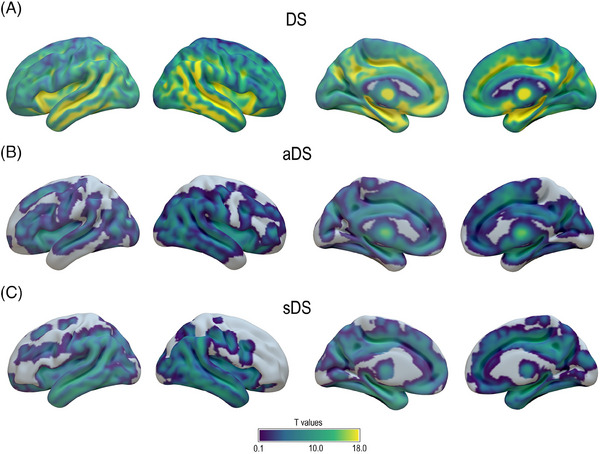
Voxelwise associations between white matter hyperintensities and gray matter volume. Results of voxelwise regression model showing gray matter volume negatively associated with WMH in (A) whole DS cohort and (B) aDS and (C) sDS subgroups. The significance of the results (T values) is shown using a threshold pFWE < 0.05. Abbreviations: aDS, asymptomatic Down syndrome; DS, Down syndrome; sDS, symptomatic Down syndrome; FWE, family‐wise error.
3.6. Associations between WMH volume and GM volume
Finally, we also investigated voxelwise associations between total WMH volume and GM volumes within the DS population. These analyses revealed significant associations across the entire cortex for the whole cohort (Figure 5A). Specifically, the pattern predominated in the medial and lateral temporo‐parietal regions, medial prefrontal and orbitofrontal cortex, anterior insula, hippocampi, and thalamus. The stratification of DS by clinical status revealed a very similar pattern in both aDS and sDS, although slightly less widespread and significant than in the whole population (Figure 5B,C).
4. DISCUSSION
Our results highlighted an increase in the presence and severity of WMHs with age in DS, as well as with the incidence of both AD symptoms and AD pathology. We further demonstrated that lesions are mostly located in periventricular regions, especially in the frontal, parietal, and occipital lobes. WMHs expand into more cortical layers throughout the AD continuum in DS, suggesting a distinct underlaying pathophysiology compared to the general population. Together, these findings underscore that WMHs represent a core neuroimaging feature in DS, and provide novel insight into the interplay between these lesions and AD processes.
Consistent with previous studies, 17 we observed that WMH volume gradually increased with age in both DS and euploid populations. This age‐related increase is stronger in DS individuals. This finding expands previous evidence from our group obtained in a smaller sample of DS and using the Fazekas visual scale. 16 We further demonstrated that WMHs emerge at a younger age in DS than HC, with CI no longer overlapping at the age of 43, around 10 years before symptom onset. 5 Interestingly, a recent study on PSEN1 mutation carriers also reported a sharp increase in WMHs at 43 years, 15 and WMHs were found to increase approximately 6.6 years before estimated symptom onset in mutation carriers of AD‐causing genes. 14 The presence of WMHs at an early age across distinct genetically determined AD forms suggests that AD‐related mechanisms contribute, at least partially, to these lesions. Indeed, in population‐based studies, WMHs typically increased at a later age, 10 , 32 starting from 55 years. 33
We mapped WMH topography and found different associations in both populations. Associations with age predominated in the frontal lobe in HC, as previously described in elderly populations. 34 By contrast, associations in DS involved all brain lobes, particularly the fronto–parieto–occipital regions. This aligns with previous evidence showing a widespread age‐related pattern of WMHs in DS, driven by frontal, parietal, and occipital lobes, 17 as well as the typical parieto–occipital AD pattern. 12 Further, correlation coefficients were strongest in the periventricular region, showing earlier age‐related changes than total WMH volume, and progressively decreased in deep and juxtacortical areas. These results add to a body of work identifying different WMH topography in AD compared to age‐related WMHs. 12
Other sociodemographic and genetic factors may also contribute to WMHs. In line with previous studies, 16 , 17 we did not evidence an effect of sex or APOE haplotype on total WMH volume in DS. The specific sex effect on AD in DS is still debated, with a recent study showing no impact on AD biomarkers. 35 Concerning the APOE ε4 allele, it was found to influence AD processes in DS, 36 but this effect might be too subtle to be identified in WMHs. The link between APOE ε4 and WMHs is also inconsistent in the general population and sporadic AD, with some studies suggesting a gene–dose effect on WMHs 37 , 38 and others reporting opposite or no effect. 39 , 40 , 41 , 42
Surprisingly, greater ID was associated with higher WMH volumes. However, sensitivity analyses suggested that this effect might result from the different proportion of symptomatic cases across the different ID groups, that is, a higher proportion of moderate level in dDS. Two recent studies examining the effect of ID on AD trajectory in DS revealed no differences in AD biomarkers, cognitive impairments, 43 or decline. 44 Together these results suggest that ID has a limited impact on AD and vascular changes in DS. However, further research is warranted to confirm this result and assess if differences in lifestyle factors across ID can lead to differences in WMHs.
