

Abstract
Molecular studies have an important role in the elucidation of the mechanisms involved in Glioblastoma multiforme (GBM) development. The occurrence of FHIT gene alterations, which has an important role in different cancers, has not yet been studied well in GBM. We aimed to investigate the occurrence of alterations of FHIT gene sequence and protein expression in the GBMs. Sequence alterations in exons 5–9 of the FHIT gene were screened in 63 GBMs using the single-strand conformational polymorphism method, followed by DNA sequencing. Additionally, the level of Fhit protein expression in tissues of 48 tumors was assessed by immunohistochemistry (IHC). In our investigation, FHIT gene alterations in the coding region were detected in 11 of the 63 GBM cases (17.5%). Two different sequence variants were determined: one novel missense variant (G→C transition at codon 49) and one previously described silent alteration (C→T transition at codon 88). Using web-based programs, such as SIFT and ESEfinder, it was determined that both alterations might have caused significant modification on protein function. In addition, we identified a previously reported an intronic polymorphism (T→A transition at IVS8-17) in 47.5% of cases as a similar rate (45%) in the control group. Moreover, it was observed that Fhit protein expression was reduced in 87.5% of tumors. In conclusion, the reduction or loss of Fhit protein expression by genetic alterations or epigenetic mechanisms in GBM might be associated with brain tumorigenesis.
Keywords: Glioblastoma multiforme, FHIT gene, Sequence alterations, SSCP, IHC
Introduction
Brain tumors are among the 10 most common malignancies in Turkey (http://www.saglik.gov.tr/istatistikler). Glioblastoma multiforme (GBM) is the most malignant form of astrocytic tumor and the most common primary brain tumor in adults (Jansen et al. 2004). Survival varies from a couple of months to a couple of years, with a median survival time of less than 1 year despite current treatment strategies (Curran et al. 1993; Barker et al. 1996). Biological markers of prognostic and/or diagnostic value are available for tumor progression and development, and several oncogenes and tumor suppressor genes involved in the development of human GBM have been identified (Ohgaki et al. 2004; Ichimura et al. 2004; Collins 2004; Batchelor et al. 2004; Hill et al. 2003; Ohgaki and Kleihues 2005; Tunca et al. 2007). Cloning and characterization of the tumor suppressor genes have contributed to understanding the biological mechanisms involved in the development of GBM.
Numerous previous studies have shown that carcinoma cell lines and primary tumors exhibit chromosomal aberrations with endpoints within the fragile regions, especially within the most active fragile region, FRA3B, encompassed by the FHIT gene (Huebner and Croce 2001). In our previous studies, we have been interested in the role of genes at fragile sites in cancer development and genetic predisposition to cancer and have assessed several carcinomas for the expression of fragile sites (Egeli et al. 1997, 2000; Cecener et al. 1998; Tunca et al. 2000, 2002; Karadag et al. 2002). In particular, our previous findings showed that FRA3B expression was more frequent in normal lymphocytes of cancer patients and their relatives than in healthy individuals. FHIT, encompassing FRA3B, was cloned and characterized in 1996 as a tumor suppressor gene and was the first fragile gene identified (Sozzi et al. 1996). The FHIT gene is among the most commonly altered genes in human cancers (Pekarsky et al. 2002). It is well known that loss of heterozygosity and large deletions of the FHIT tumor suppressor gene play an important role in cancer development. However, there have been few reports on the importance of point sequence alterations or germ line variants in the FHIT gene (Ahmadian et al. 1997; Kannan et al. 2000; Fong et al. 1997; Zhao et al. 2003; Gemma et al. 1997). Furthermore, FHIT gene and protein alterations in brain tumors have been examined in only a few studies (Frank et al. 1997; Tsai et al. 1999).
The aim of this study was to investigate alterations of the DNA sequence and level of protein expression of the FHIT gene in GBM, as well as to identify possible correlations with the development of GBM.
