

Summary
Mulibrey nanism (MN) is a extremely rare genetic condition first described in 1973, with around 150 cases reported worldwide. MN is characterised by growth delay and multiorgan manifestations, the most fatal being a combination restrictive-constrictive, perimyocardial heart disease that results in diastolic heart failure. We present a male toddler with MN who presented with recurrent episodes of hypoxia, feeding intolerance, and generalised swelling (anasarca) in the setting of subtle echocardiographic findings. A multidisciplinary and systematic diagnostic approach was used to determine the underlying aetiology. Invasive cardiac testing via right heart catheterisation revealed the final diagnosis of restrictive cardiomyopathy. Transplant decision-making was limited due to hepatic involvement. This case highlights the limitations of echocardiography in diagnosing restrictive cardiomyopathy, which has a preserved ejection fraction, as well the need for multidisciplinary involvement and a family-centred approach in treating patients with this rare condition.
Keywords: Genetics, Pediatrics, Heart failure
Background
Mulibrey nanism (MN), also known as Perheentupa syndrome and muscle-liver-brain-eye nanism, is an autosomal recessive disorder caused by mutations in the TRIM37 gene located on chromosome 17q.22-23.1 TRIM37 encodes a peroxisomal RING-B-box-Coiled-coil zinc protein. Mutations in this gene lead to multiorgan effects. Prenatal growth restriction and dystrophy affecting the cardiac, skeletal, hepatic, ophthalmologic and endocrine systems are characteristic features.1 Cardiac dysfunction is the primary cause of mortality, with up to 52% of patients developing heart failure (HF), often leading to death before the age of 10.2 Pathologically, mortality is often associated with various combinations of constrictive pericardial thickening and myocardial fibrosis which leads to a restrictive cardiomyopathy.2 Thus, in MN, HF typically manifests as diastolic dysfunction, with most indicators of systolic function, particularly ejection fraction, remaining within normal limits on echocardiography.2,4 Therefore, recognising clinical signs of HF in MN and distinguishing them from other diagnoses is essential. However, this requires an understanding of echocardiography’s role, and limitations, in both diagnosing diastolic HF and differentiating restrictive cardiomyopathy from constrictive pericarditis.5 6 In this report, we present a case with subtle echocardiographic findings despite frank anasarca, illustrating these points.
Case presentation
A male toddler presented to our emergency department (ED) with increased fussiness, anasarca and increased oxygen requirements. Two months prior, the patient had been admitted (Admission A) after home furosemide of 1 mg/kg given by mouth two times per day was discontinued by an outside facility. Known medical history at that time included tracheomalacia, reactive airway disease, multiple cutaneous and hepatic haemangiomas, communicating hydrocephalus and a partial deletion in the dystrophin gene. Admission A labs were notable for N-terminal pro-brain natriuretic peptide (NT-proBNP) level of 5489 pg/mL (reference range: <126 pg/mL), albumin level of 3.4 g/dL (reference range 3.0–4.3 g/dL) and alpha-fetoprotein (AFP) level of 38.9 ng/mL (reference range: <9.0 ng/mL). Chest X-ray was significant for bilateral perihilar opacities, and a right band-like opacity presumed to favour atelectasis. Hepatic ultrasound revealed multiple lesions consistent with benign haemangiomas with direct comparison to a previous CT liver. His partial deletion in the dystrophin generalised concern for cardiomyopathy underlying his presentation. Echocardiogram (Echo A) showed prominent trabeculations in the left ventricle apex/free wall but with normal biventricular size and systolic function. The patient’s oxygen requirements returned to baseline of 0.2 L/min via nasal cannula and anasarca decreased after treatment with furosemide. On discharge from Admission A, he was sent home on furosemide 1 mg/kg two times per day orally. During this same admission, rapid whole exome sequencing was obtained by paediatric genetics and in the interim results returned, revealing a diagnosis of MN (partial allelic TRIM37 deletion). Figure 1 demonstrates the timeline of presentation and subsequent investigations.
Figure 1. Timeline of initial admission, emergency department visit, and second admission. Location of echocardiograms and other imaging is provided. Timeline is provided in weeks (W). This figure was created for this report by author JAK using draw.io.

