

Abstract
This account describes our recent progress on the strategic incorporation of fluorine and organofluorine moieties into new-generation taxoid anticancer agents for medicinal chemistry and chemical biology studies.
In the case study 1, novel 3rd-generation fluorotaxoids bearing 3-OCF3 or 3-OCF2H group in the C2-benzoate moiety were designed, synthesized and examined for their anticancer activities. The potency of novel taxoids against drug-resistant cancer cell lines was 2–3 orders of magnitude higher than that of paclitaxel (PTX). Molecular modeling analysis indicated the favorable van der Waals interactions of OCF3 and OCHF2 groups in the binding site. Overall, taxoids bearing a OCHF2 group at the C2 benzoate position exhibited the highest potencies against multidrug-resistant (MDR) cancer cell lines and cancer stem cell (CSC)-enriched cell lines, indicating that the new 3rd-generation fluorotaxoids are promising candidates as chemotherapeutic agents.
In the case study 2, novel 3rd-generation 3′-difluorovinyl (DFV)-taxoids, bearing 3-CF3O or 3-CHF2O group in the C2-benzoyl moiety, were designed, synthesized, and evaluated for their potencies and pharmacological properties. These new DFV-taxoids exhibited remarkable cytotoxicity against extremely drug-resistant cancer cell lines with subnanomolar IC50 values, indicating that these new DFV-taxoids can overcome MDR caused by the overexpression of Pgp and other ABC cassette transporters. The molecular docking analysis of new DFV-taxoids revealed that the 3′-DFV moiety and the 3-CF3O/3-CHF2O group of the C2-benzoate moiety are nicely accommodated to the deep hydrophobic pocket of the PTX/taxoid binding site in the β-tubulin, enabling an enhanced binding through unique attractive interactions between F/OCF3/OCHF2 and the protein. This enhancement in binding is reflected in the remarkable high potency of new 3rd-generation DFV-taxoids.
In the case study 3.1, the therapeutic potential of new 3rd-generation DFV-taxoids in anaplastic thyroid cancer (ATC) cells was evaluated in vitro and in vivo. This study demonstrated that these new DFV-taxoids were more efficacious than PTX against ATC cell lines and tumor xenografts, as demonstrated by the efficient inhibition of cell proliferation and colony formation, induction of apoptosis via the mitotic arrest at the G2/M phase, as well as the suppression of tumorigenic potential in nude mice. Furthermore, tubulin polymerization assay and molecular docking analysis confirmed that these new DFV-taxoids promoted far more rapid polymerization of β-tubulin than PTX through stronger binding to tubulin/microtubules. Taken together, this study has indicated a promising therapeutic potential of these new DFV-taxoids against ATC.
In the case study 3.2, DFV-OTX displayed potent cytotoxicity and effective induction of β-tubulin polymerization, as well as the G2/M phase arrest, leading to apoptosis in PTX-sensitive and PTX-resistant breast cancer cells. Furthermore, DFV-OTX clearly exhibited efficacy against MCF-7R and MDA-MB-231R tumor xenografts in mouse models. Thus, DFV-OTX effectively overcame PTX-resistance in MDA-MB-231R cells and tumor xenografts, wherein the drug resistance was attributed to ABCB1/ABCG2 upregulation. DFV-OTX was also effective against MCF-7R cells and tumor xenografts, which are PTX-resistant due to different MOA. Accordingly, DFV-OTX is a promising chemotherapeutic agent for the treatment of PTX-resistant cancers.
Overall, these next-generation fluorotaxoids are promising candidates for highly potent chemotherapeutic agents, as well as payloads for tumor-targeting drug conjugates such as antibody-drug conjugates (ADCs).
Keywords: Taxane, Structure-activity relationship, Fluorine-containing, Difluorovinyltaxoid, Molecular docking analysis
1. Introduction
It has been shown that the role of fluorine is exceptionally significant in medicinal chemistry, chemical biology, and drug discovery, as evidenced by the fact that the FDA has approved many fluorine-containing compounds for medical and agricultural use [1–5]. Because of the unique properties of fluorine, e.g., small atomic radius, high electro-negativity and low polarizability of C-F bond, the selective introduction of fluorine or organofluorine moieties into medicinally active compounds often improves their pharmacological properties such as higher metabolic stability, increased binding to target molecules, and enhanced membrane permeability. Thus, fluorine is the second most utilized heteroatom (after nitrogen) in the current drug discovery and development. Therefore, it is natural to explore fluoro-analogs of current lead compounds for further optimization in the field of drug discovery and development [1,6–8]. This account presents our recent progress on the strategic incorporation of organofluorine moieties into new-generation taxoid anticancer agents by exploiting the unique nature of fluorine.
According to the American Cancer Society, nearly 2 million new cancer cases and around 0.6 million cancer deaths were projected to occur in the United States in 2021 [9]. Cancer remains one of the most challenging diseases to deal with, as conventional chemotherapy has many undesirable side effects, which deteriorate patient’s quality of life. Furthermore, a very serious issue associated with traditional chemotherapy is its inability to eradicate cancer stem cells (CSCs), which are relatively rare and highly drug-resistant, comprising a population of quiescent or slowly proliferating tumor-initiating cells [10]. It has been shown that CSCs are tumor-initiating cells and also responsible for resistance to therapeutics, metastasis and recurrence. CSCs promote the formation of a diverse set of proliferating, but slowly differentiating cells, thereby contributing to the tumor cell heterogeneity in human cancers [10]. Thus, CSCs are one of the most crucial targets in the development of next-generation anticancer agents [10–13].
The 1st-generation taxane anticancer drugs, paclitaxel (PTX, “Taxol”) and docetaxel (Fig. 1), are the most extensively used chemotherapeutic drugs in the clinic for the treatment of various cancers, e.g., ovarian, breast, and lung cancers, as well as Kaposi’s sarcoma, head/neck and other cancers [14–16]. PTX and docetaxel are often combined with one or more other anticancer agents to exhibit synergistic effects [15]. More recently, FDA approved a 1st-generation taxoid, cabazitaxel (Fig. 1), which is used in combination with prednisone for hormone-refractory metastatic prostate cancer [17]. Besides these drugs, several novel taxoids, new formulations, and new combination therapies are currently in various stages of preclinical and clinical development [15]. Despite the significant impact of these drugs on cancer chemotherapy, there are clear limitations in the use of these drugs, such as susceptibility to multi-drug resistance (MDR) caused by overexpression of ABC cassette efflux pumps, overexpression of β-III tubulin isoform, point mutations in the microtubule-binding site and CSCs, and lack of tumor specificity. These drugs do not exhibit efficacy against melanoma, colon, pancreatic, and renal cancers. For example, human colon carcinoma overexpresses ATP-binding cassette transporter proteins, e.g., P-glycoprotein (Pgp), which binds to hydrophobic anticancer drugs, such as paclitaxel and taxoids, and acts as an efflux pump, reducing their intracellular drug accumulation, leading to multidrug resistance (MDR) [18–25].
Fig. 1.
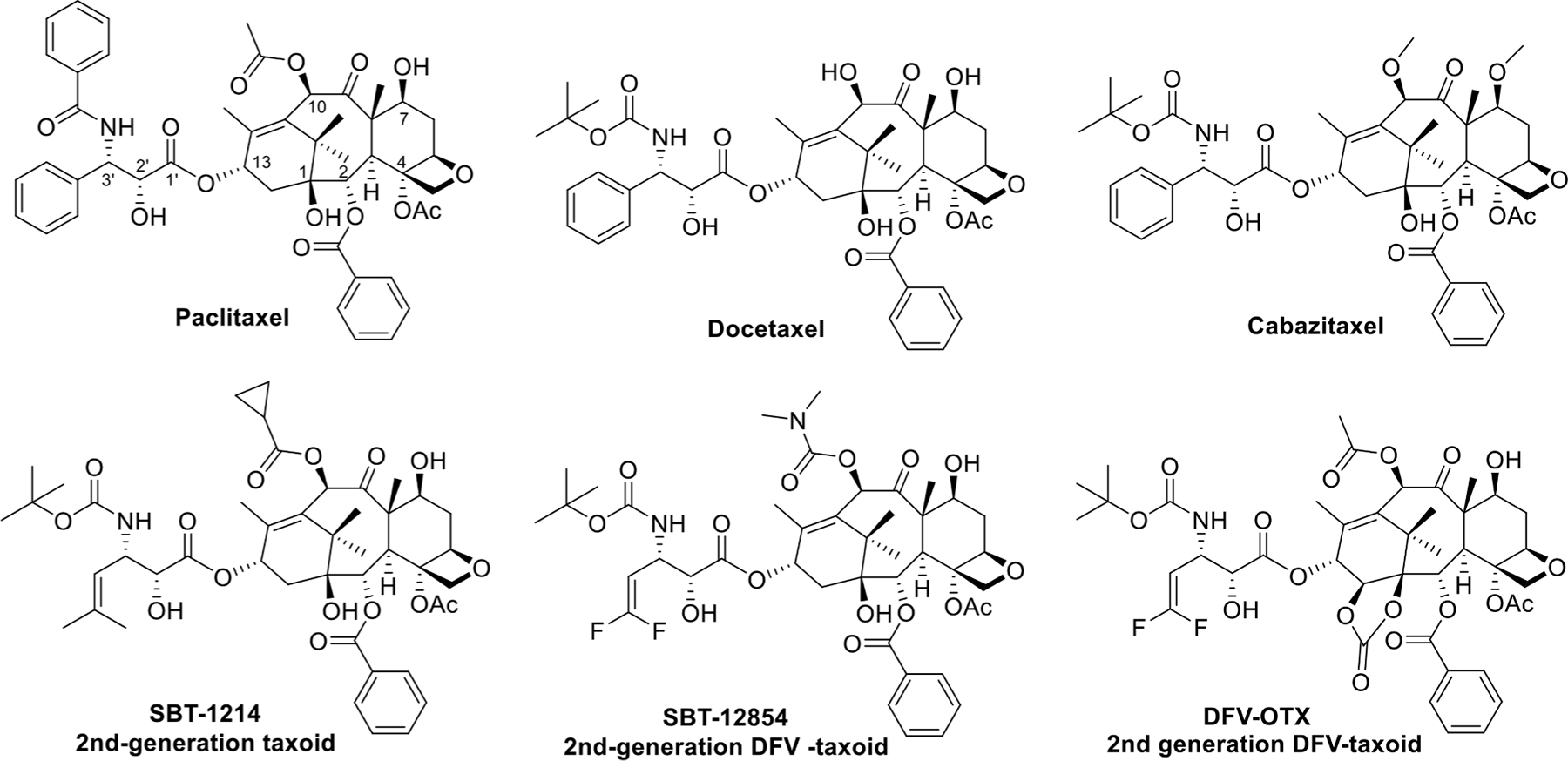
PTX, docetaxel, cabazitaxel, and 2nd-generation taxoids.