Materials and Methods
Patients and Tumor Specimens
A total of 63 (40 male and 23 female) GBM patients with an age range of 25–76 years (mean age ± SEM, 56.14 ± 1.64 years) who were scheduled for surgical treatment and whose cancers fulfilled all inclusion criteria, assessed by conventional histopathological examination, were evaluated prospectively. Approval from the Institutional Review Board and written informed consent of the patients were obtained. At the time of surgery, tissue samples were taken from the tumors. All tumors were ‘de novo’ supratentorial gliomas. At histopathological reevaluation, all of the tumors were diagnosed as GBM (WHO grade IV) according to the WHO classification of the central nervous system tumors (Kleihues et al. 1993). The tumor specimens were evaluated by DNA analyses in our Cancer Genetics Laboratory to identify FHIT gene alterations. The clinical and histopathological features of the tumors and oncology outcomes were also evaluated and correlations between these variables and the occurrence of FHIT mutations in tumors were assessed. The patients were followed up for at least 24 months or until death. The mean survival time was 8.00 + 0.77 months.
Mutation Detection
Genomic DNA was extracted using standard methods, from each tissue sample taken from the tumors at the time of surgery (Tunca et al. 2007). Each coding exon from the FHIT gene was subjected to polymerase chain reaction (PCR) analysis as previously described (Cecener et al. 2007). The high-quality amplified products were assessed with single-strand conformational polymorphism (SSCP) analysis (Bekar et al. 2007). Samples that showed one or two bands separated from the wild-type bands were identified as SSCP-positive. All the samples that contained aberrant migration patterns during gel electrophoresis were subjected to the SSCP analysis procedure at least twice to rule out contamination.
Sequencing Analysis
All samples with different SSCP bands were sequenced using the BigDye Terminator Chemistry (Applied Biosystems, Foster City, CA) and FHIT primers, which was used PCR and analyzed using an automated ABI Prism 310 Genetic Analyzer (Applied Biosystems). The results of sequencing analysis were compared with wild-type samples and normal sequencing of the FHIT gene (MIM# 601153, Gen Bank NM 002012.1).
Detection of Exonic Splicing Enhancer (ESE) Sequences
Splicing efficiencies in the normal and mutant sequences were calculated using a splice prediction program: the Splice Site Finder. The influence of base substitutions on putative ESE sites was determined with the ESE finder program (http://rulai.cshl.edu/tools/ESE/).
Predicting Deleterious Amino Acid Substitutions
The SIFT web-based program was used to calculate the probabilities of having an amino acid at a specific position relative to the most frequent amino acid at that position (http://blocks.fhcrc.org/sift/SIFT.html).
Protein Expression
The level of Fhit protein expression was determined by immunohistochemistry (IHC). Formalin-fixed paraffin-embedded tissues were available from 48 patients. IHC staining, using rabbit polyclonal antiserum to human Fhit, was performed as described previously (Guler et al. 2004). Sections of normal brain tissue served as positive controls. Briefly, after antigen retrieval in 0.01 mol/L sodium citrate buffer (pH 6.0) using a pressure cooker, primary antibody was applied at a 1/2,000 dilution. The details of immunostaining methods were described previously (Bekar et al. 2007). One section from each tumor was stained and evaluated for extent and intensity of staining. Because of the heterogeneous staining of some tumors, both staining intensity and extent of staining in neoplastic cells were taken into account in expression level scoring. Intensity was graded as strong or reduced expression. Extent of staining was classified as the fraction of stained neoplastic cells: >25, 25–50, and >50%. Cases with strong expression in more than half of the neoplastic cells were scored as high expression; cases with reduced staining intensity but in more than 50% of tumor cells or strong staining in 25–50% of neoplastic cells were scored as reduced expression; cases with reduced expression in 50% of neoplastic cells or strong staining in <25% of neoplastic cells were scored as highly reduced expression (Fig. 1).
Fig. 1.