One month later, the patient presented to genetics clinic with increased anasarca, work of breathing and oxygen requirements. ED evaluation revealed similar findings, with faint crackles on pulmonary examination. Laboratory analyses were notable for NT-proBNP level of 5,038 pg/mL (<126 pg/mL), C-reactive protein (CRP) of 0.41 mg/mL (reference range: <0.86 mg/dL); repeat chest X-ray again revealed bilateral perihilar opacities noted to be improved from previous admission. Cardiology recommended increasing oral furosemide dose to 2 mg/kg two times per day and repeating echocardiogram (Echo B) which showed mild to moderate atrial enlargement (more prominent than prior), preserved biventricular systolic function, and prominent ascites (figure 2B). He returned to baseline oxygen requirements after a single dose of intravenous 1 mg/kg of furosemide, with discharge home.
Figure 2. (A) Echo A: apical four-chamber view showing upper normal atrial enlargement with potential prominent trabeculations in the left ventricle myocardium. (B) Echo B: apical four-chamber view showing mild to moderate biatrial enlargement (increased from prior). (C) Echo C: parasternal short-axis view of the ventricles and the interventricular septum that shows flattened end-systolic septal geometry with eccentricity index of 1.4 and septal bounce. (D) Echo C: Doppler pattern tracing of the main pulmonary artery that shows notched pulmonary artery pattern concerning for elevated right-sided heart pressures and potential pulmonary hypertension.

Five days after discharge, the patient’s family observed a recurrence of anasarca, fussiness, feeding intolerance and increased oxygen requirements (0.2–0.5 L nasal cannula). They returned to ED and were readmitted (Admission B). ED evaluation revealed free fluid in the right upper quadrant on point of care ultrasound, with no frank pericardial effusion; numerous B-lines were present on lung window. Labs showed elevated NT-proBNP level of 5,555 pg/mL (reference range: <126 pg/mL), hypochloraemic hypokalaemic metabolic alkalosis and mild aspartate aminotransferase elevation.
Investigations
On admission it was important to consider the multiple causes of anasarca which include hepatic, renal, inflammatory and cardiac aetiologies. Hepatic dysfunction was ruled out through laboratory evaluation which revealed mildly elevated International normalized ratio (INR) of 1.2 (reference range: 0.9–1.1), albumin 3.2 g/dL (reference range 3.0–4.3 g/dL) and total protein of 5.8 g/dL (reference range: 5.3–6.9 g/dL). Other laboratory values were notable for fibrinogen of 184 mg/dL (reference range: 213–435 mg/dL), AFP 56.8 ng/mL (reference range: <9.0 ng/mL) and alpha-1 antitrypsin 220 mg/dL (reference range: 100–190 mg/dL). A hepatic ultrasound with Doppler was performed due to the mildly elevated transaminases which revealed stable hepatic haemangiomas, patent vasculature, and stable small to moderate volume ascites with no progression from previous admission. His underlying liver haemangiomas were felt to be a contributing factor to his overall presentation given this additional left-to-right shunt that often can lead to symptoms of HF in these patients.
Renal causes, such as a nephrotic syndrome, were considered and ruled out with a spot urine protein-to-creatinine ratio which returned at 0.747 mg/mg (nephrotic range >2 mg/mg).
Erythrocyte sedimentation rate and CRP were normal making an inflammatory or rheumatological cause less likely. Respiratory viral panel was negative, decreasing the likelihood of infection underlying presentation.
Highest on the differential was an underlying cardiac cause, now prioritised due to repeat admissions and MN diagnosis. As such, a repeat echocardiogram (Echo C) was obtained and compared with previous echocardiograms revealing continued mild to moderate biatrial enlargement with a stable patent foramen ovale. Left ventricular and right ventricular (RV) systolic function remained normal. However, a flattened end-systolic geometry with an eccentricity index of 1.4, septal bounce, and a notched pulmonary artery pattern concerning for potential signs of pulmonary hypertension were noted (figure 2C, D).