Based on extensive structure-activity relationship (SAR) studies, we have developed a series of highly potent 2nd-generation taxoids (modifications at C10, C3′ and C3′ N), including SBT-1214 as representative (Fig. 1) [26–30] and 3rd-generation taxoids (modifications at C2, C10, C3′, and C3′ N) [28,29,31]. These new-generation taxoids exhibit 2–3 orders of magnitude higher potency than PTX and docetaxel against various drug-resistant cancer cell lines, expressing the MDR phenotype, e.g., LCC6-MDR (breast), NCI-ADR (ovarian), DLD-1 (colon), and CFPAC-1 (pancreas), as well as having point mutations at the taxane-binding site in β-tubulin, e.g., 1A9PTX10 (ovarian) [32]. A 2nd-generation 3′-difluorovinyl (DFV) taxoid, SBT-12854 (Fig. 1), is highly potent against CSC-enriched HCT-116 human colon cancer cells, which is 230–33,000 times higher potency than that of conventional anticancer drugs, e.g., cisplatin, doxorubicin, methotrexate, topotecan and PTX [33]. This means that these new-generation taxoids can promote differentiation of the treated CSCs by suppressing the expression of “stemness genes”. This finding has added a new mechanism of action (MOA) to taxane anticancer agents whose major MOA is the blocking of the cell mitosis at the G2/M stage, leading to apoptosis [12]. Another 2nd-generation DFV-taxoid, DFV-ortataxel (DFV-OTX) (Fig. 1) [34], has shown promising efficacy against MDA-MB-231R taxane-resistant human breast tumor xenografts, overexpressing ABCB1/ABCG2 efflux pumps. DFV-OTX is a DFV analog of ortataxel (OTX) which was derived from 14β-baccatin III [35] and advanced to phase II clinical trials in humans [36].
The 3-position (meta position) of the C2-benzoate moiety in PTX was identified as one of the metabolism sites by cytochrome C P450 [37], as well as the site for attractive interaction with His229 in β-tubulin, enhancing the binding affinity of PTX [38,39] and taxoids [29,40]. Our SAR studies have disclosed that the modifications of this position with halogens (F, Cl), azide and methoxy groups substantially enhance the potency of 2nd-generation taxoids against MDR-cancer cell lines. This finding led to the development of 3rd-generation fluorotaxoids [28,31].
2. 3rd-generation fluorotaxoids bearing 3-CF3O and 3-CHF2O groups at the C2-benzoate position [41]
Fluorine substitution in bioactive molecules not only exerts significant effects on their binding conformations through dipolar, van der Waals and H-bonding interactions, but also on the intrinsic conformation of a molecule which would influence its binding to the target [42].
Unique effects of fluorine substitution on conformation are clearly observed in the comparison of the OCF3 group in trifluoroanisole with the OCH3 group in anisole. Although the methoxy group in anisole is preferred to be coplanar with the phenyl ring, the OCF3 group in trifluoroanisole has a strong preference to be out of the plane [2,43–45]. The conformational preference of the OCHF2 group in Ar-OCHF2 has turned out to be between the planar and the orthogonal states and thus it acts as a bioisosteric replacement for the OCH3 or OCF3 moiety, depending on its conformation [2,43,46]. Due to the unique non-spherical feature of the OCHF2 group, either one or both C-F bonds of Ar-OCHF2 can be in the anomeric position to take the endo-exo or endo-endo conformation as indicated in their X-ray crystal structures [46]. The polarity of the C-F bond is largely cancelled out in the endo-endo conformation in contrast to the three times higher polarity in the endo-exo conformation. Thus, Ar-OCHF2 could be more (endo-endo) or less (endo-exo) lipophilic than their Ar-OCF3 counterparts [2,43,46]. This unique characteristic of the OCHF2 group to different conformations in polar and nonpolar environments can provide unconventional property combinations, e.g., good aqueous solubility,cellular permeability, and low lipophilicity [46]. In addition, the matched molecular pair (MMP) analysis indicated that the OCHF2 group might be superior to the OCH3 and OCF3 groups for optimizing ADME properties, e.g., metabolic stability and passive transmembrane permeability [43]. Furthermore, the multipolar interactions of fluorine atoms with the electrophilic carbon of nearby protein amide carbonyl groups in protein-ligand binding were found to be highly significant, which is supported by the analysis of large numbers of crystal structures and biological activity data [2]. The significance of the multipolar interactions of fluorine provides a rationale for the strategic incorporation of fluorine in lead optimization [47].
Four 3rd-generation taxoids, bearing a 3-CF3O-benzoate moiety at C2, were initially examined for their potency against an extremely PTX-resistant breast cancer cell line, MCF-7/PTX, together with PTX and SBT-1214 (1c) (Fig. 1) [48]. It was found that SBT-121205 (1a-05) (Fig. 2) possessed high potency (IC50 19.0 nM), which is 121 times more potent than PTX (IC50 2,291 nM) and 4 times more potent than 1c (IC50 80.5 nM) [48]. Moreover, the MOA study of 1a-05 revealed the effective suppression of the PI3k/Akt pathway and epithelial-mesenchymal transition (EMT) by activating PTEN tumor suppressor expression [48]. This new and significant finding on MOA along with a high potency against CSCs by effectively suppressing the stemness gene expressions prompted us to investigate a series of new 3rd-generation fluorotaxoids [41].
Fig. 2.
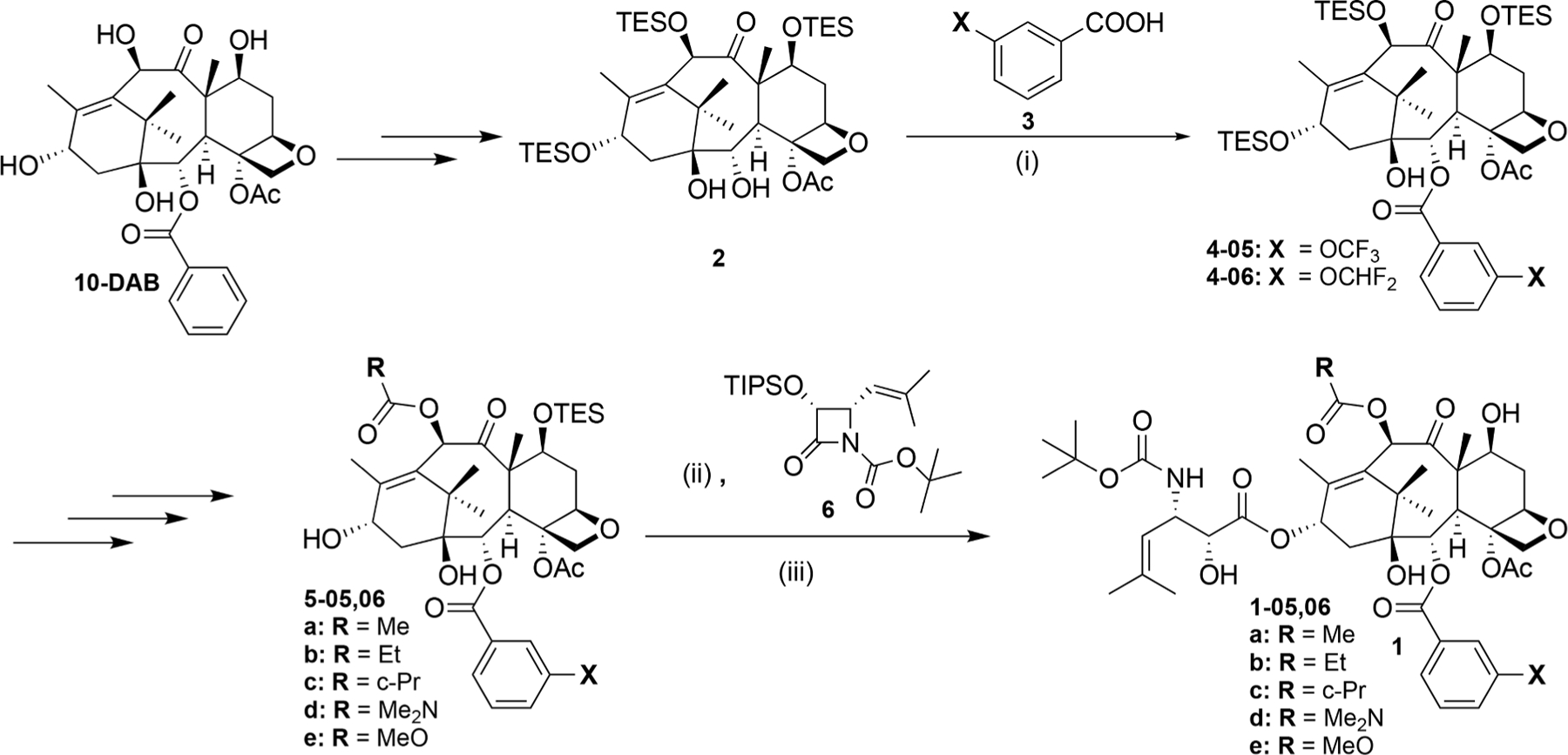
Synthesis of novel 3rd-generation fluorotaxoids bearing 3-OCF3 or 3-OCF2H at the C2-benzoate moiety. (i) 3-CF3O or 3-CHF2O-benzoic acid, DIC, DMAP, CH2Cl2, r.t; ii) LiHMDS, THF, −40°C; iii) HF/pyridine, pyridine/MeCN, 0°C-r.t.
