

Abstract
INTRODUCTION
Dementia is a multifactorial disease with Alzheimer's disease (AD) and vascular dementia (VaD) pathologies making the largest contributions. Yet, most genome‐wide association studies (GWAS) focus on AD.
METHODS
We conducted a GWAS of all‐cause dementia (ACD) and examined the genetic overlap with VaD. Our dataset includes 800,597 individuals, with 46,902 and 8702 cases of ACD and VaD, respectively. Known AD loci for ACD and VaD were replicated. Bioinformatic analyses prioritized genes that are likely functionally relevant and shared with closely related traits and risk factors.
RESULTS
For ACD, novel loci identified were associated with energy transport (SEMA4D), neuronal excitability (ANO3), amyloid deposition in the brain (RBFOX1), and magnetic resonance imaging markers of small vessel disease (SVD; HBEGF). Novel VaD loci were associated with hypertension, diabetes, and neuron maintenance (SPRY2, FOXA2, AJAP1, and PSMA3).
DISCUSSION
Our study identified genetic risks underlying ACD, demonstrating overlap with neurodegenerative processes, vascular risk factors, and cerebral SVD.
Highlights
We conducted the largest genome‐wide association study of all‐cause dementia (ACD) and vascular dementia (VaD).
Known genetic variants associated with AD were replicated for ACD and VaD.
Functional analyses identified novel loci for ACD and VaD.
Genetic risks of ACD overlapped with neurodegeneration, vascular risk factors, and cerebral small vessel disease.
Keywords: all‐cause dementia, Alzheimer's disease, cross‐ancestry, genome‐wide association study (GWAS), GWAS meta‐analysis, vascular dementia
1. BACKGROUND
Traditionally, Alzheimer's disease (AD) is considered the most common dementia subtype, followed by vascular dementia (VaD). The two conditions are considered clinically distinct. VaD is diagnosed based on the presence of stroke or extensive cerebral vascular disease, with atherosclerosis and arteriolosclerosis considered the underlying pathologies. 1 However, a wealth of evidence from recent years has emphasized a broad role for brain vascular damage, beyond that of lacunar and larger cerebral infarcts, as a major mechanism for cognitive impairment. 2 It is now increasingly recognized that a component of vascular pathology is prominent in all major dementias and acts synergistically with amyloid beta (Aβ), tau, and other neurodegenerative pathologies to affect dementia risk. 3 Moreover, a new hypothetical model of dementia dynamics suggests that damage to brain vasculature is an early process in the dementia continuum that precedes brain atrophy, neurodegeneration, and the emergence of amyloid and tau biomarker abnormalities. 4 Recent genetic studies using methods that are relatively immune to reverse causation also suggest a putative causal relationship between brain imaging markers of cerebral small vessel disease (SVD) and AD. 5
Hence there is a strong rationale to examine the “vascular contributions to cognitive impairment and dementia” (VCID), a term that includes a broad range of vascular mechanisms and phenotypes and represents the multifactorial nature of dementia and related disorders as a pathway for reducing dementia burden. 6 In particular, genetic exploration of VCID may highlight important mechanisms across the wide spectrum of pathologies, including vascular pathways, which, in turn, are considered to be a major and modifiable target for the prevention of dementia, including the Alzheimer's type. 7
Emerging evidence suggests that VCID is highly heritable. 8 Mutations in the NOTCH3 gene known to cause monogenic cerebral SVD and early cognitive impairment also influence later onset polygenic manifestations of VCID by acting through common, less pathogenic variations in the same genes. Other examples are several point mutations in the amyloid precursor protein (APP) gene that lead to cerebral amyloid angiopathy (CAA) 9 as well as mutations in HtrA Serine Peptidase 1 (HTRA1) and Collagen Type IV Alpha 1 Chain (COL4A1) or COL4A2 genes. 10 Further support for the strong genetic basis of VCID stems from heritability and genome‐wide association studies (GWASs) of cerebral SVD endophenotypes that are closely related to VCID, including ischemic stroke (IS), 11 and white matter hyperintensities (WMHs). 12 , In contrast to the over 70 loci identified as being associated with AD genetic variance, the genetic architecture of “sporadic” VCID is largely unknown. Most genetic studies of VCID have utilized a candidate gene approach, which did not yield consistent and replicable findings. 13
RESEARCH IN CONTEXT
Systematic review: While findings from genome‐wide association studies (GWASs) of Alzheimer's disease (AD) highlighted multiple genetic risk variants, the genetics of all‐cause dementia (ACD) and vascular dementia (VaD) has been rarely studied. In this meta‐analysis of unpublished GWASs, we utilized data from 21 cohorts and consortia for a total of 46,902 and 8702 cases of ACD and VaD, respectively.
Interpretation: Known genetic variants for AD were identified as risk factors for ACD and VaD. Downstream bioinformatics revealed novel genetic loci functionally associated with ACD and VaD, including SEMA4D, RBFOX1, and SPRY2.
Future directions: These results should be validated in additional datasets. Particularly, studies are warranted to explore the genetic variation of ACD and VaD in non‐European individuals.
GWASs of VCID are sparse. In 2012, a GWAS of VaD conducted among the participants of the Rotterdam Study (N = 67 cases and 5700 controls) identified a novel locus associated with VaD, located near the androgen receptor on the X chromosome 14 ; however, this finding could not be replicated. 15 More recently, a GWAS of dementia and its clinical endophenotypes was conducted as part of the GR@ACE study. 16 This study demonstrated the differential biological pathways associated with clinical AD subgroups based on the degree of vascular burden. It identified a variant near CNTNAP2 associated with probable or possible VCID (N = 373). However, this finding did not reach genome‐wide (GW) significance.
The multifactorial nature of VaD and the heterogeneity of the clinicopathological criteria used to define this entity have hampered the identification of genetic polymorphisms underlying VCID. To overcome these limitations, large‐scale studies with sufficient power to detect genetic signals specific to VCID are needed. 17 In this study, we investigate the genetic predisposition to VCID specifically. Hence, we explored the genetic variability associated with ACD as a broad phenotype, as well as VaD as an extreme phenotype of the dementia continuum characterized by increased vascular burden. Our findings were then analyzed in light of the knowledge already gained from previous large‐scale GW and sequencing studies on the genetic determinants of AD, stroke, and additional phenotypes along the VCID spectrum. 10
2. METHODS
2.1. Study population
A total of 800,597 participants from 21 cohorts and consortia contributed to 46,902 and 8702 cases of ACD and VaD, respectively. The overall sample included individuals from four different ethnicities (European, African, Asian, and Hispanic) from North America, Europe, and Asia. The mean age ranged between 54 and 80 years, with 54% to 68% females. The summary demographics are described in Table 1 (also detailed in Table S1 of Supplementary File 1). Each study obtained written informed consent from participants or, for those with substantial cognitive impairment, from a caregiver, legal guardian, or other proxy. Study protocols for all cohorts were reviewed and approved by the appropriate institutional review boards.
TABLE 1.
Demographics: Data from 17 CHARGE cohorts were included in our meta‐analysis, as were the UKBB, ADGC, and EADB for the replication of our VaD results in European ancestry.
| Study | N/Control | ACD | VaD | Percentage VaD | Age (mean) | Sex, % (percentage female) |
|---|---|---|---|---|---|---|
| European ancestry | ||||||
| 3C | 6475 | 808 | 162 | 20.1 | 74.2 | 61.0 |
| AGES | 5656 | 1501 | 118 | 7.9 | 76.1 | 61.0 |
| ARIC | 3145 | 165 | 36 | 21.8 | 75.5 | 60.0 |
| ASPREE | 12,480 | 319 | NA | NA | 75.0 | 55.0 |
| CHS | 2169 | 508 | 156 | 30.7 | 74.9 | 61.5 |
| FVG | 804 | 73 | NA | NA | 58.2 | 58.3 |
| FHS | 4175 | 679 | 167 | 24.5 | 54.6 | 54.3 |
| GRACE | 12,599 | 7516 | 1953 | 26.0 | 78.8 | 68.2 |
| GREAT‐AGE | 1504 | 138 | 7 | 5.1 | 73.7 | 50.3 |
| HUNT | 69,633 | 3982 | 681 | 17.1 | 67.7 | 57.4 |
| MEMENTO | 2050 | 263 | 36 | 13.7 | ||
| MYHAT | 865 | 50 | NA | NA | 83.7 | 59.5 |
| ROSMAP | 1335 | 626 | NA | NA | 79.8 | 69.7 |
| RS (1,2,3) | 11,390 | 1715 | 178 | 10.4 | 63.6 | 56.8 |
| ADGC‐NAJ‐2011 | 15,675 | 8309 | NA | NA | 75.4 | 59.5 |
| UKBB | 314,278 | 17,008 | 332 | NA | 66.1 | 63.1 |
| Total European | 466,606 | 44,009 | 3892 | |||
| African ancestry | ||||||
| ARIC | 905 | 101 | 31 | 30.7 | 75.5 | 60.0 |
| CHS | 514 | 194 | 65 | 33.5 | 74.9 | 61.5 |
| ADGC‐Reitz (2013) | 5896 | 1968 | NA | NA | 80.5 | 63.9 |
| Total African | 7315 | 2263 | 96 | |||
| Asian ancestry | ||||||
| HKOS | 2373 | 349 | 66 | 18.9 | 60.1 | 67.9 |
| Harmonization | 385 | 153 | 49 | 32.0 | 73.6 | 55.0 |
| Total Asian | 2758 | 502 | 115 | |||
| Hispanic ancestry | ||||||
| SALSA | 1271 | 128 | 35 | 27.3 | 68.9 | 58.6 |
| Total Stage 1 for ACD and VaD | 477,950 | 46,902 | 4138 | |||
| Replication of VaD results in EADB | ||||||
| EADB | 275,745 | NA | 4,564 | |||
| Total | 753,695 | 46,902 | 8702 | |||
Note: Overall, 800,597 individuals were included in this study, accounting for 46,902 and 8702 cases of ACD and VaD, respectively. For UKBB, we used the proxy‐AD (familial AD) for ACD analysis and assessed VaD cases using ICD10 codes (see Methods). We also used ADGC‐NAJ‐201118 and ADGC‐Reitz‐201319 for ACD in European and African ancestry, respectively, to avoid overlap with CHARGE samples. We subsequently replicated our VaD results in EADB.
Abbreviations: ACD, all‐cause dementia; ADGC, Alzheimer's Disease Genetics Consortium; EADB, the European Alzheimer Disease Biobank; UKBB, the UK Biobank.
2.2. Phenotype definition
The primary study outcomes are ACD and VaD, measured by each participating cohort as described in Supplementary File 2. Briefly, to diagnose ACD and VaD, a neurological evaluation and diagnosis based on validated criteria were required. These criteria, as shown in Table S1 of Supplementary File 1, include the use of International Statistical Classification of Diseases and Related Health Problems (ICD) codes in most cohorts, as well as additional criteria such as the Diagnostic and Statistical Manual of Mental Disorders, Third to Fifth editions (DSM‐III to V), National Institute of Neurological Disorders and Stroke‐Alzheimer's Disease and Related Disorders Association, National Institute of Neurological Disorders and Stroke‐the Association Internationale pour la Recherche et l'Enseignement en Neurosciences, and dementia by proxy for United Kingdom Biobank (UKBB). Additionally, VaD cases were included in ACD, and the proportion of ACD classified as VaD is reported in Table S1 of Supplementary File 1. Moreover, to increase the sensitivity, cohorts were asked to run separate association analyses for (a) incident ACD, (b) prevalent ACD, (c) incident VaD, (d) prevalent VaD, (e) incident probable and definite VaD, and (f) prevalent probable whenever possible. To address the overlap with AD, we included all VCID (including persons with possible VCID) and separately analyzed only cases of “pure” (probable and autopsy‐proven definite) VCID and requested that all cohorts provide the most accurate, detailed description of their diagnostic algorithm. Although VaD in UKBB was defined based on ICD‐10 codes, we used the family history of dementia GWAS (“imputed dementia”) recently published by Marioni et al. 18 for ACD. Imputed dementia was defined as individuals at least 65 years old reporting a history of dementia in one or both parents. As explained in Ghosh et al., 19 the effect sizes and standard errors of the imputed dementia GWASs were doubled to analytically correct for the use of proxy phenotypes.
2.3. Genotyping and imputation
Genotyping was performed using cohort‐specific genotyping arrays as described in Supplementary File 2. Genetic variants were imputed using 1000 Genomes Project (1KG), the Haplotype Reference Consortium (HRC), 20 and the National Heart, Lung, and Blood Institute Trans‐Omics for Precision Medicine (TOPMed). UKBB imputed the genotypes to HRC, 1KG, and UK10K. Details on study‐specific quality control (QC) filters and software used for phasing and imputation are provided as supplementary materials (Supplementary File 2). Briefly, rare variants (minor allele frequency [MAF] < 1%) and poorly imputed variants (imputation quality, Rsq < 0.3) were excluded, as were variants mapping to sex chromosomes or mitochondria. Samples with poor genotyping call rate (<95%) and Hardy–Weinberg p values < 1 × 10−6 were removed. All genetic positions are reported in genome build 37 (GRCh37, hg19). Moreover, we used HRC version 1.2 as the main reference panel, and only variants in this panel were subsequently used in the association analyses. Additional details on the genotyping and imputation methods and QC are provided in Supplementary File 2.
2.4. Genome‐wide association analyses, QC, and meta‐analysis
2.4.1. Study‐level association analyses
We conducted study and ethnicity‐specific association analyses adjusting for age, sex, sites, and population structure to test the association of each variant with VaD and ACD. Cohorts were asked to run logistic regression and Cox proportional hazard models for prevalent and incident VaD/ACD, respectively, assuming additive allelic effects and imputed dosages. The UKBB association analyses were performed with linear mixed models (LMMs) using the BOLT‐LMM software. 21 BOLT‐LMM has the advantage over other methods in that it accounts for cryptic relatedness and population structure and, thus, allows the inclusion of related individuals in models, which increases the overall sample size. Details on the methods and software used for study‐level association analyses are provided in Supplementary File 2.
