

Abstract
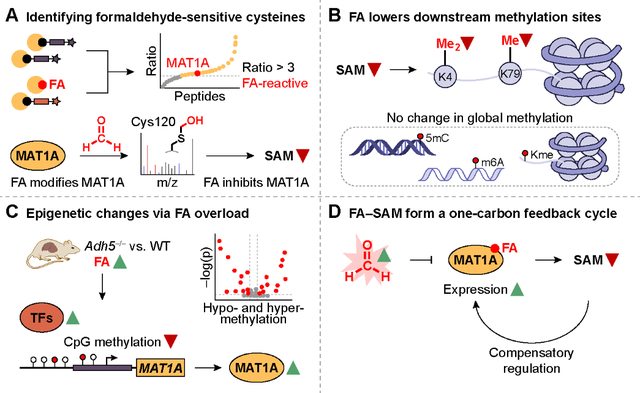
One-carbon metabolism is an essential branch of cellular metabolism that intersects with epigenetic regulation. Here, we show formaldehyde, a one-carbon unit derived from both endogenous sources and environmental exposure, regulates one-carbon metabolism by inhibiting the biosynthesis of S-adenosylmethionine (SAM), the major methyl donor in cells. Formaldehyde reacts with privileged, hyperreactive cysteine sites in the proteome, including Cys120 in S-adenosylmethionine synthase isoform type-1 (MAT1A). Formaldehyde exposure inhibited MAT1A activity and decreased SAM production with MAT-isoform specificity. A genetic mouse model of chronic formaldehyde overload showed a decrease in SAM and in methylation on selected histones and genes. Epigenetic and transcriptional regulation of Mat1a and related genes function as compensatory mechanisms for formaldehyde-dependent SAM depletion, revealing a biochemical feedback cycle between formaldehyde and SAM one-carbon units.
One Sentence Summary:
Formaldehyde influences S-adenosylmethionine biosynthesis in a one-carbon metabolism feedback cycle through epigenetic regulation.
Graphical Abstract
Introduction
The one-carbon cycle is central to cell function, where activation of one-carbon units is employed for nucleotide and amino acid biosynthesis and epigenetic regulation (1, 2). The one-carbon unit S-adenosylmethionine (SAM) is a ubiquitous co-substrate and key methyl donor for transmethylation reactions that underlie one-carbon metabolic (3) and epigenetic processes (4), including modulating mTORC1 signaling for methionine sensing and cellular growth (5). Misregulation of SAM biosynthesis and metabolism is observed in diseases such as nonalcoholic fatty liver disease (6) and cancers (1, 3, 7).
Another one-carbon unit of emerging importance is formaldehyde (8, 9). In addition to the role of formaldehyde as an environmental carcinogen, formaldehyde is endogenously produced through biological pathways, including enzymatic serine cleavage (3), oxidative folate degradation (10), and oxidative demethylation (11, 12). Previously, activity-based sensing probes have shown the substantial generation of endogenous formaldehyde (13–17) with high relative abundance compared to other reactive carbon species such as aldehydes (18), where the formaldehyde basal levels in blood is 50 μM and can exceed 500 μM in disease states (19, 20). Formaldehyde is detoxified by a two-tier mechanism involving the enzyme alcohol dehydrogenase 5 (ADH5) for formaldehyde removal and the Fanconi anemia pathway for formaldehyde-induced DNA crosslinking repair, where loss of formaldehyde scavenging promotes carcinogenesis (21, 22). Here, we report the discovery of a biochemical feedback cycle between formaldehyde and SAM one-carbon units. Specifically, we identified formaldehyde dose-dependent inhibition of SAM production through targeting S-adenosylmethionine synthase isoform type-1 (MAT1A), the terminal enzyme in SAM biosynthesis, at a privileged, isoform-specific Cys120, to decrease the overall methylation potential of the cell. Contrary to the conventional view of formaldehyde as a promiscuous electrophile, this work reveals that this one-carbon signal can participate in site-specific post-translational modifications to enable broader biochemical regulation of central metabolic functions in the cell.
Results
Activity-based protein profiling chemoproteomics reveals privileged, formaldehyde-sensitive cysteine targets in one-carbon metabolism.
Motivated by in vitro observations that formaldehyde reacts with cysteine residues on peptides to form hemithioacetal and thiazolidine adducts (23, 24), and related reactive electrophilic species emerging as key players in redox signaling and stress pathways (18), we sought to identify formaldehyde-sensitive targets across the proteome in an unbiased, high-throughput manner. We applied the isotopic tandem orthogonal proteolysis–activity-based protein profiling (isoTOP-ABPP) platform (25) to mouse liver lysates exposed to 500 μM formaldehyde (FA)—close to disease-level concentrations (20)—or vehicle control. Using the cysteine-reactive iodoacetamide (IA)-alkyne probe followed by copper-catalyzed azide-alkyne cycloaddition (CuAAC) to enable attachment of either light (vehicle-treated) or heavy (formaldehyde-treated) isotopically labeled TEV protease-cleavable biotin enrichment tags (fig. S1) (26), liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis afforded quantified ratios of light- versus heavy-modified peptides. Sites with higher incorporation of IA-alkyne on vehicle light versus formaldehyde-treated heavy labels indicated higher formaldehyde reactivity at specific cysteine residues of identified protein targets (Fig. 1A).
Fig. 1. isoTOP-ABPP identifies privileged, formaldehyde-sensitive cysteine sites across the proteome.
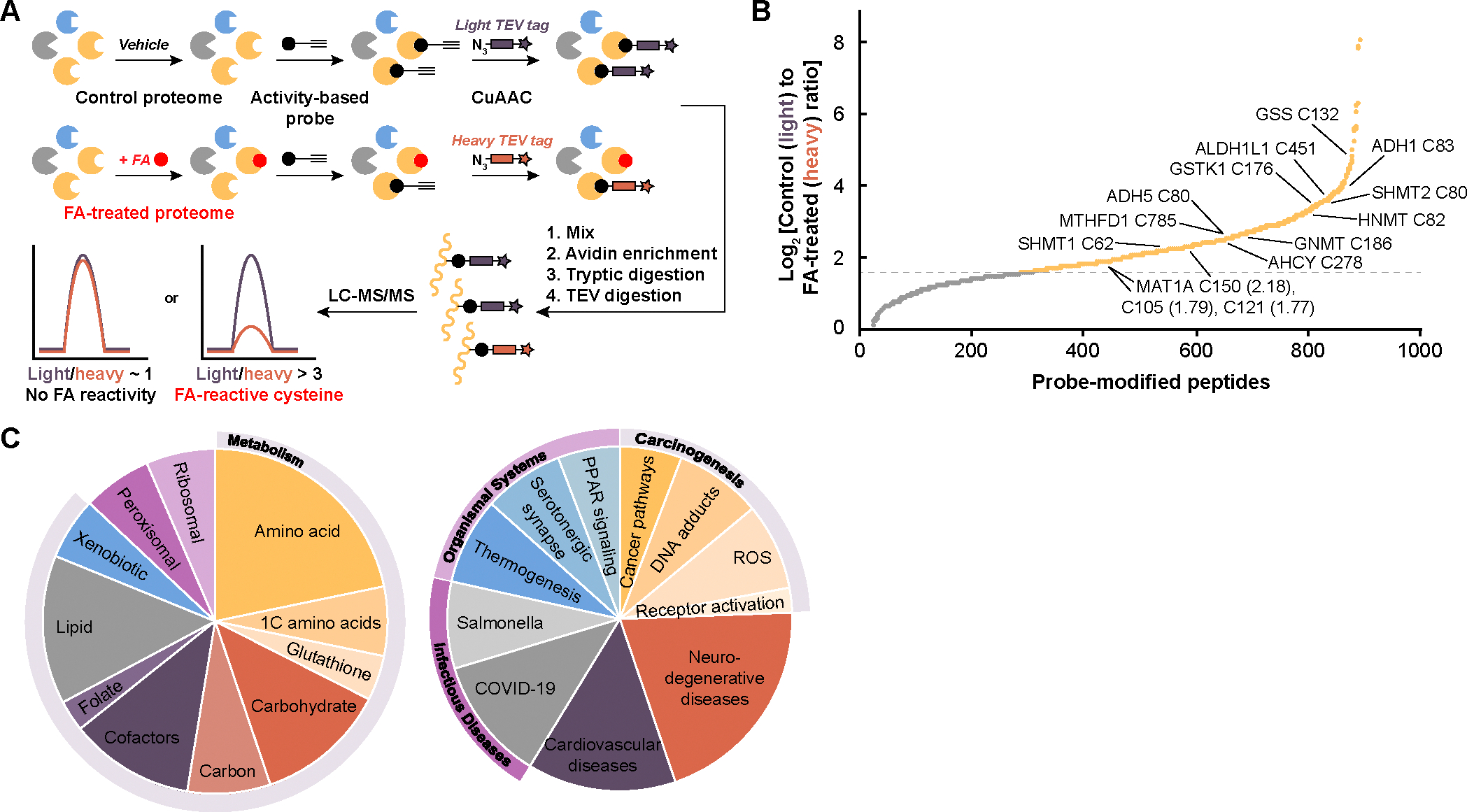
(A) Workflow for isotopic tandem orthogonal proteolysis–activity-based protein profiling (isoTOP-ABPP) analysis of formaldehyde (FA)-sensitive cysteine sites applied to whole proteomes. Mouse liver lysate was treated with vehicle or formaldehyde, followed by iodoacetamide (IA)-alkyne cysteine activity-based probe, labeling of isotopic tags with copper-catalyzed azide-alkyne cycloaddition (CuAAC), downstream digestion, and subsequent analysis of peptide fragments using LC-MS/MS. (B) Waterfall plot of light/heavy ratios of formaldehyde (FA)-sensitive cysteine sites from isoTOP-ABPP (yellow), revealing a pattern of privileged targets for this one-carbon unit. Important enzymes involved in one-carbon and formaldehyde metabolism are labeled with the cysteine residue and MAT1A ratios in parentheses. Gray dotted line represents log2 ratio of 1.58, equivalent to a ratio of 3. Targets were filtered for appearing in at least two technical replicates. (C) Pie chart summarizing the KEGG pathways most abundant with enzymes with ratios greater than 3 for formaldehyde-sensitive cysteines. Pathways were separated into two pie charts by metabolism and cellular processes (308 proteins, left) and organismal systems and diseases (172 proteins, right).