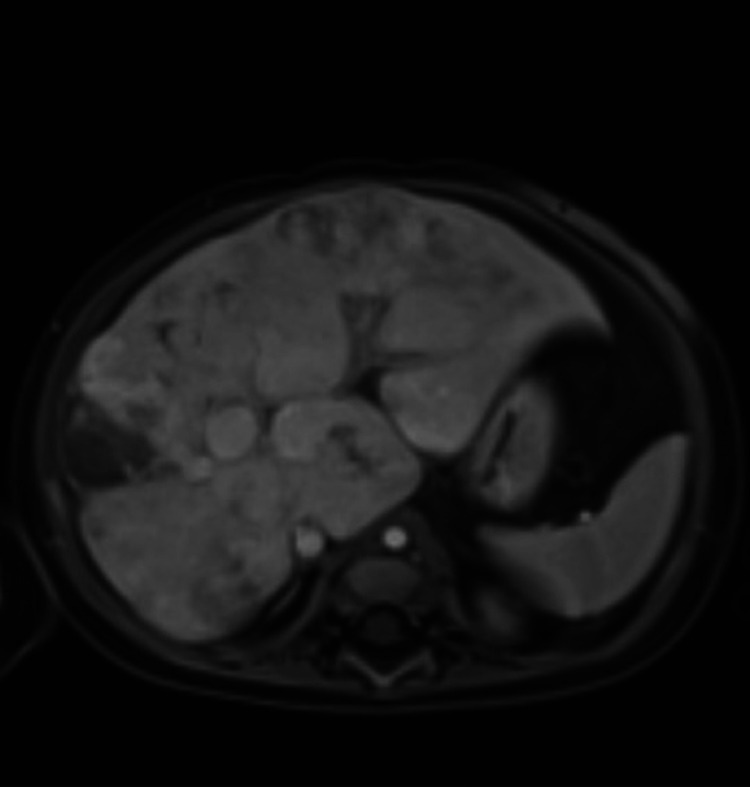
Given these findings and his underlying pulmonary comorbidities, a CT chest and cardiac catheterisation were ordered for further characterisation. CT chest showed diffuse ground glass opacities with multiple areas of probable air trapping along with new scattered areas of band-like opacities favoured to represent an acquired aetiology such as chronic lung disease of immaturity (figure 3). Hepatic MRI was also obtained (figure 4).
Figure 3. Chest CT reveals scattered diffuse ground-glass opacities, multiple areas of increased lucency consistent with air trapping and multifocal band-like opacities within the middle and lower lobes representing areas of suspected subsegmental atelectasis.
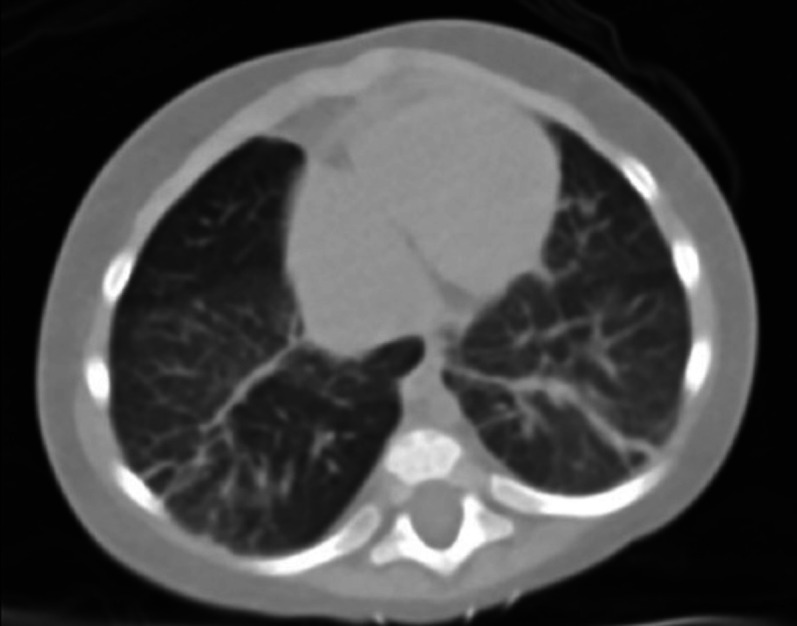
Figure 4. Hepatic MRI demonstrates numerous round lesions with increased T2 signal, arterial enhancement, and peripheral pooling with central filling on delayed contrast phases. The bile ducts are not dilated or grossly irregular.
The patient was scheduled for cardiac catheterisation and concurrent bronchoscopy to rule out intrabronchial lesions. In the interim, oral furosemide 2 mg/kg was increased from two to three times per day with initiation of daily oral spironolactone at 1 mg/kg. On the night of hospital day 12, the patient developed a run of ectopic atrial tachycardia and was sent to the paediatric intensive care unit (PICU), as this did not convert after administration of adenosine. An esmolol drip was started, with subsequent return of normal sinus rhythm. The patient stabilised and was transferred back to the step-down unit prior to cardiac catheterisation.