2.4.2. QC of study‐level summary statistics
We performed a stringent QC check of the summary statistics from each cohort using EasyQC. 22 We mapped each variant from the non‐European ancestry (EA) cohort to the appropriate 1KG project phase 3 reference panel and all EA to HRC (details in Table S1). Then the following steps were performed to ensure proper QC of each file before the meta‐analysis: (a) remove all structural variants and INDELs; (b) filter out variants with missing or unusual values (p value < 0 or > 1, effect size > 10, effect allele frequency < 0 or > 1, imputation quality < 0 or > 1); (c) filter out variants with effective allele count (EAC, 2 × minor allele frequency × N × imputation quality) < 10; (d) filter out variants with low imputation quality (eg, INFO scores reported by the imputation software); (e) filter out variants with MAF < 1%; (f) align variants to the main reference panel (HRC for EA, and ethnicity‐specific 1KG for others); remove variants with absolute difference between its allele frequencies in the cohort and reference panel greater than 0.2. All variants were assigned a unique identifier as a combination of the chromosome, position, reference, and alternative alleles separated by semi‐colons (CHR:POS:REF:ALT) to avoid issues with chromosomal positions mapping to multiple marker IDs. The foregoing steps were repeated until satisfactory results were obtained after visual inspection of the different diagnostic QC plots (AF, P‐Z, Q‐Q, and SE plots) generated by EasyQC as explained in Winkler et al. 22
2.4.3. Meta‐analysis of GWAS results
Ancestry‐specific meta‐analysis
The meta‐analyses were conducted using the fixed‐effect inverse variance‐weighted method implemented in METAL. 23 Post‐analysis results were filtered to retain only variants present in more than 40% of the overall cohorts and the effective sample size greater than 40% of the study sample size. We evaluated the heterogeneity across cohorts using the I 2 statistic provided by METAL, which represents the percentage of variation across studies that is due to heterogeneity rather than chance. We used the standard p value thresholds for GW significance, p < 5e‐8, and suggestive p < 1e‐6. Since there was no evidence of genomic inflation in the cohort summary statistics (lambda 0.98 to 1.06), no genomic control was applied during the meta‐analysis. Genomic loci were defined as the region ±500 kb around the single nucleotide polymorphism (SNP) with the lowest p value, considered as the index SNP. We assessed the heterogeneity across studies using the I 2 statistics of METAL (HetPVal output), which represents the percentage of variation across cohorts that is due to genetic heterogeneity rather than chance. Except for the APOE region (defined as SNPs located on chromosome 19 between positions 45,000,000 and 45,800,000 base pairs according to GRCh37 [hg19]), for which the HetPVal was >1e‐8, significant SNPs were selected with HetPVal > 0.01. We conducted ancestry‐specific meta‐analyses of VaD and ACD for EA, African ancestry (AA), Asian ancestry (SA), and Hispanic ancestry (HA). In addition, we used linkage disequilibrium (LD) score regression to quantify the contribution of true polygenicity and biases such as cryptic relatedness and population stratification of the meta‐analysis results.
Cross‐ancestry meta‐analysis
We performed cross‐ancestry meta‐analyses to assess whether the increase in sample size could lead to adequate power to identify additional GW significant loci associated with ACD and VaD. To this end, we used Meta‐Regression of Multi‐Ancestry Genetic Association (MR‐MEGA) software, which has proven more efficient than others when dealing with genetic heterogeneity. 24 MR‐MEGA uses a matrix of mean pairwise allele frequency differences to quantify the genetic similarity between studies and estimate the effect of each SNP after adjusting for ancestry principal components. We applied study‐specific filters, as previously described in the QC section, with EAC > 20, for studies with small sample sizes to reduce the amount of noise in the results‐driven rare SNPs in small cohorts. We fitted three principal components, as suggested by MR‐MEGA authors, which proved sufficient to separate the cohorts into self‐reported ancestry groups (Figure S1). As in the ancestry‐specific meta‐analysis, we retained only SNPs that were present in over 40% of cohorts, with >40% total sample size. GW significant SNPs had p < 5e‐8 and showed evidence of allelic heterogeneity across populations (MR‐MEGA P‐Het > 1e‐5).
2.5. Shared genetic susceptibility with complex disease traits
A gene‐based association test was conducted using MAGMA, 25 with p < 2.8e−6 as a genome‐wide significance threshold. Gene regions with SNPs not reaching GW significance for ACD or VaD in the primary GWAS analysis and additionally not in LD (r 2 < 0.10) with the lead SNP were considered novel.
We first explored the association of lead risk variants with related vascular, neurological traits and metabolic traits, excluding the APOE region. For each related trait, association statistics of SNPs falling in a window of ±250 kb around each lead SNP were queried, 26 and SNPs satisfying the GW significance threshold in the original study were retained. Leveraging the polygenicity of ACD (mean chi‐squared = 1.1) and VaD (mean chi‐squared = 1.06), we systematically explored the genetic overlap of ACD and VaD (in European‐only analysis) with (i) neurological and neurodegenerative traits (any stroke [AS], IS, small vessel stroke [SVS], large artery stroke [LAS], cardioembolic stroke [CES], general cognitive function [GCF], and Alzheimer‐type dementia [AD]); (ii) common magnetic resonance imaging (MRI) marker of cerebral SVD (WMHs) 5 ; and (iii) vascular risk factors (systolic blood pressure [SBP], diastolic blood pressure [DBP], pulse pressure [PP], high‐density lipoprotein [HDL], low‐density lipoprotein [LDL]). 27 We acquired summary statistics of the largest European‐only GWAS for these traits.
Using LD score regression (LDSR) analysis, 28 genetic correlation estimates between ACD/VaD and the aforementioned complex traits were obtained. A similar and potentially powerful approach called genetic covariance analyzer (GNOVA) 29 was additionally used to study the shared genetic covariance across the genome between a given pair of complex traits. LDSR and GNOVA compute genetic correlation and covariance, respectively, while adjusting for potential sample overlap and accounting for the LD of genetic variants. Though LDSR and GNOVA are substantially similar, differences in the minor allele frequency thresholds may influence genetic correlation estimates and significance to some extent. A p value < 8.3e‐3 correcting for six independent phenotypes was considered significant. Additionally, for the traits with significant genetic overlap, we performed causal inference analysis in the Mendelian randomization (MR) framework with ACD/VaD as the outcome. Using the MR‐LAP method, 30 we addressed potential bias in the causal effects due to sample overlap between the exposure and the outcome variables. Briefly, MR‐LAP utilizes the LDSR intercept estimates – a measure of the degree of sample overlap, polygenic architecture, and the heritability of the genetic instruments of the exposures – to account for the sample overlap bias and other biases (weak instrument and winner's curse bias) that push the causal estimates toward the null.
Since GW correlation estimates may miss significant correlations at the regional level (balancing effect), 31 a Bayesian pairwise GWAS approach (GWAS‐PW) was applied. 32 GWAS‐PW identifies trait pairs with high posterior probability of association (PPA) with a shared genetic variant (Model 3, PPA3 ≥ 0.90). To ensure that PPA3 is unbiased by sample overlap, fgwas version 0.3.6 was run on each pair of traits, and the correlation estimated from regions with null association evidence (PPA < 0.20) was used as a correction factor. 32 We then calculated Spearman's rank correlation for regions showing PPA3 > 0.90, approximating the direction of effect.
Finally, using a Bayesian method – ashR 33 – we studied the effect‐size distribution for ACD and VaD and related risk factors. Briefly, ashr tests the probability of non‐zero effect conferred by SNPs as a function of LD score, measuring the true effect size that is not zero and the underlying polygenic background. Using MTAG, 34 traits falling in similar polygenic profile to ACD or VaD are jointly analyzed in a bivariate scheme leveraging the pairwise trait genetic correlation to boost power to discover new loci. The significance threshold in the MTAG analysis is determined based on the number of traits sharing a similar polygenic profile and was additionally restricted to SNPs that also had nominal significance (p < 0.05) for each phenotype separately in the pre‐existing univariate GWAS.
2.6. Transcriptome‐wide association study and colocalization
We performed transcriptome‐wide association studies (TWASs) using the association statistics from the ACD and VaD (European‐only) and weights from 22 publicly available gene expression reference panels from blood (Netherlands Twin Registry [NTR], Young Finns Study [YFS]), arterial (genotype‐tissue expression [GTEx]), brain (GTEx, CommonMind Consortium [CMC]), and peripheral nerve tissues (GTEx). For each gene in the reference panel, precomputed SNP‐expression weights in the 1‐Mb window were obtained, including the highly tissue‐specific splicing quantitative trait loci (sQTLs) information on gene isoforms in the dorsolateral prefrontal cortex (DLPFC) derived from the CMC. TWAS‐Fusion 35 was used to estimate the TWAS z‐score (association statistic between predicted expression and ACD or VaD), derived from the SNP‐expression weights, SNP‐trait effect estimates, and the SNP correlation matrix. Transcriptome‐wide (TW) significant genes (eGenes) and the corresponding QTLs (expression QTLs [eQTLs]) were determined using Bonferroni correction in each reference panel, based on the average number of features (4235 genes) tested across all the reference panels. 35 eGene regions with eQTLs not reaching GW significance in association with ACD or VaD and not in LD (r 2 < 0.01) with the lead SNP for GW significant risk loci were considered novel. Finally, a colocalization analysis (COLOC) 36 was carried out at each locus to estimate the posterior probability of a shared causal variant (PP4 ≥ 0.75) between the gene expression and trait association, using a prior probability of 1.1 × 10−5. Furthermore, functional validation of the eGenes was performed by testing for positional overlap of the best eQTLs from TWAS with enhancer (H3K4me1, H3K27ac) and/or promoter (H3K4me3/H3K9ac) elements across a broad category of relevant tissue types (blood [BLD], brain/neurological [BRN]) using Haploreg version 4.1. 37
2.7. Identification of independent case–case loci with case–case GWAS
Leveraging summary statistics from our GWAS of ACD and VaD, as well as from publicly available existing GWASs of AD 38 and stroke, 39 we examined genetic uniqueness between these highly correlated though distinct disorders using case–case GWAS (CC‐GWAS), a method that tests for differences in allele frequency between cases of two disorders without individual‐level data. 40 By allowing for sample overlap between the two case‐control GWASs, CC‐GWAS can increase the power to detect signals otherwise missed in case‐control GWASs. We used a LD threshold of 0.2 (r 2 < 0.2) to distinguish CC‐GWAS‐specific loci from genome‐wide significant variants identified in the input case‐control GWAS.
3. RESULTS
Our analysis included 800,597 individuals comprising 46,902 and 8702 cases of ACD and VaD, respectively. They were recruited from the 19 Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) cohorts, the Alzheimer's Disease Genetics Consortium (ADGC), the European Alzheimer Disease Biobank (EADB), and the UK Biobank (UKBB), encompassing four different reported ancestries: European (98.5%), African (1.0%), Asian (0.4%), and Hispanics/Latino (0.1%). Association analyses were performed in each cohort following a predefined analysis plan, using logistic regression and Cox proportional hazards models for prevalent and incident cases, respectively. We performed study‐specific QC of the summary statistics data, followed by ancestry‐specific meta‐analyses and cross‐ancestry meta‐analyses of ACD and VaD, as described in the Methods section. For each cohort, a description, association analysis method, QC parameters, and cutoffs are provided in Supplementary File 2.
3.1. Meta‐analyses of ACD and VaD GWAS in European ancestry populations replicated known AD loci
We conducted fixed‐effects inverse variance‐weighted meta‐analyses of the 14 European ancestry cohorts (N = 466,606, N ACD = 44,009, N VaD = 3892) from the CHARGE consortium, ADGC, and the UKBB. Furthermore, we replicated significant and suggestive signals from our VaD GWAS in the EADB consortium VCID data (N = 275,745, N VaD = 4564). The complete list of cohorts included in this study is provided in Table S1.
A total of 11,596,629 and 9,878,961 SNPs passed the study‐level QC criteria and were tested for association with ACD and VaD, respectively. After post‐meta‐analysis QC, we identified 10 GW significant loci associated with ACD (GWS, p < 5 × 10−8), all of which had been previously associated with AD (Table 2, extended results in Table S2). Significant loci associated with ACD included signals in or around known AD genes such as APOE, BIN1, MS4A6A, PICALM, CR1, CD2AP, ABCA7, PILRB, SLC24A4, and ACE. For VaD, only one variant located near the APOE gene reached GW significance (Table 3, extended results in Table S3). The genomic inflation coefficients (lambda) were 1.05 and 1.07 for ACD and VaD, respectively. The lambda intercept computed with the LDSC software was 1.01 for both analyses, suggesting no systematic inflations of association statistics. The Manhattan and quantile‐quantile (QQ) plots for both analyses are provided in Figures 1, 2, 3 (Forest and locusZoom plots for prominent signals are provided in Figures S2 to S65).
TABLE 2.
Genome‐wide significant (p < 5 × 10−8) and suggestive (p < 1 × 10−6) variants associated with all‐cause dementia in European populations.
| rsID | Nearest gene | CHR | POS | EA/NEA | EAF | BETA | p value | HetISq | HetChiSq | HetPVal |
|---|---|---|---|---|---|---|---|---|---|---|
| rs429358 | APOE | 19q13.32 | 45411941 | C/T | 0.1515 | 1.1728 | 6.87E‐305 | 98.3 | 1104.478 | 2.534E‐222 |
| rs4663105 | BIN1 | 2q14.3 | 127891427 | C/A | 0.4151 | 0.156 | 5.856E‐34 | 63.4 | 51.892 | 0.00006868 |
| rs12453 | MS4A6A | 11q12.2 | 59945745 | C/T | 0.3998 | −0.0933 | 1.116E‐16 | 13.2 | 23.037 | 0.287 |
| rs10792832 | PICALM | 11q14.2 | 85867875 | A/G | 0.3585 | −0.0857 | 8.234E‐14 | 61.8 | 52.41 | 0.0000992 |
| rs10948367 | CD2AP | 6p12.3 | 47585615 | G/A | 0.2712 | 0.0886 | 4.041E‐13 | 4.2 | 20.88 | 0.4042 |
| rs4295 | ACE | 17q23.3 | 61556298 | C/G | 0.3897 | −0.0784 | 1.014E‐11 | 10.8 | 22.43 | 0.3176 |
| rs4844610 | CR1 | 1q32.2 | 207802552 | A/C | 0.1846 | 0.0961 | 1.245E‐11 | 71.4 | 66.468 | 3.519E‐07 |
| rs2906644 | PILRB | 7q22.1 | 99956290 | G/C | 0.1294 | −0.1016 | 7.627E‐09 | 0 | 8.219 | 0.9616 |
| rs3764650 | ABCA7 | 19p13.3 | 1046520 | G/T | 0.0957 | 0.1141 | 1.125E‐08 | 35 | 24.604 | 0.07712 |
| rs9323877 | SLC24A4 | 14q32.12 | 92934269 | G/A | 0.2521 | 0.0699 | 3.804E‐08 | 0 | 19.881 | 0.4654 |
| rs1532278 | CLU | 8p21.1 | 27466315 | T/C | 0.3819 | −0.0629 | 6.262E‐08 | 66.2 | 59.185 | 9.516E‐06 |
| rs7912495 | USP6NL | 10p14 | 11718713 | G/A | 0.4612 | 0.0684 | 6.774E‐08 | 0 | 15.343 | 0.7006 |
| rs17125924 | FERMT2 | 14q22.1 | 53391680 | G/A | 0.0928 | 0.1009 | 9.38E‐08 | 2.6 | 19.508 | 0.4247 |
| rs11767557 | EPHA1 | 7q34 | 143109139 | C/T | 0.1998 | −0.0742 | 9.721E‐08 | 31.2 | 29.052 | 0.08675 |
| rs1854554 | SEMA4D | 9q22.2 | 92155871 | A/G | 0.409 | 0.0598 | 1.096E‐07 | 4 | 20.83 | 0.4072 |
| rs7118826 | ANO3 | 11p14.2 | 26195535 | G/C | 0.4585 | 0.0686 | 1.238E‐07 | 18.7 | 23.368 | 0.2215 |
| rs79832570 | SPATC1 | 8q24.3 | 145097720 | C/T | 0.086 | 0.1433 | 1.385E‐07 | 37.3 | 19.147 | 0.08504 |
| rs13010870 | RBM43 | 2q23.3 | 151765163 | C/T | 0.2162 | −0.0732 | 1.649E‐07 | 0 | 18.774 | 0.5366 |
| rs1354106 | CD33 | 19q13.41 | 51737991 | G/T | 0.3441 | −0.0614 | 1.673E‐07 | 6.4 | 21.373 | 0.3755 |
| rs6014724 | CASS4 | 20q13.2 | 54998544 | G/A | 0.0874 | −0.1071 | 1.863E‐07 | 0 | 18.603 | 0.4825 |
| rs897150 | TRIB1 | 8q24.13 | 126576702 | A/G | 0.3016 | −0.0634 | 2.33E‐07 | 4 | 20.842 | 0.4065 |
| rs11168036 | HBEGF | 5q31.3 | 139707439 | T/G | 0.4933 | 0.0572 | 2.336E‐07 | 0 | 18.21 | 0.5735 |
| rs17269688 | NCK2 | 2q12.2 | 106469267 | G/A | 0.025 | −0.1937 | 2.389E‐07 | 41.8 | 27.478 | 0.03647 |
| rs677649 | RNU6‐11P | 7 | 123439244 | T/G | 0.1756 | 0.0851 | 2.505E‐07 | 9.9 | 19.987 | 0.3336 |
| rs8081878 | ZNF652 | 17q21.32 | 47436812 | T/A | 0.4634 | 0.057 | 2.609E‐07 | 9.9 | 22.19 | 0.3303 |
| rs4654450 | RP1‐37J18.2 | 1 | 4667378 | G/A | 0.3314 | −0.0697 | 2.684E‐07 | 16.2 | 22.679 | 0.2518 |
| rs11218343 | SORL1 | 11q24.1 | 121435587 | C/T | 0.0364 | −0.1658 | 2.909E‐07 | 20.7 | 21.432 | 0.2076 |
| rs2297508 | SREBF1 | 17p11.2 | 17715317 | C/G | 0.3659 | −0.0585 | 4.604E‐07 | 31.9 | 29.367 | 0.08078 |
| rs442495 | ADAM10 | 15q21.3 | 59022615 | C/T | 0.3245 | −0.0599 | 4.722E‐07 | 41 | 33.895 | 0.02684 |
| rs13316744 | AC091493.2 | 3 | 16742711 | C/G | 0.4544 | −0.0549 | 7.609E‐07 | 0 | 15.72 | 0.7338 |
| rs7068231 | ANK3 | 10q21.2 | 61784928 | T/G | 0.4005 | −0.0579 | 7.833E‐07 | 11.1 | 22.503 | 0.3139 |
| rs834398 | GABRB2 | 5q34 | 160528276 | G/A | 0.1971 | 0.0793 | 8.951E‐07 | 0 | 11.885 | 0.8905 |
| rs62013908 | RBFOX1 | 16p13.3 | 5991314 | G/C | 0.2547 | 0.0724 | 9.422E‐07 | 3.9 | 19.767 | 0.4087 |
Note: Genome‐wide significant variants are highlighted in orange.
