

Abstract
Spinal Muscular Atrophy (SMA) is a neurodegenerative disease that is caused by deletion of the SMN (Survival of Motor Neuron) gene. The SMN protein is essential for cell survival and co-localized with TIA-1/R and G3BP, two characteristic markers of stress granules (SGs). To further study the SMN function in stress granules and in response to stress, we generated stable cell lines with SMN knockdown. Our data indicate that suppression of SMN drastically reduces cellular ability to form stress granules in response to stress treatment. In addition, we show that SMN deficiency sensitizes cells to sodium arsenite and H2O2, two well-known stress inducers, leading to cell death at a much lower concentration of inducers in SMN knockdown cells than in control cells. Interestingly, the cell death is correlated with formation of stress granules, suggesting that involvement of SMN in formation of stress granules may play an important role in cell survival. Furthermore, rescue of SGs formation by overexpression of G3BP can reverse the defective formation of stress granules and results in partial abrogation of cell death against SMN deficiency. We deduce that modulation of stress response may be useful for potential SMN treatment.
Electronic supplementary material
The online version of this article (doi:10.1007/s10571-011-9647-8) contains supplementary material, which is available to authorized users.
Keywords: Spinal muscular atrophy (SMA), Survival motor neuron, Cell death, Stress granules
Introduction
Spinal muscular atrophy (SMA) represents one of the most common genetic diseases leading to death in childhood. SMA is characterized by the degeneration of α-motor neurons in the anterior horn of the spinal cord. This loss of motor neurons leads clinically to progressive muscular weakness, dysphagia, dyspnea, and death (Crawford and Pardo 1996; Melki 1997; Iannaccone 1998). Regardless of disease severity, over 98% of SMA patients are caused by loss-of-function mutations in the survival motor neuron (SMN1) gene on chromosome 5q11.2-13.3 (Lefebvre et al. 1995). The SMN protein is ubiquitously expressed in all tissues of metazoan organisms and distributed in both the cytoplasm and nucleus (Liu and Dreyfuss 1996; Coovert et al. 1997; Paushkin et al. 2002).
SMN oligomerizes (Lorson et al. 1998) (Young et al. 2000) and forms a stable SMN complex with a group of proteins named Gemins [Gemins 2-8 (Liu et al. 1997; Charroux et al. 1999, 2000; Baccon et al. 2002; Gubitz et al. 2002; Pellizzoni et al. 2002; Carissimi et al. 2006)]. Numerous studies have showed that SMN is mainly involved in small nuclear ribonucleoprotein (snRNP) biogenesis, pre-mRNA splicing, RNA editing, and mRNA transport (Pellizzoni et al. 1998; Hannus et al. 2000; Meister et al. 2000; Rossoll et al. 2002; Zhang et al. 2008). Aside from its roles in RNA processing, the SMN protein has also been implicated to function in axonal transport (Rossoll et al. 2003; Zhang et al. 2003; Carrel et al. 2006) and forms granules in axons (Todd et al. 2010). However, the fact that SMN protein is expressed in all tissues and provides a housekeeping function required by all cells cannot explain why low levels of SMN protein in the cells of SMA patients lead only to the specific loss of motor neurons.
Stress response is a protective cellular process induced by a variety of environmental stresses including chemical exposure, heat shock, and UV irradiation. Expression of stress response proteins such as heat shock proteins (HSPs) and formation of stress granules (SGs) are two of the major events in response to stress. Motor neurons, the primary targets in diseases such as SMA and ALS, are well known to have a high threshold for induction of HSPs in response to stress both in vivo (Manzerra and Brown 1992; Manzerra and Brown 1996) and in primary cultures (Batulan et al. 2003). Formation of stress granules (SGs), on the other hand, is not well documented in motor neurons. SGs are formed in response to physiological and environmental stressors (Nover et al. 1989), under which transcription and translation of specific stress-induced genes are prioritized while many other genes encoding “housekeeping” proteins are silenced. About 50% of total poly(A)+mRNAs are actively recruited and dynamically sorted into SGs (Kedersha et al. 1999) in response to stress-induced phosphorylation of eukaryotic initiation factor (eIF) 2α (Clemens 2001). Once the stress is released, the SGs are disassembled, and mRNAs are repacked into translationally competent mRNAs and proteins are synthesized (Nover et al. 1989). Selective recruitment of specific mRNA transcripts into SGs is thought to regulate their stability and translation to protect cells against stress (Anderson and Kedersha 2002).