Cardiac catheterisation was notable for severely elevated right-sided heart pressures with elevated distal pulmonary arterial (PA) pressures, RV pressures that were 90% of systemic, and an RV end-diastolic pressure of 22 mm Hg. He also had an elevated left ventricular end-diastolic pressure of 22 mm Hg and pressure tracings in the right and left ventricles did not vary with respiration that was felt to be more consistent with restrictive cardiomyopathy than constrictive pericarditis.6 Also of note during this catheterisation, the patient was found to have a markedly elevated pulmonary vascular resistance of 11.7 Wood units×m2 consistent with pulmonary hypertension, most likely secondary to left heart diastolic dysfunction. Cardiac output was 2.1 L/min/m2. As a result, pulmonary vasodilator testing was completed by providing 100% FiO2 and 20 ppm of inhaled nitrous oxide, with subsequent marked improvement in PA pressures and increased cardiac index with lowering of peripheral vascular resistance. This pulmonary hypertension was felt to be related to left heart diastolic dysfunction and as such pulmonary vasodilator therapy was limited so as to not worsen left atrial hypertension and pulmonary oedema that would lead to irreversible pulmonary vascular resistance. Bronchoscopy revealed no intrabronchial lesions.
Differential diagnosis
With the subtle findings on echocardiogram, a high degree of suspicion for a variable combination of restrictive cardiomyopathy and constrictive pericarditis was at the top of the differential diagnosis. Nevertheless, the complex and multisystem findings of MN required systematic consideration and rule-out of alternative causes for anasarca. After cardiac catheterisation, some variable combination of restrictive cardiomyopathy and constrictive pericarditis was suspected, complicated by pulmonary hypertension due to the restrictive cardiomyopathy and left ventricular diastolic dysfunction. It was felt that the restrictive cardiomyopathy was primary due to minimal respiratory variation in pressures during cardiac catheterisation. Some contribution to his anasarca was felt to arise from a left-to-right shunting mechanism from his liver haemangiomas.
Outcome and follow-up
Based on the patient’s hepatic haemangiomas, age, and weight he was deemed high risk for isolated heart transplant, with recommendation for concurrent heart-liver transplant. While our institution has expertise in sequential heart-liver transplant, we do not currently offer concurrent heart-liver transplant. Second opinions were sent to hospitals across the country with expertise in this transplant approach. He continued his oral furosemide with the addition of propranolol 3 mg/kg three times per day orally for both ectopic atrial tachycardia and liver haemangiomas. Outpatient follow-up with cardiology and hepatology was arranged. In the cardiology clinic, he was noted to have clinically improved, with increased activity, decreased fussiness, and better feeding comfort and intake.
However, approximately 1 week later (2 weeks after discharge from Admission B), his mother noted increased fussiness and pulse oximetry desaturations (less than 90% SpO2) with tachycardia, noting a heart rate greater than 200 beats per minute. He was evaluated at another local ED where the diagnosis was thought to be pneumonia versus aspiration versus pulmonary oedema. He was discharged with strict follow-up with cardiology as he was deemed stable. Parents then noted desaturations into 70s at home and transported him to our institution’s ED. Enroute their oxygen tank ran low, with the patient becoming pale and unresponsive. Emergency medical services were called, and he was transported to the ED. On arrival, he was found to be febrile, hypoxic, lethargic, hypoglycaemic and hypotensive requiring 12 L/min high-flow nasal cannula and vasopressors. He was admitted to the PICU for a possible respiratory infection. During the PICU stay, the patient experienced rising lactate levels, respiratory distress necessitating intubation, and instances of ectopic atrial tachycardia. There was also a bradycardic event that required chest compressions and a dose of epinephrine.
Consultation with a facility specialising in concurrent heart-liver transplants concluded that a liver transplant was not advisable due to the absence of portal hypertension, a normal spleen size, and no definitive liver cirrhosis confirmed by biopsy. Regrettably, the patient’s acute condition deteriorated while undergoing workup for transplant and he passed away in the PICU after an asystolic cardiac arrest in his family’s arms.