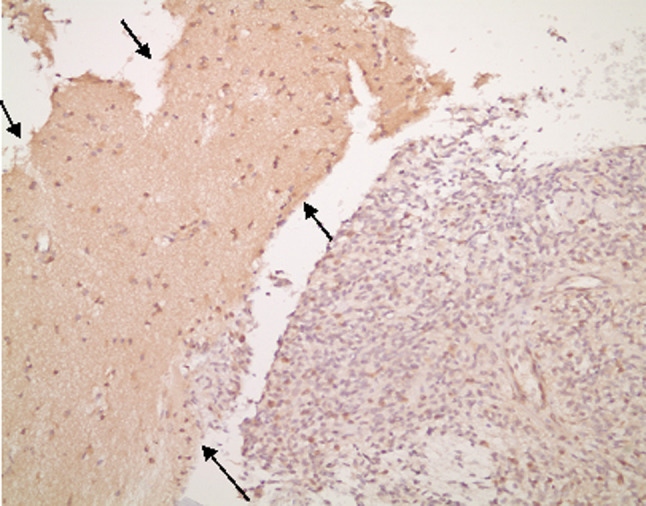
The example of reduced Fhit expression in GBM (between arrows) (Fhit IHC, ×200)
Statistical Analysis
Chi-squared tests (χ2) and logistic regression analysis were used to compare the determined FHIT gene mutations and clinical parameters. Differences with P > 0.05 were accepted as statistically non-significant. Calculations were done using SPSS for Windows, version 11.5 (SPSS, Chicago, IL, USA).
Results
Sequence Alterations
We investigated the coding region of the FHIT gene by PCR-SSCP and by sequence analysis. Our study examined sequence alterations of exons 5–9 using the intronic primer pairs in 63 glioblastomas. Variations in the coding region of the FHIT gene were detected in the tumors of 11 patients (11/63; 17.5%) by SSCP analysis. In our study, we observed two different sequence variations in the coding region of the FHIT gene in GBM in Turkish patients. One variant was novel. This novel variant was identified at codon 49 G→C transition in exon 6 (7/63; 11.1%) and one silent C→T transition type alteration was found at codon 88 (4/63; 6.4%) (Table 1, Fig. 2).
Table 1.
Summary of sequence variants in the coding region and protein expression determined in this study
| Subject no. | Exon/intron | Localization | Nucleotide transition | Codon transition | Amino acid transition | Sequence alteration type | Previously described | IHC analysis |
|---|---|---|---|---|---|---|---|---|
| 29,30,36,43,46,47,48 | Exon 6 | Codon 49 | G→C | GAC→CAC | Asp→His | Missense | − | b,c,c,c,a,b,a |
| 1,52,56,59 | Exon 7 | Codon 88 | C→T | GCC→GCT | Ala→Ala | Silent | + | a,a,c,c |
The expression rate of Fhit protein: a highly reduced, b reduced, c not materialized
Fig. 2.

a Codon 49 G to C and d codon 88 C to T (using reverse primer), g IVS8-17 T to A, c, f, i sequences of lymphocytes of same patients, b, e, h normal sequences of same areas
Polymorphism Analysis
We looked for the presence of these variations in genomic DNAs isolated from the matched lymphocytes of patients. As a result of this analysis, we identified these variants in lymphocytes of the same patients (Fig. 2). We also screened 23 of their first-degree relatives for these alterations. While the G→C transition at codon 49 was not observed in any relatives, the C→T transition at codon 88 was identified in one daughter of a patient who carried this silent alteration. These alterations were not detected in 109 healthy unrelated control individuals. Furthermore, we identified an intronic T→A variant at IVS8-17 in 47.5% of cases. This alteration was observed at a similar rate (45%) in the control group.
Detection of the ESEs
In addition, to calculate splicing efficiency and to investigate the relevance of the silent mutations, we used the ESEfinder program and searched for the serine/arginine-rich (SR) protein-specific putative ESEs motifs in the related regions. As a result of this analysis, using four different SR proteins (SF2/ASF, SC35, SRp40, and SRp55), three ESEs identified within the wild-type sequence were missing within the variant sequences of this region (Fig. 3).
Fig. 3.