A series of new fluorotaxoids, bearing a 3-OCF3 or 3-OCF2H group in the C2-benzoate moiety, was designed and synthesized, using the procedures and protocols developed for the 3rd-generation taxoids, starting from 10-deacetylbaccatin III (10-DAB) by means of the β-lactam synthon method [41,49] (Fig. 2).
2.1. Cytotoxicity of 3rd-generation fluorotaxoids in cancer cells [41]
The 3rd-generation fluorotaxoids were evaluated against various drug-sensitive and drug-resistant cancer cell lines for their potencies as shown in Table 1, wherein PTX and SBT-1214 (1c) were also included for comparison.
Table 1.
Cytotoxicity (IC50 nM) of 3rd-generation fluorotaxoids.
| Taxoid | MCF-7 | NCI/ADR | LCC6-WT | LCC6-MDR | DLD-1 | CFPAC- |
|---|---|---|---|---|---|---|
| PTX | 0.9 | 130 | 1.81 | 619 | 364 | 60 |
| SBT-1214 (1c) | 0.74 | 0.88 | 0.91 | 3.19 | 3.4 | 0.71 |
| 1a-05 | 0.98 | 0.83 | 0.51 | 2.20 | 3.0 | 0.57 |
| 1b-05 | 0.37 | 0.43 | 0.71 | 2.40 | 3.1 | 0.75 |
| 1c-05 | 0.56 | 0.42 | 0.58 | 2.11 | 3.2 | 0.79 |
| 1d-05 | 0.28 | 0.66 | 0.74 | 2.15 | 3.3 | 0.69 |
| 1e-05 | 0.32 | 0.66 | 0.85 | 1.96 | 3.1 | 0.51 |
| 1a-06 | 0.34 | 0.45 | 0.42 | 2.01 | 2.9 | 0.48 |
| 1b-06 | 0.60 | 0.26 | 0.56 | 1.64 | 3.0 | 0.42 |
| 1c-06 | 0.37 | 0.36 | 0.45 | 1.80 | 3.1 | 0.39 |
| 1d-06 | 0.56 | 0.42 | 0.40 | 1.77 | 2.9 | 0.44 |
| 1e-06 | 0.41 | 0.39 | 0.67 | 1.75 | 3.0 | 0.49 |
MCF7: human breast cancer cell line.
NCI/ADR: drug-resistant (Pgp+) human ovarian cancer cell line.
LCC6-WT: human breast cancer cell line.
LCC6-MDR: drug-resistant (Pgp+) human breast cancer cell line.
DLD-1: drug-resistant (Pgp+) human colon cancer cell line.
CFPAC-1: human pancreatic cancer cell line.
As Table 1 shows, all 3rd-generation fluorotaxoids possess two orders of magnitude greater potency than PTX against different drug-resistant human cancer cell lines, NCI/ADR (ovarian), LCC6/MDR (breast), and DLD-1 (colon), which clearly indicates that these fluorotaxoids can overcome MDR caused by the overexpression of Pgp and other ABC cassette transporters [41]. Also, all these fluorotaxoids exhibit subnanomolar IC50 values against MCF7 and LCC6-WT drug-sensitive breast cancer cell lines, NCI/ADR drug-resistant ovarian cancer cell line, as well as CFPAC-1 pancreatic cancer cell line, which is moderately resistant to PTX.
2.2. Dose-response analysis of 3rd-generation fluorotaxoids in drugresistant cancer cell lines [41]
PTX is essentially inactive against NCI/ADR, LCC6-MDR, and DLD-1 drug-resistant cancer cell lines (IC50 130 nM, 619 nM and 364 nM respectively, Table 1, Fig. 3). The 3rd-generation fluorotaxoids possess significantly higher potency (Fig. 3) than PTX and better dose-response than 2nd-generation taxoid 1c. It is noteworthy that 10% of cells were still alive in the NCI/ADR and LCC6-MDR cell lines after 100 nM PTX treatment, while only <3% of cells were alive in the NCI/ADR and LCC6-MDR cell lines after 12.5 nM of 3rd-generation fluorotaxoids treatment. In the NCI/ADR drug-resistant ovarian cancer cell line, 1d-05, 1e-05, 1d-06 and 1e-06 are highly potent (< 10% cell viability) at 2.5 nM concentration and especially, 1d-06 and 1d-05 display impressive dose-response (kill) curves as compared to that of PTX and 1c. In the LCC6-MDR drug-resistant breast cancer cell line, taxoids 1c-05, 1d-05, 1e-05, and 1a-06 are highly potent (< 15% cell viability) at 2.5 nM concentration and show impressive dose-response (kill) curves as compared to those of PTX and 1c. In the DLD-1 drug-resistant colon cancer cell line, 80% of the cancer cells were alive at 12.5 nM PTX concentration, while less than 10% of cancer cells were alive after treatment with 3rd-generation fluorotaxoids at the same concentration. Furthermore, in DLD-1 drug resistant colon cancer cell line, taxoids 1a-06, 1d-06, and 1e-06 possess high potency (< 15% cell viability) at 5 nM concentration, wherein 1d-06 and 1e-06 exhibit the most impressive dose-response (kill) curves as compared to that of PTX and 1c. Interestingly, the 1–05 series of taxoids did not show distinct dose response (kill) curves compared to that of 1c against this drug-resistant cell line.
Fig. 3.
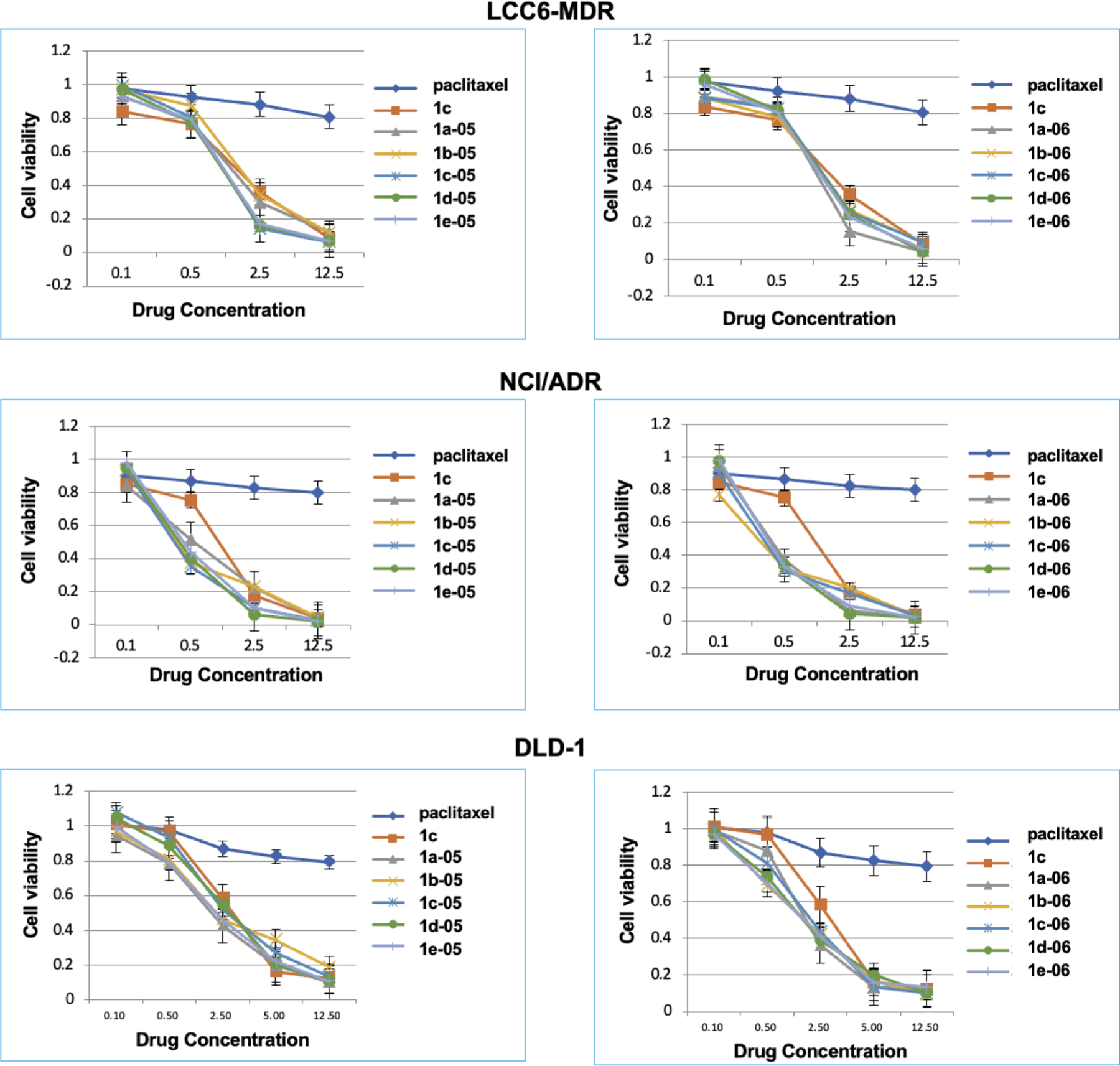
Dose-response curves of new 3rd-generation fluorotaxoids in LCC6-MDR breast cancer, NCI/ADR ovarian cancer and DLD-1 colon cancer cell lines. Adapted from Ref. [41] with permission.