WMH volumes increased linearly with AD stage (aDS < pDS < dDS), consistent with previous studies in DS, 16 , 17 and investigations in early‐ 45 and late‐onset sporadic AD 10 , 46 and autosomal dominant AD (ADAD), 14 , 47 all of which showed a gradual increase in WMH burden with disease severity. Topographically, WMHs increased locally and expanded to new layers as the disease progressed. Mainly circumscribed to the periventricular region at the asymptomatic stage, the lesions extended into deep and juxtacortical areas, including the frontal, parietal, and occipital lobes at the prodromal stage, and further increased in the same regions at the dementia stage. Similar topographical progression has been previously described in sporadic AD, 48 where periventricular WMHs were common at the mild cognitive impairment (MCI) stage, and in the AD group expanded outward, particularly to affect the corpus callosum. The frequency map further showed that posterior corona radiata, posterior thalamic radiation, and splenium of the corpus callosum were the most affected areas in DS. These structures were previously found vulnerable to AD processes, 49 and associated with Aβ pathology. 50
Compared to HC, the aDS group presented increased WMH volume only in the temporal lobe. Previous studies in preclinical AD revealed larger WMH total volume in Aβ‐positive compared to Aβ‐negative individuals. 51 In ADAD, asymptomatic carriers present with increased WMHs in parieto‐occipital regions. 14 Taken together, these results suggest that WMHs emerge at an early preclinical stage of AD. The different topographic patterns may reflect specificities of each presentation or differences in the preclinical or control groups. For instance, our aDS cohort included adults ranging from 19 to 63 years, with 32.9% at > 20 years from the estimated age of symptom onset (i.e., 53.8 years 5 ).
Relationships between WMHs and CSF AD biomarkers revealed robust associations in DS with both Aβ and pTau181 concentrations. In the literature, the link between WMHs and AD pathology is inconsistent. Several studies in the euploid population reported no association between WMHs and Aβ‐ 52 and tau positron emission tomography (PET) 53 load. In DS, WMHs were also not significantly associated with Aβ PET. 17 However, some evidence supported a relationship with Aβ PET in non‐demented individuals 12 , 53 and tau PET in preclinical ADAD. 45 Moreover, Aβ in CSF, but not tau, was found related to WMHs in ADAD, 14 , 54 , 55 MCI, and sporadic AD. 14 , 54 , 55 , 56 , 57 These discrepant results might be explained by the modality to measure AD pathology, or the composition of the cohort studied. Indeed, when we stratified our analyses by disease stage, we found significant associations at the asymptomatic but not symptomatic stages, aligning with findings in the AD continuum. 57
Associations between WMHs and neurodegenerative markers were also strong. Consistent with previous observations in cognitively normal, 54 MCI, 54 , 58 , 59 sporadic AD, 59 , 60 and ADAD, 61 we found that NfL levels in CSF were associated with WMHs. Indeed, NfL showed i) the strongest association with total WMH volume, ii) was significantly related to WMH volume at different disease stages, and iii) was the only CSF biomarker to remain significantly associated with WMHs in the multivariate analyses. This strong link between WMHs and NfL is not surprising because WM mainly consists of myelinated axons and NfL, an axonal cytoskeleton protein particularly expressed in long myelinated axons, is released upon axonal damage. 62
To further understand WMHs and neurodegeneration, we explored the association with GM volume. There were significant associations that encompassed the entire cortex, but were more pronounced in lateral and medial temporo‐parietal regions. This pattern overlaps with the typical AD regions showing tau accumulation 63 and atrophy. 64 Similar associations between WMH burden and lower GM volume 9 and cortical thickness 65 were previously reported in sporadic AD.