Proteomic ratio analyses produced a list of 576 probe-modified peptides with an isotopic ratio greater than 3, equivalent to a log2 ratio greater than 1.58 (Fig. 1B and table S1). In contrast to the conventional view of formaldehyde as an indiscriminate electrophile, the data revealed privileged formaldehyde-sensitive cysteine sites, similar to other reactive species such as hydrogen peroxide, nitric oxide, and methylglyoxal (27, 28), and provides foundational information on formaldehyde-cysteine reactivity across the proteome in a systematic manner. Importantly, modified cysteines were found enriched in protein families spanning formaldehyde-dependent pathways (e.g., carbon metabolism, glutathione) and one-carbon metabolism (e.g., one-carbon amino acids, folate) rather than demonstrating proteome-wide, indiscriminate reactivity (Fig. 1C). Included are targets such as a key enzyme in formaldehyde detoxification alcohol dehydrogenase 5 (class III) (ADH5) (21), a known regulator of methyl unit balance through the biosynthesis of sarcosine from glycine, glycine-N-methyltransferase (GNMT) (29), and serine hydroxymethyltransferase 1 and 2 (SHMT1 and 2) that regulate serine/glycine pools in the folate cycle. Additional targets identified in amino acid metabolism, methyl transfer and transsulfuration chemistry, and carbohydrate metabolism point to the specificity with which formaldehyde engages enzymes known to regulate methyl units and the one-carbon cycle in a variety of cellular contexts. Of particular interest is S-adenosylmethionine synthase isoform type-1 (MAT1A), a key enzyme in SAM biosynthesis that regulates the flux of transmethylation reactions. isoTOP-ABPP analysis identified three specific cysteine residues, out of the ten total in MAT1A, that were hyperreactive towards formaldehyde: murine MAT1A Cys105, Cys121, and Cys150 with ratios of 3.45, 3.40, and 4.54, respectively.
Formaldehyde makes covalent adducts with MAT1A.
We next sought to directly identify sites of formaldehyde modification on human MAT1A through mass spectrometry. We treated purified MAT1A protein with excess formaldehyde (5 mM) for 4 hours at 37 °C and then digested overnight into peptides. When added in large excess, formaldehyde forms modifications on arginine, lysine, tryptophan, histidine, and cysteine in simple in vitro contexts of model peptides and free amino acids (23, 24). However, LC-MS/MS quantification of MAT1A modified peptides and an unbiased mass search revealed limited formaldehyde reactivity with nucleophilic amino acid residues beyond cysteine (Fig. 2A). Only the three hyperreactive cysteine residues identified by isoTOP-ABPP (human Cys104, Cys120, Cys149, analogous to murine Cys105, Cys121, Cys150) were modified by excess formaldehyde in vitro, along with a fourth solvent-accessible cysteine residue (human Cys376), indicating high selectivity of formaldehyde towards these privileged cysteine sites compared to other nucleophilic amino acids (Fig. 2, B to E). Modification of Cys104 indicated a mass-to-charge ratio (m/z) change of +12 Da, corresponding to nucleophilic addition to formaldehyde by the cysteine thiol to form a hemithioacetal and cyclization to form a thiazolidine (fig. S2) (24). In contrast, Cys120 (fig. S3), Cys149 (fig. S4), and Cys376 (fig. S5) showed a m/z change of +30 Da, corresponding to a hemithioacetal modification (24). The labeling pattern observed in vitro with purified protein further establishes that formaldehyde is not a global, indiscriminate electrophile and validates the isoTOP-ABPP chemoproteomics method in identifying privileged cysteine sites that are selective to formaldehyde.
Fig. 2. Formaldehyde forms site-specific covalent cysteine modifications on MAT1A.
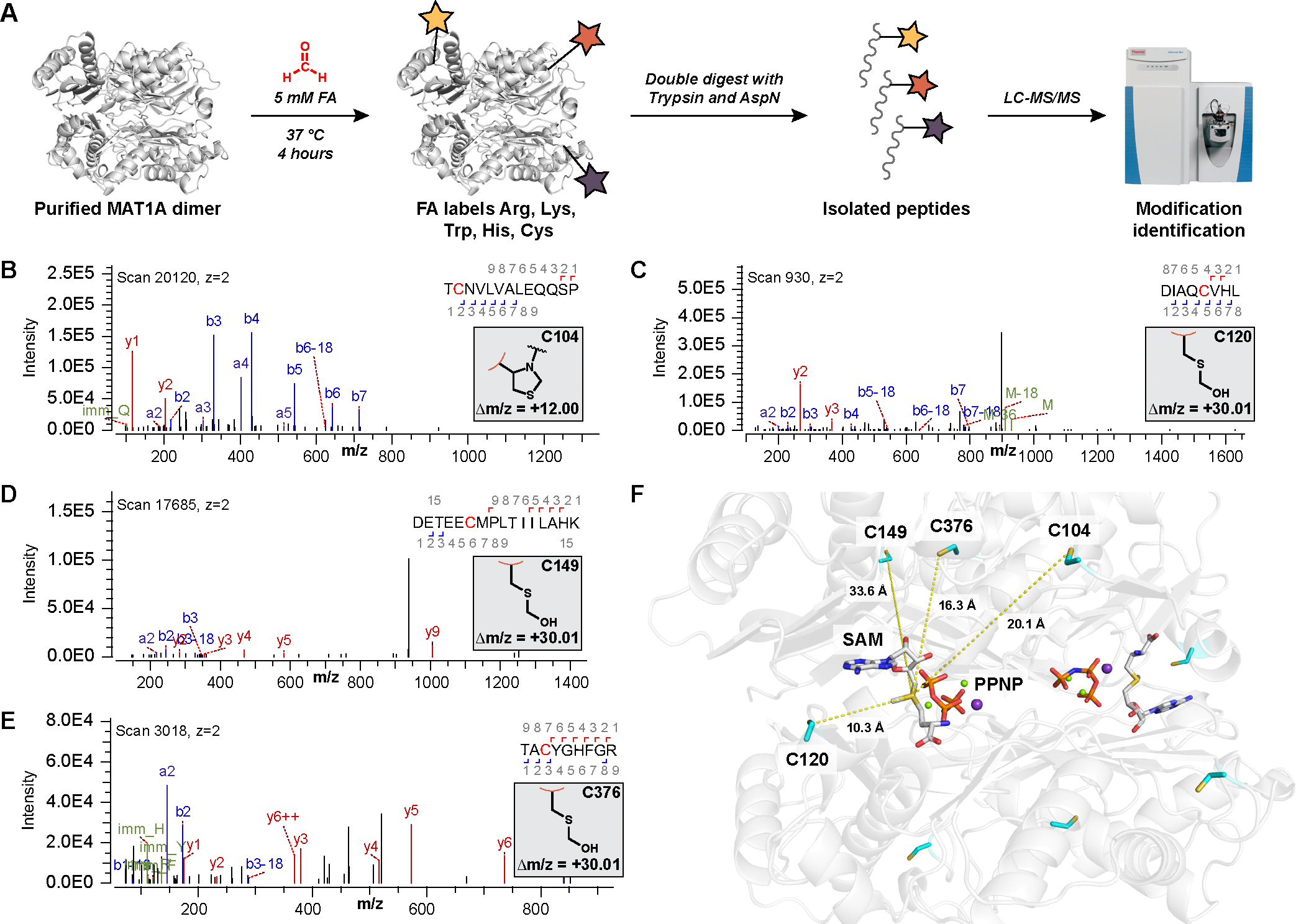
(A) Workflow for the identification of formaldehyde (FA)-dependent covalent modifications on purified MAT1A protein in vitro. (B to E) Representative MS/MS spectra of formaldehyde modification of (B) Cys104, (C) Cys120, (D) Cys149, and (E) Cys376 upon fragmentation and sequencing to generate b (blue) and y (red) ions to indicate a (B) thiazolidine (Δm/z = +12.00) and (C to E) hemithioacetal (Δm/z = +30.01) modifications, respectively. (F) Crystal structure showing the position of all four identified cysteine residues relative to the SAM-bound active site of MAT1A. The angstrom distance between each cysteine residue and SAM are noted. SAM and imidotriphosphate (PPNP), substitute for ATP, are labeled.
To determine if modification was selective for formaldehyde over other aldehydes, we performed the modification identification on purified MAT1A with excess acetaldehyde (5 mM) (30). Quantification of the same peptide coverage as with formaldehyde revealed no discernable modifications of any amino acid residue with acetaldehyde. The lack of acetaldehyde modification is supported by data showing that formaldehyde reacts the fastest and forms the most stable product with cysteine compared to other carbonyl compounds (24), which supports the specificity of formaldehyde as a privileged electrophile for protein post-translational modifications.
Attempts to characterize covalent formaldehyde-MAT1A modifications by protein crystallography have not yet been successful, in part due to the susceptibility of formaldehyde adducts to long-term hydrolysis (24). We were able to obtain a crystal structure of MAT1A at 2.00-Å resolution, which reveals SAM bound in the active site along with additional cofactors not mapped in previous structures (31), including imidotriphosphate (PPNP), Mg2+ ions, and K+ ions. Cys120 lies closest in proximity to the MAT1A active site, presaging this residue as the functional site of formaldehyde reactivity (Fig. 2F, fig. S6, and table S2). Additionally, previous studies have reported Cys120 as a target of hydrogen peroxide and nitric oxide inhibition of rat MAT1A (32–34).