2.3. Potencies of 3rd-generation fluorotaxoids against CSC-enriched colon and patient-derived prostate cancer cells [41]
The potency of selected 3rd-generation fluorotaxoids was evaluated against a CSC-enriched colon cancer cell line, HCT116CSC, and a patient-derived prostate CSCs, PPT2, which express a high level of CD133, CD44, CD44v6, EpCAM, CD49f, and CD166 genes. Since CSC-enriched cancer cells with CD133+ and CD44+ are highly resistant to traditional chemotherapeutic drugs, it is important to examine the potency of select 3rd-generation fluorotaxoids against those cells. As HCT116CSC formed 3D spheroids, those cells did not attach firmly to the bottom of the wells. However, after 72 h treatment with 1d-05, 1c-05, and 1c at 10 μM concentration, much fewer floating 3D spheroids were observed as compared to the control. The cell death rate with 10 μM of 1d-05 and 1c-05 (72 h) was found to be appreciably higher than that of cabazitaxel which is the most potent taxane approved by the FDA (Fig. 1). After treatment with select 3rd-generation fluorotaxoids, 32–40% of HCT116CSC cells survived. However, those survived cells showed distinct morphological abnormalities, e.g., enlarged size, severe vacuolization, knobby projections, prolonged size, and multiple nuclei, which are characteristic of seriously damaged CSCs, losing pluripotency and ability to form secondary spheroids, as reported previously for 1c [50].
In the patient-derived prostate CSCs PPT2, 1b-06 exhibited the best potency (IC50 23 nM) among the twelve fluorotaxoids examined, wherein the order of potency was 1–06 (CF2HO) > 1–05 (CF3O) in all variations at C10 at 50 nM and 250 nM concentrations. Overall, 1e-06 exhibited the best potency among all the fluorotaxoids examined, which caused 62% cell death at 50 nM concentration and 78% cell death at 250 nM concentration.
2.4. Molecular modeling analysis of new 3rd-generation taxoids [41]
The molecular docking analysis of 1a-05 and 1a-06 with β-tubulin indicated that the OCF3 and OCF2H groups of 1a-05 and 1a-06 make favorable van der Waals interactions with hydrophobic amino acid residues, Leu230 and Leu275, in the proximal binding site of β-tubulin (Fig. 4). Interestingly, the 1–05 and 1–06 series of fluorotaxoids orient the OCF3 and OCF2H groups to the proximal site, while the corresponding 3rd-generation taxoids with 3-Me- and 3-MeO-benzoate groups place the Me and OMe groups to the distal binding site of β-tubulin. This finding indicates the presence of favorable interactions with OCF3/OCF2H groups over Me/OMe groups in the proximal site, which was supported by a clear difference in docking energy scores [41].
Fig. 4.
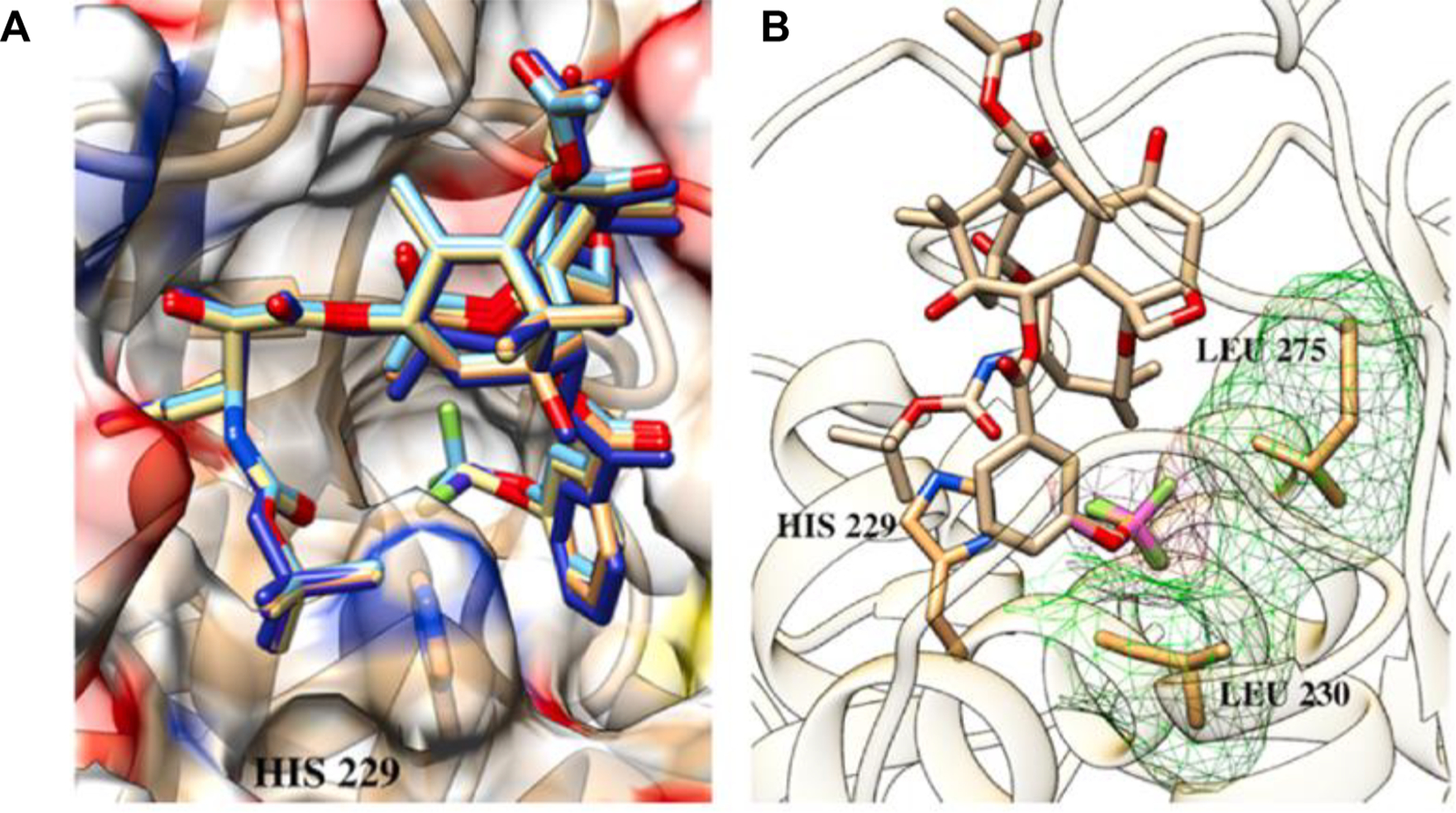
A: Overlay of 1a-05 and 1a-06 in the proximal binding site of β-tubulin; B: Favorable van der Waals interactions of the OCF3 and OCF2H residues in the proximal binding site (Overlay of 1a-05 and 1a-06 as an example). Adapted from Ref. [41] with permission.
In summary, new 3rd-generation fluorotaxoids, bearing 3-OCF3 or 3-OCF2H at the C2 benzoate moiety, exhibited higher potency than PTX against drug-sensitive cancer cell lines and drug-resistant cancer cell lines. Furthermore, the 1–06 series of taxoids, bearing an OCHF2 group at the C2 benzoate position, exhibited the highest potencies against MDR-cancer cell lines and CSC-enriched cancer cell line, HCT-116CSC (colon), as well as patient-derived PPT2 CSCs (prostate). Molecular modeling analysis further confirmed the benefit of having 3-OCF3 or 3-OCF2H groups at the C2 benzoate position through favorable van der Waals interactions of OCF3 and OCF2H groups with hydrophobic amino acid residues in the β-tubulin binding site.
3. 3rd-generation DFV-taxoids bearing 3-OCF3 or 3-OCF2H group at the C2-benzoate moiety and DFV group at the C3′ position [33]
The metabolism study of 2nd-generation taxoids revealed that a major human metabolic enzyme, CYP 3A4, of the cytochrome P450 family [40], metabolized these taxoids through hydroxylation primarily at the two allylic methyl groups of the 3′-isobutenyl moiety [51]. Thus, the replacement of the isobutenyl group at the C3′ position of 2nd-generation taxoids with a 2,2-difluorovinyl (DFV) group successfully blocked the hydroxylation of allylic methyl groups, as well as further oxidations of the taxoids [29,51]. Computational analysis of 3′-DFV-taxoids in the taxane binding site of β-tubulin indicated that the 2,2-DFV group would nicely mimic an isobutenyl group despite the difference in size and electronic nature between the two groups [32].
Based on the observed highly beneficial effects of the DFV group at the C3′ position [24] and the 3-OCF3/OCHF2 groups at the C2-benzoyl moiety in the 2nd- and 3rd-generation taxoids [41], it is natural to assume that some extraordinarily potent taxane anticancer agents could be developed through the combination of these two modifications in the 3rd-generation taxoids. As anticipated, novel 3rd-generation DFV-taxoids with a 3-CF3O-/CHF2O-benzoate moiety at the C2 position, indeed, exhibited a remarkable activity against drug-sensitive and drug-resistant human cancer cell lines, which are superior to the 2nd-generation DFV-taxoids, as well as the 3rd-generation fluorotaxoids bearing a 3-CF3O/CHF2O-benzoyl moiety at the C2 position [33].
The novel 3rd-generation DFV-taxoids were synthesized using the procedures in a manner similar to those employed for the synthesis of 3rd-generation fluorotaxoid described above, by means of the β-lactam synthon method [49] through the Ojima-Holton coupling of 2,10-modified baccatins with (3S,4R)-1-t-Boc-3-TIPSO-4-DFV-β-lactam 9 (Fig. 5) [33].