We had previously shown that SMN was distributed in granule-like structures in cytoplasm and co-localized with TIA-1/R and G3BP, two protein assemblers of SGs (Hua and Zhou 2004a, b). We concluded that SMN granules are SGs. To further examine the functions of SMN granules, we have knocked down SMN expression in several neural cell lines (P19 and PC12) and primary motor neurons. We demonstrate that the threshold of SG formation is elevated in cells with SMN deficiency, resulting in sensitization of cells to stressors. On the other hand, over-expression of G3BP partially overcomes the loss of SMN, leading to recovery of cellular ability to form SGs for protection against stressors. Our results suggest that modulation of SG formation may be potentially targeted for SMA treatment.
Methods
Cell Culture
P19 embryonic carcinoma cells and HEK293T cells were cultured in DMEM supplemented with 10% fetal calf serum. PC12 cells were cultured in DMEM supplemented with 10% horse serum and 5% fetal calf serum. Cortical neurons were prepared from embryonic day 18 rats as previously described (Sahin et al. 2005). Briefly, dissected cortices were placed into papain solution, and cells were mechanically dissociated by trituration. Neurons were plated on Poly-d-lysine-coated culture dishes in Neurobasal media (Invitrogen) supplemented with 2% B27 (Invitrogen), glutamine, and Penicillin/Streptomycin, and transduced with lentiviral particles containing either SMN shRNA or control shRNA sequence (see below).
Lentiviral Vector Carrying shRNA Against SMN
The pSP-108, pCMVD 8.2, and pMD2G plasmids were obtained from Dr. Y-X Wang at UMass Medical School (Yang et al. 2004). Oligonucleotides corresponding to SMN shRNA (5′-CCT TTA AGC ATG CTC TAA AGA ACG G-3′) and control shRNA (5′-CGT AAG CTA CGT AAT CGA ATC GAT C-3′) were synthesized. Two complementary oligonucleotides were annealed, digested, and then cloned into the pSP-108 plasmid. To produce recombinant lentiviruses (Zufferey et al. 1997), subconfluent HEK293T cells grown on 10 cm plates were cotransfected with 15 μg shRNA plasmid (Control shRNA or SMN shRNA), 6 μg pMD2G, and 10 μg pCMVD 8.2 using calcium phosphate. At 72 h after transfection, the medium containing lentiviral particles was collected and stocked at −80°C.
Generation of P19 and PC12 Stable Cell Lines Expressing SMN shRNA
Cells were transduced with lentiviral particles containing either SMN shRNA or control shRNA sequence. Forty eight hours after transduction, cells were selected for stable integrants using a complete medium containing puromycin. Individual clones were picked and expanded. Efficiency of stable RNA interference-mediated knockdown of SMN expression was determined by RT-PCR and Western blot analysis.
MTT Assay and Trypan Blue Exclusion Assay
To determine the cell viability, MTT assay was used. In brief, the MTT (Roche) was added to cultures at a final concentration of 0.5 mg/ml for 4 h at 37°C, and the resulting purple colored formazan product was solubilized with MTT solubilization solution (Roche). The dissolved reaction product was spectrophotometrically quantified at 570 nm and normalized against blank controls without cells. The relative cell viability was calculated as follows:
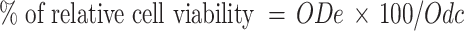 |
“ODe” is the experimental absorbance and “ODc” is the absorbance of untreated controls. The means and standard deviations were calculated based on results from three independent experiments.
In addition to the MTT assay, trypan blue exclusion assay was carried out to determine the cell death by staining the cells with 0.4% trypan blue solution (Gibco). This method relies on the alteration of membrane integrity as determined by the uptake of dye by dead cells that turn blue. The percentage of cell death was calculated by the following formula:
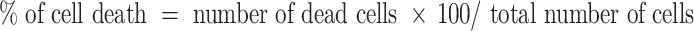 |
The means and standard deviations were calculated based on results from three independent experiments.
RNA Extraction and Reverse Transcription-PCR
Total RNA was isolated from culture cells using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. First-strand cDNA was reverse transcribed from 1 μg of total RNA using a Promega RT kit. Amplification of different genes by PCR was carried out with the following primers (SMN: 5′-primer: 5′-TTC CCC GAC CTG TGA AGT AG-3′; 3′-primer: 5′-GGG ATT ATT GGT GGT CCT GA-3′. GADPH: 5′- primer: 5′-GGC TGT GTG TCC CTG TAT-3′; 3′-primer: 5′-CCG CTC ATT GCC GAT AGT G-3′). All PCR products were visualized on ethidium bromide-stained 1.5% agarose gels.