Formaldehyde inhibits MAT1A but not MAT2A activity in vitro at Cys120.
To characterize the biochemical effects of formaldehyde-modified cysteine sites on MAT1A and to establish the role of formaldehyde in SAM biosynthesis, we investigated the effects of formaldehyde on the activity of both MAT1A and MAT2A isoforms. Notably, an isoform switch from MAT1A to MAT2A occurs during hepatocellular carcinogenesis, resulting in intracellular SAM depletion in tumor environments (35, 36). The cancer-dependent isoform switch, coupled with the high binding affinity of SAM to MAT2A compared to MAT1A, makes MAT2A a promising therapeutic target for blocking tumor growth by increasing the bioavailability of this one-carbon unit (37).
We purified catalytically active forms of MAT1A (tetramer) and MAT2A (dimer). SAM biosynthesis occurs through three genes (MAT1A, MAT2A, and MAT2B) encoding MAT enzymes, which catalyze the nucleophilic addition of L-methionine to adenosine triphosphate (ATP) (38). MAT1A encodes for expression of tetramer (MAT1A) and homodimer (MATIII), which are primarily expressed in healthy liver tissue, while MAT2A and MAT2B encode for expression of MAT2A and MATβ regulatory subunit, respectively, which are expressed in all other cell types, fetal liver tissue, and liver cancer (38). Whereas MAT1A and MAT2A share high sequence homology (84%) (39), they differ in substrate affinity and product inhibition, affecting their levels of SAM production (Supplementary Discussion) (40, 41).
To determine a physiologically relevant range of formaldehyde concentrations, we measured basal formaldehyde levels in HepG2 cells using a ratiometric fluorescent probe previously developed in our laboratory, RFAP-1 (16). Standard addition from exogenously added formaldehyde allowed us to obtain a cellular formaldehyde concentration of 49 ± 10 μM in HepG2 (Fig. 3A) comparable to reported measurements (19, 20, 22, 42). To probe the effects of formaldehyde on MAT1A activity, we performed kinetic enzyme assays to monitor SAM production in vitro using triple quadrupole (QqQ) mass spectrometry (37). Enzymatic activity of MAT1A decreased 44% in response to 100 μM formaldehyde and 60% in response to 500 μM formaldehyde (Fig. 3B). In contrast, MAT2A retains full catalytic activity in the presence of even large excess of formaldehyde at 100 and 500 μM (Fig. 3B). Formaldehyde thus selectively inhibits MAT1A activity in an isoform-specific manner.
Fig. 3. Formaldehyde inhibits SAM biosynthesis of MAT1A at Cys120 on purified protein in a dose- and isoform-dependent manner, lowering levels of SAM in cells.
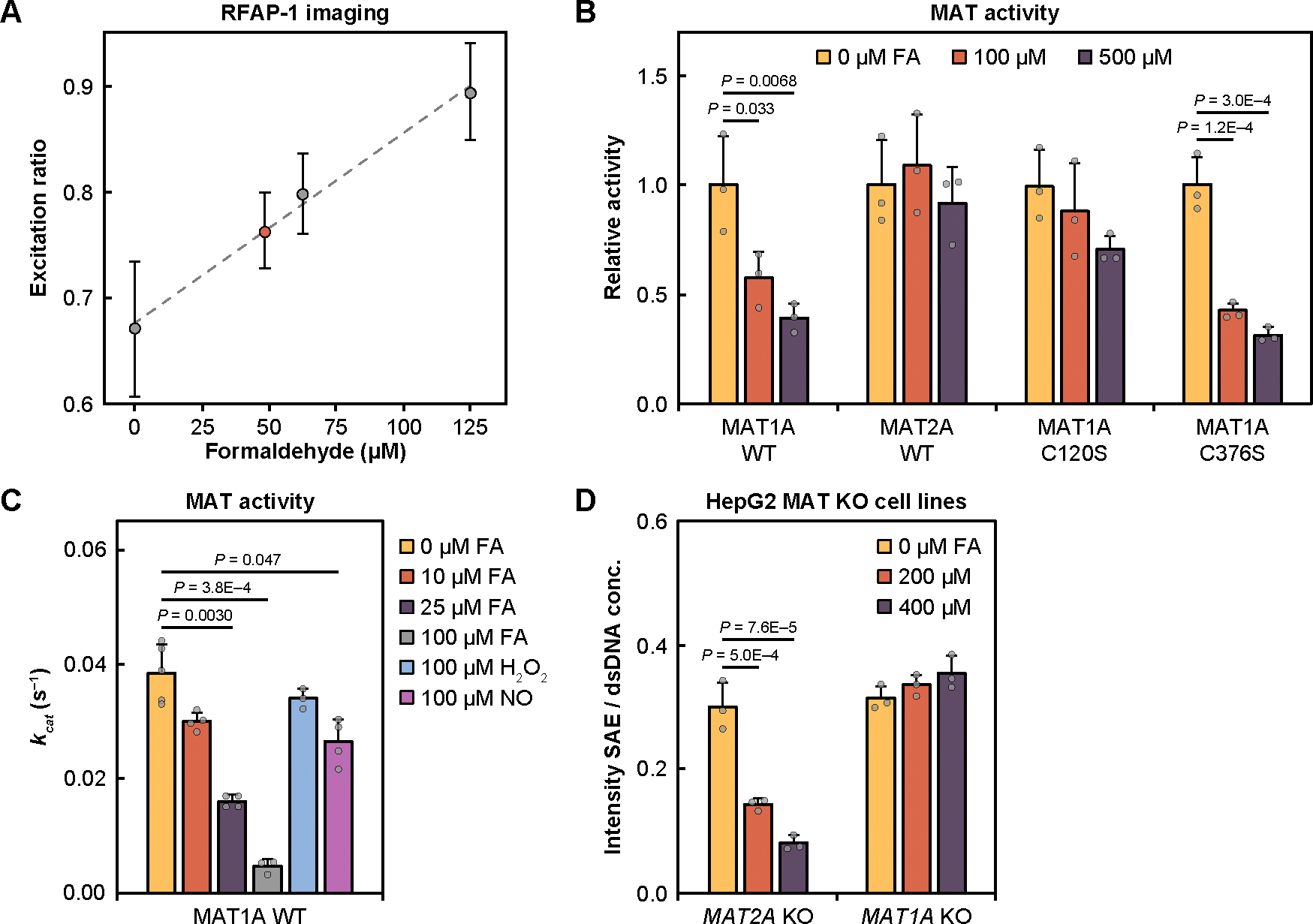
(A) Standard addition curve of formaldehyde in HepG2 by RFAP-1 fluorescent probe. Error bars are SD (n = 4) of technical cell replicates for each concentration. (B) Relative activity of MAT1A WT, MAT2A WT, MAT1A C120S, and MAT1A C376S enzymes in response to 0 (yellow), 100 (orange), or 500 μM (purple) formaldehyde (FA). Error bars denote SD (n = 3) of independent protein aliquots. (C) kcat of MAT1A WT untreated or treated with 10 μM, 25 μM, 100 μM of formaldehyde, and 100 μM of reactive species (H2O2 or NO). Michaelis-Menten kinetic analysis and KM values are reported in table S3. Error bars are SD (n = 3 or 4) of independent protein aliquots. (D) Measurements of SAE levels in CRISPR-generated HepG2 cell models showing that MAT1A-positive MAT2A KO cells are sensitive to formaldehyde and lead to SAE depletion whereas SAE levels in MAT2A-positive MAT1A KO cells are unaffected by formaldehyde exposure. Error bars are SD (n = 3) of technical cell replicates. Statistical significance (B to D) was determined with one-way ANOVA and P-values are from Tukey’s HSD post hoc analyses.
Cys104 and Cys149 are common to both MAT isoforms, whereas Cys120 and Cys376 are present only in MAT1A. To identify which of the cysteine residues imparts sensitivity to formaldehyde-dependent catalytic activity inhibition, we purified MAT1A C120S and MAT1A C376S mutants. MAT1A C120S activity displayed no significant reduction in activity in the presence of 100 and 500 μM formaldehyde (Fig. 3B), indicating a functional role of Cys120 in formaldehyde-sensitivity. In contrast, MAT1A C376S activity displayed comparable activity to that of wildtype MAT1A with 58% inhibition from 100 μM formaldehyde and 68% inhibition from 500 μM formaldehyde (Fig. 3B). A MAT2A G120C mutant was created and analyzed with no significant reduction in activity with 100 μM formaldehyde and slightly decreased activity with 500 μM formaldehyde (fig. S7A), indicating additional factors around the Cys120/Gly120 residue (fig. S7B) (39) may drive the role of formaldehyde modification in MAT2A.
Analyzing MAT1A activity at lower formaldehyde concentrations, even 10 μM formaldehyde was sufficient to reduce the apparent kcat by 22%, with treatments of 25 and 100 μM formaldehyde further reducing kcat by 59% and 88%, respectively, showing dose-dependent MAT1A inhibition in a physiologically relevant 10–100 μM range (Fig. 3C, fig. S7C, and table S3). Formaldehyde treatments also reduced substrate binding affinity as reflected by increasing KM values for formaldehyde (fig. S7C and table S3), though to a lesser extent, showing that MAT1A inhibition by formaldehyde is largely kcat driven. We also assessed the selectivity of formaldehyde for MAT1A inhibition over hydrogen peroxide (H2O2), nitric oxide (NO), and acetaldehyde. There was only modest inhibition of MAT1A upon treatment with 100 μM H2O2 (12% reduction) and 100 μM NO (31% reduction) (Fig. 3C, fig. S7D, and table S3), at concentrations which represents a large excess of these species, typically found in the nanomolar range (18). We also did not observe significant change in kcat with 100 μM acetaldehyde (fig. S7, E and F, and table S3), show that MAT1A inhibition is formaldehyde selective.