Fig. 5.
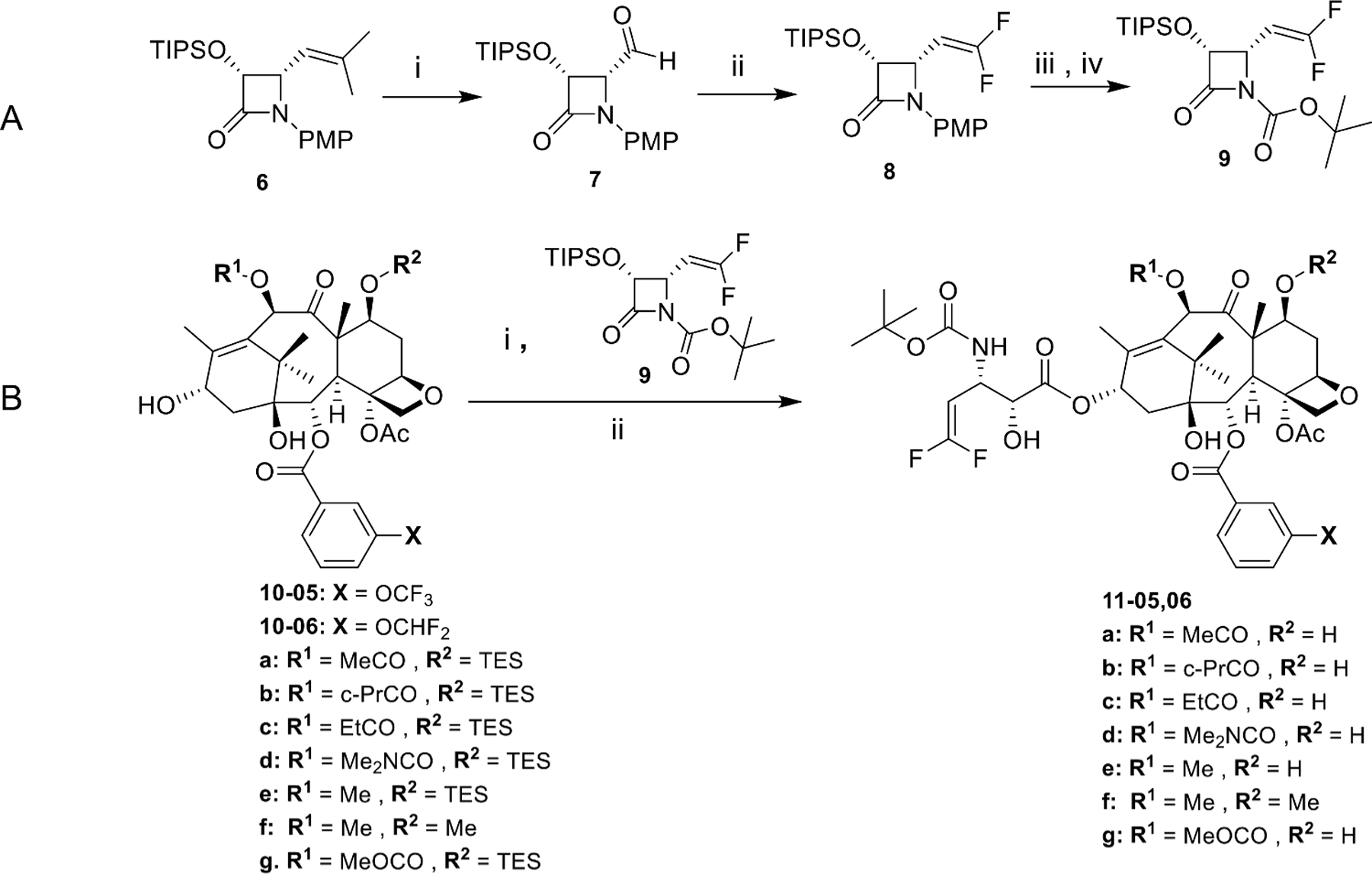
Synthesis of 3rd-generation DFV-taxoids bearing 3-OCF3 or 3-OCF2H at the C2-benzoate moiety. A: (i) O3, MeOH/CH2Cl2, −78°C, Me2S; ii) ClF2CCO2Na, Ph3P, DMF, 90°C; iii) CAN, MeCN/H2O, −10°C; iv) Boc2O, Et3N, DMAP, CH2Cl2, r.t; B: (i) LiHMDS, THF, −40°C; ii) HF/pyridine, pyridine/MeCN, 0°C-r.t.
3.1. Cytotoxicity of new 3rd-generation DFV-taxoids [33]
The cytotoxicity of the 3rd-generation DFV-taxoids was evaluated and summarized in Table 2. All the 3rd-generation DFV-taxoids exhibit remarkable cytotoxicity against human breast (MCF7, LCC6-MDR), lung (A549), colon (HT29, DLD-1), pancreatic (PANC-1), and prostate (Vcap, PC3) cancer cell lines, with subnanomolar IC50 values against drug-sensitive cell lines, A549, HT29, Vcap, and PC3, as well as PANC-1. Furthermore, these taxoids possess 2–4 orders of magnitude greater potency against extremely drug-resistant cancer cell lines, LCC6-MDR and DLD-1, as compared to PTX, indicating that these new DFV-fluorotaxoids can overcome MDR caused by the overexpression of Pgp and other ABC cassette transporters. New DFV-fluorotaxoids bearing a 3-OCHF2 group at the C2-benzoate moiety (11–06 series) possess slightly higher cytotoxicity than those bearing a 3-CF3O-benzoate group at C2 (11–05 series). It should be noted that 11f-06, a DFV-mimic of cabazitaxel, exhibited the highest potency against Vcap, MCF7, PANC-1, and PC3 cancer cell lines, as well as the second highest potency (IC50 89 pM) against LCC6-MDR.
Table 2.
Cytotoxicity (IC50 nM) of 3rd-generation DFV-taxoids.
| Taxoid | A549 | HT29 | Vcap | PC3 | MCF7 | PANC-1 | DLD-1 | LCC6-MDR |
|---|---|---|---|---|---|---|---|---|
| PTX | 1.94 | 2.59 | 4.52 | 3.05 | 2.94 | 2.89 | 428 | 503 |
| Docetaxel | 0.42 | 0.65 | 1.05 | 0.94 | 0.50 | 0.55 | ND | 118 |
| SBT-1214 | 0.28 | 0.40 | 0.40 | 0.48 | 0.20 | 0.33 | 4.00 | 2.59 |
| 11a-05 | 0.14 | 0.22 | 0.39 | 0.45 | 0.23 | 0.41 | 0.168 | 0.163 |
| 11b-05 | 0.22 | 0.38 | 0.41 | 0.44 | 0.14 | 0.27 | 0.079 | 0.951 |
| 11c-05 | 0.17 | 0.28 | 0.37 | 0.33 | 0.12 | 0.34 | 0.564 | 0.565 |
| 11d-05 | 0.26 | 0.32 | 0.42 | 0.29 | 0.18 | 0.62 | 0.211 | 0.490 |
| 11e-05 | 0.17 | 0.34 | 0.26 | 0.22 | 0.07 | 0.08 | 0.073 | 0.644 |
| 11f-05 | 0.30 | 0.22 | 0.22 | 0.23 | 0.15 | 0.33 | 0.089 | 0.132 |
| 11g-05 | 0.42 | 0.34 | 0.50 | 0.37 | 0.20 | 0.63 | 0.110 | 0.771 |
| 11a-06 | 0.14 | 0.15 | 0.23 | 0.22 | 0.08 | 0.22 | 0.177 | 0.904 |
| 11b-06 | 0.10 | 0.18 | 0.27 | 0.39 | 0.10 | 0.43 | 0.34 | 0.392 |
| 11c-06 | 0.12 | 0.21 | 0.20 | 0.22 | 0.08 | 0.29 | 0.093 | 0.248 |
| 11d-06 | 0.14 | 0.24 | 0.32 | 0.26 | 0.15 | 0.18 | 0.242 | 0.816 |
| 11e-06 | 0.19 | 0.24 | 0.36 | 0.25 | 0.13 | 0.19 | 0.073 | 1.280 |
| 11f-06 | 0.25 | 0.21 | 0.21 | 0.05 | 0.06 | 0.03 | 0.159 | 0.089 |
| 11g-06 | 0.10 | 0.24 | 0.34 | 0.26 | 0.13 | 0.43 | 0.024 | 0.034 |
A549: Human lung (NSCL) cancer cell line.
HT29: Human colon cancer cell line.
Vcap: Human prostate cancer cell line.
PC3: Human metastatic prostate cancer cell line.
MCF7: Human breast cancer cell line.
PANC-1: Human pancreatic cancer cell line.
DLD-1: MDR (Pgp +) human colon cancer cell line.
LCC6-MDR: MDR (Pgp +) human breast cancer cell line.
3.2. Dose-response analysis of 3rd-generation DFV-taxoids in drugresistant cancer cell lines [33]
As discussed in Section 2.1, PTX is essentially ineffective (IC50 503 nM) against the LCC6-MDR drug-resistant breast cancer cell line since the 20 nM PTX treatment only killed 20% of the cancer cells. In contrast, less than 10% of cancer cells were viable after the 20 nM treatment with any of the 3rd-generation DFV-taxoids (Fig. 6), and all of the new DFV-taxoids exhibit impressive dose-response (kill) curves as compared to PTX and 1c. Among all 3rd-generation DFV-taxoids, 11f-06 and 11g-06 display the most effective dose-responses.
Fig. 6.