The link between WMHs and both AD pathology and neurodegeneration can be explained by different non‐exclusive hypotheses. 12 , 66 First, it is possible that WMHs, in the AD context, are caused by Wallerian degeneration secondary to neurofibrillary tangles. 67 , 68 , 69 Indeed, disruption of microtubules containing tau could lead to axonal degeneration, resulting in WM damage observed as WMHs on MRI. 70 Second, WMHs might be induced by subtle hypoperfusion or other cerebrovascular diseases, which in turn contribute to tau pathology 71 and/or the disruption of WM tracts affecting the cortical regions. 11 For instance, cerebral amyloid angiopathy is frequently found in AD 72 and DS, 16 and likely causes some posterior WM damage. 53 , 73 Another hypothesis is that neuroinflammation, which is a key pathogenic factor of both AD and vascular pathologies, might also participate in the genesis of part of WMHs, 74 , 75 as suggested by the association with GFAP and YKL‐40. Finally, we cannot exclude that the associations between these different biomarkers are partially driven by disease progression.
The current study has some limitations. First, the WMH segmentation algorithm and templates to parcellate the WM were not specifically designed for the DS population. Although a thorough visual quality control demonstrated that the segmentation and WM parcellation were correctly performed, with no visible impact of DS brain anatomy, we cannot discard a reduced sensitivity to detect some lesions. Second, the cross‐sectional design of this study does not allow for the assessment of causality, such as the relationships among WMHs, AD pathology, and neurodegeneration. Future longitudinal studies will undoubtedly provide new insight into the etiology of WMHs in DS.
In summary, this study offers a comprehensive understanding of the development and distribution of WMHs in DS. Our findings revealed that WMHs are associated with AD clinical stages, manifesting even at the asymptomatic phase. The parcellation of the WM revealed the predominance of WMHs in periventricular regions of frontal, parietal, and occipital lobes, and their expansion into cortical layers with AD progression. These results demonstrate that WMHs are a core imaging feature in DS and shed light on their relationship with AD pathological processes.
AUTHOR CONTRIBUTIONS
Alejandra O. Morcillo‐Nieto and Sara E. Zsadanyi contributed equally to this work and share first authorship. Alejandra O. Morcillo‐Nieto, Sara E. Zsadanyi, and Alexandre Bejanin conceived and designed the study. Jose E. Arriola‐Infante, Maria Carmona‐Iragui, Victor Montal, Jordi Pegueroles, Mateus Rozalem Aranha, Lídia Vaqué‐Alcázar, Concepción Padilla, Bessy Benejam, Laura Videla, Isabel Barroeta, Laura Videla, Miren Altuna, Sandra Giménez, Sofía González‐Ortiz, Núria Bargalló, Laia Ribas, Javier Arranz, Soraya Torres, Maria Florencia Iulita, Olivia Belbin, Valle Camacho, Daniel Alcolea, Alberto Lleó, and Juan Fortea acquired and interpreted the data. Alejandra O. Morcillo‐Nieto performed the statistical analysis. Alejandra O. Morcillo‐Nieto, Sara E. Zsadanyi, and Alexandre Bejanin drafted the manuscript, which all authors critically reviewed for important intellectual content.
CONFLICT OF INTEREST STATEMENT
MRA has provided paid consultancy for Veranex, and is a partner and director of production at Masima—Soluções em Imagens Médicas LTDA. MFI is now a paid employee of Altoida Inc. and may hold stock options in the company. The work in this manuscript only relates to the reported affiliation. DA reported receiving personal fees for advisory board services and/or speaker honoraria from Fujirebio‐Europe, Roche, Nutricia, Krka Farmacéutica, Lilly, Zambon S.A.U., Grifols, and Esteve, outside the submitted work. AL has served as a consultant or on advisory boards for Fujirebio‐Europe, Roche, Biogen, Grifols, Novartis, Eisai, Lilly, and Nutricia, outside the submitted work. JF reported serving on the advisory boards, adjudication committees, or speaker honoraria from Roche, NovoNordisk, Esteve, Biogen, Laboratorios Carnot, Adamed, LMI, Novartis, Lundbeck, Roche, AC Immune, Alzheon, Zambon, Lilly, Spanish Neurological Society, T21 Research Society, Lumind foundation, Jérôme‐Lejeune Foundation, Alzheimer's Association, National Institutes of Health USA, and Instituto de Salud Carlos III. DA, AL, and JF report holding a patent for markers of synaptopathy in neurodegenerative disease (licensed to ADx, EPI8382175.0). No other competing interests were reported. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants and/or their legally authorized representatives gave written informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank all study participants, their families, and caregivers from the DABNI and SPIN cohort for their support of, and dedication to, this research. We also would acknowledge the Fundació Catalana Síndrome de Down (https://fcsd.org/) for global support. This study was funded by the Fondo de Investigaciones Sanitario, Instituto de Salud Carlos III and co‐funded by the European Union (PI18/00335 to MCI, PI18/00435 to DA, PI14/1561, PI20/01330 to AL, PI20/01473 to JF, PI22/00307 to AB), the CIBERNED Program 1, the National Institutes of Health (NIH; 1R01AG056850‐01A1; 3RF1AG056850‐01S1; AG056850, R21AG056974, and R01AG061566 to JF), the Departament de Salut de la Generalitat de Catalunya, Fundación Tatiana Pérez de Guzmán el Bueno (IIBSP‐DOW‐2020‐151 to JF), the European Union's Horizon 2020, “MES‐CoBraD” (H2020‐SC1‐BHC‐2018‐2020 / GA 965422 to JF), Brightfocus, and Life Molecular Imaging (LMI) to JF. JEAI was supported by Instituto de Salud Carlos III through the Río Hortega Fellowship “CM22/00219” and co‐funded by the European Union. VM acknowledges support from the Instituto de Salud Carlos III through the Predoctoral grant “FI18/00275” and co‐funded by the European Union, and Juan de la Cierva fellowship (JDC2022‐048492‐I) from the Ministry of Science of the Spanish Government. MCI acknowledges support from the Alzheimer's Association and Global Brain Health Institute (GBHI_ALZ‐18‐543740), the Jérôme Lejeune Foundation (#1913 Cycle 2019B), the Societat Catalana de Neurologia (Premi Beca Fundació SCN 2020). MRA acknowledges support from Alzheimer's Association Research Fellowship to Promote Diversity (AARFD‐21‐852492). MRA has provided paid consultancy for Veranex. MRA is a partner and director of production at Masima—Soluções em Imagens Médicas LTDA. LVA was supported by Margarita Salas junior postdoctoral fellowship (UNI/551/2021, NextGenerationUE). CP was supported by Instituto de Salud Carlos III through the Sara Borrell Postdoctoral Fellowship “CD20/00133” and co‐funded by the European Union. JA was supported by Instituto de Salud Carlos III through the Río Hortega Fellowship “CM21/00243” and co‐funded by the European Union. AB acknowledges support from Instituto de Salud Carlos III through the Miguel Servet grant “CP20/00038” and co‐funded by the European Union, and the Alzheimer's Association “AARG‐22‐923680.” The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit for publication.
Morcillo‐Nieto AO, Zsadanyi SE, Arriola‐Infante JE, et al. Characterization of white matter hyperintensities in Down syndrome. Alzheimer's Dement. 2024;20:6527–6541. 10.1002/alz.14146
Maria Florencia Iulita affiliation's during article conception and development.
Alejandra O. Morcillo‐Nieto and Sara E. Zsadanyi contributed equally to this work and share first authorship.
DATA AVAILABILITY STATEMENT
The authors may share de‐identified data that underlie the results reported in this article. Data will be available upon receipt of a request detailing the study hypothesis and statistical analysis plan. All requests should be sent to the corresponding authors. The steering committee of this study will discuss all requests and decide, based on the novelty and scientific rigor of the proposal, whether data sharing is appropriate. All applicants will be asked to sign a data access agreement.