Distribution of ESEs motifs on normal sequence of region determined silent alteration (a) and mutated sequence of this region (b) according to ESE finder program used four different SR proteins (SF2/ASF, SC35, SRp40, and SRp55). Arrow indicates the nucleotide effected from silent alteration. White columns represent ESEs motifs related with SF2/ASF proteins. Light gray columns represent ESEs motifs related with SC35 protein. Dark gray columns represent ESEs motifs related with SRp40 protein. Black columns represent ESEs motifs related with SRp55 protein
Relationship Between FHIT Alterations and Clinical Parameters
To study the potential influence of clinical features, statistical analysis was performed. There were no significant differences in the clinical parameters between cases with or without the FHIT alterations (P > 0.05).
Protein Expression
In the IHC analysis, only 6 (12.5%) tumors showed strong Fhit expression. In 13 tumors (27.1%), there was reduced Fhit expression and in 29 (60.4%) tumors, highly reduced Fhit expression was detected. Drastically reduced Fhit expression was observed in 87.5% of all tumors. Fhit expression was assessed by IHC in 6 out of 11 tumors exhibiting coding sequence variation, where the level of Fhit protein expression was reduced in all 6 of these.
Discussion
The FHIT gene has been widely studied in almost all types of carcinomas (Huebner and Croce 2001; Le Beau et al. 1998). In spite of strong evidence of FHIT tumor suppressor function, our knowledge of the specific molecular mechanisms involving the suppressor activity in cancer development is limited. In addition, it is well known that inactivation of Fhit protein via LOH, as well as large deletions and promoter methylation, has an important role in cancer development (Iliopoulos et al. 2006); however, searches for a role for FHIT sequence variations in the loss of protein expression have not been extensively reported. Although point mutations are an alternative mechanism of the inactivation of tumor suppressor genes in cancer, only a few studies have identified variations in the small FHIT exons (Kannan et al. 2000; Fong et al. 1997; Ohta et al. 1996; Yoshino et al. 1998; Druck et al. 1997). The evidence of such alterations is available from only two studies with brain tumors (Frank et al. 1997; Tsai et al. 1999). Earlier studies failed to detect effects of genetic or epigenetic mechanisms for inactivation of the Fhit protein in GBM. Therefore, this study used more GBM tumor samples to investigate the level of Fhit protein expression and the presence and roles of the FHIT gene alterations in GBM patients.
Several rates of FHIT gene sequence alterations have been reported in primary gastric (2.50%), lung (3.92%), and cervical (39.29%) cancers (Ohta et al. 1996; Fong et al. 1997; Yoshino et al. 1998). By using PCR-SSCP and sequence analysis in the present study, we were able to provide evidence for the presence of sequence variants in the coding region of FHIT gene in 17.5% (11/63) of the tumors and matched lymphocytes. We detected two different gene variations in this region (Table 1). One novel missense variant was a G→C transition at codon 49 in exon 6 and one silent C→T transition was determined at codon 88 in exon 7.
The G→C transition at codon 49 observed in our study led to the replacement of aspartic acid (GAC) with histidine (CAC) (7/63; 11.1%). A new protein arising from missense variants contains only a single amino acid substitution, and it often possesses some of the biological activity of the original protein (Watson and Hopkins 1987). Notably, the importance of the mutated amino acid depends on its functional role and inclusion in the protein’s active site. We do not know whether this alteration point is associated with the Fhit active site. For this reason, we used a web-based tool, SIFT, to calculate the probability of having an amino acid at a specific position relative to the most frequent amino acid at that position. A cutoff for these probabilities is used to classify the mutations as tolerated and non-tolerated (Mathe et al. 2006). As a result of SIFT analysis, we determined that histidine amino acid was localized at this position, instead of aspartic acid which was not tolerated. Four out of seven tumors with this variation were examined by IHC and the levels of protein expression were reduced in all of them. Therefore, we believe that this alteration might be important in GBM formation. The second alteration we detected was a silent nucleotide change (C→T transition) at codon 88 in 6.4% (4/63) of GBM patients. This alteration did not lead to a change in the amino acid sequence (GCT→GCC, Ala→Ala). It has been described previously in various cancer-derived cell lines (Druck et al. 1997) and has been reported as a SNP according to the ensemble site. Translationally silent alterations were normally classified as polymorphisms and were considered neutral (Wang et al. 2005). Nevertheless, there are a variety of ways in which the functionality of a gene product can be affected without requiring an amino acid change in the protein. In addition, in our previous study of breast cancer patients, we identified three ESE motifs within the region of the wild-type sequence affected by this silent variation. However, no ESE was identified on the mutated sequence of this region by an ESEs finder program used to predict the location of the SR protein specific for the putative ESEs (Cecener et al. 2007). In the present study, the level of Fhit protein expression was evaluated in two of four tumors carrying this silent variation and highly reduced Fhit expression was detected in both tumors. This situation was supported previously by results of the ESEs finder program (Cecener et al. 2007), and it was shown that this alteration had an effect on protein formation.