MAT1A-positive, but not MAT2A-positive cells, respond to formaldehyde by decreasing cellular SAM biosynthesis.
To evaluate the effects of formaldehyde-dependent inhibition of MAT1A activity in a cellular context, we used CRISPR-Cas9-mediated knockout of MAT1A and MAT2A in hepatoblastoma-derived HepG2 cells. HepG2 cells endogenously express both MAT1A and MAT2A, making them ideally suited to create a pair of matched cellular models expressing only MAT1A or MAT2A. We successfully created two such cell lines: MAT2A knockout (KO) HepG2 cells expressing only MAT1A (MAT1A-positive) and MAT1A KO HepG2 cells expressing only MAT2A (MAT2A-positive) (fig. S8A).
Based on the collective data from proteomics and biochemical experiments, we reasoned that formaldehyde-dependent inhibition of MAT1A would result in a decrease in cellular SAM levels and methylation of downstream targets, whereas the methylation potential of MAT2A-dependent cells would be insensitive to formaldehyde. To test this hypothesis, we measured levels of SAM production for MAT activity in response to varying doses of formaldehyde. We applied ethionine supplementation as a MAT substrate to create the more stable cellular product S-adenosylethionine (SAE) (37), which cannot be used by downstream methyltransferases, resulting in an intracellular buildup of SAE that can be quantified through mass spectrometry. Cells were also incubated in a one-carbon unit restriction (1CR) media lacking serine, glycine, and methionine, which results in depletion of methyl units (43). After validating the use of SAE and 1CR (fig. S8B), results showed a dose-dependent decrease in SAE production in MAT1A-positive cells in response to 0, 200, and 400 μM formaldehyde and no change in SAE production in MAT2A-positive cells (Fig. 3D), confirming that MAT2A activity is not sensitive to formaldehyde. To determine the biological relevance of MAT1A inhibition on SAM levels, we determined a SAM/SAH ratio of 23.1 in MAT1A-positive cells compared to a ratio of 20.5 in MAT1A-positive cells with 200 μM formaldehyde, 18.4 in HepG2 WT, and 7.6 in MAT2A-positive cells (fig. S8B), showing that even when compensatory mechanisms and one-carbon units are available, inhibition by formaldehyde and the more extreme full knockout of MAT1A resulted in a decrease in cellular SAM.
Genetic mouse models of chronic formaldehyde overload show deficiencies in MAT1A-dependent SAM production and methylation on privileged targets.
The collective data identify a reciprocal feedback cycle in which formaldehyde exposure and elevation inhibits SAM biosynthesis in a dose-dependent and isoform-specific manner. Given that formaldehyde-related diseases arise from chronic rather than acute exposure (44–47), we sought to test this model in vivo to help decipher the physiological relationships between formaldehyde and SAM pools and their effects on downstream methylation pathways (Fig. 4A). We investigated a previously developed Adh5−/− mouse that has been used as a model of chronic formaldehyde stress (Supplementary Discussion) (10, 48, 49). Adh5−/− mice were determined to have elevated formaldehyde and N2-hydroxymethyl-deoxyguanosine, a major formaldehyde adduct on DNA (50), compared to their wildtype counterparts (10, 21, 22). Formaldehyde is detoxified through two clearance mechanisms relying on ADH5 and ALDH2; the double knockout mice are visibly smaller than individual knockout mice, and the rare survivors did not live longer than 47 weeks (22).
Fig. 4. Genetic mouse model of chronic formaldehyde overload has reduced methylation potential that specifically targets histone methyl sinks.
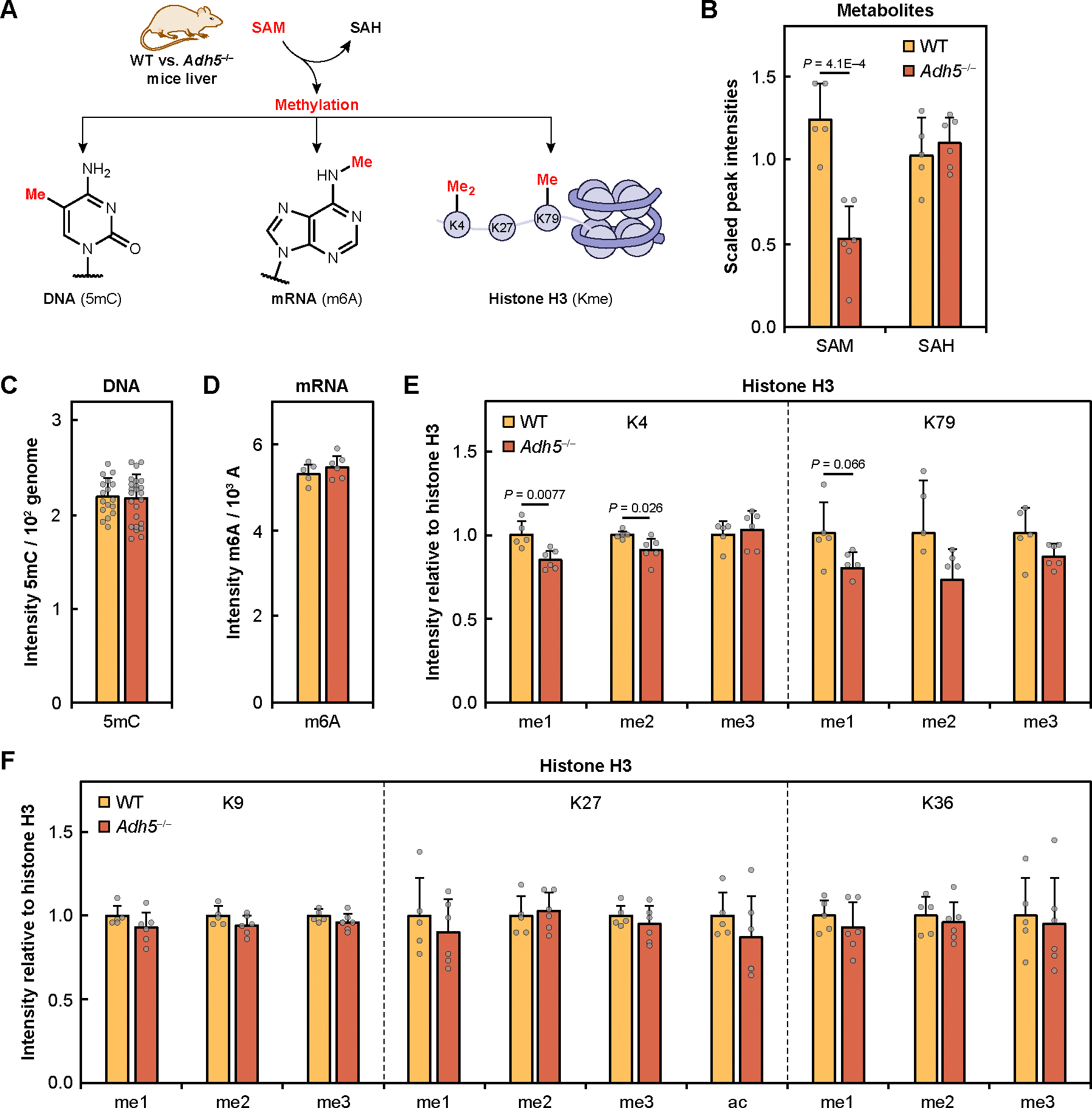
(A) Schematic of SAM and its downstream methylation that were measured in WT and Adh5−/− mice liver. (B) SAM and SAH measured by mass spectrometry in WT and Adh5−/− liver. (C) Global DNA methylation measured by 5-methyldeoxycytidine (5mC) and normalized by deoxycytidine (dC). (D) Global RNA methylation measured by N6-methyladenosine (m6A) and normalized by adenosine (A). (E) Histone H3 methylation measured by immunoblotting for K4 and K79 mono- (me1), di- (me2), and trimethylation (me3). (F) Histone H3 methylation measured by immunoblotting for K9, K27, and K36 mono-, di-, and trimethylation and K27 acetylation (ac). All histone blots were normalized by total histone H3. WT (n = 5, yellow) and Adh5−/− (n = 6, orange) of biological replicates. Error bars represent SD for all graphs. Statistical significance was determined with two-tailed t-test.
We first assessed the ratio of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) as a measure of cellular methylation potential (51), hypothesizing that formaldehyde-dependent inhibition of MAT1A would result in a decreased SAM/SAH ratio in Adh5−/− mice liver compared to WT. Metabolomic data quantifying extracted metabolites indeed showed a significant decrease in SAM in Adh5−/− liver compared to WT with no change in SAH (Fig. 4B). We next examined if lowered SAM/SAH ratios would be reflected in inhibition of SAM-dependent methylation of DNA and mRNA. However, quantification of 5-methyldeoxycytidine (5mC) as a measure of global DNA methylation (Fig. 4C) and N6-methyladenosine (m6A) as a measure of global mRNA methylation (Fig. 4D) by mass spectrometry displayed no significant changes between WT and Adh5−/− mice liver samples. These results aligned with previous observations that Adh5−/− mice are physiologically comparable to their WT counterparts and may be able to sustain methylation through compensatory aldehyde dehydrogenase activity (22). In addition to DNA and mRNA, we also examined histone methylation, with particular interest in monitoring mono-, di-, and trimethylation of H3K4, K36, and K79 histone that were reported as methyl sinks (52) associated with transcriptional activation, elongation (53), and chromatin structure (54). Indeed, we observed that in histones purified from Adh5−/− mice liver, K4 and K79 show statistically significant decreases in methylation in mono- and dimethylation (Fig. 4E and fig. S9, A and C). In contrast, K9, K27, and K36 methylation and K27 acetylation were not found to be significantly changed in Adh5−/− histones (Fig. 4F and fig. S9, B and C). The data are in line with the highly prioritized and compartmentalized nature of one-carbon metabolism pathways (4), showing privileged specificity for certain methyl sinks that are affected by chronic elevations in cytosolic formaldehyde pools despite a global decrease in SAM.