Discussion
In this case, the presence of HF secondary to restrictive cardiomyopathy and/or constrictive pericarditis was notable, despite subtle findings of atrial enlargement on previous echocardiograms. This underscores the limitations of echocardiography in diagnosing diastolic dysfunction in paediatric patients with cardiomyopathy, as highlighted by Dragulescu et al.7,11 Typical markers of diastolic function in adults lack transferability to the paediatric population, suggesting that cardiac catheterisation or MRI is necessary to properly diagnose restrictive cardiomyopathy.12 While biatrial enlargement, a very subtle finding which likely predisposed to the development of ectopic atrial tachycardia, can suggest restrictive cardiomyopathy, additional testing is necessary.5 6 Specific to this case, providers may have overlooked such subtle biatrial enlargement as they were primed to focus on systolic function, especially given his partial dystrophin deletion which may have been thought to predict dilated cardiomyopathy. This illustrates the critical role of medical genetics in MN diagnosis and management.
Despite this case’s complexity, a structured anasarca workup guided final diagnosis, reducing the anchoring bias that may have played a role in Admission A. This demonstrates the importance of considering diastolic HF in any paediatric patient with anasarca, especially one with MN, even in the absence of overt echocardiographic findings as they lack sensitivity. While MN has mild effects on systolic function, most of its pathological changes lead to diastolic HF, the leading cause of mortality which cannot be missed.2
The therapeutic decision regarding pericardiectomy versus transplantation was challenging due to remaining diagnostic uncertainty. While it was felt that restrictive cardiomyopathy was the most likely diagnosis, it was difficult to fully rule out some component of constrictive pericarditis, for which pericardiectomy has shown functional improvement.2 Others have aptly noted that in one-third of these cases, pericardiectomy is not curative as it merely prolongs onset of diastolic HF from restrictive cardiomyopathy, bolstering the argument for early cardiac transplantation.13 Nevertheless, with no prospective-controlled comparisons of transplant versus pericardiectomy there is a lack of consensus, with some arguing that benefit from pericardiectomy depends on time to total pericardial adhesion.14 Regarding hepatic involvement, this case is unique in that transplantation decision-making was complicated due to a high-risk profile for isolated heart transplant. To our knowledge, no previous MN case has required concurrent liver transplant. Similar bridging fibrosis and nodularity as our case was reported in two patients, a 2 and 3.5-year-old, as part of a large study of MN liver pathology.15 As these authors suggest, with sufficient right-sided pressures, congestive hepatopathy can develop rapidly, with characteristic fibrosis.15 Critically, a case of a 32-year-old woman, paired with multiple searches of biobank engines, suggested congestive hepatopathy is the primary determinant of liver cirrhosis in MN.16 Therefore, it may be reasonable to consider delaying liver transplant in those eligible for an isolated heart transplant, provided there is an expectation that their haemodynamics will improve.13
This highlights the urgent need for research of transplant strategies in MN, particularly those that include prospective comparisons to early pericardiectomy. Crucially, this case illustrates that without such data, collaborative multidisciplinary decision-making is difficult. Regardless, it remains critical to effectively address MN’s clinical complexity via early diagnosis of diastolic HF which should not be based solely on echocardiographic parameters.
Learning points.
Consider diastolic heart failure in paediatrics with anasarca, especially those with Mulibrey nanism, even in the absence of echocardiographic evidence of reduced systolic function.
Any diagnostic uncertainty pertaining to diastolic heart failure necessitates consideration of alternative diagnostic modalities such as cardiac catheterisation or MRI.
The decision regarding pericardiectomy versus transplantation in Mulibrey nanism is complex, requiring shared multidisciplinary decision-making until further research is conducted.
Acknowledgements
We would like to acknowledge the review and approval of the content of this manuscript by Cesar Lopez Angel, the supervising senior resident present during the case. Additionally, this case would not be possible without the involvement of the patient and their family.
Footnotes
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Consent obtained from parent(s)/guardian(s)
Contributor Information
Judah Andrew Kreinbrook, Email: judah.kreinbrook@duke.edu.
Laura Izzo, Email: laura.meissner@duke.edu.
Christopher Atkins, Email: christopher.atkins@duke.edu.
Samrat Das, Email: samrat.das@duke.edu.