TABLE 3.
Genome‐wide significant (p < 5 × 10−8) and suggestive (p < 1 × 10−6) variants associated with vascular dementia in European populations.
| rsID | Nearest gene | CHR | POS | EA/NEA | EAF |
BETA CHARGE |
p value CHARGE |
p value EADB |
p value COMBINED |
Direction |
|---|---|---|---|---|---|---|---|---|---|---|
| rs429358 | APOE | 19q13.32 | 45411941 | C/T | 0.1549 | 0.8794 | 2.67E‐86 | 5.66E‐113 | 2.9E‐196 | ++ |
| rs11911 | SPRY2 | 13q31.1 | 80910851 | C/A | 0.3756 | −0.1466 | 2.60E‐06 | 0.0653 | 0.0000335 | − |
| rs7101996 | GALNT18 | 11 | 11259298 | T/C | 0.4198 | −0.1362 | 3.06E‐06 | 0.48 | 0.00452 | −+ |
| rs2845990 | LINC02113 | 5 | 98907502 | C/T | 0.352 | −0.1383 | 3.24E‐06 | 0.872 | 0.00131 | −+ |
| rs117904289 | FOXA2 | 20p11.21 | 22782154 | G/A | 0.0858 | 0.2516 | 3.35E‐06 | 0.987 | 0.00112 | +− |
| rs838941 | SCARB1 | 12q24.31 | 125183316 | A/G | 0.4261 | 0.1344 | 3.56E‐06 | 0.33 | 0.0000654 | ++ |
| rs17418160 | ERBB4 | 2q34 | 213119022 | C/T | 0.0396 | 0.3341 | 3.58E‐06 | 0.882 | 0.00171 | +− |
| rs77542509 | TRPC6 | 11q22.1 | 101415824 | C/T | 0.058 | −0.3038 | 4.77E‐06 | 0.137 | 0.0000213 | − |
| rs6127311 | DOK5 | 20q13.2 | 53501017 | C/T | 0.0532 | −0.3141 | 4.83E‐06 | 0.294 | 0.0144 | +− |
| rs35945091 | LCN1P2 | 9 | 136185411 | C/T | 0.2227 | 0.164 | 5.15E‐06 | 0.855 | 0.00176 | +− |
| rs17059857 | ZNF236 | 18q23 | 74469493 | C/T | 0.0403 | 0.358 | 5.41E‐06 | 0.417 | 0.00934 | +− |
| rs143750890 | AJAP1 | 1p36.32 | 4602505 | C/T | 0.0273 | 0.4283 | 5.56E‐06 | 0.163 | 0.0000334 | ++ |
| rs9510987 | SPATA13 | 13 | 24575243 | G/T | 0.2525 | 0.1466 | 6.28E‐06 | 0.291 | 0.0000744 | − |
| rs55709546 | PHACTR3 | 20q13.32 | 58261107 | C/A | 0.0484 | 0.3044 | 6.53E‐06 | 0.645 | 0.000496 | ++ |
| rs12667855 | TMEM106B | 7p21.3 | 12124166 | T/G | 0.0981 | 0.225 | 6.59E‐06 | 0.542 | 0.000299 | ++ |
| rs281219 | SEMA6D | 15q21.1 | 47711652 | A/G | 0.1954 | 0.1641 | 6.65E‐06 | 0.316 | 0.0129 | +− |
| rs138352554 | GBP1 | 1p22.2 | 89517105 | G/A | 0.0359 | 0.3761 | 6.95E‐06 | 0.573 | 0.000406 | ++ |
| rs16967121 | RASGRP1 | 15q14 | 38923007 | G/A | 0.0658 | 0.2605 | 7.31E‐06 | 0.916 | 0.00126 | ++ |
| rs11007123 | WAC | 10p12.1 | 28763005 | C/T | 0.2804 | 0.1395 | 7.70E‐06 | 0.292 | 0.0139 | +− |
| rs4794009 | GIP | 17q21.32 | 47051955 | A/G | 0.4412 | 0.1274 | 8.17E‐06 | 0.797 | 0.000746 | ++ |
| rs35448830 | PRKCE | 2p21 | 46080762 | C/T | 0.0368 | 0.3346 | 8.48E‐06 | 0.631 | 0.000579 | ++ |
| rs2233754 | PSMA3 | 14q23.1 | 58755574 | C/A | 0.07 | 0.2575 | 9.39E‐06 | 0.601 | 0.00604 | +− |
Note: The meta‐analysis includes 11 cohorts from the CHARGE consortium and the UK Biobank (UKBB) GWAS. Direction denotes the direction of association in CHARGE and EADB. The genome‐wide significant variant is highlighted in orange.
Abbreviations: ADGC, Alzheimer's Disease Genetics Consortium; CHARGE, Cohorts for Heart and Aging Research in Genomic Epidemiology; EADB, European Alzheimer Disease Biobank; UKBB, UK Biobank.
FIGURE 1.
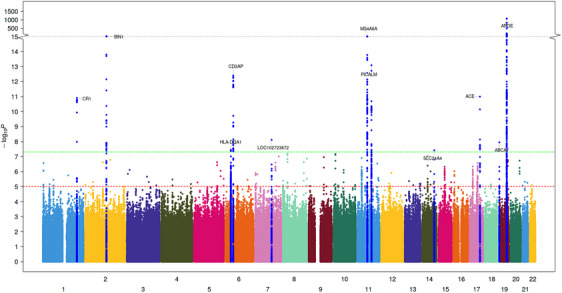
Manhattan plot of ACD GWAS. In addition to variants in APOE region, we identified five new genetic loci associated with VaD. Blue and red lines correspond to p value of 5e−7 and 5e−8 for genome‐wide suggestive and significant SNPs, respectively. Manhattan plots for the cross‐ancestry meta‐analysis. Each dot represents a SNP, the x‐axis shows the chromosomes where each SNP is located, and the y‐axis shows −log10 p value of the association of each SNP with ACD in the cross‐ancestry meta‐analysis. The red horizontal line shows the genome‐wide significant threshold (p value = 5e‐8; −log10 p value = 7.30). The nearest gene to the most significant SNP in each locus has been labeled.
FIGURE 2.
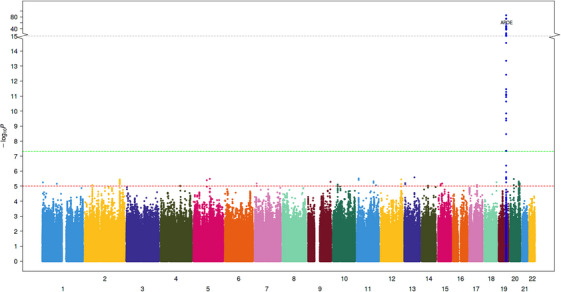
Manhattan plot of VaD GWAS. In addition to variants in the APOE region, we identified five new genetic loci associated with VaD. Blue and red lines correspond to a p value of 5e−7 and 5e−8 for genome‐wide suggestive and significant SNPs, respectively. Manhattan plots for cross‐ancestry meta‐analysis. Each dot represents a SNP, the x‐axis shows the chromosomes where each SNP is located, and the y‐axis shows the −log10 p value of the association of each SNP with VaD in the cross‐ancestry meta‐analysis. The red horizontal line shows the genome‐wide significant threshold (p value = 5e‐8; −log10 p value = 7.30). The gene closest to the most significant SNP in each locus has been labeled.
FIGURE 3.
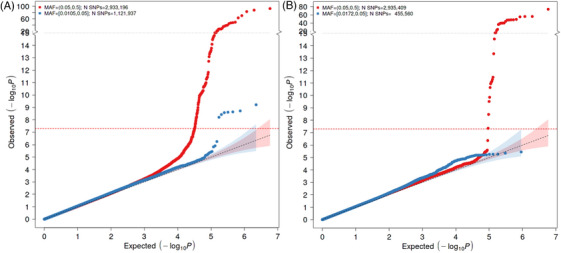
Q‐Q plots of ACD (left) and VaD (right) GWASs. The expected p values (x‐axis) are plotted against the observed p values (y‐axis). The units of the axes are the −log10 of the p value. The red and blue curves represent the plots with MAF ≥ 0.05 and 0.01, respectively. The diagonal line of the null hypothesis and its 95% confidence interval are plotted in gray based on the p values without the previously reported SNPs. The red dotted line represents the cutoff for genome‐wide significance. MAF, minor allele frequency.
For the VaD trait, we selected all variants with a p value less than 1 × 10−5 and meta‐analyzed with EADB summary results using a weighted sum of z‐scores approach. Only one variant near the APOE gene was statistically significant and had the same direction of effect in both studies (Table 3).
The meta‐analyses of ACD and VaD GWAS in African, Asian, and Hispanic/Latino ancestries did not provide new GW significant variants.
We replicated our VaD signals using EADB data (Table 3). Overall, we replicated an association within the APOE region. The suggestive variant near SPRY2 also has the lowest p value in the EADB GWAS with the same direction of effect in both studies.
3.2. Cross‐ancestry meta‐analysis of ACD and VaD GWAS
Next, we performed a cross‐ancestry meta‐analysis using MR‐MEGA, first to assess whether the increased sample size could lead to the identification of additional loci associated with ACD and VaD and to identify loci that are relevant in other ancestries. Most of the cross‐ancestry meta‐analyses included individuals of European ancestry and smaller samples from African, Asian, and Hispanic/Latino ancestries. The total number of variants included was 17,054,226 and 11,595,061 for ACD and VaD, respectively. The Manhattan plots of the SNP‐wide meta‐analyses for both traits are provided in Figures S66 and S67. Significant and suggestive signals for ACD and VaD are presented in Tables 4 and 5, and the extended results are in Tables S4 and S5. We identified novel signals reaching GW significance at 20q11.21 (CHD6, an oxidative DNA damage response factor previously associated with neurological phenotype), 41 2q14.1 (DAW1, involved in cerebrospinal fluid circulation and cilia motility during development), 42 and 15q15.1 (PWRN2, previously associated with tauopathy and Prader–Willi syndrome) 43 for ACD and 17q21.1 (MARCHF10) for VaD.
TABLE 4.
Genome‐wide significant (p < 5 × 10−8) variants associated with all‐cause dementia in cross‐ancestry meta‐analysis.
| rsID | Nearest gene | Chr | Pos | EA/NEA | P value | MAF | Beta | SE |
|---|---|---|---|---|---|---|---|---|
| rs10402524 | BCAM | 19p11 | 45329344 | T/C | 1.21E‐17 | 0.2336 | −0.168 | 0.045 |
| rs744373 | BIN1 | 2q14.3 | 127894615 | A/G | 1.90E‐17 | 0.358 | −0.139 | 0.031 |
| rs2278867 | MS4A6A | 11q13.1 | 59943109 | A/T | 1.72E‐15 | 0.2897 | 0.113 | 0.020 |
| rs10792832 | PICALM | 11q13.1 | 85867875 | A/G | 3.77E‐12 | 0.3135 | −0.074 | 0.036 |
| rs10948367 | CD2AP | 6q14.3 | 47585615 | A/G | 1.67E‐11 | 0.2328 | −0.042 | 0.017 |
| rs1408077 | CR1 | 1q11.1 | 207804141 | A/C | 4.75E‐10 | 0.1412 | 0.088 | 0.055 |
| rs4295 | ACE | 17q21.1 | 61556298 | C/G | 1.60E‐09 | 0.3666 | −0.066 | 0.018 |
| rs2208524 | CHD6 | 20q11.21 | 40423299 | T/C | 1.66E‐09 | 0.1268 | −0.103 | 0.027 |
| rs11691153 | DAW1 | 2q14.1 | 228780072 | T/C | 1.83E‐09 | 0.1536 | 0.099 | 0.025 |
| rs6853262 | LPHN3 | 4q22.1 | 61221892 | C/T | 6.22E‐09 | 0.06989 | 0.208 | 0.112 |
| rs2677386 | PWRN2 | 15q15.1 | 24432053 | T/C | 7.18E‐09 | 0.3612 | −0.083 | 0.017 |
| rs7006786 | ARHGEF10 | 8q13.2 | 1792639 | G/A | 8.81E‐09 | 0.08986 | 0.097 | 0.045 |
| rs35483531 | DEGS2 | 14q21.3 | 100653772 | C/T | 1.35E‐08 | 0.2993 | −0.004 | 0.024 |
| rs170084 | PMFBP1 | 16q11.2 | 72178483 | T/A | 2.79E‐08 | 0.107 | −0.068 | 0.029 |
| rs10940421 | SNX18 | 5q14.3 | 54036059 | A/G | 3.34E‐08 | 0.372 | 0.040 | 0.017 |
| rs138908633 | EPB41L4A | 5q14.3 | 111649017 | G/A | 3.76E‐08 | 0.03095 | −0.029 | 0.050 |
| rs74435987 | DUSP6 | 12q14.1 | 89152253 | G/T | 4.20E‐08 | 0.08766 | 0.078 | 0.130 |
| rs11225924 | DDI1 | 11q13.1 | 103493165 | C/T | 4.27E‐08 | 0.1034 | 0.152 | 0.105 |
| rs113747850 | MAPK9 | 5q14.3 | 179710663 | T/C | 4.93E‐08 | 0.123 | 0.071 | 0.024 |
Note: The meta‐analysis includes European, African, Asian, and Hispanic/Latino ancestries. Three new variants at 20q11.21, 2q14.1, and 15q15.1 reached genome‐wide significance (highlighted in orange).