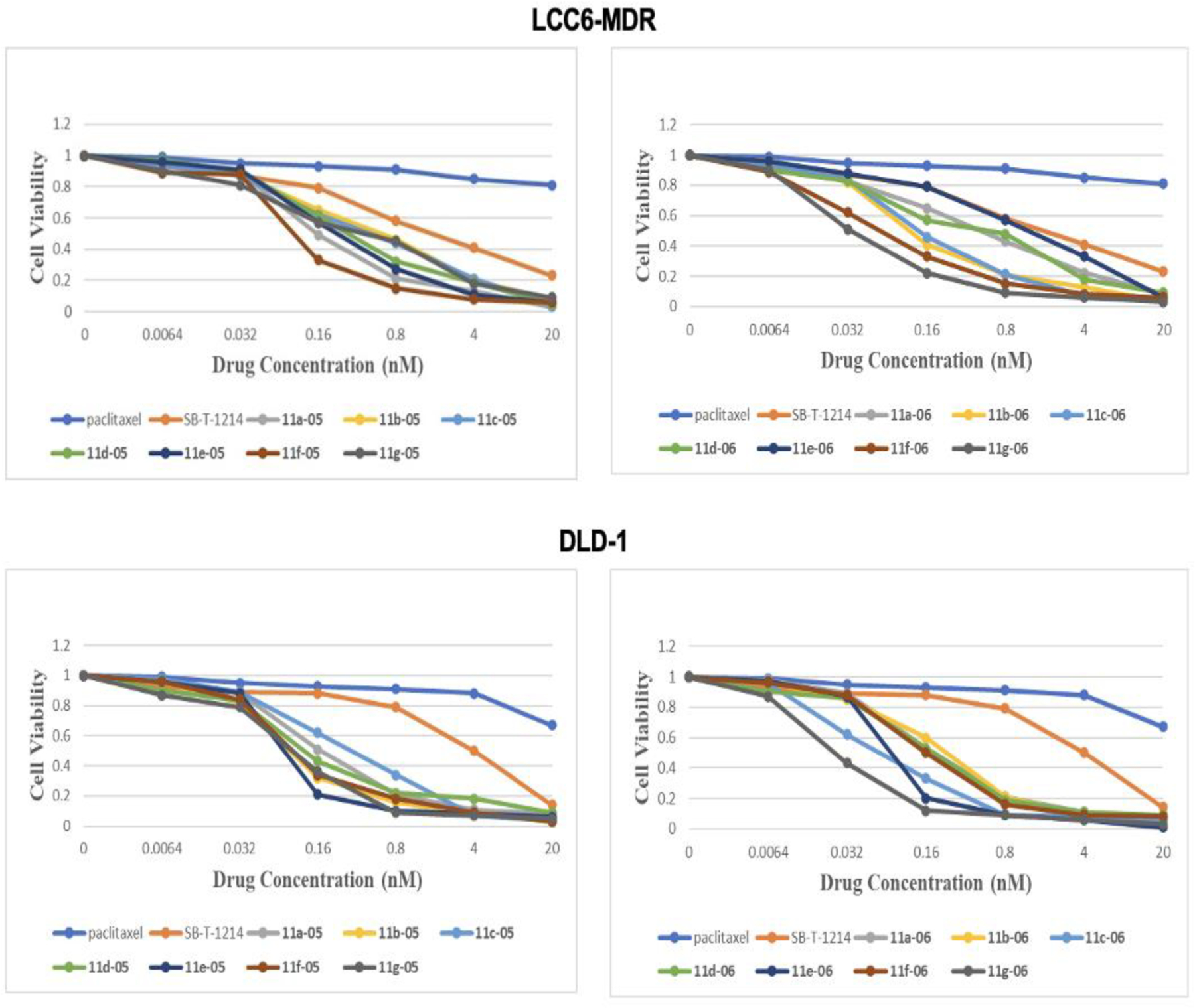
Dose-response of new 3rd-generation fluorotaxoids in LCC6-MDR breast cancer and DLD-1 colon cancer cells. Adapted from Ref. [33] with permission.
PTX is again essentially ineffective (IC50 428 nM) against the DLD-1 drug-resistant colon cancer cell line, wherein 90% and 50% of DLD-1 cells were viable at 4 nM doses of PTX and 1c, respectively (Fig. 6). In sharp contrast, less than 10% of cancer cells were viable at 4 nM dose for most of the 3rd-generation DFV-taxoids. Among all of the new DFV-taxoids, 11e-05 and 11g-06 exhibited the most effective dose-responses. Notably, 11g-06 was able to kill ca. 90% of the cancer cells even at 0.16 nM dose and eradicated the cancer cells at 20 nM dose.
3.3. Molecular modeling analysis of 3rd-generation DFV-taxoids [33]
The molecular docking analysis of 3rd-generation DFV-taxoids bound to β-tubulin shows that the 3′-DFV moiety and the 3-OCF3/3-OCHF2 group of the C2-benzoate moiety are nicely accommodated in the proximal binding site in β-tubulin. In the case of the most potent compounds, 11e-05 and 11f-06, one of the most notable features is the attractive interactions of fluorine substituents with the aromatic π-system of Phe272 and the H-bonding of the exo-fluorine of the DFV group with the hydroxyl group of Ser236 (Fig. 7A, B) [33,52]. In addition, the OCF3/OCHF2 moiety makes favorable van der Waals contacts with hydrophobic residues, Leu217, Leu230, and Leu275, in the proximal binding site in the same manner as that of 3rd-generation fluorotaxoids mentioned above (Section 2.4.) [33,41,53]. It should be noted that the 3-OCF3 group of 11f-05 is shown to have a unique H-bonding interaction with (π)N3-H of His229 (OF2C-F—H-N) (Fig. 7C) [33,44,52], while the 3-OCHF2 group of 11f-06 is oriented to form another type of H-bond to the (π)N3 of His229 (OF2C-H—N) (Fig. 7D) [33,54], by taking advantage of facile tautomerization in the imidazole moiety of His229. These unique H-bonding interactions clearly enhance the affinity of these two DFV-fluorotaxoids to β-tubulin. In spite of the difference in size between fluorine and methyl, the DFV group mimics the binding mode of the isobutenyl group of the 2nd- and 3rd-generation taxoids. Moreover, the smaller DFV group is positioned in closer proximity to Phe272 and Ser236 than the isobutenyl group, which enables the attractive interaction of fluorine with the aromatic π-system, enhancing the affinity [33].
Fig. 7.
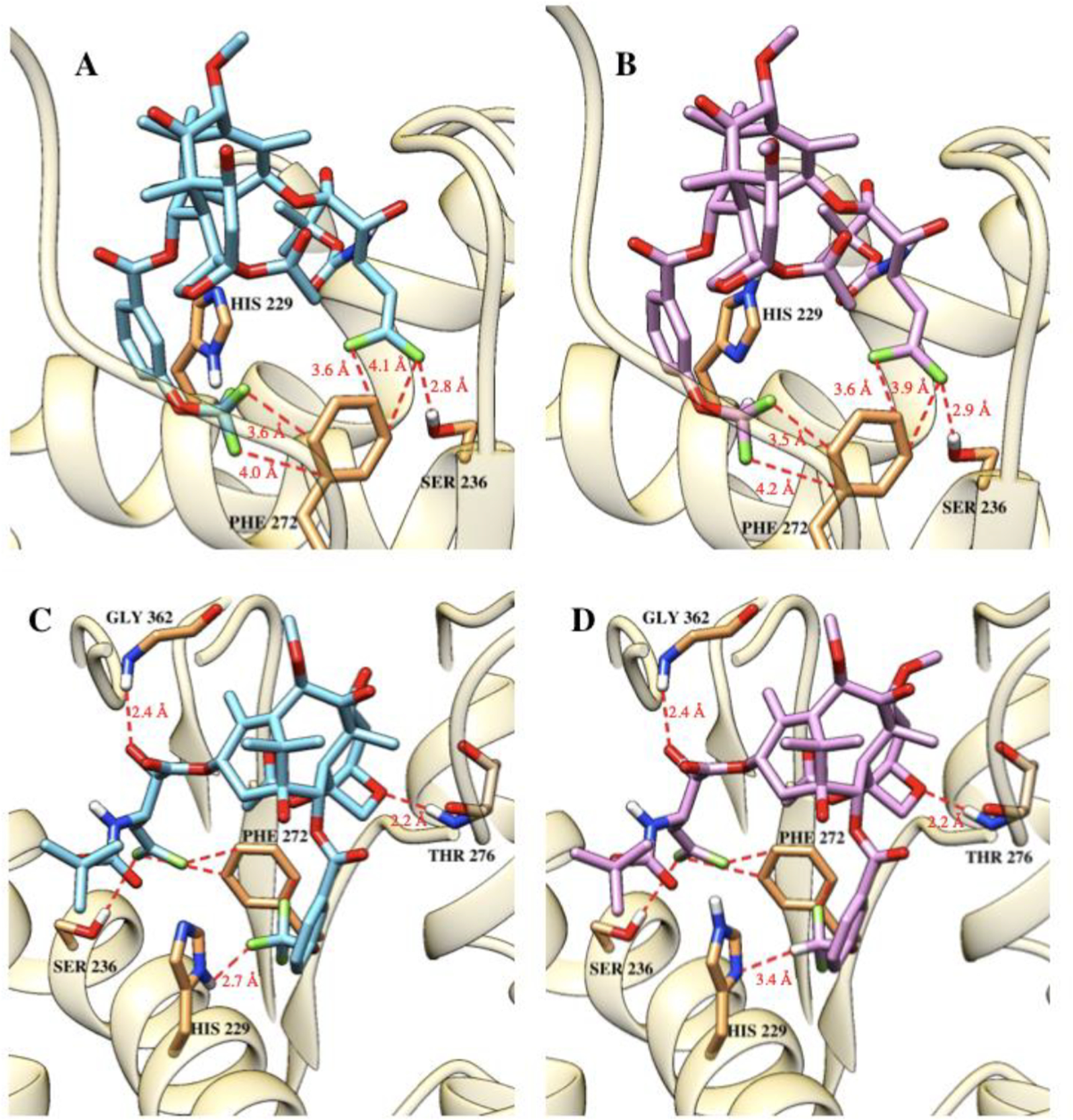
3rd-Generation DFV-taxoids in the β-tubulin binding site. The fluorine interactions of the DFV group with the π-system of Phe272 and H-bonding with Ser236 for (A) 11e-05 and (B) 11f-06. H-bonding interactions of the OCF3 group of 11e-05 (C) and the OCF2H group of 11f-06 (D) with His229. Adapted from Ref. [33] with permission.