Formaldehyde-inhibited SAM production leads to isoform-specific compensatory increased MAT1A expression through genetic and epigenetic mechanisms.
During periods of chronic liver stress, such as in hepatocellular carcinoma, an isoform switch from MAT1A to MAT2A leads to an overall reduction in basal levels of SAM production (36, 37). As such, we sought to test how chronic formaldehyde elevations in the genetic Adh5−/− mouse model may influence overall MAT expression to compensate for the sustained loss of MAT1A activity (Fig. 5A). We first measured MAT1A and MAT2A protein expression levels in Adh5−/− mice liver lysate; immunoblotting showed that protein expression for MAT1A was elevated 2.8-fold in Adh5−/− liver over WT, but MAT2A expression was not significantly changed (Fig. 5B and fig. S10A). Therefore, in this in vivo context, SAM deficiency caused by chronic formaldehyde overload is indeed a result of inhibition of MAT1A activity rather than an isoform switch to MAT2A, with the observed increase in MAT1A expression as a potential compensatory mechanism to combat this deficiency.
Fig. 5. Chronic elevations in formaldehyde induce compensatory increases in MAT1A expression by decreasing promoter methylation.
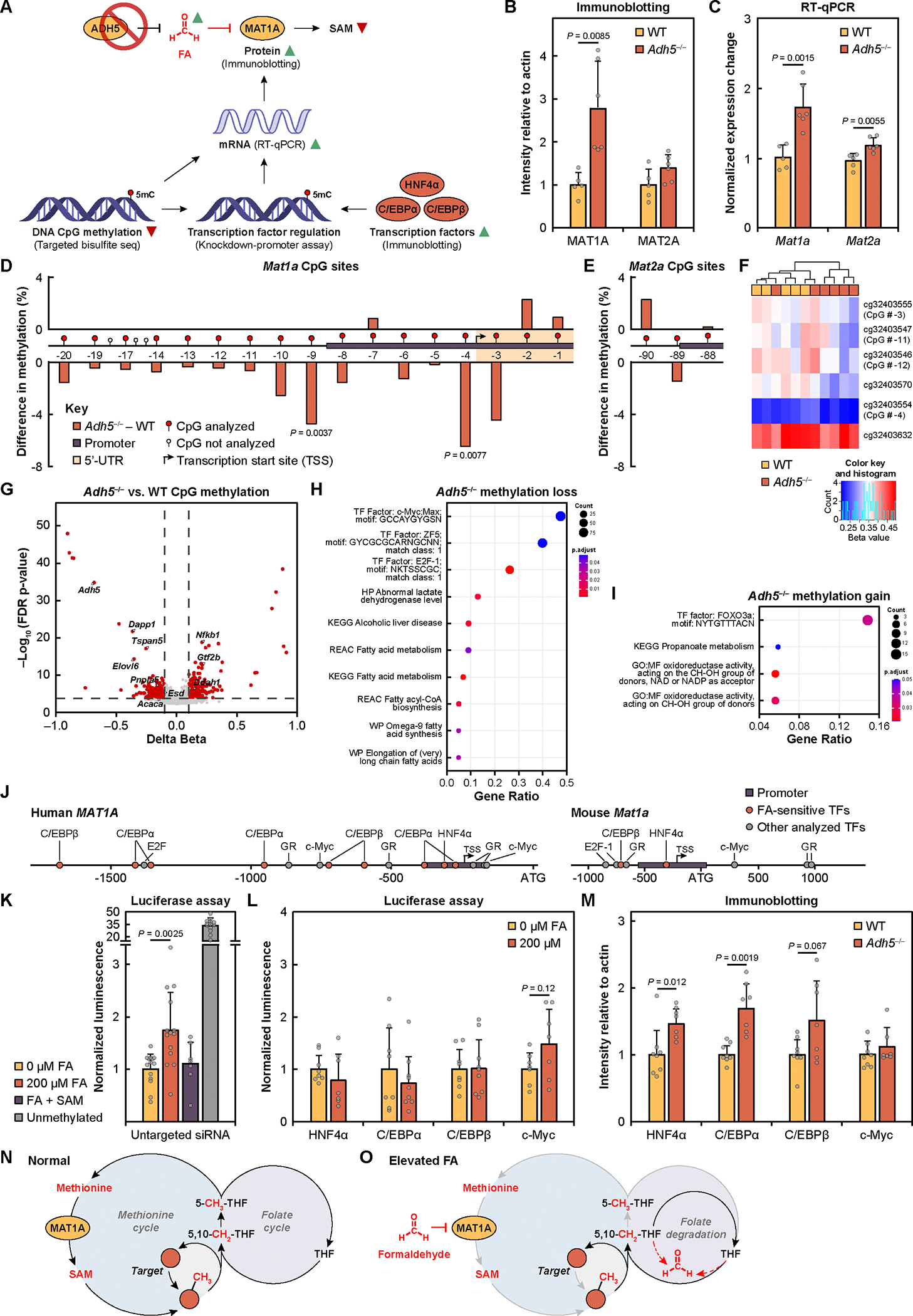
(A) Schematic and summary of results of MAT1A regulation in response to elevated formaldehyde (FA) in Adh5−/− mice liver. (B) MAT1A and MAT2A protein expression measured by immunoblotting for WT (n = 5, yellow) and Adh5−/− liver (n = 6, orange) of biological replicates. (C) Mat1a and Mat2a mRNA transcript levels measured by RT-qPCR. (D to E) Difference in percent methylation of (D) Mat1a and (E) Mat2a DNA CpG sites between Adh5−/− and WT liver. X-axis shows schematic of Mat1a and Mat2a promoters for CpG sites with greater than 2% methylation. Sites are numbered sequentially relative to the ATG site. P-values are determined with two-way t-test. (F) Heatmap of CpG beta values for MAT1A sites analyzed in the genome-wide CpG analysis. Beta values represent percent methylation from 0 (fully unmethylated) to 1 (fully methylated). (G) Volcano plot of the delta between Adh5−/− compared to WT for genome-wide CpG beta values. One-carbon and fatty acid metabolism gene hits of interest are labeled. (H) Pathway enrichment analysis of methylation loss in Adh5−/−. (I) Pathway enrichment analysis of methylation gain in Adh5−/−. (J) Schematic of human MAT1A and mouse Mat1a promoter with transcription factors (TFs) studied in this work labeled. Transcription factors that were deemed as important for MAT1A promoter sensitivity for formaldehyde are in orange. (K) Luciferase assay of HepG2 human MAT1A methylated and unmethylated promoters with 0 or 200 μM formaldehyde and 5 mM SAM treatments. Data is normalized to untreated control. 0 and 200 μM formaldehyde treatment (n = 14) technical cell replicates from four experiments, formaldehyde with SAM treatment (n = 7) technical cell replicates from two experiments, unmethylated promoter (n = 13) technical cell replicates from four experiments. (L) Luciferase assay of HepG2 siRNA-mediated transcription factor knockdown with human MAT1A methylated promoter with 0 or 200 μM formaldehyde. Data is normalized to each untreated control. HepG2 (n = 7 to 9) of technical cell replicates from two experiments. (M) Expression of several MAT1A-associated transcription factors in WT (n = 8) versus Adh5−/− (n = 7) of biological replicates. Error bars represent SD for all graphs. Statistical significance was determined with two-tailed t-test. (N) Proposed model of normal one-carbon metabolism in cells through MAT1A-catalyzed production of SAM and demethylation reactions, preserving the carbon unit through folate-mediated synthesis of methionine. (O) Proposed model of formaldehyde (FA)-dependent regulation of one-carbon metabolism in cells under situations of formaldehyde overload, showing decrease of SAM biosynthesis from isoform-specific formaldehyde-inhibition of MAT1A (gray arrows), disruption of the folate cycle due to hypermethioninemia, and further formaldehyde elevation from spontaneous folate degradation (dashed red arrows).
To decipher the mechanism by which MAT1A expression was altered by chronic formaldehyde elevation, we quantified the normalized expression change of mRNA transcripts for Adh5−/− compared to WT. We found that the trends for both Mat1a and Mat2a mRNA levels paralleled the trends in protein expression (Fig. 5C), indicating that compensation is mediated at the transcriptional level. In this context, the MAT1A-to-MAT2A expression switch has been reported to be regulated by DNA methylation, where hypo- or unmethylated MAT1A and MAT2A promoters are known to induce expression of the respective isoform (40, 55, 56). To investigate CpG methylation of Mat1a and Mat2a CpG sites, targeted next-generation bisulfite sequencing was analyzed in the mouse Mat1a and Mat2a promoters and gene bodies to identify changes in methylation that are potentially regulating enzyme expression (57–59). We observed select CpG sites in the Mat1a promoter that showed reduced methylation in Adh5−/− compared to WT liver (Fig. 5D). The observed decreases in methylation of the Mat1a promoter with concomitant increases in mRNA transcripts and protein expression are in line with reports showing that hypomethylation of the Mat1a promoter results in an increase in expression (55, 56). In contrast, we did not observe formaldehyde-dependent methylation changes of CpG sites in Mat2a (Fig. 5E). These results support the model that SAM deficiency upon formaldehyde overload is regulated by MAT1A activity in an isoform-specific manner, with DNA hypomethylation as one of the potential compensatory mechanisms that elevate MAT1A expression and thus help maintain SAM levels in response to this deficiency.