Abbreviation: MAF, minor allele frequency.
TABLE 5.
Genome‐wide significant (p < 5 × 10−8) and suggestive (p < 1 × 10−6) variants associated with vascular dementia in cross‐ancestry meta‐analysis.
| rsID | Nearest gene | Chr | Pos | EA/NEA | p value | MAF | Beta | SE |
|---|---|---|---|---|---|---|---|---|
| rs10119 | TOMM40 | 19 | 45406673 | G/A | 1.21E‐17 | 0.2476 | −0.327 | 0.054 |
| rs4380108 | MARCHF10 | 17 | 60893485 | C/T | 9.59E‐09 | 0.3127 | −0.172 | 0.031 |
| rs55747619 | ITSN2 | 2 | 24530447 | C/G | 8.05E‐08 | 0.08706 | −0.384 | 1.969 |
| rs9379092 | CAGE1 | 6 | 7344531 | G/A | 9.80E‐08 | 0.1172 | −0.336 | 0.077 |
| rs3757193 | RPS6KA2 | 6 | 166923463 | C/T | 1.09E‐07 | 0.08347 | 2.151 | 0.636 |
| rs3871399 | CMTM7 | 3 | 32496413 | C/G | 2.61E‐07 | 0.124 | 0.550 | 0.640 |
| rs17315346 | BRINP2 | 1 | 177282235 | C/T | 2.67E‐07 | 0.01538 | −2.412 | 5.17 |
| rs1738249 | DNAH8 | 6 | 38753960 | C/T | 2.86E‐07 | 0.3013 | −0.050 | 0.040 |
| rs12095469 | OSBPL9 | 1 | 52206082 | G/A | 3.60E‐07 | 0.05292 | 3.654 | 3.513 |
| rs4820650 | ADRBK2 | 22 | 25925358 | T/C | 3.82E‐07 | 0.2468 | 0.050 | 0.054 |
| rs61859886 | MGMT | 10 | 131353192 | T/G | 4.45E‐07 | 0.1528 | −0.274 | 0.057 |
| rs9857196 | RYK | 3 | 133830660 | T/A | 5.09E‐07 | 0.01997 | 3.438 | 2.670 |
| rs637924 | PCDH7 | 4 | 31465610 | T/C | 6.77E‐07 | 0.2564 | −0.056 | 0.051 |
| rs35810115 | ZNF675 | 19 | 23780763 | C/T | 6.81E‐07 | 0.04992 | −1.169 | 2.339 |
| rs115331896 | CRBN | 3 | 3204942 | T/G | 6.95E‐07 | 0.01218 | 3.139 | 2.706 |
| rs4401880 | SLC18A1 | 8 | 19946066 | C/T | 7.20E‐07 | 0.3249 | 0.019 | 0.051 |
| rs4823298 | FBLN1 | 22 | 45915987 | T/C | 7.96E‐07 | 0.4581 | 0.029 | 0.0439 |
| rs17335455 | NXPH1 | 7 | 8853946 | T/G | 8.20E‐07 | 0.1633 | −0.096 | 0.042 |
| rs517484 | RP11‐6N13.1 | 5 | 104490130 | T/C | 8.51E‐07 | 0.1965 | −0.120 | 0.046 |
| rs12814413 | RBMS2 | 12 | 56916614 | T/C | 8.77E‐07 | 0.3514 | 0.050 | 0.051 |
| rs4665372 | CGREF1 | 2 | 27325837 | T/A | 9.19E‐07 | 0.3948 | −0.104 | 0.043 |
Note: The meta‐analysis includes European, African, Asian, and Hispanic/Latino ancestries. Genome‐wide significant variants are highlighted in orange.
Abbreviation: MAF, minor allele frequency.
3.3. Functional characterization of GW suggestive signals for ACD and VaD meta‐analyses
3.3.1. Shared genetic susceptibility with complex disease traits
The substantial shared genetic susceptibility of ACD and VaD with risk factors and complex disease traits is evident across different genomic scales (single variant, regional, and the global level). ACD exhibits genetic pleiotropy with vascular risk factors (hypertension, WMH burden), hematological traits (neutrophil, lymphocyte count), and blood‐based biomarkers indicative of inflammation (C‐reactive protein levels), hemostasis (fibrinogen, factor‐VII levels), and neurodegeneration (soluble TREM2 levels) (Table S6). This shared genetic susceptibility is primarily driven by the MS4A gene family (membrane‐spanning 4A; MS4A6A, MS4A4A). The sharing of common genetic variation between ACD and vascular risk factors (blood pressure traits [DBP, SBP, PP], and T2D) at the ACE and PILRB locus (Figure S68, Table S6) is further supported by our regional Bayesian pairwise (GWAS‐PW) analysis highlighting the high probability of harboring a shared causal variant (Table S7). Interestingly, the GWAS‐PW approach additionally reveals the shared genetic susceptibility of VaD with IS and WMH at the PRPF8 and PRDM6 locus. In support, global‐level genetic overlap analysis (excluding the APOE region) using GNOVA showed statistically robust evidence for the association of increased levels of WMH with increased risk of VaD (Table S26, Figure 4). Additionally, we observed an inverse association of high levels of HDL (protective) with ACD risk and high levels of DBP and LDL with VaD risk. As expected, a strong genetic correlation between poorer cognitive performance (GCF) and ACD was also observed. Our causal inference analysis, using MR‐LAP, confirmed the putative causal association of increased DBP and WMH levels with VaD risk. However, the genetic correlation between AD and related risk factors using Kunkle 2019 GWAS did not show this causal association (Tables S26–S27, Figure 4).
FIGURE 4.
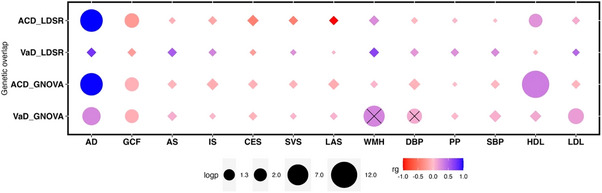
Shared genetic contribution between ACD/VaD and related risk factors. Contributions determined by LD score regression analysis (LDSR) (top), and Genetic Covariation Analyzer (GNOVA) (bottom). Effect sizes (rg) and significance levels (logp) are represented by color and symbol size. AD, Alzheimer's disease; GCF, general cognitive function; all stroke (AS) and its subtypes (ischemic, IS; cardioembolic, CES; small vessel, SVS; large artery, LAS); WMH, white matter hyperintensity burden; DBP, diastolic blood pressure; SBP, systolic blood pressure; PP, pulse pressure; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein. Diamond shape: non‐significant. Cross‐significant causal effect estimates from MR‐LAP analysis.
3.4. Polygenicity and multi‐trait analysis
To identify additional SNPs conferring susceptibility to ACD and acting through related risk factors, we jointly studied the GW distribution of genetic effects for ACD and its closely related traits. We first prioritized those traits that have a polygenic background similar to ACD using a Bayesian approach (ashR). The ashR analysis showed that certain traits (ACD, stroke and its subtypes, WMH, coronary artery disease [CAD]) had specific, possibly overlapping, pathophysiological processes compared to other ACD risk factors (SBP, smoking [SMK], body mass index) that involved multiple biological pathways (Figure S69, Table S9). Next, using multitrait GWAS analysis (MTAG, see Methods) on ACD and the prioritized traits (CAD, stroke, WMH), we identified intronic SNPs in SMG6 and ABCG8 to be GW significant (pMTAG < 1.67E‐08, for three phenotypes) for ACD (Table S10). Interestingly, SMG6 may also have a role in tau biology. 44 Finally, we explored the genetic difference between ACD/VaD and related disorders using case–case GWAS (CC‐GWAS, see Methods). Specifically, we compared (1) ACD/VaD with AD and (2) ACD with stroke. VaD was not compared with stroke because the two disorders are highly correlated. Here we report signals that were not GW significant in both respective case‐control GWASs. For AD, we identified two loci associated with ACD‐AD status, including the known APOE region and the IQUB gene (p < 2e‐08) on chr7 (Table S11). No GW significant loci were associated with VaD‐AD status, although we observed some suggestive association (Table S12). For ACD‐stroke, we identified 56 variants mapping to 10 genes on chromosomes 17, 8, 11, 15, 4, and 12, most of which are located at the SREBF1/TOM1L2 locus (p < 1e‐10) on chr17 (Table S13).
3.5. Functional prioritization using molecular profile (gene expression)
To functionally characterize and prioritize individual ACD and VaD genomic risk loci, we performed TWASs using TWAS‐Fusion, ACD, and VaD association statistics and weights from 23 gene‐expression reference panels from blood, arterial, and brain tissues (see Methods). We identified 29 trait‐associated (ACD/VaD) SNPs functioning as eQTLs, regulating the expression of 22 genes (eGenes) in disease‐relevant tissue types (Table S14). To explore whether the observed associations are real or merely reflect the random overlap between eQTLs and non‐causal risk variants for the dementia traits, a colocalization analysis was performed at each significant locus estimating the posterior probability of a shared causal variant (PP4 ≥ 75%) between the gene expression and trait association. Overall, 30% of the eQTL‐eGene satisfied the colocalization threshold for a shared causal variant between the ACD or VaD and gene expression. In addition to fine mapping functional genes (RP11‐385F7.1, CR1, MS4A6A, ACE, APOC4) in the loci exhibiting GW association with ACD/VaD, the TWASs identified putative novel (CLU‐ACD, PIKFYVE‐VaD, SH3D21‐ACD) genes satisfying transcriptome‐wide significance threshold (pTWAS < 1.18E‐05) and the colocalization probability threshold. Most (91%) of the eGenes are supported by the positional overlap of corresponding eQTLs with regulatory marks (enhancer and promoter binding sites) for active transcription in relevant tissue types.
3.6. Protein–protein interaction (PPI) evidenced SEMA4D, RBFOX1, and SPRY2 as hub genes for ACD and VaD
To determine the functional interactome of genes near genome‐wide significant (excluding APOE region) and suggestive loci (p < 1e‐6) associated with ACD and VaD, we performed a PPI analysis using the STRING database. The analysis comprised 82 ACD and 21 VaD GW significant and suggestive genes that were successfully mapped to the human genome. Evidence of interaction between proteins was based on “experiments,” “co‐occurrence,” “database,” and “co‐expression,” with a minimum score of 0.15. Non‐connected proteins were removed from the network. To further determine how suggestive genes will fit in the network of known AD genes, we used kmeans to cluster the proteins based on validated interaction. ACD genes formed two main clusters (Figure S70). The first cluster was enriched in known AD genes, including BIN1, CLU, ABCA7, and CR1, but also suggestive genes, including SEMA4D, CHD18, and APH1B, with more than two types of connection evidence. RBFOX1 appears to be a major hub gene for the second cluster, which includes other suggestive genes like AJAP1, ANO3, and TRIB1. RBFOX1 and SEMA4D strongly (>2 evidence of connection) interact with known AD genes, suggesting their potential role in ACD. The PPI network of VaD (Figure S71) genes highlights the potential role of SPRY2 as it functionally connects other genes, including ERBB4, RASGRP1, and FOXA2.
3.7. Pathway and functional enrichment analysis
We conducted several analyses (pathways, gene ontology, disease enrichment) to obtain functional and biological contexts of genes (near variants with p < 1e‐6, excluding the APOE region) associated with ACD and VaD.
3.7.1. Pathway analysis
Pathway analyses (Tables S15 and S16) revealed enrichment in several pathways, including “SREBF and miR33 in cholesterol and lipid homeostasis,” “Hypertrophy model,” and “Cholesterol metabolism with Bloch and Kandutsch‐Russell pathways” for ACD.
3.7.2. Gene Ontology (GO) analysis
GO analysis for ACD (Figure S72 and Table S17) focusing on the biological processes (GO‐BP) were enriched in terms related to amyloid‐beta, “amyloid‐beta metabolic process,” “amyloid precursor protein catabolic process,” and “negative regulation of amyloid precursor protein catabolic process” for ACD. For VaD (Figure S73 and Table S18), GO‐BP analysis was enriched in several terms, including “response to glucose,” “response to hexose,” “response to monosaccharide,” “mesenchymal cell differentiation,” and “response to carbohydrate.”
3.7.3. Disease enrichment and association analysis
(Figures S74 and S75, Tables S19–S25) revealed that ACD genes were previously connected to AD, tauopathy, nephritis, and central nervous system disease. It also highlighted previous associations of SEMA4D and RBFOX1 with diseases of the central nervous system. Besides the AD connection, VaD genes were previously related to cancer, diabetes, and colorectal carcinoma. Finally, we used Framingham Heart Study data to estimate the heritability of VaD and the genetic correlation with ACD. We found the heritability of VaD to be 6.1%, with a 95% confidence interval of [3.2%, 21%]. The genetic correlation of VaD and ACD was 0.48 (SE = 0.84).
4. DISCUSSION
Our findings expand the current knowledge base of dementia genetics by focusing on both ACD and VaD. Our GWAS of ACD replicated several genes previously associated with AD, and GWAS of VaD identified SNPs in the APOE region. Using functional PPI and TW analyses, we identified novel genes underlying ACD that have been implicated in recovery from vascular injury and in neurotrophin signaling. On the basis of LD score regression analysis, we suggest that certain vascular risk factors may not have a causal role not in both ACD and VaD pathogenesis.