Moreover, we have attempted to discover whether these were germ line variants. To this end, we identified these variations in the genomic DNAs isolated from the lymphocytes of patients with matching alterations in their tumor tissue (17.5%). We confirmed that both variants identified in our study were present in the germ line of cancer patients. Moreover, because we have identified a C→T transition at codon 88 in one first-degree relative, it is possible that this variant is associated with genetic predisposition associated with GBM carcinogenesis; or it may be a very rare polymorphism. Because these sequence variants were not detected in the control group, we suggest that they might be related to the GBM tumorigenesis.
In addition, we identified a T→A IVS8-17 variant in 47.5% of cases. This variation was also observed at a similar rate (45%) in the control group. It was located in the intron portion of the gene and was previously detected in breast cancer (Kannan et al. 2000). The variant has been reported as a single nucleotide polymorphism (SNP) according to the ensemble site (www.ensemble.org/Homo_sapiens/genesnpview?db=core;gene=ENSG00000189283). This is also a polymorphism for the Turkish population. We believe that this polymorphism does not affect function, because it is located in the intron portion of the gene and is present in many individuals.
In our study, 87.5% of GBMs showed reduced levels of the Fhit protein expression by IHC analysis. This result demonstrates that the loss of Fhit protein is significant for GBM development, as well as for the other tumor types. In addition, since our findings revealed that 17.5% of sequence variations in the GBM patients were in the coding region of the FHIT gene, we gained new knowledge about the FHIT gene variant types and frequencies. Moveover, all of the cases with coding sequence variants that were tested for protein expression by IHC showed reduced Fhit expression. Therefore, our findings point out that present sequence variations, as well as other genetic and epigenetic mechanisms, may affect the Fhit protein expression. Although only a few previously reported brain tumor studies have demonstrated that large deletions play a role in the loss of Fhit protein function, our results indicate that base variations may also play an important role in the loss of Fhit protein function in the development of GBM tumors.
In conclusion, the present study reveals new and important information regarding the reduced level of Fhit protein expression in GBM formation. Furthermore, this is the first study that has identified FHIT gene alterations in the GBM patients. We believe that these types of studies will help to clarify the molecular mechanisms of GBM.
Acknowledgments
We would like to thank Professor Dr. Kay Huebner in the Comprehensive Cancer Center, Ohio State University for her suggestions and kindly providing Fhit antiserum. We thank Dr. Ediz in the Biostatistics Department, Medical Faculty of Uludag University for his expert advice. We also thank Prizma and Elips Ltd for their support in dealing with experimental equipment.