REFERENCES
- 1. Lott IT, Head E. Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol. 2019;15:135‐147. 2019 153. doi: 10.1038/s41582-018-0132-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fortea J, Vilaplana E, Carmona‐Iragui M, et al. Clinical and biomarker changes of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet. 2020;395:1988‐1997. doi: 10.1016/S0140-6736(20)30689-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fortea J, Zaman SH, Hartley S, Rafii MS, Head E, Carmona‐Iragui M. Alzheimer's disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. 2021;20:930‐942. doi: 10.1016/S1474-4422(21)00245-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCarron M, McCallion P, Reilly E, Dunne P, Carroll R, Mulryan N. A prospective 20‐year longitudinal follow‐up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2017;61:843‐852. doi: 10.1111/JIR.12390 [DOI] [PubMed] [Google Scholar]
- 5. Iulita MF, Garzón Chavez D, Klitgaard Christensen M, et al. Association of Alzheimer disease with life expectancy in people with Down syndrome. JAMA Netw Open. 2022;5:e2212910‐e2212910. doi: 10.1001/JAMANETWORKOPEN.2022.12910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carmona‐Iragui M, Videla L, Lleó A, Fortea J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: the complex triangle of brain amyloidosis. Dev Neurobiol. 2019;79:716‐737. doi: 10.1002/DNEU.22709 [DOI] [PubMed] [Google Scholar]
- 7. Draheim CC, Geijer JR, Dengel DR. Comparison of intima‐media thickness of the carotid artery and cardiovascular disease risk factors in adults with versus without the Down syndrome. Am J Cardiol. 2010;106:1512‐1516. doi: 10.1016/J.AMJCARD.2010.06.079 [DOI] [PubMed] [Google Scholar]
- 8. Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18:684‐696. doi: 10.1016/S1474-4422(19)30079-1 [DOI] [PubMed] [Google Scholar]
- 9. Gaubert M, Lange C, Garnier‐Crussard A, et al. Topographic patterns of white matter hyperintensities are associated with multimodal neuroimaging biomarkers of Alzheimer's disease. Alzheimer's Res Ther. 2021;13:1‐11. 2021 131. doi: 10.1186/S13195-020-00759-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11:157‐165. 2015 113. doi: 10.1038/nrneurol.2015.10 [DOI] [PubMed] [Google Scholar]
- 11. ter Telgte A, van Leijsen EMC, Wiegertjes K, Klijn CJM, Tuladhar AM, de Leeuw F‐E. Cerebral small vessel disease: from a focal to a global perspective. Nat Rev Neurol. 2018;14:387‐398. 2018 147. doi: 10.1038/s41582-018-0014-y [DOI] [PubMed] [Google Scholar]
- 12. Garnier‐Crussard A, Cotton F, Krolak‐Salmon P, Chételat G. White matter hyperintensities in Alzheimer's disease: beyond vascular contribution. Alzheimer's Dement. 2023;19:3738‐3748. doi: 10.1002/ALZ.13057 [DOI] [PubMed] [Google Scholar]
- 13. Garnier‐Crussard A, Bougacha S, Wirth M, et al. White matter hyperintensity topography in Alzheimer's disease and links to cognition. Alzheimer's Dement. 2022;18:422‐433. doi: 10.1002/ALZ.12410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee S, Viqar F, Zimmerman ME, et al. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79:929‐939. doi: 10.1002/ANA.24647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schoemaker D, Zanon Zotin MC, Chen K, et al. White matter hyperintensities are a prominent feature of autosomal dominant Alzheimer's disease that emerge prior to dementia. Alzheimer's Res Ther. 2022;14:1‐11. doi: 10.1186/S13195-022-01030-7/FIGURES/3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carmona‐Iragui M, Balasa M, Benejam B, et al. Cerebral amyloid angiopathy in Down syndrome and sporadic and autosomal‐dominant Alzheimer's disease. Alzheimer's Dement. 2017;13:1251‐1260. doi: 10.1016/J.JALZ.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lao PJ, Gutierrez J, Keator D, et al. Alzheimer‐related cerebrovascular disease in Down syndrome. Ann Neurol. 2020;88:1165‐1177. doi: 10.1002/ANA.25905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alcolea D, Clarimón J, Carmona‐Iragui M, et al. The Sant Pau Initiative on Neurodegeneration (SPIN) cohort: a data set for biomarker discovery and validation in neurodegenerative disorders. Alzheimer's Dement Transl Res Clin Interv. 2019;5:597‐609. doi: 10.1016/J.TRCI.2019.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hon J, Huppert FA, Holland AJ, Watson P. Neuropsychological assessment of older adults with Down's Syndrome: an epidemiological study using the Cambridge Cognitive Examination (CAMCOG). Br J Clin Psychol. 1999;38:155‐165. doi: 10.1348/014466599162719 [DOI] [PubMed] [Google Scholar]