In our ACD analysis of European ancestry, we identified 10 GW significant loci, including APOE, BIN1, MS4A6A, PICALM, CR1, CD2AP, ABCA7, PILRB, SLC24A4, and ACE, all of which have been linked with AD risk in prior studies. 45 In addition, our analyses highlighted 24 suggestive risk loci, of which 13 are novel. Among them are variants located near ANO3, a gene that encodes anoctamin‐3, a transmembrane protein that belongs to a family of calcium‐activated chloride channels and is implicated in focal dystonia, particularly craniocervical. 46 Another suggestive locus was located near SEMA4D, a gene that encodes Semaphorin 4D and is known to modulate various processes related to neuroinflammation and neurodegeneration, including the initiation of inflammatory microglial activation. 47 Indeed, SEMA4D is critical in regulating the transition between homeostatic and reactive states of various types of glial cells. Antibody blockade of SEMA4D is being explored as a potential disease‐modifying strategy to slow cognitive decline in patients with early Huntington's disease 48 and may be beneficial in other ACD. We have also identified a prominent signal near RBFOX1, a gene that encodes the RNA binding fox‐1, which has been shown to have a role in alternative splicing of the amyloid precursor protein. Genetic variation in this gene has been associated with brain amyloid burden in preclinical and early AD and with the risk of clinical AD in African Americans. 49 This gene may also impact dementia risk through non‐amyloidogenic pathways as it additionally regulates neuron development and neuronal excitability, including brain‐derived neurotrophic factor (BDNF)‐dependent long‐term potentiation in the hippocampus and has been implicated in brain development, essential tremor, and schizophrenia. 50
Other suggestive loci are located in the ZNF652 gene, a transcriptional repressor involved in nucleic acid binding that has diverse effects, including determining the risk of hypertension. Hypertension is the most important risk factor for stroke and WMH and may be the most important modifiable risk factor for population prevention of dementia. 51 We additionally identified a variant near Heparin Binding EGF like growth factor (HBEGF), a growth factor implicated in the pathobiology of cerebral autosomal dominant arteriopathy with sub‐cortical infarcts and leukoencephalopathy (CADASIL), 52 the major Mendelian prototype of VaD. HBEGF also has an effect on angiogenesis, expression of vascular endothelial growth factor A (VEGF‐A), inflammation, and oxidative stress and has been implicated in hydrocephalus. 53
APOE was strongly associated with both ACD and VaD in our meta‐analysis. While AD could drive the association of APOE with ACD, the relationship with VaD is less established but has been demonstrated in some population studies and candidate‐gene analyses 54 and in a recent GWAS among the GR@ACE project participants. 16 The link of APOE with VaD is in line with recent literature suggesting that the pathogenesis of APOE extends beyond Aβ peptide aggregation and clearance. 55 Indeed, APOE also influences microglia and the blood‐brain barrier (BBB) 56 and is associated with intracranial atherosclerosis, 57 WMH burden, and the presence of cerebral microbleeds, 58 as well as with cerebral hypertensive angiopathy, which is common in individuals with VaD. 59
In addition to a significant association of APOE with VaD in our sample, we identified several suggestive variants also associated with VaD. These include variants near the SPPRY2 protein‐coding gene as well as GALNT8, FOXA1, ERBB4, PSMA3, and SEMA6D with consistency across samples in the direction of effect and many SNPs in LD with the lead SNP. Our downstream analyses supported a highly plausible causal link between variants, including SEMA4D, HBEGF, PIKFYVE, and RBFOX1 with ACD and SPRY2 with VaD. These genes collectively emphasize a possible role for novel pathological mechanisms in ACD and VaD. Our findings highlight a crucial mechanism underlying ACD: recovery after vascular injury. For example, SEMA4D, a member of the semaphorin family, is upregulated in the neurovascular unit after IS, where it exerts multiple neuroprotective effects. 60 Moreover, this gene has been additionally highlighted in our PPI analysis as strongly associated with known AD genes. Another example is SPRY2, highlighted in our study as a suggestive gene for VaD, with strong functional associations with known AD and related dementias genes.
In the replication analysis of VaD signals in the EADB dataset, SPRY2 has the lowest p value and a consistent direction of association. This gene has also been suggested as a possible pharmacological target for stroke patients, as it promotes angiogenesis and glial scarring around the ischemic injury, preventing an increase in lesion size and secondary damage to brain tissue. 61 Also, SPRY2 may exert neuroprotective effects as its expression regulates BDNF‐induced signaling pathways. 62 Similarly, PIKFYVE is an essential regulator of platelet lysosome homeostasis, which in turn may promote recovery after IS. 63 Another hub gene in our analyses is RBFOX1, which, in addition to having a role in amyloid accumulation as discussed earlier, mediates ischemic damage by enhancing neuronal survival and BBB integrity after stroke. 64 This gene is a neuron‐specific splicing factor implicated in intellectual disability, epilepsy, autism, and Parkinson's disease. Its downregulation has been associated with destabilizing mRNAs encoding for synaptic transmission proteins, which may contribute to the loss of synaptic function in AD. 65 Furthermore, RBFOX1 upregulation was shown to influence neuronal expression levels of the BDNF receptor, TrkB, which in turn may affect the risk for ACD. 66
We found that the MS4A gene cluster drove genetic pleiotropy that involves vascular risk factors, inflammation, hemostasis, and soluble TREM2 levels. These findings align with preclinical studies 67 and emphasize the critical role and multifactorial contribution of this gene cluster to ACD pathogenesis. Although previous literature pointed to an association of ACE with AD but not VaD, 68 we herein show that this gene underlies both ACD and vascular risk factors. A recent study supports this finding by showing that overexpression of ACE on macrophages reduces vascular amyloid and GFAP+ astroglial reactivation, indicating its role in the protection of the neurovascular unit. 69 Moreover, our pairwise analysis highlighted a locus at the PRDM6 that explained a shared genetic susceptibility of VaD with IS and WMH. Low levels of leukocyte DNA methylation of the PRDM6 gene have been associated with an increased risk of IS and worse outcomes 3 months after an IS. 70 Moreover, PRDM6 acts as an epigenetic regulator of vascular smooth muscle cell plasticity. 71
Despite evidence showing an inverse relationship between plasma HDL levels and risk of incident AD, results are conflicting, with some studies pointing to higher dementia risk in individuals with high HDL levels, as was also the case in our study. 72 It should be acknowledged that HDL represents a class of lipoproteins that are heterogeneous in structure and function, which is not reflected by a simple measurement of HDL plasma levels. High HDL levels can be deleterious under certain conditions. 73 Vascular risk factors and the presence of cardiovascular disease can alter HDL functionality by changing the structure of HDLs and converting them into pro‐inflammatory, pro‐oxidant, prothrombotic, and proapoptotic compounds. Our observation aligns with recent Mendelian randomization data implicating an elevated HDL in risk of AD. 74
The following limitations should be considered when interpreting the results of this study. First, the multifactorial nature and heterogeneous clinical manifestations of ACD and VaD have led to various attempts to develop diagnostic criteria, which were differentially applied across the participating cohorts. ACD has been ascertained using DSM‐IV in some studies. In contrast, others have used ICD‐9/10 codes alone or in combination with autopsy or death certificate information, which can result in a varying proportion of persons identified as having dementia. The various cohorts also used different diagnostic criteria to define VaD. In all cohorts, a key requirement for VaD diagnosis remains the demonstration of a cognitive deficit and the presence of cerebrovascular disease, consistent with the most recent consensus criteria for VCID. 75 Whereas these criteria differ in sensitivity and specificity, thereby introducing statistical noise, this heterogeneity does not diminish the importance of the loci identified despite the constraints. A second limitation is the limited power to identify associations with VaD in ancestries other than European.
Our study identified several putative genetic variants and biological pathways associated with ACD and VaD and added additional support for the involvement of vascular mechanisms in dementia pathogenesis.
CONFLICT OF INTEREST STATEMENT
Agustin Ruiz and Itziar de Rojas acknowledge research support from Grifols SA (Spain), Fundacion Bacaria LaCaixa (Spain), Instituto de Salud Carlos III Ministry of Health (Spain), Roche, and Janssen. Agustin Ruiz received consulting fees and honoraria from Landsteiner Genmed SL, Grifols SA, and Janssen; support for attending meetings from Grifols SA; and stock options from Landsteiner Genmed SL. All authors report no conflicts of interest. Additional author disclosures are available in the supporting information (Supplementary File 2).
CONSENT STATEMENT
All participants provided written informed permission, or, for those with substantial cognitive impairment, consent was provided by a caregiver, legal guardian, or other proxy. Author disclosures are available in the supporting information.
SOFTWARE AVAILABILITY
Gene expression weights for TWAS: http://gusevlab.org/projects/fusion/
Radial‐MR: https://github.com/WSpiller/RadialMR
Magma.Celltyping: https://github.com/NathanSkene/MAGMA_Celltyping
DEFINITIONS
GWAS: Genome‐Wide Association Analysis Study
ACD: All‐cause dementia
VaD: Vascular dementia
VCID: Vascular Cognitive Impairment and Dementia
CHARGE: Cohorts for Heart and Aging Research in Genomic Epidemiology
ADGC: Alzheimer's Disease Genetics Consortium
UKBB: UK Biobank
EADB: European Alzheimer Disease DNA BioBank
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We thank the many study participants, researchers, and staff for collecting and contributing to the data. This project was conducted within the neurology working group of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. The CHARGE cohorts are supported in part by the National Heart, Lung, and Blood Institute (NHLBI) infrastructure grants R01HL105756 (Psaty), RC2HL102419 (Boerwinkle) and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193. Additional funding sources include the UT Health San Antonio Center for Biomedical Neuroscience (CBN) and grants from the NIA (AG059421, AG054076, AG049607, AG033090, AG066524, P30 AG066546, 5P30AG059305‐03, RF1 AG061729A1, 5U01AG052409‐04) and NINDS (NS017950, UF1NS125513, K01NS126489). Funding sources for each cohort are listed in Supplementary File 2.
1. APPENDIX COLLABORATORS
Bernard Fongang1,2,3,#,*, Muralidharan Sargurupremraj1,3,*, Xueqiu Jian1,3, Aniket Mishra4, Vincent Damotte5, Itziar de Rojas6,7, Olivia Skrobot8, Joshua C. Bis9, Kang‐Hsien Fan10, Erin Jacobsen11, Gloria Hoi‐Yee Li12, Jingyun Yang13, Bizzarro Alessandra14, Lauria Alessandra14, Saima Hilal15,16, Joyce Ruifen Chong15, Yuek Ling Chai15, M. J. Knol17, Maria Pina Concas18, Girotto Giorgia18,19, Moeen Riaz20, Chenglong Yu20, Alexander Guojonsson21, Paul Lacaze20, Adam C Naj22, Monica Gireud‐Goss1, Yannick N. Wadop1, Aicha Soumare4, Vincent Bouteloup4,23, Vilmundur Gudnason21,24, Petronilla Battista25, Aurora Santin19, Beatrice Spedicati19, Rodolfo Sardone26,27, Lenore Launer28, Jan Bressler29, Rebecca F Gottesman30, Quentin Le Grand31, Ilana Caro31, Gennady V. Roshchupkin32,33, Hampton L. Leonard34,35,36, Chaojie Yang37,38, Traci M. Bartz39,40, Constance Bordes31, Paul M. Ridker41,42, Mirjam I. Geerlings43,44,45,46, Natalie C. Gasca40, Ani Manichaikul37, Mike A. Nalls34,35,36, Stephen S. Rich37, Carsten O. Schmidt47, Stella Trompet48,49, Jessica van Setten50, Marion van Vugt50, Hans J. Grabe51,52, J Wouter Jukema49,53,54, Ina L. Rissanen55, Sylvia Wassertheil‐Smoller56, M. Arfan Ikram32, Eleanor M. Simonsick57, W T. Longstreth58,59, Daniel I. Chasman41,42, Jerome I. Rotter60, Naveed Sattar61, David J Stott62, Eric J Shiroma63, Sigurdur Sigurdsson24, Mohsen Ghanbari32, Ulf Schminke64, Eric Boerwinkle29,65, Hugo J Aparicio66,67, Alexa S Beiser66,68, Jose R Romero66,67, Vasileios Lioutas66,69, Ruiqi Wang66,68, Chloe Sarnowski70,71, Alexander Teumer51,72, Uwe Völker72,73, Thomas H. Mosley74, Marta Marquié6,7, Pablo García‐González6,7, Clàudia Olivé6, Raquel Puerta6, Amanda Cano6,7, Oscar Sotolongo‐Grau6,7, Sergi Valero6,7, Vanesa Veronica Pytel6, Maitée Rosende‐Roca6,7, Montserrat Alegret6,7, Lluís Tàrraga6,7, Mercè Boada6,7, Ángel Carracedo75,76, Emilio Franco‐Macías7,77, Gerard Piñol‐Ripoll78,79, Guillermo Garcia‐Ribas7,80,81,82, Jordi Pérez‐Tur7,82, Jose Luís Royo83, Jose María García‐Alberca84, Luis Miguel Real85,86, María Eugenia Sáez87, María J. Bullido7,88,89,90, Miguel Calero7,91,92, Miguel Medina7,93, Pablo Mir7,94,95, Pascual Sánchez‐Juan7,96, Pau Pastor97,98, Victoria Álvarez99,100, Benjamin Grenier‐Boley5, Fahri Küçükali101,102,103, Sven Van der Lee104,105,106, Oliver Peters107,108, Anja Schneider109,110, Martin Dichgans111,112,113, Dan Rujescu114, Jürgen Deckert115, Emrah Düzel116,117, Jens Wiltfang118,119,120, Michael Wagner121,122, Timo Grimmer123, Nikolaos Scarmeas124,125, Fermin Moreno7,126,127, Raquel Sánchez‐Valle128, Luis M Real85,129, Eloy Rodriguez‐Rodriguez7,130, Adolfo Lopez de Munain7,126,131, Alexandre de Mendonça132, Jakub Hort133,134, Caroline Graff135, Goran Papenberg136, Vilmantas Giedraitis137, Børge G. Nordestgaard138,139, Hilkka Soininen140, Miia Kivipelto141,142,143,144,145, Annakaisa Haapasalo146, Gael Nicolas147, Florence Pasquier148, Olivier Hanon149, Edna Grünblatt150,151,152, Daniela Galimberti153,154, Beatrice Arosio155,156, Patrizia Mecocci157, Alessio Squassina158, Lucio Tremolizzo159, Innocenzo Rainero160, Davide Seripa161, Julie Williams162, Philippe Amouyel163, Frank Jessen109,164,165, Tsolaki Magda166, Ruth Frikke‐Schmidt167,168, Kristel Sleegers101,102,169, Sebastiaan Engelborghs170,171, Rik Vandenberghe172,173, Martin Ingelsson174,175,176, Giacomina Rossi177, Mikko Hiltunen178, Rebecca Sims162, Magdalena Gugała‐Iwaniuk179, Mitchell K. P. Lai15, Venketasubramanian N180, Boon‐Yeow Tan181, Angelo Baldassare Cefalù182, Nicola J Armstrong183, Roberta Baschi184,185, Regis bordet 186,187, Anne‐Marie Bordet186,187, Henry Brodaty188, Srdjan Djurovic189,190, Grazia D'Onofrio191, Margaret Esiri192, Patrick Gelé186,187, Teresa Juarez‐Cedillo193, Raj Kalaria194,195, Pekka Karhunen196, Jan LACZO133, Ondrej LERCH133,134, Carlo Masullo197, Karen A Mather188,198, Vaclav MATOSKA199, Susanna Melkas200, Roberto Monastero184,185, Katya Numbers188, Francesco Panza201,202,203, Tuomo M Polvikoski195,204, Joe Quinn205, Arvid Rongve206,207, Perminder S Sachdev188,208, Michela Scamosci209, Anbupalam Thalamuthu188, Anne Tybjærg‐Hansen210, Martin VYHNALEK133,134, Shawn K. Westaway211, Amy E Martinsen212,213,214, Anne Heidi Skogholt214, Cristen J Willer215, Eystein Stordal216,217, Geir Bråthen218,219,220, Jonas Bille Nielsen214,215, Lars G Fritsche221, Laurent F Thomas214,222,223,224, Linda M Pedersen212, Maiken E Gabrielsen214, Ole Kristian Drange216,225, Sigrid Botne Sando214,218,226, Tore Wergeland Meisingset218,226, Genevieve Chene4,23, Wei Zhou227,228, Christophe Tzourio4,229, Adrienne Tin230, Oscar L Lopez231, Haan Mary232, Allison E Aiello233, Sigrid Børte213,214,234, Ingunn Bosnes216,217, Cornelia van Duijn235,236,237, Ching‐Lung Cheung238, David A Bennett13, Christopher Chen15, M. Ilyas Kamboh10, Claudia Satizabal1,3, M. Kamran Ikram17,239, Hieab Adams240,241,242, Yang Qiong68, Gerard D. Schellenberg22, Geir Selbæk213,243,244, Kristian Hveem214,245,246, Ole A Andreassen247,248, Alfredo Ramirez109,249,250,251, Carole Dufouil4,23, Wiesje van der Flier252, John‐Anker Zwart212,213,214, Stéphanie Debette4,253, Myriam Fornage29,254, Bendik Winsvold214,255,256, Jean‐Charles Lambert5, Agustin Ruiz6,7, Patrick G. Kehoe257, Galit Weinstein258,#, and Sudha Seshadri1,259,260,261,#