Consequently, we have identified attractive interactions between 3rd-generation DFV-taxoids and β-tubulin through OF2C-F—HN, OF2C-H—N and CH=C(F)-F—HO-R H-bonding, as well as of C-F—Ar π-system in this molecular docking analysis, as shown in Fig. 8. These unique attractive interactions involving fluorine collectively enable an enhanced binding of the 3rd-generation DFV-taxoids with β-tubulin beyond those of 1st-generation taxane drugs (i.e., PTX and docetaxel), cabazitaxel and new-generation taxoids without bearing organofluorine groups. These unique features are reflected in the remarkable potency of novel DFV-fluorotaxoids 11–05 and 11–06.
Fig. 8.
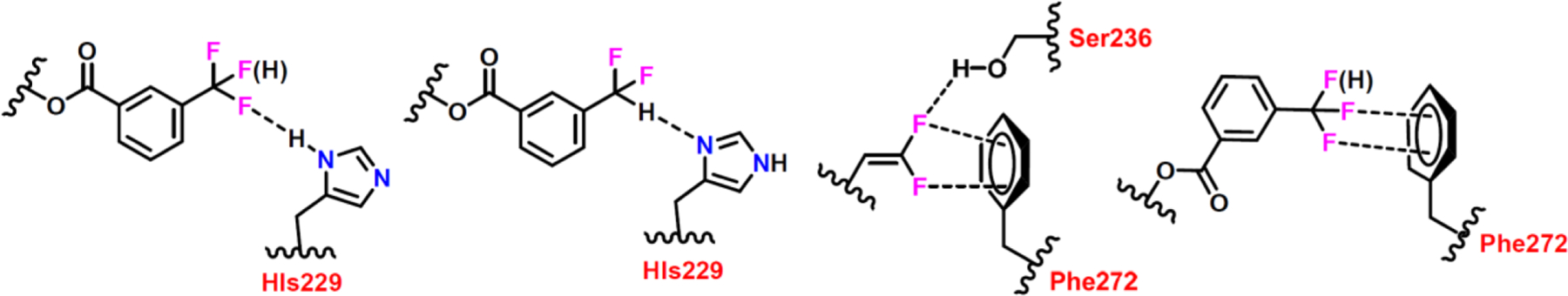
Unique attractive interactions of 3rd-generation DFV-taxoids, involving OF2C-F—HN, OF2C-H—N and CH=C(F)-F—HO-R H-bonding, as well as C-F—Ar π-system, in the β-tubulin binding site.
3.3.1. Current understanding of C-F—Ar π-system interactions
The stabilizing interaction between halogens and aromatic groups (C-X—π) has been discussed in the literature over the past two decades and more intensively in the last several years. In 2016, Holl et al. reported that C-F—π interactions can bring >1,500-fold increase in the relative rate of an electrophilic substitution reaction by promoting activation of the aromatic rings [53]. Traditionally, fluorine as an aromatic substituent is deactivating for electrophilic aromatic substitution reactions based on the inductive effect. However, Holl et al. observed a through-space stabilization of the Wheland intermediate that ultimately increased the nitration reaction rate (Fig. 9A). A non-covalent interaction (NCI) analysis predicted a slightly stabilizing interaction between the fluorine and arene moieties [55]. Finally, ab initio calculations of the complexes predicted greater stabilizing interactions when the fluorine was directed toward the arene, as compared to away from the arene or without a fluorine present (Fig. 9B).
Fig. 9.
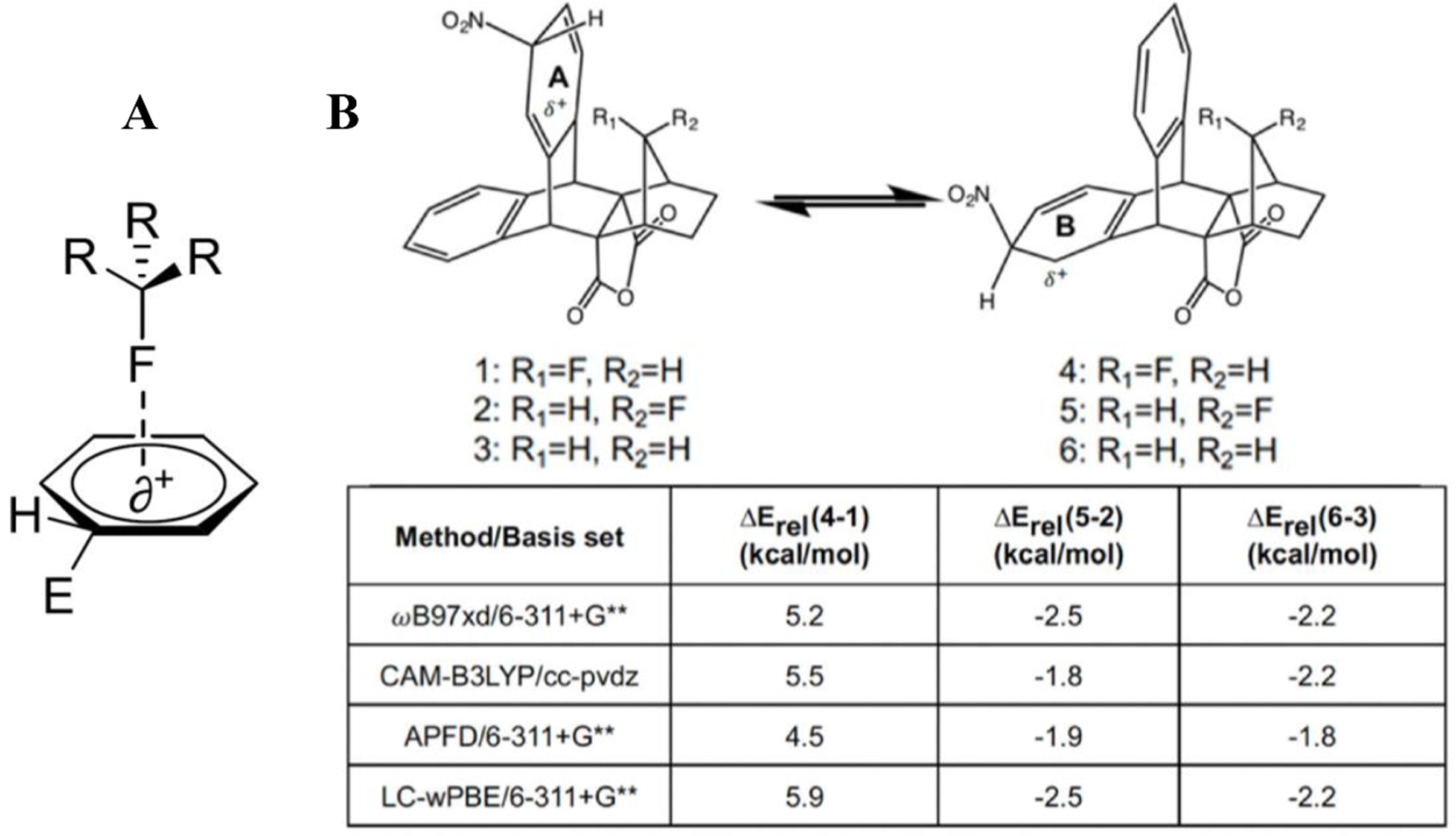
(A) Activation of arene ring by perpendicular through-space electron-donating fluorine interaction, as opposed to traditional deactivation by covalently bound fluorine. (B) Ab initio results indicating that the fluorine substituent stabilizes the Wheland intermediate. Adapted from Ref. [53] with permission.
The C-F—π interactions have also been structurally resolved in a biochemical context. In 2003, Saraogi and co-workers reported a statistical investigation on the role of C-X—π interaction in protein structures using the Brookhaven Protein Data Bank, analyzing halogen interactions with histidine, phenylalanine, tyrosine and tryptophan [56]. The probability for the formation of a C-X—π interaction was higher in the case of fluorine compared to other halogens. Additionally, the average interaction distance between the halogen and the aromatic ring was very close to the van der Waals radius, hinting that the size of fluorine may play an important role in the interaction. Although the sample size was not statistically significant, the authors noted a directional preference for the C-X—π interaction that was later confirmed in computational work [57]. In 2004, Kawahara et al. observed a dependence of the interaction energy on the electron deficiency of the aromatic system through ab initio potential energy scans of the interaction coordinate [58]. Notably, Kawahara et al. also noticed an underestimation of attractive interactions and an overestimation of repulsive interactions with smaller basis sets. The authors concluded that the C-F—π interaction involving benzene as the aromatic component was very weakly repulsive, while the hexafluorobenzene resulted in an attractive interaction. The authors noted that for each interaction studied, the electron-correlation energy was the largest contributor to the overall interaction energy. Lastly, the electron repulsion term was much smaller in the hexafluorobenzene calculations than in the benzene calculations, indicating that the relative electropositive nature of the hexafluorobenzene ring, and subsequent increase in the “hole” for halogen interaction, is an important part of the C-F—π interaction (Fig. 10).
Fig. 10.