To further support whether the observed changes in Mat1a promoter CpG methylation and MAT1A expression were a direct consequence of elevated formaldehyde levels, rather than disruption from formate and other one-carbon metabolite sources upon Adh5–/–, we utilized cell models with acute formaldehyde treatment to monitor CpG methylation and MAT1A and MAT2A expression. We exposed HepG2 cells to 200 μM formaldehyde and analyzed CpG methylation and protein expression at 2- to 10-hour timepoints relative to 10-hour vehicle control. Similar to what was observed in the Adh5−/− mouse model, we observed an increase in MAT1A expression (0.047-fold change/hour) and mRNA transcripts (0.026-fold change/hour) over time, but MAT2A expression and transcripts remain largely unchanged (fig. S10B and fig. S11, A and B). A similar experiment with 5 mM SAM supplementation displayed no significant change in MAT1A expression (fig. S11, C and D), suggesting that the increase in MAT1A expression from formaldehyde treatment is SAM-dependent. Analysis of targeted bisulfite sequencing CpG sites with higher than 2% methylation revealed, aside from CpG # −15, that the assayed MAT1A CpG sites were either unchanged or had a negative rate of methylation percent change per hour over the 10-hour formaldehyde treatment; MAT2A sites instead increased over time (fig. S11, E and F). Together, the cell and mouse model data are consistent with formaldehyde overload triggering a compensatory, isoform-specific increase in MAT1A expression through genetic and epigenetic regulation mechanisms.
Chronic formaldehyde elevation alters genome-wide methylation and transcriptional regulation.
We next explored effects of chronic formaldehyde elevation on genome-wide gene regulation. Indeed, in this context it has been shown that SAM treatment in HepG2 cells leads to differential hypo- and hypermethylation of select DNA CpG sites (60). As such, we performed genome-wide CpG methylation analysis in liver samples from Adh5−/− mice compared to WT controls. Mat1a and Mat2a CpG DNA methylation were analyzed in the genome-wide dataset to support the results from the targeted bisulfite sequencing. We observed a trend of hypomethylation in Adh5−/− samples in 4 of the 6 analyzed Mat1a CpG sites (Fig. 5F). No change was observed for the 5 CpG sites for Mat2a (fig. S12A). Similar to a previous report on SAM-dependent DNA methylation changes (60), we observed both hypo- and hypermethylation of CpG sites in Adh5−/− compared with WT (Fig. 5G, fig. S12B, and table S4). Pathway analysis showed that there was selective hypomethylation of CpGs present in metabolic genes involved in alcoholic liver disease, fatty acid metabolism, and affecting genes containing the binding sites motifs of c-Myc, ZF5, and E2F1 in Adh5−/− mice (Fig. 5H and fig. S12C). In addition, a gain of methylation was observed in genes involved in propanoate metabolism and in oxidoreductase activity on alcohol group of donors (Fig. 5I and fig. S12D). The genes containing binding site motifs for FOXO3a presented a significant gain of methylation in Adh5−/− compared to WT. Remarkably, most of the genes showing loss or gain of methylation are involved in metabolic pathways, suggesting formaldehyde might drive a metabolic reprogramming through selectively altering CpG methylation. The expression of genes controlled by c-Myc or FOXO3a might be significantly altered by endogenous formaldehyde. Subsequently, analysis of TF binding profiles with the JASPAR database (61) revealed 11 TF binding profiles with significant enrichment (P < 0.01) among the hypomethylated CpG sites and 37 TF binding profiles among the hypermethylated CpG sites (fig. S13, A and B, and table S5). Focusing on CpG sites located within promoter regions identified 2 TF binding profiles (CEBPB and NFIX) for CpG hypomethylation and 3 TF binding profiles (ZNF707, PAX3, FOSB:JUNB) for CpG hypermethylation (fig. S13, C and D, table S5), exhibiting TFs with potential key roles in formaldehyde-dependent gene regulation.
To provide functional support for the effect of formaldehyde on MAT1A expression through regulation of the MAT1A promoter (Fig. 5J), we performed a luciferase assay with a MAT1A promoter-luciferase plasmid (55). In vitro methylation was performed to obtain a pair of fully unmethylated active and fully methylated inactive MAT1A plasmids. HepG2 cells were transfected with methylated MAT1A promoter-luciferase plasmid, and luminescence was measured, with the unmethylated MAT1A promoter plasmid as a positive control for promoter activity. Cells treated with 200 μM formaldehyde for 10 hours showed a 1.7-fold change increase in luciferase activity compared to untreated samples (Fig. 5K). The observed increase in formaldehyde-triggered promoter activity was negated by 5 mM SAM, supporting that the effect of formaldehyde is SAM-dependent.
Since the changes in Mat1a promoter hypomethylation are modest and alone likely do not fully explain the large compensatory increase in MAT1A expression upon formaldehyde exposure, we investigated other potential regulatory mechanisms. To this end, MAT1A is known to be regulated by several transcription factors (TFs), including but not limited to HNF4α, C/EBPα, C/EBPβ, GR, c-Myc, and E2F1 (Fig. 5J) (40). To determine the potential contributions of these TFs on formaldehyde-dependent regulation of MAT1A expression, we applied the luciferase MAT1A promoter assay after individual knockdown of the 6 TFs or an untargeted control siRNA (fig. S14A). Upon knockdown of hepatocyte nuclear factor 4-alpha (HNF4α), CCAAT/enhancer-binding protein alpha (C/EBPα), and beta (C/EBPβ), we observed that promoter activity was insensitive to formaldehyde treatment (Fig. 5L), showing that these TFs are involved in formaldehyde-dependent MAT1A regulation. C/EBPβ was of particular interest as its binding profile was enriched in promoters in our TFs analysis (fig. S13C). In contrast, upon knockdown of Myc proto-oncogene protein (c-Myc), increases in formaldehyde-dependent promoter activity were comparable to that of untargeted siRNA control (Fig. 5, K and L).
The functional cellular assays are supported by immunoblotting data showing that levels of HNF4α, C/EBPα, and C/EBPβ protein, but not c-Myc, are higher in Adh5−/− mice relative to WT (Fig. 5M and fig. S14B). These results are in line with reports showing that these TFs can positively regulate MAT1A expression (57, 62). Although its putative binding motif was found to be enriched in differentially methylated positions (DMPs) in our genome-wide CpG analysis, c-Myc expression was unchanged in Adh5−/− mice. The transcription factors GR and E2F1 were also analyzed (fig. S14, B to D), but their expression increases were not correlated with significance in formaldehyde-sensitivity, suggesting more complex factors, including localization, binding, and activity, are in play. There may also be a difference in mechanism between human hepatoblastoma HepG2 cells and normal mice liver as the human and murine MAT1A promoters differ (Fig. 5J). Together with our data showing MAT1A promoter sensitivity to formaldehyde with these TFs, the observed increases of HNF4α, C/EBPα, and C/EBPβ expression in Adh5−/− mouse model of chronic formaldehyde elevation establish their participation in formaldehyde/SAM-dependent regulation of MAT1A expression.
Conclusions
Activity-based protein profiling of formaldehyde-reactive cysteine residues reveals that formaldehyde is not an indiscriminate electrophile, but instead serves as a regulatory post-translational modifier that can target a privileged class of hyperreactive cysteine residues in the proteome. By focusing on its contributions in one-carbon metabolism, we characterized an isoform-specific inhibition of MAT1A activity by formaldehyde at a key, conserved cysteine site. We identified a biochemical feedback cycle between two ubiquitous one-carbon units, where formaldehyde can regulate the biosynthetic production of SAM, the major methyl donor of the cell. Using an Adh5−/− mouse model of chronic formaldehyde overload, we established the physiological significance of this one-carbon crosstalk by observing that formaldehyde-dependent inhibition of SAM biosynthesis decreases the methylation potential of the cell in a highly selective manner, despite a global decrease in SAM levels. Specifically, we observed methylation changes in privileged K4 and K79 histone methylation sites as methyl sinks, rather than ubiquitous, global changes to DNA, RNA, and histone methylation targets. Additionally, we uncovered a compensatory feedback pathway for formaldehyde-dependent SAM deficiency, where chronic and acute formaldehyde overload results in selective increase in MAT1A expression. A formaldehyde-MAT1A-SAM feedback cycle is mediated through formaldehyde-dependent MAT1A inhibition and a combination of epigenetic regulation via altered methylation at select transcription factor binding sites and increased expression of select transcription factors to trigger with compensatory upregulation of MAT1A expression.
The collective data support a model where in healthy cells, when formaldehyde homeostasis is tightly maintained, the methyl unit on methionine is converted to SAM for use in downstream methyltransferase writers that install one-carbon post-translational modifications on proteins, nucleic acids, and other metabolic substrates (Fig. 5N) (63). In turn, a number of formaldehyde-generating demethylase enzyme erasers for these post-translational modifications, such as lysine-specific demethylase, possess folate binding pockets that sequester the released formaldehyde into 5,10-methylenetetrahydrofolate (5,10-me-THF), which is then converted into 5-methyl-THF (5-Me-THF) to methylate homocysteine and produce methionine (64). As such, cells achieve a biochemical conservation of one-carbon units within the one-carbon cycle by balancing SAM biosynthesis and methylation/demethylation reactions. In cells with elevated levels of formaldehyde—owing to toxic exposure, metabolic imbalances, and/or disease progression—dysregulation of carbon units stems from the decreased biosynthesis of SAM through MAT1A regulation and results in hypomethylation of downstream disease targets (Fig. 5O). This dysregulation can lead to epigenetic dysregulation and genome instability. Indeed, hypermethioninemia occurs from decreased MAT1A activity (65), leading to a buildup of THF intermediates that we have previously shown to spontaneously degrade to release formaldehyde (10). In turn, this elevated formaldehyde production further inhibits the biosynthesis of SAM, leading to a global depletion of one-carbon units in the cell. Owing to the universal nature of one-carbon metabolism in all organisms and the central roles of SAM-dependent biochemical processes in the cell, the identification of a molecular interplay between one-carbon formaldehyde and SAM biosynthesis sets the stage for further investigations of how these reactive carbon species contribute to biological function.