Abbreviations
- GBM
Glioblastoma multiforme
- IHC
Immunohistochemistry
- PCR
Polymerase chain reaction
- SSCP
Single strand conformational polymorphism
- WHO
World Health Organization
References
- Ahmadian M, Wistuba II, Fong KM et al (1997) Analysis of the FHIT Gene and FRA3B region in sporadic breast cancer, preneoplastic lesions, and familial breast cancer probands. Cancer Res 57:3664–3668 [PubMed] [Google Scholar]
- Barker FG, Prados MD, Chang SM et al (1996) Radiation response and survival time in patients with glioblastoma multiforme. J Neurosurg 84:442–448 [DOI] [PubMed] [Google Scholar]
- Batchelor TT, Betensky RA, Esposito JM, Pham LD, Dorfman MV, Piscatelli N, Jhung S, Rhee D, Louis DN (2004) Age-dependent prognostic effects of genetic alterations in glioblastoma. Clin Cancer Res 10:228–233 [DOI] [PubMed] [Google Scholar]
- Bekar A, Cecener G, Tunca B, Guler G, Egeli U, Tolunay S (2007) Investigation of mutations and expression of the FHIT gene in Turkish patients with brain metastases derived from non-small cell lung cancer. Tumori 93:604–607 [DOI] [PubMed] [Google Scholar]
- Cecener G, Egeli Ü, Taşdelen İ, Tunca B, Duman H, Kızıl A (1998) Common fragile sites expression and genetic predisposition to breast cancer. Teratog Carcinog Mutagen 18:279–291 [DOI] [PubMed] [Google Scholar]
- Cecener G, Egeli U, Tunca B, Tasdelen I, Tolunay S, Bilgel N (2007) Importance of novel sequence alterations in the FHIT gene on formation of breast cancer. Tumori 93:597–603 [DOI] [PubMed] [Google Scholar]
- Collins VP (2004) Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry 75:2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran WJ Jr, Scott CB, Horton J et al (1993) Recursive partitioning analysis of prognostic factors in three radiation therapy oncology group malignant glioma trials. J Natl Cancer Inst 85:704–710 [DOI] [PubMed] [Google Scholar]
- Druck T, Hadaczek P, Fu TB et al (1997) Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res 57:504–512 [PubMed] [Google Scholar]
- Egeli Ü, Karadağ M, Tunca B, Özyardımcı N (1997) The expression of common fragile sites and genetic predisposition to squamous cell lung cancers. Cancer Genet Cytogenet 95:153–158 [DOI] [PubMed] [Google Scholar]
- Egeli U, Özkan L, Tunca B, Kahraman S, Cecener G, Ergül E, Engin K (2000) The relationship between genetic susceptibility to head and neck cancer with the expression of common fragile sites. Head Neck 22:591–598 [DOI] [PubMed] [Google Scholar]
- Fong KM, Biesterveld EJ, Virmania A et al (1997) FHIT and FRA3B 3p14.2 Allele loss are common in lung cancer and preneoplastic bronchial lesions and are associated with cancer related FHIT cDNA splicing aberrations. Cancer Res 57:2256–2267 [PubMed] [Google Scholar]
- Frank S, Muller J, Plaschke J, Hahn M, Hampl J, Hampl M, Pistorius S, Schackert G, Schackert HK (1997) The putative tumor suppressor gene FHIT at 3p14.2 is rarely affected by loss of heterozygosity in primary human brain tumors. Cancer Res 57:2638–2641 [PubMed] [Google Scholar]
- Gemma A, Hagiwarn K, Ke Y, Burke LM, Khan MA, Nagashima M, Bennett WP, Harris CC (1997) FHIT mutations in human primary gastric cancer. Cancer Res 57:1435–1437 [PubMed] [Google Scholar]
- Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, McCue P, Huebner K (2004) The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer 100:1605–1614 [DOI] [PubMed] [Google Scholar]
- Hill C, Hunter SB, Brat DJ (2003) Genetic markers in glioblastoma: prognostic significance and future therapeutic implications. Adv Anat Pathol 10:212–217 [DOI] [PubMed] [Google Scholar]
- Huebner K, Croce C (2001) FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer 1:214–221 [DOI] [PubMed] [Google Scholar]
- Ichimura K, Ohgaki H, Kleihues P, Collins VP (2004) Molecular pathogenesis of astrocytic tumours. J Neurooncol 70:137–160 [DOI] [PubMed] [Google Scholar]