1 Glenn Biggs Institute for Alzheimer's & Neurodegenerative Diseases, University of Texas Health Science Center, San Antonio, TX, USA
2 Department of Biochemistry and Structural Biology, University of Texas Health Science Center, San Antonio, TX, USA
3 Department of Population Health Sciences, University of Texas Health Science Center, San Antonio, TX, USA
4 University of Bordeaux, Inserm, Bordeaux Population Health Research Center, UMR 1219, F‐33000 Bordeaux, France
5 Univ. Lille, Inserm, CHU Lille, Institut Pasteur de Lille, U1167‐RID‐AGE facteurs de risque et déterminants moléculaires des maladies liés au vieillissement, Lille, France
6 Research Center and Memory Clinic, ACE Alzheimer Center Barcelona. Universitat Internacional de Catalunya, Spain
7 Network Center for Biomedical Research in Neurodegenerative Diseases (CIBERNED), Instituto de Salud Carlos III, Madrid, Spain
8 Population Health Sciences, Bristol Medical School, University of Bristol, Bristol, UK
9 Cardiovascular Health Research Unit, Department of Medicine, University of Washington, Seattle, WA
10 Department of Human Genetics, School of Public Health, University of Pittsburgh, Pittsburgh, PA, USA
11 Department of Psychiatry and Neurology, School of Medicine, University of Pittsburgh, Pittsburgh, PA, USA
12 Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hung Hom, Hong Kong
13 Rush Alzheimer's Disease Center and Department of Neurological Sciences, Rush University Medical Center, Chicago, IL, USA
14 Geriatrics Unit, Policlinico Universitario Fondazione Agostino Gemelli IRCCS, Largo a Gemelli, 8−00168 Rome, Italy
15 Department of Pharmacology, National University of Singapore, Singapore
16 Saw Swee Hock School of Public Health, National University of Singapore and National University Health System, Singapore
17 Department of Epidemiology, Erasmus MC, University Medical Center, Rotterdam, the Netherlands
18 Institute for Maternal and Child Health, IRCCS Burlo Garofolo, 34127 Trieste, Italy
19 Department of Medicine, Surgery and Health Sciences, University of Trieste, 34139 Trieste, Italy
20 Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, VIC, Australia
21 Faculty of Medicine, University of Iceland, Reykjavik, Iceland
22 Department of Biostatistics and Epidemiology/Center for Clinical Epidemiology and Biostatistics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA
23 Pôle de Santé Publique Centre Hospitalier Universitaire (CHU) de Bordeaux, Bordeaux, France
24 Icelandic Heart Association, Kopavogur, Iceland
25 Istituti Clinici Scientifici Maugeri IRCCS, Laboratory of Neuropsychology, Bari Institute, Italy
26 Department of Translational Biomedicine and Neuroscience, University of Bari “Aldo Moro,” Bari, Italy
27 Unit of Statistics and Epidemiology, Local Healthcare Authority of Taranto, Taranto, Italy
28 Laboratory of Epidemiology and Population Sciences, Intramural Research Program, National Institute of Aging, National Institutes of Health, Bethesda, MD, USA
29 Human Genetics Center, School of Public Health, The University of Texas Health Science Center at Houston, Houston TX, USA
30 Stroke Branch, National Institute of Neurological Disorders and Stroke Intramural Program, National Institutes of Health, Bethesda, MD, USA
31 University of Bordeaux, Inserm, Bordeaux Population Health Research Center, team ELEANOR, UMR 1219, F‐33000 Bordeaux, France
32 Department of Epidemiology, Erasmus MC University Medical Center Rotterdam, Rotterdam, the Netherlands
33 Department of Radiology and Nuclear Medicine, Erasmus MC University Medical Center, the Netherlands
34 Center for Alzheimer's and Related Dementias, National Institutes of Health, Bethesda, MD, USA
35 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD, USA
36 Data Tecnica International LLC, Glen Echo, MD, USA
37 Center for Public Health Genomics, University of Virginia, Charlottesville, VA, USA
38 Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA, USA; Department of Biochemistry and Molecular Genetics, University of Virginia, Charlottesville, VA, USA
39 Cardiovascular Health Research Unit, Department of Medicine, University of Washington, Seattle, WA, USA
40 Department of Biostatistics, University of Washington, Seattle, WA, USA
41 Division of Preventive Medicine, Brigham and Women's Hospital, Boston, MA, USA
42 Harvard Medical School, Boston, MA, USA
43 Department of General Practice, Amsterdam UMC, location University of Amsterdam, Meibergdreef 9, Amsterdam, the Netherlands
44 Amsterdam Public Health, Aging & Later Life and Personalized Medicine, Amsterdam, the Netherlands
45 Amsterdam Neuroscience, Neurodegeneration and Mood, Anxiety, Psychosis, Stress, and Sleep, Amsterdam, the Netherlands
46 Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht and Utrecht University, Utrecht, the Netherlands
47 University Medicine Greifswald, Institute for Community Medicine, SHIP/KEF, Greifswald, Germany
48 Department of Internal Medicine, Section of Gerontology and Geriatrics, Leiden University Medical Center, Leiden, the Netherlands
49 Department of Cardiology, Leiden University Medical Center, Leiden, the Netherlands
50 Division Heart & Lungs, Department of Cardiology, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands
51 Department of Psychiatry and Psychotherapy, University Medicine Greifswald, Germany
52 German Center for Neurodegenerative Diseases (DZNE), Site Rostock/Greifswald, Rostock, Germany
53 Netherlands Heart Institute, Utrecht, the Netherlands
54 Einthoven Laboratory for Experimental Vascular Medicine, LUMC, Leiden, the Netherlands
55 Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands
56 Department of Epidemiology and Population Health, Albert Einstein College of Medicine, New York, NY, USA
57 Longitudinal Studies Section, Translational Gerontology Branch, National Institute on Aging, Baltimore, MD, USA
58 Department of Epidemiology, University of Washington, Seattle, WA, USA
59 Department of Neurology, University of Washington, Seattle, WA, USA
60 Institute for Translational Genomics and Population Sciences, Department of Pediatrics, Lundquist Institute for Biomedical Innovation at Harbor‐UCLA Medical Center, Los Angeles, CA, USA
61 BHF Glasgow Cardiovascular Research Centre, Faculty of Medicine, Glasgow, UK
62 Institute of Cardiovascular and Medical Sciences, College of Medical, Veterinary and Life Sciences, University of Glasgow, UK
63 Laboratory of Epidemiology and Population Sciences—National Institutes of Health, Bethesda, MD, USA
64 University Medicine Greifswald, Department of Neurology, Greifswald, Germany
65 Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA
66 Framingham Heart Study, Framingham, MA, USA
67 Department of Neurology, Boston University School of Medicine, Boston, MA, USA
68 Department of Biostatistics, Boston University School of Public Health, Boston, MA, USA
69 Department of Neurology, Beth Israel Deaconess Medical Center, Boston, MA, USA
70 Department of Epidemiology, Human Genetics and Environmental Sciences, University of Texas Health Science Center at Houston, School of Public Health, Houston, TX, USA
71 Department of Epidemiology, Human Genetics and Environmental Sciences, The University of Texas School of Public Health, Houston, Texas, USA
72 DZHK (German Centre for Cardiovascular Research), Partner Site Greifswald, Greifswald, Germany
73 Interfaculty Institute for Genetics and Functional Genomics, University Medicine Greifswald, Greifswald, Germany
74 Memory Impairment and Neurodegenerative Dementia (MIND) Center and Department of Medicine, University of Mississippi Medical Center, Jackson, MS, USA
75 Grupo de Medicina Xenómica, CIBERER, CIMUS. Universidade de Santiago de Compostela, Santiago de Compostela, Spain
76 Fundación Pública Galega de Medicina Xenómica‐IDIS, Santiago de Compostela, Spain
77 Unidad de Demencias, Servicio de Neurología y Neurofisiología. Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain
78 Unitat Trastorns Cognitius, Hospital Universitari Santa Maria de Lleida, Lleida, Spain
79 Institut de Recerca Biomedica de Lleida (IRBLLeida), Lleida, Spain
80 Hospital Universitario Ramon y Cajal, IRYCIS, Madrid, Spain
81 Unitat de Genètica Molecular, Institut de Biomedicina de València‐CSIC, Valencia, Spain
82 Unidad Mixta de Neurologia Genètica, Instituto de Investigación Sanitaria La Fe, Valencia, Spain
83 Departamento de Especialidades Quirúrgicas, Bioquímica e Inmunología. School of Medicine. University of Málaga, Málaga, Spain
84 Alzheimer Research Center & Memory Clinic, Instituto Andaluz de Neurociencia, Málaga, Spain
85 Unidad Clínica de Enfermedades Infecciosas y Microbiología, Hospital Universitario de Valme, Seville, Spain
86 Departamento de Especialidades Quirúrgicas, Bioquímica e Inmunología, Facultad de Medicina, Universidad de Málaga, Málaga, Spain
87 CAEBI, Centro Andaluz de Estudios Bioinformáticos, Sevilla, Spain
88 Centro de Biología Molecular Severo Ochoa (UAM‐CSIC)
89 Instituto de Investigacion Sanitaria “Hospital la Paz” (IdIPaz), Madrid, Spain
90 Universidad Autónoma de Madrid
91 CIEN Foundation/Queen Sofia Foundation Alzheimer Center/Instituto de Salud Carlos III
92 UFIEC, Instituto de Salud Carlos III, Madrid, Spain
93 CIEN Foundation/Queen Sofia Foundation Alzheimer Center, Madrid, Spain
94 Unidad de Trastornos del Movimiento, Servicio de Neurología y Neurofisiología. Instituto de Biomedicina de Sevilla (IBiS), Hospital Universitario Virgen del Rocío/CSIC/Universidad de Sevilla, Seville, Spain
95 Departamento de Medicina, Facultad de Medicina, Universidad de Sevilla, Seville, Spain
96 Alzheimer's Centre Reina Sofia‐CIEN Foundation, Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED), Madrid, Spain
97 Unit of Neurodegenerative Diseases, Department of Neurology, Hospital Germans Trias i Pujol, Badalona, Barcelona, Spain
98 Neurodegenerative Diseases Research Laboratory, Germans Trias i Pujol Research Laboratory, Badalona, Barcelona, Spain
99 Laboratorio de Genética, Hospital Universitario Central de Asturias, Oviedo, Spain
100 Instituto de Investigación Sanitaria del Principado de Asturias (ISPA), Oviedo, Spain
101 Complex Genetics of Alzheimer's Disease Group, VIB Center for Molecular Neurology, VIB, Antwerp, Belgium
102 Laboratory of Neurogenetics, Institute Born—Bunge, Antwerp, Belgium
103 Department of Biomedical Sciences, University of Antwerp, Neurodegenerative Brain Diseases Group, Center for Molecular Neurology, VIB, Antwerp, Belgium
104 Alzheimer Center Amsterdam, Neurology, Vrije Universiteit Amsterdam, Amsterdam UMC Location VUmc, Amsterdam, the Netherlands
105 Amsterdam Neuroscience, Neurodegeneration, Amsterdam, the Netherlands
106 Section Genomics of Neurodegenerative Diseases and Aging, Human Genetics, Vrije Universiteit Amsterdam, Amsterdam UMC location VUmc, Amsterdam, the Netherlands
107 German Center for Neurodegenerative Diseases (DZNE), Berlin, Germany
108 Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt‐Universität zu Berlin, and Berlin Institute of Health, Institute of Psychiatry and Psychotherapy, Hindenburgdamm 30, 12203 Berlin, Germany
109 German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
110 Department for Neurodegenerative Diseases and Geriatric Psychiatry, University Hospital Bonn, Venusberg‐Campus 1, 53127 Bonn, Germany
111 Institute for Stroke and Dementia Research (ISD), University Hospital, LMU Munich, Munich, Germany
112 German Center for Neurodegenerative Diseases (DZNE), Munich, Germany
113 Munich Cluster for Systems Neurology (SyNergy), Munich, Germany
114 Martin‐Luther‐University Halle‐Wittenberg, University Clinic and Outpatient Clinic for Psychiatry, Psychotherapy and Psychosomatics, Halle (Saale), Germany
115 Department of Psychiatry, Psychosomatics and Psychotherapy, Center of Mental Health, University Hospital of Würzburg, Germany
116 German Center for Neurodegenerative Diseases (DZNE), Magdeburg, Germany
117 Institute of Cognitive Neurology and Dementia Research (IKND), Otto‐von‐Guericke University, Magdeburg, Germany
118 Department of Psychiatry and Psychotherapy, University Medical Center Goettingen, Goettingen, Germany
119 German Center for Neurodegenerative Diseases (DZNE), Goettingen, Germany
120 Medical Science Department, iBiMED, Aveiro, Portugal
121 Department of Neurodegenerative Diseases and Geriatric Psychiatry, University of Bonn, Bonn, Germany
122 German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
123 Technical University of Munich, School of Medicine, Klinikum rechts der Isar, Department of Psychiatry and Psychotherapy
124 Taub Institute for Research in Alzheimer's Disease and the Aging Brain, The Gertrude H. Sergievsky Center, Department of Neurology, Columbia University, New York, NY, USA