- 20. Esteba‐Castillo S, Dalmau‐Bueno A, Ribas‐Vidal N, Vilá‐Alsina M, García Alba J, Novell R. Adaptation and validation of CAMDEX‐DS (Cambridge Examination for Mental Disorders of Older People with Down's Syndrome and others with intellectual disabilities) in Spanish population with intellectual disabilities [in Spanish]. Rev Neurol. 2013;57:337‐346. ISSN 0210‐0010, Vol 57, No 8, 2013, Págs 337‐346. [PubMed] [Google Scholar]
- 21. Fortea J, Carmona‐Iragui M, Benejam B, et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer's disease in adults with Down syndrome: a cross‐sectional study. Lancet Neurol. 2018;17:860‐869. doi: 10.1016/S1474-4422(18)30285-0 [DOI] [PubMed] [Google Scholar]
- 22. Regier DA, Kuhl EA, Kupfer DJ. The DSM‐5: classification and criteria changes. World Psychiatry. 2013;12:92‐98. doi: 10.1002/WPS.20050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gaser C, Dahnke R, Thompson PM, Kurth F, Luders E, Initiative ADN . CAT – a computational anatomy toolbox for the analysis of structural MRI data. Biorxiv. 2022:11.495736. doi: 10.1101/2022.06.11.495736. 2022.06.. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage. 2011;54:2033‐2044. doi: 10.1016/J.NEUROIMAGE.2010.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt P, Bayesian inference for structured additive regression models for large‐scale problems with applications to medical imaging 2017. (accessed April 16, 2022) https://edoc.ub.uni‐muenchen.de/20373/
- 26. Sudre CH, Gomez Anson B, Davagnanam I, et al. Bullseye's representation of cerebral white matter hyperintensities. J Neuroradiol. 2018;45:114‐122. doi: 10.1016/J.NEURAD.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiménez‐Balado J, Corlier F, Habeck C, Stern Y, Eich T. Effects of white matter hyperintensities distribution and clustering on late‐life cognitive impairment. Sci Rep. 2022;12:1‐13. 2022 121. doi: 10.1038/s41598-022-06019-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orlhac F, Eertink JJ, Cottereau AS, et al. A guide to ComBat harmonization of imaging biomarkers in multicenter studies. J Nucl Med. 2022;63(2):172‐179. doi: 10.2967/JNUMED.121.262464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carmona‐Iragui M, Santos T, Videla S, et al. Feasibility of lumbar puncture in the study of cerebrospinal fluid biomarkers for Alzheimer's disease in subjects with Down syndrome. J Alzheimer's Dis. 2017;55:1489‐1496. doi: 10.3233/JAD-160827 [DOI] [PubMed] [Google Scholar]
- 30. Alcolea D, Pegueroles J, Muñoz L, et al. Agreement of amyloid PET and CSF biomarkers for Alzheimer's disease on Lumipulse. Ann Clin Transl Neurol. 2019;6:1815‐1824. doi: 10.1002/ACN3.50873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subirana I, Sanz H, Vila J. Building Bivariate Tables: the compareGroups Package for R. J Stat Softw. 2014;57:1‐16. doi: 10.18637/JSS.V057.I12 25400517 [DOI] [Google Scholar]
- 32. Garnier‐Crussard A, Bougacha S, Wirth M, et al. White matter hyperintensities across the adult lifespan: relation to age, Aβ load, and cognition. Alzheimer's Res Ther. 2020;12:1‐11. 2020 121. doi: 10.1186/S13195-020-00669-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hopkins RO, Beck CJ, Burnett DL, Weaver LK, Victoroff J, Bigler ED. Prevalence of white matter hyperintensities in a young healthy population. J Neuroimaging. 2006;16:243‐251. doi: 10.1111/J.1552-6569.2006.00047.X [DOI] [PubMed] [Google Scholar]
- 34. Holland CM, Smith EE, Csapo I, et al. Spatial distribution of white‐matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke. 2008;39:1127. doi: 10.1161/STROKEAHA.107.497438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iulita MF, Bejanin A, Vilaplana E, et al. Association of biological sex with clinical outcomes and biomarkers of Alzheimer's disease in adults with Down syndrome. Brain Commun. 2023;5:14. doi: 10.1093/BRAINCOMMS/FCAD074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bejanin A, Iulita MF, Vilaplana E, et al. Association of apolipoprotein E ɛ4 allele with clinical and multimodal biomarker changes of Alzheimer disease in adults with Down syndrome. JAMA Neurol. 2021;78:937‐947. doi: 10.1001/JAMANEUROL.2021.1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Operto G, Cacciaglia R, Grau‐Rivera O, et al. White matter microstructure is altered in cognitively normal middle‐aged APOE‐ε4 homozygotes. Alzheimers Res Ther. 2018;10(1):48. doi: 10.1186/S13195-018-0375-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rojas S, Brugulat‐Serrat A, Bargalló N, et al. Higher prevalence of cerebral white matter hyperintensities inhomozygous APOE‐ɛ4 allele carriers aged 45‐75: results from theALFA study. J Cereb Blood Flow Metab. 2018;38:250. doi: 10.1177/0271678x17707397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Habes M, Erus G, Toledo JB, et al. White matter hyperintensities and imaging patterns of brain ageing in the general population. Brain. 2016;139:1164. doi: 10.1093/BRAIN/AWW008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar D, Yatawara C, Wang B, et al. APOE4 and confluent white matter hyperintensities have a synergistic effect on episodic memory impairment in prodromal dementia. J Alzheimer's Dis. 2022;87:1103‐1114. doi: 10.3233/JAD-215556 [DOI] [PubMed] [Google Scholar]