Real-Time PCR
Real-time PCR to quantify the expression of the HRI gene was carried out on an Eppendorf mastercycler realplex4 real-time PCR system using SYBR Green master (Roche) with the following primers (5′-primer: 5′-CGA TGC CAA GTC AGA TAT GT-3′; 3′-primer: 5′-CAG TTG TTT GAA AAA GCT CAC-3′). The mRNA level for ®-actin was used as an internal control. The β-actin primers were as follows: 5′-primer: 5′- TCC CTG GAG AAG AGC TAC G-3′; 3′- primer: 5′- GTA GTT TCG TGG ATG CCA CA-3′. Relative changes of mRNA levels were calculated based on the ∆Ct method (Livak and Schmittgen 2001). Samples from at least three independent experiments were analyzed and the data were expressed as the averages ± S.E.
Western Blot Analysis
SDS-PAGE and western blot were performed as previously described (Zou et al. 2006). The transblots were probed with primary antibodies against SMN (1:5000, Transduction laboratories), G3BP (1:1000, Transduction laboratories), eIF2α and Phospho-eIF2α (1:1000, Cell Signaling), HRI (1:5000, Millipore) followed by appropriate secondary HRP-conjugated antibodies. In all cases, the blots were stripped with stripping buffer [62.5 mM Tris-HCl (pH 6.7), 2% SDS, and 90 mM 2-mercaptoethanol] and reprobed with anti-tubulin antibody (EMD) for loading controls. The signal was detected by enhanced chemiluminescence (Pierce). To quantify the level of target proteins, the blots were scanned and analyzed by densitometric analysis using NIH image software.
Immunofluorescence
P19, PC12 cells or primary cortical neurons cultured on poly l-lysine-coated glass cover slips were treated with sodium arsenite or heat-shock (42°C for 30 min). The cells were fixed with 4% paraformaldehyde for 30 min, washed 3 times with PBS buffer, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked with 5% goat serum in PBS for 30 min. The cells were incubated with primary antibodies against TIA-1/R (mouse monoclonal, 1:100, Santa Cruz, California) and/or against SMN (H195, rabbit polyclonal, 1:100, Santa Cruz, California) overnight at 4°C and washed three times with PBS. Secondary antibodies, goat anti-mouse IgG (Alexa fluor 568) and/or goat anti-rabbit IgG (Alexa fluor 488) (1:100, Molecular Probes, Oregeon), were applied for 1 h at room temperature, followed by three 5 min washes in PBS. Cell nuclei were counterstained with DAPI. The distribution of proteins was visualized under a confocal fluorescence microscopy (Leica). Images were acquired with a SPOT digital camera.
For SGs positive cells quantification, 100 cells were counted every sample. The percentage of SGs positive cells was calculated by the following formula:
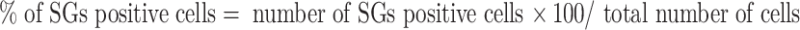 |
The means and standard deviations were calculated based on results from three independent experiments.
Statistical Analysis
All data, expressed as Mean ± SEM, were analyzed with the student t-test. Differences were considered statistically significant at P < 0.05.
Results
shRNA Efficiently Suppresses SMN Expression in P19 Cells
Although the functions of SMN protein in the biogenesis of ribonucleoprotein complexes, RNA processing and axonal transport have been reported (Pagliardini et al. 2000; Meister et al. 2002; Rossoll et al. 2002; Zhang et al. 2008), it is not completely understood how the loss of SMN protein contributes to the degeneration of motor neurons and SMA pathogenesis.
We have previously described that SMN is co-localized with TIA-1/R and G3BP in stress granules in response to stress (Hua and Zhou 2004a, b). To further address the potential role of SMN in cell survival under stressed conditions, we generated neuronal cell lines with decreased SMN expression. Lentiviral particles with SMN shRNA constructs were produced, and transduced into P19 cells. Stable P19 cell lines with RNA interference mediated knockdown of SMN were established. RT-PCR analysis of SMN knockdown P19 cell lines exhibited significant reduction of SMN mRNA (18.25% of control; Fig. 1a) as well as protein (33.09% of control; Fig. 1b). In contrast, expression of SMN in P19 cells transduced with lentivirus containing control shRNA was not affected. The cells with SMN knockdown are more sensitized to cell death inducers (Fig. 3).
Fig. 1.

RNAi-mediated knockdown of SMN. P19 cells were transduced with lentiviral particles of either shRNA against SMN (SMNi) or control shRNA (Con). Stable cell lines were established and analyzed for SMN mRNA levels by semi-quantitative RT-PCR with primers against SMN or control GADPH (a) and SMN protein expression by Western blotting with antibodies against SMN or Tubulin (b). RT-PCR products and Western blot bands were analyzed and quantified by ImageJ software. The mean values ± SEM (n = 3) are plotted
Fig. 3.