125 1st Department of Neurology, Aiginition Hospital, National and Kapodistrian University of Athens, Medical School, Greece
126 Department of Neurology, Hospital Universitario Donostia, San Sebastian, Spain
127 Neurosciences Area. Instituto Biodonostia. San Sebastian, Spain
128 Alzheimer's Disease and Other Cognitive Disorders Unit, Service of Neurology, Hospital Clinic of Barcelona, Institut d'Investigacions Biomèdiques August Pi i Sunyer, University of Barcelona, Barcelona, Spain
129 Depatamento de Especialidades Quirúrgicas, Bioquímica e Inmunología, Facultad de Medicina, Universidad de Málaga, Málaga, Spain
130 Neurology Service, Marqués de Valdecilla University Hospital (University of Cantabria and IDIVAL), Santander, Spain
131 Department of Neurosciences, Faculty of Medicine and Nursery, University of the Basque Country, San Sebastián, Spain
132 Faculty of Medicine, University of Lisbon, Portugal
133 Memory Clinic, Department of Neurology, Charles University, Second Faculty of Medicine and Motol University Hospital, Czech Republic
134 International Clinical Research Center, St. Anne's University Hospital Brno, Brno, Czech Republic
135 Unit for Hereditary Dementias, Theme Aging, Karolinska University Hospital‐Solna, 171 64 Stockholm, Sweden
136 Aging Research Center, Department of Neurobiology, Care Sciences and Society, Karolinska Institutet and Stockholm University, Stockholm, Sweden
137 Department of Public Health and Carins Sciences/Geriatrics, Uppsala University, Sweden
138 Department of Clinical Biochemistry, Copenhagen University Hospital – Herlev Gentofte, Denmark
139 Department of Clinical Medicine, University of Copenhagen, Denmark
140 Institute of Clinical Medicine—Neurology, University of Eastern Finland, Finland
141 Division of Clinical Geriatrics, Center for Alzheimer Research, Care Sciences and Society (NVS), Stockholm, Sweden
142 Karolinska Institutet, Stockholm, Sweden
143 Institute of Public Health and Clinical Nutrition, University of Eastern Finland, Kuopio, Finland
144 Neuroepidemiology and Ageing Research Unit, School of Public Health, Imperial College London, London, UK
145 Stockholms Sjukhem, Research & Development Unit, Stockholm, Sweden
146 A.I. Virtanen Institute for Molecular Sciences, University of Eastern Finland, Kuopio, Finland
147 University of Rouen Normandy, Normandy University, Inserm U1245 and CHU Rouen, Department of Genetics and CNRMAJ, Rouen, France
148 University of Lille Inserm 1171, CHU Clinical and Research Memory Research Centre (CMRR) of Distalz, Lille, France
149 Université de Paris, EA 4468, APHP, Hôpital Broca, Paris, France
150 Department of Child and Adolescent Psychiatry and Psychotherapy, University Hospital of Psychiatry Zurich, University of Zurich, Zurich, Switzerland
151 Neuroscience Center Zurich, University of Zurich and ETH Zurich, Switzerland
152 Zurich Center for Integrative Human Physiology, University of Zurich, Switzerland
153 Neurodegenerative Diseases Unit, Fondazione IRCCS Ca’ Granda, Ospedale Policlinico, Milan, IT
154 Department of Biomedical, Surgical and Dental Sciences, University of Milan, Milan, Italy
155 Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy
156 Geriatric Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
157 Institute of Gerontology and Geriatrics, Department of Medicine and Surgery, University of Perugia, Italy
158 Department of Biomedical Sciences, University of Cagliari, Cagliari, Italy
159 Neurology, “San Gerardo” Hospital, Monza and University of Milano‐Bicocca, Italy
160 Department of Neuroscience “Rita Levi Montalcini,” University of Turin, Turin, Italy
161 Department of Hematology and Stem Cell Transplant, Vito Fazzi Hospital, Lecce, Italy
162 Division of Psychological Medicine and Clinical Neuroscience, School of Medicine, Cardiff University, Wales, UK
163 Univ. Lille, Inserm, CHU Lille, Institut Pasteur de Lille, U1167‐RID‐AGE—LabEx DISTALZ, facteurs de risque et déterminants moléculaires des maladies liés au vieillissement, Lille, France
164 Department of Psychiatry and Psychotherapy, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
165 Cluster of Excellence Cellular Stress Responses in Aging‐Associated Diseases (CECAD), University of Cologne, Cologne, Germany
166 1st Department of Neurology, Medical school, Aristotle University of Thessaloniki, Thessaloniki, Makedonia, Greece
167 Department of Clinical Biochemistry, Copenhagen University Hospital—Rigshospitalet, Copenhagen, Denmark
168 Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark
169 Department of Biomedical Sciences, University of Antwerp, Antwerp, Belgium
170 Department of Neurology, Universitair Ziekenhuis Brussel and NEUR (Neuroprotection & Neuromodulation) Research Group, Center for Neurosciences, Vrije Universiteit Brussel (VUB), Brussels, Belgium
171 Reference Center for Biological Markers of Dementia (BIODEM), Institute Born‐Bunge, University of Antwerp, Antwerp, Belgium
172 Laboratory for Cognitive Neurology, Department of Neurosciences, University of Leuven, Leuven, Belgium
173 Neurology Department, University Hospitals Leuven, Leuven, Belgium
174 Department of Public Health and Carins Sciences/Geriatrics, Uppsala University, Sweden
175 Krembil Brain Institute, University Health Network, Toronto, Canada
176 Tanz Centre for Research in Neurodegenerative Diseases, Departments of Medicine and Laboratory Medicine & Pathobiology, University of Toronto, Toronto, Canada
177 Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
178 Institute of Biomedicine, University of Eastern Finland, Kuopio, Finland
179 Institute of Psychiatry and Neurology, First Department of Neurology, Warsaw, Poland
180 Raffles Neuroscience Center, Raffles Hospital, Singapore
181 St Luke's Hospital, Singapore, Singapore
182 Department of Health Promotion Sciences, Maternal and Infant Care (PROMISE), University of Palermo, Palermo, Italy
183 Department of Mathematics and Statistics, Curtin University, Perth, Australia
184 Department of Biomedicine, Neuroscience and Advanced Diagnostics (BIND), University of Palermo, Palermo, Italy
185 Dementia and Parkinson's disease Center, University Hospital, “Paolo Giaccone,” Palermo, Italy
186 Univ Lille, Inserm, CHU Lille, France
187 Lille Neuroscience & Cognition, Lille, France
188 Centre for Healthy Brain Ageing, Discipline of Psychiatry & Mental Health, School of Clinical Medicine, Faculty of Medicine and Health, University of New South Wales, Sydney, Australia
189 NORMENT Centre, University of Bergen, Bergen, Norway
190 Dept of Medical Genetics, Oslo University Hospital, Oslo, Norway
191 Clinical Psychology Service, Health Department, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG), Italy
192 Nuffield Department of Clinical Neurosciences, Oxford University, Oxford, UK
193 Unidad de Investigación en Epidemiología y Servicos de Salud Área Envejecimiento, Centro Medico Nacional Siglo XXI, Instituto Mexicano del Seguro Social. Ciudad de Mexico, Mexico
194 Translational and Clinical Research Institute, Newcastle University
195 Campus for Ageing and Vitality, Newcastle upon Tyne NE4 5PL, UK
196 Faculty of Medicine and Health Technology, Tampere University, and Department of Clinical Chemistry, Fimlab Laboratories. Tampere, Finland
197 Institute of Neurology, Catholic University of the Sacred Heart, School of Medicine, Largo Agostino Gemelli, Rome, Italy
198 Neuroscience Research Australia, Sydney, Australia
199 Department of Clinical Biochemistry, Hematology and Immunology, Na Homolce Hospital, Prague, Czech Republic
200 Helsinki University Hospital, University of Helsinki, Helsinki, Finland
201 Neurodegenerative Disease Unit, Department of Basic Medicine, Neuroscience, and Sense Organs, University of Bari Aldo Moro, Policlinico, Piazza Giulio Cesare 11, 70124 Bari, Italy
202 Geriatric Unit & Laboratory of Gerontology and Geriatrics, Department of Medical Sciences, IRCCS “Casa Sollievo della Sofferenza,” San Giovanni Rotondo, Viale Cappuccini 1, 71013 San Giovanni Rotondo, Foggia, Italy
203 Unit of Research Methodology and Data Sciences for Population Health, National Institute of Gastroenterology Saverio de Bellis, Research Hospital, Castellana Grotte, Bari, Italy
204 Translational and Clinical Research Institute, Newcastle University, Newcastle, UK
205 Department of Neurology, Oregon Health & Science University, Portland, Oregon, USA
206 Department of Research and Innovation, Helse Fonna, Haugesund, Norway
207 Department of Clinical Medicine (K1), University of Bergen, Bergen, Norway
208 Neuropsychiatric Institute, Euroa Centre, Prince of Wales Hospital, Sydney, Australia
209 Institute of Gerontology and Geriatrics, Department of Medicine, University of Perugia, Perugia, Italy
210 Department of Clinical Biochemistry, Copenhagen University Hospital – Rigshospitalet, Copenhagen Denmark & Department of Clinical Medicine, Copenhagen, Denmark
211 Department of Neurology, Oregon Health & Science University, Portland, Oregon, USA
212 Department of Research and Innovation, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway
213 Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway
214 K. G. Jebsen Center for Genetic Epidemiology, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
215 Department of Internal Medicine, Division of Cardiovascular Medicine, University of Michigan, Ann Arbor, MI, USA
216 Department of Mental Health, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
217 Department of Psychiatry, Hospital Namsos, Nord‐Trøndelag Health Trust, Namsos, Norway
218 Department of Neuromedicine and Movement Science, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
219 Department of Neurology and Clinical Neurophysiology, St. Olav's Hospital, Trondheim University Hospital, Trondheim, Norway
220 K G Jebsen Centre for Alzheimer's Disease. Kavli Institutes of Systems Neuroscience, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
221 Center for Statistical Genetics, Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA
222 Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
223 BioCore—Bioinformatics Core Facility, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
224 Clinic of Laboratory Medicine, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
225 Division of Mental Health Care, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
226 Department of Neurology and Clinical Neurophysiology, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
227 Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA
228 Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA
229 Bordeaux University Hospital, Department of Medical Informatics, Bordeaux, France
230 Memory Impairment and Neurodegenerative Dementia (MIND) Center and Department of Medicine, University of Mississippi Medical Center, Jackson, MS, USA
231 Department of Neurology, School of Medicine, University of Pittsburgh, PA, USA
232 Department of Epidemiology & Biostatistics, University of California, San Francisco, CA, USA
233 Department of Epidemiology, Carolina Population Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
234 Research and Communication Unit for Musculoskeletal Health (FORMI), Department of Research and Innovation, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway
235 Department of Epidemiology, Erasmus Medical Center, Rotterdam, the Netherlands
236 Nuffield Department of Population Health, University of Oxford, Old Road Campus, Headington, Oxford, UK
237 Big Data Institute, Li Ka Shing Centre for Health Information and Discovery, Old Road Campus, Headington, Oxford, UK
238 Department of Pharmacology and Pharmacy, Centre for Genomic Sciences, University of Hong Kong, Hong Kong
239 Department of Neurology, Erasmus University Medical Centre, Rotterdam, the Netherlands
240 Department of Clinical Genetics, Erasmus MC, Rotterdam, the Netherlands
241 Department of Radiology and Nuclear Medicine, Erasmus MC, Rotterdam, the Netherlands
242 Department of Psychology, Latin American Brain Health (BrainLat), Universidad Adolfo Ibáñez, Santiago, Chile
243 Norwegian National Advisory Unit on Ageing and Health, Vestfold Hospital Trust, Tønsberg, Norway
244 Department of Geriatric Medicine, Oslo University Hospital, Oslo, Norway
245 HUNT Research Center, Department of Public Health and Nursing, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
246 Department of Research, Innovation and Education, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway
247 Division of Mental Health and Addiction, Oslo University Hospital, Oslo, Norway
248 NORMENT, University of Oslo, Oslo, Norway
249 Division of Neurogenetics and Molecular Psychiatry, Department of Psychiatry and Psychotherapy, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
250 Department of Neurodegenerative diseases and Geriatric Psychiatry, University Hospital Bonn, Medical Faculty, Bonn, Germany
251 Department of Psychiatry & Glenn Biggs Institute for Alzheimer's and Neurodegenerative Diseases, San Antonio, TX, USA
252 Alzheimer Center Amsterdam, Department of Neurology, Amsterdam Neuroscience, Vrije Universiteit Amsterdam, Amsterdam UMC, Amsterdam, the Netherlands
253 CHU de Bordeaux, Department of Neurology, Institute for Neurodegenerative Diseasese, Bordeaux, France
254 Institute of Molecular Medicine, McGovern Medical School, The University of Texas Health Science Center at Houston, Houston, TX, USA
255 Department of Research, Innovation and Education, Division of Clinical Neuroscience, Oslo University Hospital, Oslo, Norway
256 Department of Neurology, Oslo University Hospital, Oslo, Norway
257 Translational Health Sciences, Bristol Medical School, University of Bristol, Bristol, UK
258 School of Public Health, Faculty of Social Welfare and Health Sciences, University of Haifa, Haifa, Israel
259 Framingham Heart Study, Framingham, MA, USA
260 Department of Neurology, University of Texas Health San Antonio, San Antonio, TX, USA
261 Department of Neurology, Boston University School of Medicine, Boston, MA, USA
The Mega Vascular Cognitive Impairment and Dementia (MEGAVCID) consortium . A genome‐wide association meta‐analysis of all‐cause and vascular dementia. Alzheimer's Dement. 2024;20:5973–5995. 10.1002/alz.14115
Please contact Bernard Fongang for correspondence at Long School of Medicine, UT Health San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229, USA. Email: fongang@uthscsa.edu
[Correction added on July 26, 2024, after first online publication: The byline was revised to list only the consortium name. Bernard Fongang is acting as the contact for correspondence for the consortium and had been incorrectly listed in the byline.]