Materials and Methods Summary
Full details of this study’s methods are described in the Supplementary Materials. Methods are briefly listed here. Isotopic tandem orthogonal proteolysis–activity-based protein profiling (isoTOP-ABPP) and proteomics data analyses was performed as previously described (66) with mouse liver tissue lysate and 500 μM formaldehyde treatment. Site-directed mutagenesis was performed on MAT1A and MAT2A WT plasmids to obtain MAT1A C120S, MAT1A C376S, and MAT2A G120C mutants. His6-tagged MAT proteins were purified for biochemical analyses and protein crystallography; the structure was processed and refined using HKL2000 (67), Phenix (68), and COOT (69). Formaldehyde modifications on MAT1A peptides were analyzed by Q Exactive mass spectrometry. MAT activity assays were performed with formaldehyde treatment, addition of methionine and ATP substrates, quenching over multiple timepoints, and analysis with Enzkin (70) or Prism. CRISPR-mediated knockout (KO) of MAT1A and MAT2A was performed in HepG2 human hepatoblastoma cells to demonstrate isoform-specificity of formaldehyde treatment through the measurement of S-adenosylethionine (SAE) as a stable proxy of S-adenosylmethionine (SAM). To quantify methylation potential in HepG2 WT and KO cell lines upon formaldehyde treatment, SAM and S-adenosylhomocysteine (SAH) were extracted by adapting a previously reported protocol (71). Metabolites were measured by triple quadrupole LC-MS. An endogenous elevated formaldehyde model, Adh5−/− mice, was then studied in comparison to WT. Untargeted metabolomics was performed by Metabolon in mouse liver tissue as previously described (72). 5-methyldeoxycytidine (5mC) DNA methylation in the same cohort of mice as Mulderrig et al. (49), and N6-methyladenosine (m6A) mRNA methylation were measured by mass spectrometry. Histone methylation was analyzed in purified histones by immunoblotting with modification-specific antibodies, with specificity insight from the histone antibodies database (73). MAT protein expression were analyzed by immunoblotting, and mRNA transcripts were analyzed by RT-qPCR and the 2–ΔΔCT quantification method (74). CpG DNA methylation was analyzed by targeted bisulfite sequencing, and global DNA methylation was analyzed using Infinium Mouse Methylation BeadChip Array. Enrichment of transcription factor binding profiles was analyzed with JASPAR database (61, 75). Transcription factor siRNA knockdown in HepG2 was performed was followed by transfection of luciferase MAT1A promoter plasmid, provided by the Lu lab (55). Luciferase assay was performed to demonstrate the effects of formaldehyde on MAT1A promoter activity.
Supplementary Material
Acknowledgments
We thank Dr. Alison Killilea, Carissa Tasto, Molly Fischer, and Willie Hercule of the UC Berkeley Cell Culture Facility for cell culture support. We thank Dr. Jorge Marchand, Dr. Monica Neugebauer, Dr. Kersh Thevasundaram, Douglas Miller, Max Sosa, and Prof. Michelle Chang for use of and technical assistance with their QqQ mass spectrometer. We thank Metabolon, Dr. Kimberly Jackson, and Dr. Priya Ramamoorthy for their work on performing the untargeted global metabolomics. We thank EpigenDx and Austin Hopkins for their work on designing and performing the CpG methylation analysis. We thank Dr. Maria L. Tomasi and Dr. Shelly C. Lu for providing their pGL3 basic luciferase plasmid with the MAT1A promoter.
Funding
We thank the NIH (ES28096 and ES4705 to C.J.C. and D.K.N. and GM139245 to C.J.C.) for research support. K.J.B., V.N.P., and C.C.W. thank the National Science Foundation for graduate research fellowships, and C.C.W. thanks the NIH F31 for a graduate fellowship. V.N.P. and C.C.W. were partially supported by the NIH Chemical Biology Interface Training Grant T32 GM066698. J.D.W.T. thanks A*STAR for a postdoctoral fellowship. F.A.D., C.L.M., and K.J.P. were funded by the Medical Research Council. F.A.D. was supported by CRUK (C42693/A23273), C.L.M. by the Wellcome Trust (217267/Z/19/Z), and K.J.P. by the Medical Research Council (MC_UU_00029/1). L.B.P. is supported by MCIN/AEI /10.13039/501100011033 and European Union NextGenerationEU/PRTR (Ayuda RYC2021–032395-I); and Departament de Recerca i Universitats / Generalitat de Catalunya (2021 SGR 01309). M.E. is supported by MCIN/AEI /10.13039/501100011033/ and the European Regional Development Fund, ‘A way to make Europe’ ERDF (project PID2021–125282OB-I00); and Departament de Recerca i Universitats / Generalitat de Catalunya (2021 SGR 01494). M.C.P.-H. and N.T.I. were supported by the UC Cancer Research Coordinating Committee CRN-19–585450. For the purpose of Open Access, the authors have applied a CC-BY copyright license to the Author Accepted Manuscript.
Footnotes
Competing interests
D.K.N. is a co-founder, shareholder, and adviser for Artris Therapeutics and Frontier Medicines. All other authors declare no competing interests.
Data and materials availability
The raw mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via PRIDE with the dataset identifier PXD018251. The MAT1A crystal structure has been deposited to RCSB PDB as 8SWA. HepG2 MAT1A and MAT2A knockout cell lines are available upon request. The raw DNA methylation data has been deposited to the Gene Expression Omnibus database repository with the dataset ID GSE242266 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242266).
References
- 1.Locasale JW, Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 13, 572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ducker GS, Rabinowitz JD, One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu SC, Mato JM, S-adenosylmethionine in Liver Health, Injury, and Cancer. Physiol. Rev. 92, 1515–1542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tibbetts AS, Appling DR, Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, Sabatini DM, SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 358, 813–818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noureddin M, Mato JM, Lu SC, Nonalcoholic fatty liver disease: update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp. Biol. Med. 240, 809–820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang M, Vousden KH, Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer. 16, 650–662 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ, The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 318, 1469–1472 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hopkinson RJ, Schofield CJ, Deciphering functions of intracellular formaldehyde: linking cancer and aldehyde metabolism. Biochemistry. 57, 904–906 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, Pontel LB, Rosado IV, Brewer TF, Cordell RL, Monks PS, Chang CJ, Vazquez A, Patel KJ, Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature. 548, 549–554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi C, Yang C-G, He C, A non-heme iron-mediated chemical demethylation in DNA and RNA. Acc. Chem. Res. 42, 519–529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walport LJ, Hopkinson RJ, Schofield CJ, Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 16, 525–534 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Ohata J, Bruemmer KJ, Chang CJ, Activity-based sensing methods for monitoring the reactive carbon species carbon monoxide and formaldehyde in living systems. Acc. Chem. Res. 52, 2841–2848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brewer TF, Chang CJ, An Aza-Cope Reactivity-Based Fluorescent Probe for Imaging Formaldehyde in Living Cells. J. Am. Chem. Soc. 137, 10886–10889 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Bruemmer KJ, Walvoord RR, Brewer TF, Burgos-Barragan G, Wit N, Pontel LB, Patel KJ, Chang CJ, Development of a General Aza-Cope Reaction Trigger Applied to Fluorescence Imaging of Formaldehyde in Living Cells. J. Am. Chem. Soc. 139, 5338–5350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brewer TF, Burgos-Barragan G, Wit N, Patel KJ, Chang CJ, A 2-aza-Cope reactivity-based platform for ratiometric fluorescence imaging of formaldehyde in living cells. Chem. Sci. 8, 4073–4081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruemmer KJ, Green O, Su TA, Shabat D, Chang CJ, Chemiluminescent Probes for Activity-Based Sensing of Formaldehyde Released from Folate Degradation in Living Mice. Angew. Chemie. 130, 7630–7634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parvez S, Long MJC, Poganik JR, Aye Y, Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 118, 8798–8888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Y, Wang M, Liu H, Niu Y, Wang S, Zhang F, Liu H, Simultaneous determination of seven endogenous aldehydes in human blood by headspace gas chromatography–mass spectrometry. J. Chromatogr. B. 1118, 85–92 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Tong Z, Luo W, Wang Y, Yang F, Han Y, Li H, Luo H, Duan B, Xu T, Maoying Q, Tan H, Wang J, Zhao H, Liu F, Wan Y, Tumor Tissue-Derived Formaldehyde and Acidic Microenvironment Synergistically Induce Bone Cancer Pain. PLoS One. 5, e10234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pontel LB, V Rosado I, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L, Swenberg JA, Crossan GP, Patel KJ, Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell. 60, 177–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dingler FA, Wang M, Mu A, Millington CL, Oberbeck N, Watcham S, Pontel LB, Kamimae-Lanning AN, Langevin F, Nadler C, Cordell Rebecca L., Monks PS, Yu R, Wilson NK, Hira A, Yoshida K, Mori M, Okamoto Y, Okuno Y, Muramatsu H, Shiraishi Y, Kobayashi M, Moriguchi T, Osumi T, Kato M, Miyano S, Ito E, Kojima S, Yabe H, Yabe M, Matsuo K, Ogawa S, Göttgens B, Hodskinson MRG, Takata M, Patel KJ, Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Mol. Cell. 80, 996–1012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metz B, Kersten GFA, Hoogerhout P, Brugghe HF, Timmermans HAM, De Jong AD, Meiring H, ten Hove J, Hennink WE, Crommelin DJA, Jiskoot W, Identification of formaldehyde-induced modifications in proteins reactions with model peptides. J. Biol. Chem. 279, 6235–6243 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Kamps JJAG, Hopkinson RJ, Schofield CJ, Claridge TDW, How formaldehyde reacts with amino acids. Commun. Chem. 2, 1–14 (2019). [Google Scholar]
- 25.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 468, 790–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weerapana E, Speers AE, Cravatt BF, Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)—a general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2, 1414–1425 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Couvertier SM, Zhou Y, Weerapana E, Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta (BBA)-Proteins Proteomics. 1844, 2315–2330 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Coukos JS, Lee CW, Pillai KS, Liu KJ, Moellering RE, Widespread, Reversible Cysteine Modification by Methylglyoxal Regulates Metabolic Enzyme Function. ACS Chem. Biol. 18, 91–101 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y-H, Chiang E-PI, Dietary Glycine Alters One-Carbon Metabolic kinetics in vivo. FASEB J. 30, 272–274 (2016). [Google Scholar]