Downregulation of SMN sensitizes P19 cells to sodium arsenite and hydrogen peroxide, leading to cell death. a P19 cells were treated with Sodium Arsenite (SA) at different concentrations (100, 200, 500 and 1000 μM) for 24 h and the cell viability was measured by MTT. b P19 cells were cultured in medium with 1, 2 and 4 mM hydrogen peroxide for 24 h. The cell viability was determined by MTT. SMN deficiency leads to more cell death in P19-SMNi cells than in P19-Con cells in stress situation (* P < 0.05)
SMN Deficiency Delays and Reduces SG Formation Under Stress
SMN facilitates formation of stress granules in response to stress or when SMN is overexpressed in HeLa cells (Hua and Zhou 2004a, b). To further elucidate the function of SMN in response to stress, formation of SGs was examined in stable P19 cells with reduced SMN expression (Fig. 2). Stable SMN shRNA (P19-SMNi) and control shRNA (P19-Con) P19 cells were exposed to 200 μM of sodium arsenite for a time-course experiment. Distribution of SMN and TIA-1/R was determined by immunofluorescence (Fig. 2a). We found that only a few loose SGs began to emerge 1 h after sodium arsenite treatment in a portion of both P19-SMNi and P19-Con cells, and these cytoplasmic granules are positive for both SMN and TIA-1/R proteins. However, the number of cells that contained SGs in P19-Con cells (55.67 ± 4.04%) is significantly higher than that in P19-SMNi cells (20.67 ± 3.51%). Upon treatment with sodium arsenite for 2 h, we observed that almost all of the P19-Con cells formed SGs (TIA-1/R positive) (92.33 ± 2.52%) in contrast to P19-SMNi cells (51.01 ± 3.02%) of which about 50% were SG positive (Fig. 2b). Similar results were obtained when P19-Con and P19-SMNi cells were exposed to heat shock at 42°C for 30 min (Supplementary Fig. 1), indicating that SMN is necessary for SG formation under different conditions of stress. Consistent with these results, we also observed defective formation of SGs with SMN deficiency in PC12 cells (Supplementary Fig. 2). We found that the number of cells containing positive SGs in SMN shRNA PC12 cells (~25%) was significantly lower than that in the control PC12 cells (~75%), suggesting that suppression of SMN markedly reduces the cellular ability to form stress granules under stress conditions. In addition, we observed that the ability of primary neurons to form SGs in response to arsenite was significantly reduced when SMN was suppressed (Supplementary Fig. 3). Overexpression of SMN by transfecting a GFP-SMN plasmid into SMN shRNA p19 stable cells reversed the effects of shRNA on formation of SGs, indicating the specificity of SMN shRNA (data not shown).
Fig. 2.

Formation of SGs in response to sodium arsenite was delayed and reduced in P19 cells with SMN deficiency. P19 cells (Control and SMNi) were treated with 200 μM of sodium arsenite for 1 and 2 h. a Double immunofluorescence experiments were performed using mouse anti-TIA-1/R antibody and rabbit anti-SMN antibody, and secondary antibodies against rabbit (Alexa Fluor 488, green) and mouse (Alexa Fluor 568, red). Localization of proteins was monitored by a confocal microscopy. Yellow represents co-localization. The arrows point to stress granules (SGs). b Cells containing SGs after sodium arsenite treatment were counted. The number of cells with positive SGs is significantly lower in P19-SMNi cells than in P19-control cells. Three independent experiments were performed for each time point (*P < 0.01). Scale bar: 20 μm
SMN Deficiency Sensitizes Neural Cells to Stressors
One of the hallmarks of SMA disease is the death of motor neurons. To investigate how defective expression of SMN may induce cell death, P19-SMNi and P19-Con cells were exposed to sodium arsenite and H2O2, two well-known stressors that induce cellular stress response and cell death. P19 cells were treated with different amounts of sodium arsenite (100–1000 μM) or H2O2 (1–4 mM) for 24 h. Cell viability was then determined using MTT methods. As expected, we found that concentrations as low as 100uM of sodium arsenite or 1 mM of H2O2 significantly reduced cell viability in both control and SMNi P19 cells (Fig. 3). However, it is apparent that identical concentrations of either sodium arsenite or H2O2 induced more cell death in SMNi P19 cells than in control cells. For instance, 200 μM of sodium arsenite induced 50% of cell death in SMNi P19 cells but only 35% of cell death in the control P19 cells (Fig. 3a). Similarly, 1 mM of H2O2 induced death in more than 50% of SMNi cells while only 30% of P19-Con cells were killed by this treatment (Fig. 3b). The P19-con cellular viability is significantly higher than that of SMNi cells under the same treatment, suggesting that P19-SMNi cells were sensitized to stress treatment. Interestingly, we found that arsenite-induced P19 cell death between cells with and without SMN knockdown can be eliminated by emetine (Supplementary Fig. 4), a chemical that stabilizes polysomes and inhibits the formation of SGs (Kedersha et al. 2000). This finding is consistent with the hypothesis that the increased cell death in P19-SMNi cells treated with arsenite compared to control P19 cells is likely due to defects in SG formation.