- Iliopoulos D, Guler G, Han SY, Druck T, Ottey M, McCorkell KA, Huebner K (2006) Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett 232:27–36 [DOI] [PubMed] [Google Scholar]
- Jansen M, de Witt Hamer PC, Witmer AN, Troost D, van Noorden CJ (2004) Current perspectives on antiangiogenesis strategies in the treatment of malignant gliomas. Brain Res 45:143–163 [DOI] [PubMed] [Google Scholar]
- Kannan K, Krishnamurthy J, Feng J, Nakajima T, Tsuchida N, Shanmugam G (2000) Mutation profile of the p53, FHIT, p16INK4a/p19ARF and H-ras genes in Indian breast carcinomas. Int J Oncol 17:1031–1035 [DOI] [PubMed] [Google Scholar]
- Karadağ M, Tunca B, Cecener G, Egeli Ü, Özyardımcı N, Ege E, Gözü O (2002) Chromosomal fragile sites and relationship between genetic predisposition to small cell lung cancer. Teratog Carcinog Mutagen 22:31–40 [DOI] [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Scheithauer BW (1993) The new WHO classification of brain tumours. Brain Pathol 3:255–268 [DOI] [PubMed] [Google Scholar]
- Le Beau MM, Drabkin H, Glover TW, Gemmill R, Rassool FV, McKeithan TW, Smith DI (1998) An FHIT tumor suppressor gene? Genes Chromosomes Cancer 21:281–289 [DOI] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV (2006) Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res 34:1317–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P (2005) Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 64:479–489 [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Dessen P, Jourde B et al (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64:6892–6899 [DOI] [PubMed] [Google Scholar]
- Ohta M, Inoue H, Cotticelli MG et al (1996) The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 84:587–597 [DOI] [PubMed] [Google Scholar]
- Pekarsky Y, Zanesi N, Palamarchuk A, Huebner K, Croce CM (2002) FHIT: from gene discovery to cancer treatment and prevention. Lancet Oncol 3:748–754 [DOI] [PubMed] [Google Scholar]
- Sozzi G, Veronese M, Negrini M et al (1996) The FHIT Gene At 3p14.2 is abnormal in lung cancer. Cell 85:17–26 [DOI] [PubMed] [Google Scholar]
- Tsai TC, Yang HM, Wu YL, Chi CW, Chou MD, Lee LS, Chang TJ (1999) Abnormal transcripts of FHIT gene in Chinese brain tumors. Oncol Rep 6:345–348 [PubMed] [Google Scholar]
- Tunca B, Egeli Ü, Zorluoğlu A, Yılmazlar T, Yerci Ö, Kızıl A (2000) The expression frequency of common fragile sites and genetic predisposition to colon cancer. Cancer Genet Cytogenet 119:139–145 [DOI] [PubMed] [Google Scholar]
- Tunca B, Cecener G, Gebitekin C, Egeli U, Ediz B, Ercan I (2002) Investigation of genetic susceptibility to non-small cell lung cancer by fragile site expression. Teratog Carcinog Mutagen 22:205–215 [DOI] [PubMed] [Google Scholar]
- Tunca B, Bekar A, Cecener G, Egeli U, Vatan O, Tolunay S, Kocaeli H, Aksoy K (2007) Impact of novel PTEN mutations in Turkish patients with glioblastoma multiforme. J Neurooncol 82:263–269 [DOI] [PubMed] [Google Scholar]
- Wang J, Smith PJ, Krainer AR, Zang MQ (2005) Distribution of SR protein exonic splicing enhancer motifs human protein-coding genes. Nucleic Acids Res 33:5053–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, Hopkins NH (1987) The genetic code. In: Watson JD, Hopkins NH (eds) Molecular biology of the gene, 4th edn. The Benjamin/Cummings Publishing Company, California, p 444 [Google Scholar]
- Yoshino K, Enomoto T, Nakamura T, Nakashima R, Wada H, Saitoh J, Noda K, Murata Y (1998) Aberrant FHIT transcripts in squamous cell carcinoma of the uterine cervix. Int J Cancer 76:176–181 [DOI] [PubMed] [Google Scholar]
- Zhao XR, Kang LC, Zhou YS et al (2003) Mutations of fragile histidine triad gene in Peutz-Jeghers syndrome and canceration. Ai Zheng 22:50–54 [PubMed] [Google Scholar]