Contributor Information
The Mega Vascular Cognitive Impairment and Dementia (MEGAVCID) consortium:
Bernard Fongang, Muralidharan Sargurupremraj, Xueqiu Jian, Aniket Mishra, Vincent Damotte, Itziar de Rojas, Olivia Skrobot, Joshua C. Bis, Kang‐Hsien Fan, Erin Jacobsen, Gloria Hoi‐Yee Li, Jingyun Yang, Bizzarro Alessandra, Lauria Alessandra, Saima Hilal, Joyce Ruifen Chong, Yuek Ling Chai, M. J. Knol, Maria Pina Concas, Girotto Giorgia, Moeen Riaz, Chenglong Yu, Alexander Guojonsson, Paul Lacaze, Adam C Naj, Monica Gireud‐Goss, Yannick N. Wadop, Aicha Soumare, Vincent Bouteloup, Vilmundur Gudnason, Petronilla Battista, Aurora Santin, Beatrice Spedicati, Rodolfo Sardone, Lenore Launer, Jan Bressler, Rebecca F Gottesman, Quentin Le Grand, Ilana Caro, Gennady V. Roshchupkin, Hampton L. Leonard, Chaojie Yang, Traci M. Bartz, Constance Bordes, Paul M. Ridker, Mirjam I. Geerlings, Natalie C. Gasca, Ani Manichaikul, Mike A. Nalls, Stephen S. Rich, Carsten O. Schmidt, Stella Trompet, Jessica van Setten, Marion van Vugt, Hans J. Grabe, J Wouter Jukema, Ina L. Rissanen, Sylvia Wassertheil‐Smoller, M. Arfan Ikram, Eleanor M. Simonsick, W T. Longstreth, Daniel I. Chasman, Jerome I. Rotter, Naveed Sattar, David J Stott, Eric J Shiroma, Sigurdur Sigurdsson, Mohsen Ghanbari, Ulf Schminke, Eric Boerwinkle, Hugo J Aparicio, Alexa S Beiser, Jose R Romero, Vasileios Lioutas, Ruiqi Wang, Chloe Sarnowski, Alexander Teumer, Uwe Völker, Thomas H. Mosley, Marta Marquié, Pablo García‐González, Clàudia Olivé, Raquel Puerta, Amanda Cano, Oscar Sotolongo‐Grau, Sergi Valero, Vanesa Veronica Pytel, Maitée Rosende‐Roca, Montserrat Alegret, Lluís Tàrraga, Mercè Boada, Ángel Carracedo, Emilio Franco‐Macías, Gerard Piñol‐Ripoll, Guillermo Garcia‐Ribas, Jordi Pérez‐Tur, Jose Luís Royo, Jose María García‐Alberca, Luis Miguel Real, María Eugenia Sáez, María J. Bullido, Miguel Calero, Miguel Medina, Pablo Mir, Pascual Sánchez‐Juan, Pau Pastor, Victoria Álvarez, Benjamin Grenier‐Boley, Fahri Küçükali, Sven Van der Lee, Oliver Peters, Anja Schneider, Martin Dichgans, Dan Rujescu, Jürgen Deckert, Emrah Düzel, Jens Wiltfang, Michael Wagner, Timo Grimmer, Nikolaos Scarmeas, Fermin Moreno, Raquel Sánchez‐Valle, Luis M Real, Eloy Rodriguez‐Rodriguez, Adolfo Lopez de Munain, Alexandre de Mendonça, Jakub Hort, Caroline Graff, Goran Papenberg, Vilmantas Giedraitis, Børge G. Nordestgaard, Hilkka Soininen, Miia Kivipelto, Annakaisa Haapasalo, Gael Nicolas, Florence Pasquier, Olivier Hanon, Edna Grünblatt, Daniela Galimberti, Beatrice Arosio, Patrizia Mecocci, Alessio Squassina, Lucio Tremolizzo, Innocenzo Rainero, Davide Seripa, Julie Williams, Philippe Amouyel, Frank Jessen, Tsolaki Magda, Ruth Frikke‐Schmidt, Kristel Sleegers, Sebastiaan Engelborghs, Rik Vandenberghe, Martin Ingelsson, Giacomina Rossi, Mikko Hiltunen, Rebecca Sims, Magdalena Gugała‐Iwaniuk, Mitchell K. P. Lai, N Venketasubramanian, Boon‐Yeow Tan, Angelo Baldassare Cefalù, Nicola J Armstrong, Roberta Baschi, Regis bordet, Anne‐Marie Bordet, Henry Brodaty, Srdjan Djurovic, Grazia D'Onofrio, Margaret Esiri, Patrick Gelé, Teresa Juarez‐Cedillo, Raj Kalaria, Pekka Karhunen, Jan LACZO, Ondrej LERCH, Carlo Masullo, Karen A Mather, Vaclav MATOSKA, Susanna Melkas, Roberto Monastero, Katya Numbers, Francesco Panza, Tuomo M Polvikoski, Joe Quinn, Arvid Rongve, Perminder S Sachdev, Michela Scamosci, Anbupalam Thalamuthu, Anne Tybjærg‐Hansen, Martin VYHNALEK, Shawn K. Westaway, Amy E Martinsen, Anne Heidi Skogholt, Cristen J Willer, Eystein Stordal, Geir Bråthen, Jonas Bille Nielsen, Lars G Fritsche, Laurent F Thomas, Linda M Pedersen, Maiken E Gabrielsen, Ole Kristian Drange, Sigrid Botne Sando, Tore Wergeland Meisingset, Genevieve Chene, Wei Zhou, Christophe Tzourio, Adrienne Tin, Oscar L Lopez, Haan Mary, Allison E Aiello, Sigrid Børte, Ingunn Bosnes, Cornelia van Duijn, Ching‐Lung Cheung, David A Bennett, Christopher Chen, M. Ilyas Kamboh, Claudia Satizabal, M. Kamran Ikram, Hieab Adams, Yang Qiong, Gerard D. Schellenberg, Geir Selbæk, Kristian Hveem, Ole A Andreassen, Alfredo Ramirez, Carole Dufouil, Wiesje van der Flier, John‐Anker Zwart, Stéphanie Debette, Myriam Fornage, Bendik Winsvold, Jean‐Charles Lambert, Agustin Ruiz, Patrick G. Kehoe, Galit Weinstein, and Sudha Seshadri
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. https://ctg.cncr.nl/software/summary_statistics, https://pubmed.ncbi.nlm.nih.gov/36180795/
REFERENCES
- 1. Román G. Vascular dementia: a historical background. Int Psychogeriatr. 2003;15(suppl 1):11‐13. [DOI] [PubMed] [Google Scholar]
- 2. Dichgans M, Leys D. Vascular cognitive impairment. Circ Res. 2017;120(3):573‐591. [DOI] [PubMed] [Google Scholar]
- 3. Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672‐2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rajeev V, Fann DY, Dinh QN, et al. Pathophysiology of blood brain barrier dysfunction during chronic cerebral hypoperfusion in vascular cognitive impairment. Theranostics. 2022;12(4):1639‐1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sargurupremraj M, Suzuki H, Jian X, et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat Commun. 2020;11(1):6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69(24):2197‐2204. [DOI] [PubMed] [Google Scholar]
- 7. James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA. 2012;307(17):1798‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mönkäre S, Kuuluvainen L, Schleutker J, et al. Genetic analysis reveals novel variants for vascular cognitive impairment. Acta Neurol Scand. 2022;146(1):42‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011;7(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Markus HS, Schmidt R. Genetics of vascular cognitive impairment. Stroke. 2019;50(3):765‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malik R, Chauhan G, Traylor M, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alber J, Alladi S, Bae HJ, et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): knowledge gaps and opportunities. Alzheimers Dement. 2019;5:107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guerreiro R, Gibbons E, Tábuas‐Pereira M, Kun‐Rodrigues C, Santo GC, Bras J. Genetic architecture of common non‐Alzheimer's disease dementias. Neurobiol Dis. 2020;142:104946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schrijvers EM, Schurmann B, Koudstaal PJ, et al. Genome‐wide association study of vascular dementia. Stroke. 2012;43(2):315‐319. [DOI] [PubMed] [Google Scholar]
- 15. Kim Y, Kong M, Lee C. Association of intronic sequence variant in the gene encoding spleen tyrosine kinase with susceptibility to vascular dementia. World J Biol Psychiatry. 2013;14(3):220‐226. [DOI] [PubMed] [Google Scholar]
- 16. Moreno‐Grau S, de Rojas I, Hernández I, et al. Genome‐wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer's disease and three causality networks: the GR@ACE project. Alzheimers Dement. 2019;15(10):1333‐1347. [DOI] [PubMed] [Google Scholar]
- 17. Ikram MA, Bersano A, Manso‐Calderón R, et al. Genetics of vascular dementia – review from the ICVD working group. BMC Medicine. 2017;15(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer's disease. Transl Psychiatry. 2018;8(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh A, Hartge P, Kraft P, et al. Leveraging family history in population‐based case‐control association studies. Genet Epidemiol. 2014;38(2):114‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loh P‐R, Tucker G, Bulik‐Sullivan BK, et al. Efficient Bayesian mixed‐model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winkler TW, Day FR, Croteau‐Chonka DC, et al. Quality control and conduct of genome‐wide association meta‐analyses. Nat Protoc. 2014;9(5):1192‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mägi R, Horikoshi M, Sofer T, et al. Trans‐ethnic meta‐regression of genome‐wide association studies accounting for ancestry increases power for discovery and improves fine‐mapping resolution. Hum Mol Genet. 2017;26(18):3639‐3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI‐EBI GWAS Catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005‐D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evangelou E, Warren HR, Mosen‐Ansorena D, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bulik‐Sullivan B, Finucane KH, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu Q, Li B, Ou D, et al. A Powerful approach to estimating annotation‐stratified genetic covariance via GWAS summary statistics. Am Hum Genet. 2017;101(6):939‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mounier N, Kutalik Z. Bias correction for inverse variance weighting Mendelian randomization. Genet Epidemiol. 2023;47(4):314‐331. [DOI] [PubMed] [Google Scholar]
- 31. Shi H, Mancuso N, Spendlove S, Pasaniuc B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am J Hum Genet. 2017;101(5):737‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pickrell JK, Berisa T, Liu JZ, Segurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48(7):709‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18(2):275‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turley P, Walters RK, Maghzian O, et al. Multi‐trait analysis of genome‐wide association summary statistics using MTAG. Nat Genet. 2018;50(2):229‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gusev A, Ko A, Shi H, et al. Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet. 2016;48(3):245‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930‐D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51(3):404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mishra A, Malik R, Hachiya T, et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature. 2022;611(7934):115‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peyrot WJ, Price AL. Identifying loci with different allele frequencies among cases of eight psychiatric disorders using CC‐GWAS. Nat Genet. 2021;53(4):445‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mossink B, Negwer M, Schubert D, Kasri NN. The emerging role of chromatin remodelers in neurodevelopmental disorders: a developmental perspective. Cell Mol Life Sci. 2021;78(6):2517‐2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bearce EA, Irons ZH, Craig SB, et al. Daw1 regulates the timely onset of cilia motility during development. Development. 2022;149(12):dev200017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chung MS, Langouët M, Chamberlain SJ, Carmichael GG. Prader‐Willi syndrome: reflections on seminal studies and future therapies. Open Biology. 2020;10(9):200195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zuniga G, Levy S, Ramirez P, et al. Tau‐induced deficits in nonsense‐mediated mRNA decay contribute to neurodegeneration. Alzheimers Dement. 2022;19(2):405‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bellenguez C, Grenier‐Boley B, Lambert JC. Genetics of Alzheimer's disease: where we are, and where we are going. Curr Opin Neurobiol. 2020;61:40‐48. [DOI] [PubMed] [Google Scholar]
- 46. Lange LM, Junker J, Loens S, et al. Genotype‐phenotype relations for isolated dystonia genes: MDSGene Systematic Review. Mov Disord. 2021;36(5):1086‐1103. [DOI] [PubMed] [Google Scholar]
- 47. Taniguchi Y, Amazaki M, Furuyama T, et al. Sema4D deficiency results in an increase in the number of oligodendrocytes in healthy and injured mouse brains. J Neurosci Res. 2009;87(13):2833‐2841. [DOI] [PubMed] [Google Scholar]
- 48. Feigin A, Evans EE, Fisher TL, et al. Pepinemab antibody blockade of SEMA4D in early Huntington's disease: a randomized, placebo‐controlled, phase 2 trial. Nat Med. 2022;28(10):2183‐2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kunkle BW, Schmidt M, Klein HU, et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta‐analysis. JAMA Neurol. 2021;78(1):102‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomassoni‐Ardori F, Fulgenzi G, Becker J, et al. Rbfox1 up‐regulation impairs BDNF‐dependent hippocampal LTP by dysregulating TrkB isoform expression levels. eLife. 2019;8:e49673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gaussoin SA, Pajewski NM, Chelune G, et al. Effect of intensive blood pressure control on subtypes of mild cognitive impairment and risk of progression from SPRINT study. J Am Geriatr Soc. 2022;70(5):1384‐1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dabertrand F, Kroigaard C, Bonev AD, et al. Potassium channelopathy‐like defect underlies early‐stage cerebrovascular dysfunction in a genetic model of small vessel disease. Proc Natl Acad Sci USA. 2015;112(7):E796‐E805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim S, Subramanian V, Abdel‐Latif A, Lee S. Role of heparin‐binding epidermal growth factor‐like growth factor in oxidative stress‐associated metabolic diseases. Metab Syndr Relat Disord. 2020;18(4):186‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu X, Li L, Liu F, et al. ApoE gene polymorphism and vascular dementia in Chinese population: a meta‐analysis. J Neural Transm. 2012;119(3):387‐394. [DOI] [PubMed] [Google Scholar]
- 55. Wisniewski T, Drummond E. APOE‐amyloid interaction: therapeutic targets. Neurobiol Dis. 2020;138:104784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Serrano‐Pozo A, Das S, Hyman BT. APOE and Alzheimer's disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20(1):68‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Duong MT, Nasrallah IM, Wolk DA, Chang CCY, Chang TY. Cholesterol, atherosclerosis, and APOE in vascular contributions to cognitive impairment and dementia (VCID): potential mechanisms and therapy. Front Aging Neurosci. 2021;13:647990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schilling S, DeStefano AL, Sachdev PS, et al. APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta‐analysis. Neurology. 2013;81(3):292‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mahley RW. Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med. 2016;94(7):739‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carulli D, de Winter F, Verhaagen J. Semaphorins in adult nervous system plasticity and disease. Front Synaptic Neurosci. 2021;13:672891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Klimaschewski L, Sueiro BP, Millan LM. siRNA mediated down‐regulation of Sprouty2/4 diminishes ischemic brain injury. Neurosci Lett. 2016;612:48‐51. [DOI] [PubMed] [Google Scholar]
- 62. Gross I, Armant O, Benosman S, et al. Sprouty2 inhibits BDNF‐induced signaling and modulates neuronal differentiation and survival. Cell Death Differ. 2007;14(10):1802‐1812. [DOI] [PubMed] [Google Scholar]
- 63. Shaik NF, Regan RF, Naik UP. Platelets as drivers of ischemia/reperfusion injury after stroke. Blood Adv. 2021;5(5):1576‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yan H, Kanki H, Matsumura S, et al. MiRNA‐132/212 regulates tight junction stabilization in blood‐brain barrier after stroke. Cell Death Discov. 2021;7(1):380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alkallas R, Fish L, Goodarzi H, Najafabadi HS. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer's disease. Nat Commun. 2017;8(1):909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weinstein G, Beiser AS, Choi SH, et al. Serum brain‐derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol. 2014;71(1):55‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mattiola I, Mantovani A, Locati M. The tetraspan MS4A family in homeostasis, immunity, and disease. Trends Immunol. 2021;42(9):764‐781. [DOI] [PubMed] [Google Scholar]
- 68. Liu H, Liu M, Li W, et al. Association of ACE I/D gene polymorphism with vascular dementia: a meta‐analysis. J Geriatr Psychiatry Neurol. 2009;22(1):10‐22. [DOI] [PubMed] [Google Scholar]
- 69. Koronyo‐Hamaoui M, Sheyn J, Hayden EY, et al. Peripherally derived angiotensin converting enzyme‐enhanced macrophages alleviate Alzheimer‐related disease. Brain. 2020;143(1):336‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang H, Mo X, Wang A, et al. Association of DNA methylation in blood pressure‐related genes with ischemic stroke risk and prognosis. Front Cardiovasc Med. 2022;9:796245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Davis CA, Haberland M, Arnold MA, et al. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol. 2006;26(7):2626‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kjeldsen EW, Nordestgaard LT, Frikke‐Schmidt R. HDL cholesterol and non‐cardiovascular disease: a narrative review. Int J Mol Sci. 2021;22(9):4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Van Lenten BJ, Hama SY, de Beer FC, et al. Anti‐inflammatory HDL becomes pro‐inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995;96(6):2758‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Luo J, Thomassen JQ, Bellenguez C, et al. Genetic associations between modifiable risk factors and Alzheimer disease. JAMA Netw Open. 2023;6(5):e2313734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Skrobot OA, Black SE, Chen C, et al. Progress toward standardized diagnosis of vascular cognitive impairment: guidelines from the Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement. 2018;14(3):280‐292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. https://ctg.cncr.nl/software/summary_statistics, https://pubmed.ncbi.nlm.nih.gov/36180795/