Transfection of G3BP Partially Compensates for the Loss of SMN in Formation of SGs and Survival
If SMN defects induce cell death via impaired formation of SGs, stimulation of SG formation may be potentially targeted to compensate for the loss of SMN protein. To address this question, GFP-G3BP and control GFP were transfected into stable P19 cells with SMN shRNA. RasGAP-associated endoribonuclease (G3BP) is an effector molecule promoting SG assembly (Kedersha et al. 1999; Tourriere et al. 2003). As expected, transfection of GFP-G3BP but not the GFP control into P19-SMNi cells significantly increased the number of SG positive cells (Fig. 4a, c), indicating that GFP-G3BP restored cellular ability to form SGs. To examine if recovery of SG formation by G3BP compensates for the loss of SMN on cell survival against stresses, we performed trypan blue staining in P19 SMNi cells treated with sodium arsenite for 2, 8, and 24 h. We found a higher survival rate in P19-SMNi cells transfected with GFP-G3BP than those transfected with GFP at 8 and 24 h (Fig. 4d). This result suggests that formation of SGs plays an important role in cell survival in response to stress.
Fig. 4.

G3BP partially protects cells against SMN defect. a GFP-G3BP (left) and GFP (right) were transfected into P19 cells in which SMN is suppressed. Cells were then treated with sodium arsenite. Immunofluorescence analysis indicates that GFP-G3BP, but not GFP alone, restores cellular ability to form SGs. b G3BP protein expression in P19 cells transfected with GFP and GFP-G3BP was analyzed by Western blotting with antibodies against G3BP and α-tubulin. c. Quantitative analysis of SG positive cells indicates that the number of P19-SMNi cells with SGs is much higher in G3BP transfected cells than in GFP transfected cells. D. P19-SMNi cells that were transfected with G3BP or GFP were treated with sodium arsenite for 2–24 h. Cell death was determined by trypan blue exclusion assay. Overexpression of GFP-G3BP statistically reduced cell death in P19-SMNi cells (* P < 0.05). Scale bar: 10 μm
Loss of SMN Gene Reduces eIF2-α Phosphorylation in Response to Cellular Stress
In mammalian cells, the assembly of SGs is initiated by the phosphorylation of eIF2α, a translation initiation factor that loads the initiator tRNA onto the small ribosomal subunit (Berlanga et al. 1998; Gray and Wickens 1998; Kedersha et al. 1999). We have demonstrated that SMN facilitates formation of SGs and that loss of SMN reduces SGs in response to a sublethal dose of arsenite. To examine the mechanism, we analyzed eIF2α phosphorylation. P19 cells were treated with 200 μM of sodium arsenite for 2 h. As expected, western blotting showed that eIF2α phosphorylation in treated P19-con cells was significantly increased compared to untreated P19-con cells (Fig. 5a). However, suppression of SMN in P19-SMNi cells dramatically decreased eIF2α phosphorylation in response to sodium arsenite although the total eIF2α level remained similar between P19-con cells and P19-SMNi cells regardless of treatment (Fig. 5a). These results suggest that SMN influences eIF2α phosphorylation, resulting in facilitation of SG assembly in response to stress.
Fig. 5.

SMN defect reduces eIF-2α phosphorylation and HRI mRNA levels in response to arsenite treatment. a P19-Con and P19-SMNi cells were treated with 200 μM of arsenite for 2 h and total extract was collected. After protein separation by SDS-PAGE and transfer to PVDF membrane. The blots were probed with antibodies against eIF-2α, phosph-eIF-2α, HRI and SMN, α-tubulin was used as a loading control. b Total RNA was isolated from control and SMNi P19 cells with or without SA treated. HRI mRNA level was quantitated by real-time RT-PCR (* P < 0.01)
There are four protein kinases involved in the process of eIF2α phosphorylation in mammalian cells: the double-stranded RNA activated protein kinase PKR, the mammalian homolog of yeast GCN2 (Dey et al. 2005), the endoplasmic reticulum (ER)-localized eIF2α kinase PERK/PEK (Zhang et al. 2002), and the heme-regulated inhibitor kinase HRI (Zhan et al. 2002). Although these four kinases have homologous function on eIF2α phosphorylation, they each have response to different stressor depend on special regulatory domains. In response to arsenite-induced stress in mammalian cells, HRI is expressed and is required for eIF2α phosphorylation to induce SG formation and inhibit protein synthesis (McEwen et al. 2005). To assess whether, we carried out real-time PCR measurements for HRI mRNA level in P19 cells. In nonstressed cells, SMN deficiency had no effect on HRI mRNA level. After treated by SA, there was significant increase in HRI mRNA level. Comparing to nearly 4-fold increase in P19-con cells, the HRI mRNA was only 2-fold upregulated in response to arsenite stimulation (Fig. 5b). To determine if a similar change in HRI protein occurs upon SMN deficiency after SA treatment, we used western blot. In close parallel to the reduction in mRNA level, HRI protein level was reduced in P19-SMNi cells compared to P19-con cells (Fig. 5a).