- 30.Waris S, Patel A, Ali A, Mahmood R, Acetaldehyde-induced oxidative modifications and morphological changes in isolated human erythrocytes: an in vitro study. Environ. Sci. Pollut. Res. 27, 16268–16281 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Komoto J, Yamada T, Takata Y, Markham GD, Takusagawa F, Crystal structure of the S-adenosylmethionine synthetase ternary complex: a novel catalytic mechanism of S-adenosylmethionine synthesis from ATP and Met. Biochemistry. 43, 1821–1831 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Sánchez-Góngora E, Ruiz F, Mingorance J, An W, Corrales FJ, Mato JM, Interaction of liver methionine adenosyltransferase with hydroxyl radical. FASEB J. 11, 1013–1019 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Avila MA, Mingorance J, Martínez-Chantar ML, Casado M, Martín-Sanz P, Boscá L, Mato JM, Regulation of rat liver S-adenosylmethionine synthetase during septic shock: role of nitric oxide. Hepatology. 25, 391–396 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Pérez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM, Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J. Biol. Chem. 274, 17075–17079 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Lu SC, Methionine adenosyltransferase and liver disease: it’s all about SAM. Gastroenterology. 114, 403–407 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Frau M, Feo F, Pascale RM, Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 59, 830–841 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Quinlan CL, Kaiser SE, Bolaños B, Nowlin D, Grantner R, Karlicek-Bryant S, Feng JL, Jenkinson S, Freeman-Cook K, Dann SG, Wang X, Wells PA, Fantin VR, Stewart AE, Grant SK, Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 13, 785–792 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Maldonado LY, Arsene D, Mato JM, Lu SC, Methionine adenosyltransferases in cancers: Mechanisms of dysregulation and implications for therapy. Exp. Biol. Med. 243, 107–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shafqat N, Muniz JRC, Pilka ES, Papagrigoriou E, von Delft F, Oppermann U, Yue WW, Insight into S-adenosylmethionine biosynthesis from the crystal structures of the human methionine adenosyltransferase catalytic and regulatory subunits. Biochem. J. 452, 27–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mato JM, Corrales FJ, Lu SC, Avila MA, S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 16, 15–26 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Ramani K, Mato JM, Lu SC, Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers (Basel) 3, 1480–1497 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heck H. d’A., White EL, Casanova-Schmitz M, Determination of formaldehyde in biological tissues by gas chromatography/mass spectrometry. Biomed. Mass Spectrom. 9, 347–353 (1982). [DOI] [PubMed] [Google Scholar]
- 43.Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH, Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell. 61, 210–221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Tang X, Rothman N, Vermeulen R, Ji Z, Shen M, Qiu C, Guo W, Liu S, Reiss B, Freeman Laura Beane, Ge Y, Hubbard AE, Hua M, Blair A, Galvan N, Ruan X, Alter BP, Xin KX, Li S, Moore LE, Kim S, Xie Y, Hayes RB, Azuma M, Hauptmann M, Xiong J, Stewart P, Li L, Rappaport SM, Huang H, Fraumeni JF, Smith MT, Lan Q, Occupational exposure to formaldehyde, hematotoxicity, and leukemia-specific chromosome changes in cultured myeloid progenitor cells. Cancer Epidemiol. Biomarkers Prev. 19, 80–88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips RV, Wei L, Cardenas A, Hubbard AE, McHale CM, Vermeulen R, Wei H, Smith MT, Zhang L, Lan Q, Rothman N, Epigenome-wide association studies of occupational exposure to benzene and formaldehyde. Epigenetics. 17, 2259–2277 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersen ME, Clewell HJ, Bermudez E, Dodd DE, Willson GA, Campbell JL, Thomas RS, Formaldehyde: integrating dosimetry, cytotoxicity, and genomics to understand dose-dependent transitions for an endogenous compound. Toxicol. Sci. 118, 716–731 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Liu Q, Yang L, Gong C, Tao G, Huang H, Liu J, Zhang H, Wu D, Xia B, Hu G, Kunpeng Wang Z Zhuang, Effects of long-term low-dose formaldehyde exposure on global genomic hypomethylation in 16HBE cells. Toxicol. Lett. 205, 235–240 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS, Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 116, 617–628 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Mulderrig L, Garaycoechea JI, Tuong ZK, Millington CL, Dingler FA, Ferdinand JR, Gaul L, Tadross JA, Arends MJ, O’Rahilly S, Gerry P Crossan, M. R. Clatworthy, K. J. Patel, Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature. 600, 158–163 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Moeller BC, Lu K, Doyle-Eisele M, McDonald J, Gigliotti A, Swenberg JA, Determination of N 2-hydroxymethyl-dG adducts in the nasal epithelium and bone marrow of nonhuman primates following 13CD2-formaldehyde inhalation exposure. Chem. Res. Toxicol. 24, 162–164 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffman DR, Marion DW, Cornatzer WE, Duerre JA, S-Adenosylmethionine and S-adenosylhomocystein metabolism in isolated rat liver. Effects of L-methionine, L-homocystein, and adenosine. J. Biol. Chem. 255, 10822–10827 (1980). [PubMed] [Google Scholar]
- 52.Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP, A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell. 66, 180–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kouzarides T, Chromatin modifications and their function. Cell. 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Choi J, Ryoo ZY, Cho D-H, Lee H-S, Ryu H-Y, Trans-tail regulation-mediated suppression of cryptic transcription. Exp. Mol. Med. 53, 1683–1688 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomasi ML, Li TWH, Li M, Mato JM, Lu SC, Inhibition of human methionine adenosyltransferase 1A transcription by coding region methylation. J. Cell. Physiol. 227, 1583–1591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang H, Huang Z-Z, Zeng Z, Chen C, Selby RR, Lu SC, Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am. J. Physiol. Liver Physiol 280, G184–G190 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Ikeda R, Nishida T, Watanabe F, Shimizu-Saito K, Asahina K, Horikawa S, Teraoka H, Involvement of CCAAT/enhancer binding protein-ß(C/EBPß) in epigenetic regulation of mouse methionine adenosyltransferase 1A gene expression. Int. J. Biochem. Cell Biol. 40, 1956–1969 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D, The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, Billis K, Boddu S, Charkhchi M, Cummins C, Da Rin Fioretto L, Davidson C, Dodiya K, El Houdaigui B, Fatima R, Gall A, Garcia Giron C, Grego T, Guijarro-Clarke C, Haggerty L, Hemrom A, Hourlier T, Izuogu OG, Juettemann T, Kaikala V, Kay M, Lavidas I, Le T, Lemos D, Gonzalez Martinez J, Marugán JC, Maurel T, McMahon AC, Mohanan S, Moore B, Muffato M, Oheh DN, Paraschas D, Parker A, Parton A, Prosovetskaia I, Sakthivel MP, Salam AIA, Schmitt BM, Schuilenburg H, Sheppard D, Steed E, Szpak M, Szuba M, Taylor K, Thormann A, Threadgold G, Walts B, Winterbottom A, Chakiachvili M, Chaubal A, De Silva N, Flint B, Frankish A, Hunt SE, IIsley GR, Langridge N, Loveland JE, Martin FJ, Mudge JM, Morales J, Perry E, Ruffier M, Tate J, Thybert D, Trevanion SJ, Cunningham F, Yates AD, Zerbino DR, Flicek P, Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Sun Z, Szyf M, S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget. 8, 111866–111881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Vandepoele K, Wasserman WW, Parcy F, Mathelier A, JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Q, Li Y, Gao X, Kang K, Williams JG, Tong L, Liu J, Ji M, Deterding LJ, Tong X, Locasale Jason W., Li L, Shats I, Li X, HNF4α regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat. Commun. 11, 3978 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Struck A-W, Thompson ML, Wong LS, Micklefield J, S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem. 13, 2642–2655 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Luka Z, Moss F, V Loukachevitch L, Bornhop DJ, Wagner C, Histone demethylase LSD1 is a folate-binding protein. Biochemistry. 50, 4750–4756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mudd SH, Jenden DJ, Capdevila A, Roch M, Levy HL, Wagner C, Isolated hypermethioninemia: measurements of S-adenosylmethionine and choline. Metab. Exp. 49, 1542–1547 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Chung CY-S, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, Zoncu R, Nomura DK, Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 15, 776–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Otwinowski Z, Minor W, “[20] Processing of X-ray diffraction data collected in oscillation mode” in Methods in enzymology (Elsevier, 1997), vol. 276, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 68.V Afonine P, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Cardillo G, Enzkin: a tool to estimate Michaelis-Menten kinetic parameters (2010).
- 71.Kořínek M, Šístek V, Mládková J, Mikeš P, Jiráček J, Selicharová I, Quantification of homocysteine-related metabolites and the role of betaine–homocysteine S-methyltransferase in HepG2 cells. Biomed. Chromatogr. 27, 111–121 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E, Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 81, 6656–6667 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K, Guo AH, Shanle EK, Josefowicz SZ, Fuchs SM, Allis CD, Magnuson TR, Ruthenburg AJ, Strahl BD, An interactive database for the assessment of histone antibody specificity. Mol. Cell. 59, 502–511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Mondragon JAC, Puig RR, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Pérez NM, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Vandepoele K, Wasserman WW, Parcy F, Mathelier A, JASPAR TFBS LOLA databases - Part 1 (2022), doi: 10.5281/zenodo.6860527. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via PRIDE with the dataset identifier PXD018251. The MAT1A crystal structure has been deposited to RCSB PDB as 8SWA. HepG2 MAT1A and MAT2A knockout cell lines are available upon request. The raw DNA methylation data has been deposited to the Gene Expression Omnibus database repository with the dataset ID GSE242266 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE242266).