- 41. Slattery CF, Zhang J, Paterson RW, et al. ApoE influences regional white‐matter axonal density loss in Alzheimer's disease. Neurobiol Aging. 2017;57:8. doi: 10.1016/J.NEUROBIOLAGING.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morgen K, Schneider M, Frölich L, et al. Apolipoprotein E‐dependent load of white matter hyperintensities in Alzheimer's disease: a voxel‐based lesion mapping study. Alzheimers Res Ther. 2015;7(1):27. doi: 10.1186/S13195-015-0111-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hartley SL, Fleming V, Schworer EK, et al. Timing of Alzheimer's disease by intellectual disability level in Down syndrome. J Alzheimers Dis. 2023;95:213‐225. doi: 10.3233/JAD-230200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Videla L, Benejam B, Pegueroles J, et al. Longitudinal clinical and cognitive changes along the Alzheimer disease continuum in Down syndrome. JAMA Netw Open. 2022;5:e2225573‐e2225573. doi: 10.1001/JAMANETWORKOPEN.2022.25573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eloyan A, Thangarajah M, An N, et al. White matter hyperintensities are higher among early‐onset Alzheimer's disease participants than their cognitively normal and early‐onset nonAD peers: longitudinal Early‐onset Alzheimer's Disease Study (LEADS). Alzheimer's Dement. 2023;19(9):S89‐S97. doi: 10.1002/alz.13402. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brickman AM, Zahodne LB, Guzman VA, et al. Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer's disease incidence. Neurobiol Aging. 2015;36:27‐32. doi: 10.1016/J.NEUROBIOLAGING.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walsh P, Sudre CH, Manning EN, et al. White matter hyperintensity increases are a feature of familial AD and are associated with increased brain atrophy. Alzheimer's Dement. 2020;16:e038925. doi: 10.1002/ALZ.038925 [DOI] [Google Scholar]
- 48. Yoshita M, Fletcher E, Harvey D, et al. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology. 2006;67:2192. doi: 10.1212/01.WNL.0000249119.95747.1F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mayo CD, Garcia‐Barrera MA, Mazerolle EL, Ritchie LJ, Fisk JD, Gawryluk JR. Relationship between DTI metrics and cognitive function in Alzheimer's disease. Front Aging Neurosci. 2019;10:436. doi: 10.3389/FNAGI.2018.00436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weaver NA, Doeven T, Barkhof F, et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol Aging. 2019;84:225‐234. doi: 10.1016/J.NEUROBIOLAGING.2019.08.001 [DOI] [PubMed] [Google Scholar]
- 51. Caballero MÁA, Song Z, Rubinski A, et al. Age‐dependent amyloid deposition is associated with white matter alterations in cognitively normal adults during the adult life span. Alzheimer's Dement. 2020;16:651‐661. doi: 10.1002/ALZ.12062 [DOI] [PubMed] [Google Scholar]
- 52. Roseborough A, Ramirez J, Black SE, Edwards JD. Associations between amyloid β and white matter hyperintensities: a systematic review. Alzheimer's Dement. 2017;13:1154‐1167. doi: 10.1016/J.JALZ.2017.01.026 [DOI] [PubMed] [Google Scholar]
- 53. Graff‐Radford J, Arenaza‐Urquijo EM, Knopman DS, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142:2483‐2491. doi: 10.1093/BRAIN/AWZ162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Osborn KE, Liu D, Samuels LR, et al. Cerebrospinal fluid β‐amyloid42 and neurofilament light relate to white matter hyperintensities. Neurobiol Aging. 2018;68:18‐25. doi: 10.1016/J.NEUROBIOLAGING.2018.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Waalwijk Van Doorn LJC, Ghafoorian M, Van Leijsen EMC, et al. White matter hyperintensities are no major confounder for Alzheimer's disease cerebrospinal fluid biomarkers. J Alzheimers Dis. 2021;79:163‐175. doi: 10.3233/JAD-200496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pålhaugen L, Sudre CH, Tecelao S, et al. Brain amyloid and vascular risk are related to distinct white matter hyperintensity patterns. J Cereb Blood Flow Metab. 2021;41:1162. doi: 10.1177/0271678x20957604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Walsh P, Sudre CH, Fiford CM, et al. CSF amyloid is a consistent predictor of white matter hyperintensities across the disease course from aging to Alzheimer's disease. Neurobiol Aging. 2020;91:5‐14. doi: 10.1016/J.NEUROBIOLAGING.2020.03.008 [DOI] [PubMed] [Google Scholar]