Discussion
Spinal muscular atrophy (SMA) is an autosomal recessive motor neuron disease that affects approximately 1 in 6000 live births and is a leading cause of infant mortality (Pearn 1980; Crawford and Pardo 1996). Genetic analysis and physical mapping in SMA patients led to the identification of survival of motor neurons (SMN) as the disease gene for SMA (Lefebvre et al. 1995). Although SMN is ubiquitously expressed, the mechanisms by which SMN regulates cell survival, in particular in motor neurons, are not very clear. Disruption of SMN in vivo induces massive cell death (Schrank et al. 1997; Hsieh-Li et al. 2000; Monani et al. 2000; Wang and Dreyfuss 2001). However, in vitro survival of cultured sensory neurons from dorsal root ganglion of SMN−/−; hSMN2 mice in the presence of NGF or other neurotrophic factors is not affected (Jablonka et al. 2006). In contrast, it has been shown that overexpression of SMN prolongs cell survival in PC12 cells deprived of trophic support although it is ineffective in inhibiting cell death induced by UV or etoposide treatment (Vyas et al. 2002). These findings suggest that the role of SMN in cell survival is largely dependent on environment conditions.
To investigate how external factors affect cells with SMN deficiency, we established stable cell lines (P19 and PC12) using RNA interference mediated knockdown of SMN. We show that SMN shRNA effectively suppresses expression of SMN (Fig. 1), leading to sensitization of P19 cells to sodium arsenite and H2O2, resulting in more cell death. We further demonstrate that the cell death is inversely correlated with the number of stress granules (SGs) that are formed in response to stresses. Based on these data, one would expect that the correlation between SGs and cells should be reproduced in SMA patient fibroblasts. However, we demonstrated previously that there were barely SGs in fibroblasts even with stress treatment (Hua and Zhou 2004a, b). It would be interesting to test the correlation in other cells from SMA patients when they are available.
SGs are cellular structures that were first observed in the cytoplasm of tomato cells subjected to heat shock. SGs are RNA granules that contain mRNAs encoding most cellular proteins but exclude mRNAs encoding heat shock proteins (Nover et al. 1989). Unlike other RNA granules, SGs are not seen in cells that are growing under favorable conditions but are rapidly induced in response to environmental stress, such as heat shock, chemical exposure, and UV irradiation. Several groups have previously reported that SMN was localized in granule-like structures in cytoplasm under the conditions of stress or cellular starvation (Burlet et al. 1998; Pagliardini et al. 2000; Dodds et al. 2001; Gangwani et al. 2001; Sleeman et al. 2003; Zhang, Pan et al. 2003; Hua and Zhou 2004a, b). We showed that SMN was co-localized with TIA-1/R and G3BP, two characteristic proteins in SGs and that overexpression of SMN promoted formation of SGs (Hua and Zhou 2004a, b). To further characterize SMN function in SGs, we used P19, PC12 cells and primary cortical neurons with SMN knockdown in this study. We found that suppression of SMN expression in P19, PC12 cells or primary cortical neurons significantly elevated cellular threshold to form stress granules in response to stress treatment. For instance, treatment with a relatively low concentration of 200 μM of sodium arsenite for 2 h readily stimulated formation of SGs in most P19-Con cells while only 51.01 ± 3.02% of P19-SMNi cells form SGs under similar conditions.
It is well known that a key regulatory step in response to environment stress is phosphorylation of eukaryotic initiation factor (eIF)-2α. The phosphorylation of eIF-2α leads to SG assembly by decreasing the level of eIF2-GTP-tRNAMet, the ternary complex that is normally required for loading the initiator methionine onto the 48S preinitiation complex to begin translation. The scarcity of eIF2-GTP-tRNAMet allows the RNA-binding proteins TIA-1 and TIA-R to bind the 48S complex in lieu of the ternary complex, thereby promoting SG formation (Kedersha et al. 2002). While, the mechanism by which SMN promotes the formation of SGs is still unclear, our data show that defects in the SMN gene inhibit eIF-2α phosphorylation.