References
- 1.Karlberg N, Jalanko H, Perheentupa J, et al. Mulibrey nanism: clinical features and diagnostic criteria. J Med Genet. 2004;41:92–8. doi: 10.1136/jmg.2003.014118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipsanen-Nyman M, Perheentupa J, Rapola J, et al. Mulibrey heart disease: clinical manifestations, long-term course, and results of pericardiectomy in a series of 49 patients born before 1985. Circulation. 2003;107:2810–5. doi: 10.1161/01.CIR.0000070949.76608.E2. [DOI] [PubMed] [Google Scholar]
- 3.Eerola A, Pihkala JI, Karlberg N, et al. Cardiac Dysfunction in Children with Mulibrey Nanism. Pediatr Cardiol. 2007;28:155–62. doi: 10.1007/s00246-006-0007-23. [DOI] [PubMed] [Google Scholar]
- 4.Sarkola T, Lipsanen-Nyman M, Jalanko H, et al. Pericardial Constriction and Myocardial Restriction in Pediatric Mulibrey Nanism: A Complex Disease With Diastolic Dysfunction. CJC Open . 2022;4:28–36. doi: 10.1016/j.cjco.2021.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rapezzi C, Aimo A, Barison A, et al. Restrictive cardiomyopathy: definition and diagnosis. Eur Heart J. 2022;43:4679–93. doi: 10.1093/eurheartj/ehac543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geske JB, Anavekar NS, Nishimura RA, et al. Differentiation of Constriction and Restriction. J Am Coll Cardiol. 2016;68:2329–47. doi: 10.1016/j.jacc.2016.08.050. [DOI] [PubMed] [Google Scholar]
- 7.Zile MR, Gaasch WH, Carroll JD, et al. Heart failure with a normal ejection fraction: is measurement of diastolic function necessary to make the diagnosis of diastolic heart failure? Circulation. 2001;104:779–82. doi: 10.1161/hc3201.094226. [DOI] [PubMed] [Google Scholar]
- 8.Litwin SE, Zile MR. Should We Test for Diastolic Dysfunction? How and How Often? JACC Cardiovasc Imaging. 2020;13:297–309. doi: 10.1016/j.jcmg.2019.02.029. [DOI] [PubMed] [Google Scholar]
- 9.Dal Canto E, Remmelzwaal S, Van Ballegooijen AJ, et al. Diagnostic value of echocardiographic markers for diastolic dysfunction and heart failure with preserved ejection fraction. Heart Fail Rev. 2022;27:207–18. doi: 10.1007/s10741-020-09985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dragulescu A, Mertens L, Friedberg MK. Interpretation of left ventricular diastolic dysfunction in children with cardiomyopathy by echocardiography: problems and limitations. Circ Cardiovasc Imaging. 2013;6:254–61. doi: 10.1161/CIRCIMAGING.112.000175. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki N, Garcia M, Ko HH, et al. Applicability of published guidelines for assessment of left ventricular diastolic function in adults to children with restrictive cardiomyopathy: an observational study. Pediatr Cardiol. 2015;36:386–92. doi: 10.1007/s00246-014-1018-z. [DOI] [PubMed] [Google Scholar]
- 12.Lipshultz SE, Law YM, Asante-Korang A, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation. 2019;140:e9–68. doi: 10.1161/CIR.0000000000000682. [DOI] [PubMed] [Google Scholar]
- 13.Anwer M, Bin Mahmood SU, Stawiarski K, et al. Mulibrey Nanism Syndrome: A Case for Heart Transplantation. Ann Thorac Surg. 2020;109:e115–7. doi: 10.1016/j.athoracsur.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Yasuhara J, Omori S, Maeda J, et al. Successful Total Pericardiectomy for Constrictive Pericarditis in the First Series of Japanese Patients With Mulibrey Nanism. Can J Cardiol. 2018;34:690. doi: 10.1016/j.cjca.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Sivunen J, Karlberg S, Kivisaari R, et al. Liver pathology and biochemistry in patients with mutations in TRIM37 gene (Mulibrey nanism) Liver Int. 2022;42:1369–78. doi: 10.1111/liv.15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weldy CS, Ashley EA. Mulibrey Nanism and the Real Time Use of Genome and Biobank Engines to Inform Clinical Care in an Ultrarare Disease. Circ Genom Precis Med. 2021;14:e003430. doi: 10.1161/CIRCGEN.121.003430. [DOI] [PubMed] [Google Scholar]