- 58. Walsh P, Sudre CH, Fiford CM, et al. The age‐dependent associations of white matter hyperintensities and neurofilament light in early‐ and late‐stage Alzheimer's disease. Neurobiol Aging. 2021;97:10‐17. doi: 10.1016/J.NEUROBIOLAGING.2020.09.008 [DOI] [PubMed] [Google Scholar]
- 59. Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol. 2016;73:60. doi: 10.1001/JAMANEUROL.2015.3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meeker KL, Butt OH, Gordon BA, et al. Cerebrospinal fluid neurofilament light chain is a marker of aging and white matter damage. Neurobiol Dis. 2022;166:105662. doi: 10.1016/J.NBD.2022.105662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schultz SA, Strain JF, Adedokun A, et al. Serum neurofilament light chain levels are associated with white matter integrity in autosomal dominant Alzheimer's disease. Neurobiol Dis. 2020;142:104960. doi: 10.1016/J.NBD.2020.104960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90:870‐881. doi: 10.1136/JNNP-2018-320106 [DOI] [PubMed] [Google Scholar]
- 63. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12(254):eaau5732. doi: 10.1126/SCITRANSLMED.AAU5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid‐positive individuals. Cereb Cortex (New York, NY). 2009;19:497. doi: 10.1093/CERCOR/BHN113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lao PJ, Brickman AM. Multimodal neuroimaging study of cerebrovascular disease, amyloid deposition, and neurodegeneration in Alzheimer's disease progression. Alzheimer's Dement Diagnosis, Assess Dis Monit. 2018;10:638‐646. doi: 10.1016/J.DADM.2018.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brickman AM, Rizvi B. White matter hyperintensities and Alzheimer's disease: an alternative view of an alternative hypothesis. Alzheimer's Dement. 2023;19:4260‐4261. doi: 10.1002/ALZ.13371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bozzali M, Falini A, Franceschi M, et al. White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2002;72:742. doi: 10.1136/JNNP.72.6.742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McAleese KE, Walker L, Graham S, et al. Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017;134:459‐473. doi: 10.1007/S00401-017-1738-2/FIGURES/5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McAleese KE, Firbank M, Dey M, et al. Cortical tau load is associated with white matter hyperintensities 2015. doi: 10.1186/s40478-015-0240-0 [DOI] [PMC free article] [PubMed]
- 70. Salvadores N, Gerónimo‐Olvera C, Court FA. Axonal degeneration in AD: the contribution of Aβ and Tau. Front Aging Neurosci. 2020;12:581767. doi: 10.3389/FNAGI.2020.581767/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Laing KK, Simoes S, Baena‐Caldas GP, et al. Cerebrovascular disease promotes tau pathology in Alzheimer's disease. Brain Commun. 2020;2(2):fcaa132. doi: 10.1093/BRAINCOMMS/FCAA132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844‐866. doi: 10.1016/J.NEURON.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thanprasertsuk S, Martinez‐Ramirez S, Pontes‐Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83:794‐800. doi: 10.1212/WNL.0000000000000732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Garnier‐Crussard A, Cotton F, Krolak‐Salmon P, Chételat G. White matter hyperintensities in Alzheimer's disease: beyond vascular contribution. Alzheimer's Dement. 2023;19(8):3738‐3748. doi: 10.1002/ALZ.13057 [DOI] [PubMed] [Google Scholar]
- 75. Low A, Mak E, Rowe JB, Markus HS, O'Brien JT. Inflammation and cerebral small vessel disease: a systematic review. Ageing Res Rev. 2019;53:100916. doi: 10.1016/J.ARR.2019.100916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The authors may share de‐identified data that underlie the results reported in this article. Data will be available upon receipt of a request detailing the study hypothesis and statistical analysis plan. All requests should be sent to the corresponding authors. The steering committee of this study will discuss all requests and decide, based on the novelty and scientific rigor of the proposal, whether data sharing is appropriate. All applicants will be asked to sign a data access agreement.