SG formation is an important protective response to stressed conditions. To determine if defective formation of SGs in cells with low expression of SMN plays a role in motor neuron death, we studied cell death in P19-SMNi and P19-Con cells. We found that cell death after arsenite and H2O2 treatment is inversely correlated with SMN expression levels and the number of SGs formed in these cells. Emetine, which disperses SGs in cells, eliminates the difference in sensitivity of P19-SMNi and P19-Con cells to treatment with sodium arsenite, strongly suggesting that the formation of SGs facilitated (Todd 2010)expression of SMN results in reduced formation of SGs in response to stresses, leading to cell death. Based on this hypothesis, we tested whether other proteins that promote assembly of SGs may compensate for the loss of SMN. We transfected P19-SMNi cells with G3BP, a SG component protein and an effector molecule promoting SGs assembly. As expected, we observed restoration of cellular ability to form SGs. Significantly, we find that P19-SMNi cells transfected with G3BP protein show more resistance to arsenite treatment, suggesting that modulating SG formation may be potentially targeted as an alternative therapeutic approach to treat SMA disease. However, it should be pointed out that there is a relatively high level expression of endogenous G3BP. It will be interesting to investigate how G3BP knockdown affects SMN-induced SG formation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
References
- Anderson P, Kedersha N (2002) Stressful initiations. J Cell Sci 115(Pt 16):3227–3234 [DOI] [PubMed] [Google Scholar]
- Baccon J, Pellizzoni L et al (2002) Identification and characterization of Gemin7, a novel component of the survival of motor neuron complex. J Biol Chem 277(35):31957–31962 [DOI] [PubMed] [Google Scholar]
- Batulan Z, Shinder GA et al (2003) High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 23(13):5789–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlanga JJ, Herrero S et al (1998) Characterization of the hemin-sensitive eukaryotic initiation factor 2alpha kinase from mouse nonerythroid cells. J Biol Chem 273(48):32340–32346 [DOI] [PubMed] [Google Scholar]
- Burlet P, Huber C et al (1998) The distribution of SMN protein complex in human fetal tissues and its alteration in spinal muscular atrophy. Hum Mol Genet 7(12):1927–1933 [DOI] [PubMed] [Google Scholar]
- Carissimi C, Saieva L et al (2006) Gemin8 is a novel component of the survival motor neuron complex and functions in small nuclear ribonucleoprotein assembly. J Biol Chem 281(12):8126–8134 [DOI] [PubMed] [Google Scholar]
- Carrel TL, McWhorter ML et al (2006) Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 26(43):11014–11022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charroux B, Pellizzoni L et al (1999) Gemin3: a novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol 147(6):1181–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charroux B, Pellizzoni L et al (2000) Gemin4. A novel component of the SMN complex that is found in both gems and nucleoli. J Cell Biol 148(6):1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ (2001) Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol 27:57–89 [DOI] [PubMed] [Google Scholar]
- Coovert DD, Le TT et al (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6(8):1205–1214 [DOI] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA (1996) The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis 3(2):97–110 [DOI] [PubMed] [Google Scholar]
- Dey M, Trieselmann B et al (2005) PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2alpha. Mol Cell Biol 25(8):3063–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds E, Dunckley MG et al (2001) Overexpressed human survival motor neurone isoforms, SMNDeltaexon7 and SMN+exon7, both form intranuclear gems but differ in cytoplasmic distribution. FEBS Lett 495(1–2):31–38 [DOI] [PubMed] [Google Scholar]
- Gangwani L, Mikrut M et al (2001) Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol 3(4):376–383 [DOI] [PubMed] [Google Scholar]
- Gray NK, Wickens M (1998) Control of translation initiation in animals. Annu Rev Cell Dev Biol 14:399–458 [DOI] [PubMed] [Google Scholar]
- Gubitz AK, Mourelatos Z et al (2002) Gemin5, a novel WD repeat protein component of the SMN complex that binds Sm proteins. J Biol Chem 277(7):5631–5636 [DOI] [PubMed] [Google Scholar]
- Hannus S, Buhler D et al (2000) The Schizosaccharomyces pombe protein Yab8p and a novel factor, Yip1p, share structural and functional similarity with the spinal muscular atrophy-associated proteins SMN and SIP1. Hum Mol Genet 9(5):663–674 [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG et al (2000) A mouse model for spinal muscular atrophy. Nat Genet 24(1):66–70 [DOI] [PubMed] [Google Scholar]
- Hua Y, Zhou J (2004a) Rpp20 interacts with SMN and is re-distributed into SMN granules in response to stress. Biochem Biophys Res Commun 314(1):268–276 [DOI] [PubMed] [Google Scholar]
- Hua Y, Zhou J (2004b) Survival motor neuron protein facilitates assembly of stress granules. FEBS Lett 572(1–3):69–74 [DOI] [PubMed] [Google Scholar]
- Iannaccone ST (1998) Spinal muscular atrophy. Semin Neurol 18(1):19–26 [DOI] [PubMed] [Google Scholar]
- Jablonka S, Karle K et al (2006) Distinct and overlapping alterations in motor and sensory neurons in a mouse model of spinal muscular atrophy. Hum Mol Genet 15(3):511–518 [DOI] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M et al (1999) RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol 147(7):1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR et al (2000) Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151(6):1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Chen S et al (2002) Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13(1):195–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165 [DOI] [PubMed] [Google Scholar]
- Liu Q, Dreyfuss G (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J 15(14):3555–3565 [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Fischer U et al (1997) The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell 90(6):1013–1021 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408 [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J et al (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet 19(1):63–66 [DOI] [PubMed] [Google Scholar]
- Manzerra P, Brown IR (1992) Expression of heat shock genes (hsp70) in the rabbit spinal cord: localization of constitutive and hyperthermia-inducible mRNA species. J Neurosci Res 31(4):606–615 [DOI] [PubMed] [Google Scholar]
- Manzerra P, Brown IR (1996) The neuronal stress response: nuclear translocation of heat shock proteins as an indicator of hyperthermic stress. Exp Cell Res 229(1):35–47 [DOI] [PubMed] [Google Scholar]
- McEwen E, Kedersha N et al (2005) Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280(17):16925–16933 [DOI] [PubMed] [Google Scholar]
- Meister G, Buhler D et al (2000) Characterization of a nuclear 20S complex containing the survival of motor neurons (SMN) protein and a specific subset of spliceosomal Sm proteins. Hum Mol Genet 9(13):1977–1986 [DOI] [PubMed] [Google Scholar]
- Meister G, Eggert C et al (2002) SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol 12(10):472–478 [DOI] [PubMed] [Google Scholar]
- Melki J (1997) Spinal muscular atrophy. Curr Opin Neurol 10(5):381–385 [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9(3):333–339 [DOI] [PubMed] [Google Scholar]
- Nover L, Scharf KD et al (1989) Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol Cell Biol 9(3):1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Giavazzi A et al (2000) Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet 9(1):47–56 [DOI] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK et al (2002) The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol 14(3):305–312 [DOI] [PubMed] [Google Scholar]
- Pearn J (1980) Classification of spinal muscular atrophies. Lancet 1(8174):919–922 [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Kataoka N et al (1998) A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 95(5):615–624 [DOI] [PubMed] [Google Scholar]
- Pellizzoni L, Baccon J et al (2002) Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J Biol Chem 277(9):7540–7545 [DOI] [PubMed] [Google Scholar]
- Rossoll W, Kroning AK et al (2002) Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet 11(1):93–105 [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S et al (2003) Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol 163(4):801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Greer PL et al (2005) Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron 46(2):191–204 [DOI] [PubMed] [Google Scholar]
- Schrank B, Gotz R et al (1997) Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 94(18):9920–9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman JE, Trinkle-Mulcahy L et al (2003) Cajal body proteins SMN and Coilin show differential dynamic behaviour in vivo. J Cell Sci 116(Pt 10):2039–2050 [DOI] [PubMed] [Google Scholar]
- Todd AG, Shaw DJ et al (2010) SMN and the Gemin proteins form sub-complexes that localise to both stationary and dynamic neurite granules. Biochem Biophys Res Commun 394(1):211–216 [DOI] [PubMed] [Google Scholar]
- Tourriere H, Chebli K et al (2003) The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160(6):823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vyas S, Bechade C et al (2002) Involvement of survival motor neuron (SMN) protein in cell death. Hum Mol Genet 11(22):2751–2764 [DOI] [PubMed] [Google Scholar]
- Wang J, Dreyfuss G (2001) A cell system with targeted disruption of the SMN gene: functional conservation of the SMN protein and dependence of Gemin2 on SMN. J Biol Chem 276(13):9599–9605 [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117(7):927–939 [DOI] [PubMed] [Google Scholar]
- Young PJ, Man NT et al (2000) The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self-association and SIP1 binding. Hum Mol Genet 9(19):2869–2877 [DOI] [PubMed] [Google Scholar]
- Zhan K, Vattem KM et al (2002) Phosphorylation of eukaryotic initiation factor 2 by heme-regulated inhibitor kinase-related protein kinases in Schizosaccharomyces pombe is important for resistance to environmental stresses. Mol Cell Biol 22(20):7134–7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath B et al (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22(11):3864–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Pan F et al (2003) Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 23(16):6627–6637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F et al (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133(4):585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T, Ling C et al (2006) Exogenous tissue plasminogen activator enhances peripheral nerve regeneration and functional recovery after injury in mice. J Neuropathol Exp Neurol 65(1):78–86 [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D et al (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15(9):871–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.