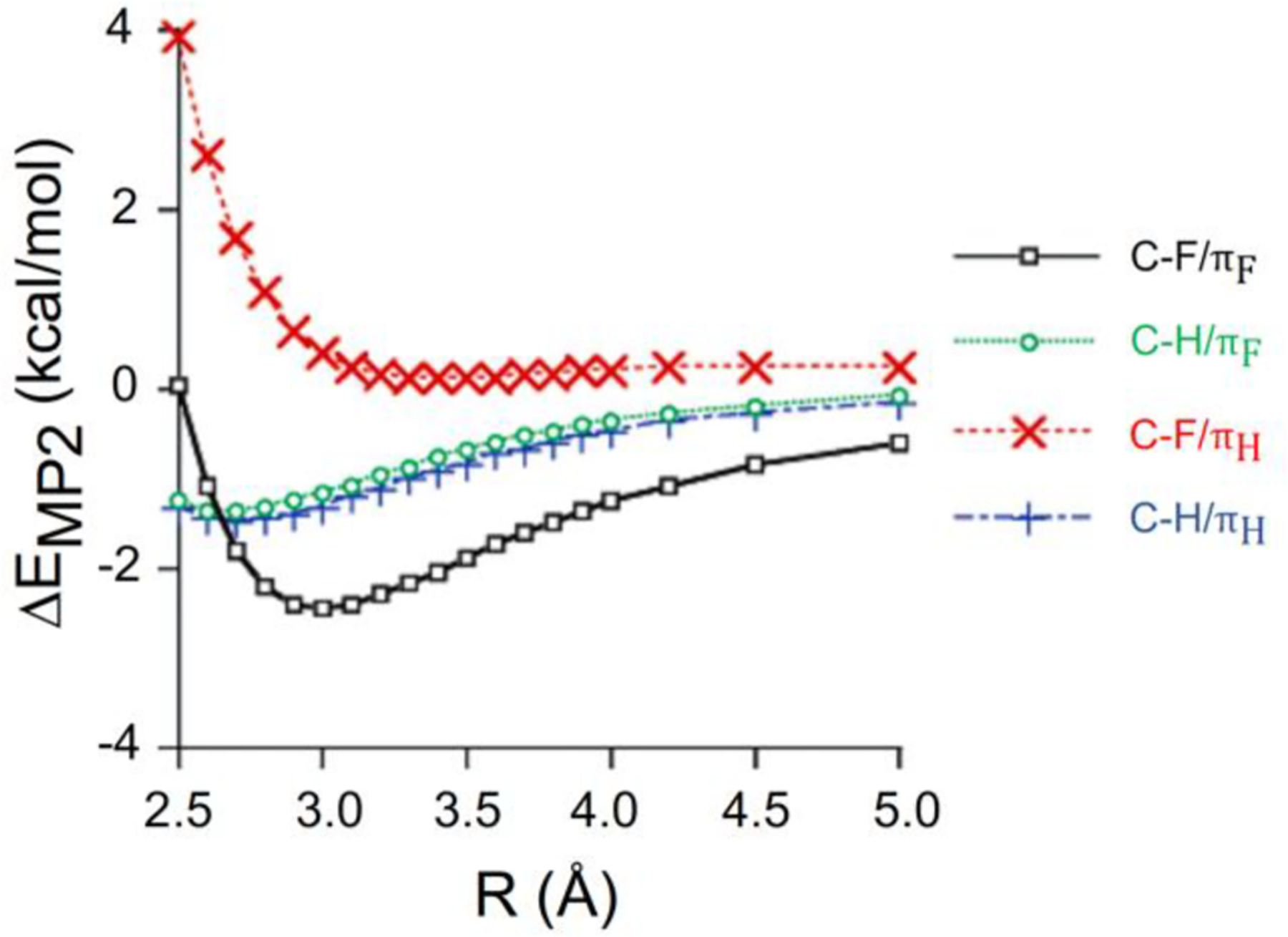
Ab initio potential energy scans of benzene (πH) and hexafluorobenzene (πF) interacting with C-H and C-F, respectively. Adapted from Ref. [58] with permission.
In 2013, Esrafili et al. employed in silico methods to describe the properties and strength of the C-X—π interactions within the complexes (X=Cl, Br; Y=H, CN, F, Cl, OH, NH2, CH3) (Fig. 11) [57]. The physical nature of the interactions was studied using symmetry-adapted perturbation theory (SAPT). The stability of the C-X—π interactions was predicted to be attributable mainly to electrostatic and dispersion effects. Even though they did not include fluorine as a halogen substituent for their studies, their natural bond order (NBO) analysis suggested the significance of the π→σ*C—X orbital interaction for the stability of the C-X—π bonded complexes.
Fig. 11.
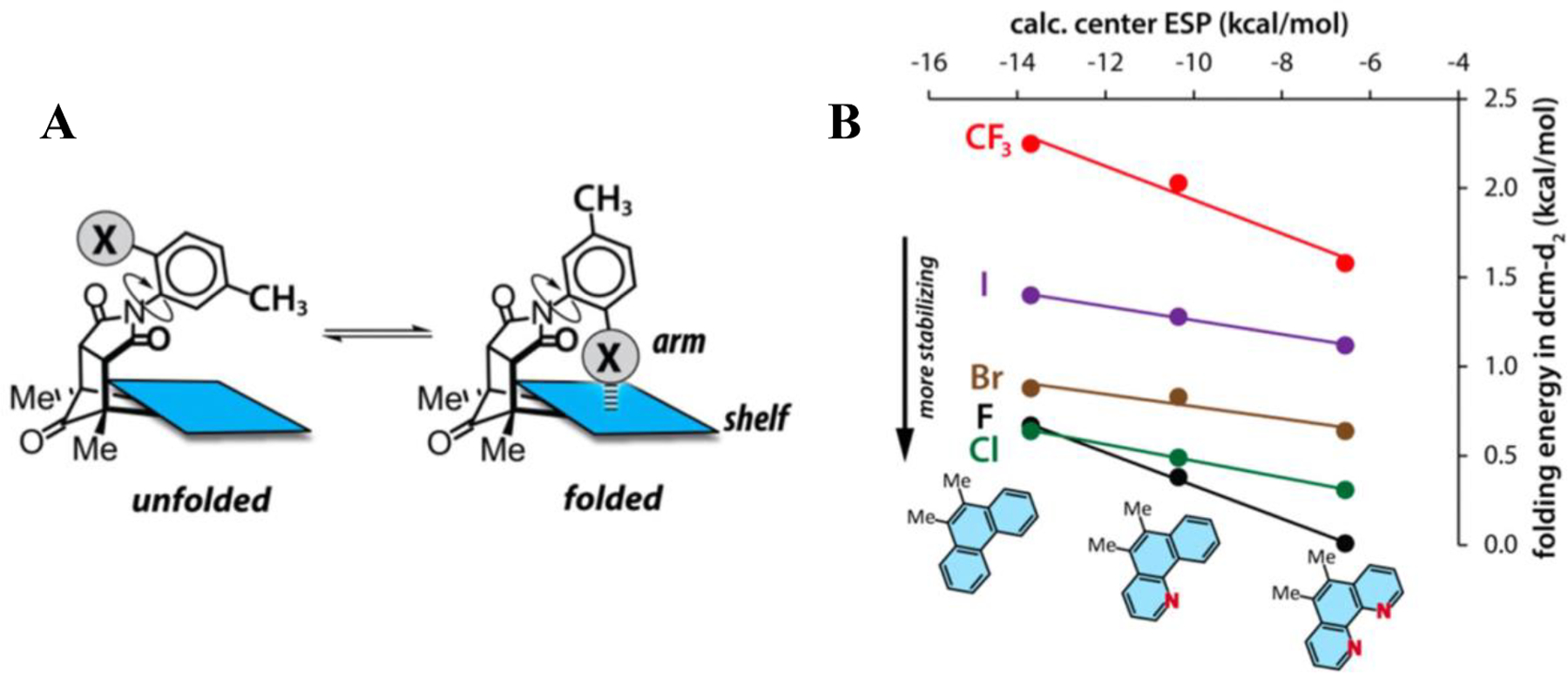
(A) General experimental scheme detailing electron-poor molecular balances. (B) Combination of NMR-derived energies and ab initio electrostatic potential (ESP) calculations. Adapted from Ref. [59] with permission.
Li et al. supported the findings of Kawahara et al. in an NMR and ab initio study of halogen interactions in electron-poor aromatic molecular balances as shown in Fig. 11 [59]. These results recapitulated the finding of Kawahara et al. in that increasing electron deficiency of the aromatic entity increases the stabilizing interaction with the halogen, but differs in the origins of the interaction energy. Whereas Kawahara et al. found the primary component was electron-correlation effects, Li et al. found that these interactions were electrostatically driven. Li et al. sampled C-F—π, C-Cl—π, C-Br—π, C-I—π, and F2C-F—π interactions and found that C-F—π interactions were most stabilizing for a given aromatic “shelf” and that more electropositive aromatic groups were more highly stabilized by halogen interactions. Similarly to Holl et al., these complexes sampled through-space intramolecular interactions measured via NMR. The authors also confirmed the structures of their compounds via X-ray crystallography and supported the perpendicular interaction between the halogen and aromatic ring previously documented in computational and structural work. Jian and coworkers similarly used molecular balances to probe C-X—π interactions [60]. Echoing the earlier work of Kawahara et al., the C-F—π interaction was found to be slightly less stabilizing than C-H—π interactions for phenyl groups. Nevertheless, fluorine far outcompeted the other halogens for the aromatic interaction, and surpassed the methyl group as well, which indicatesthat the size of the halogen group is an important part of the interaction in this system (Fig. 11B).
In summary, the results reported in the literature emphasize that the through-space stabilizing nature of the C-X—π interaction is dependent on the electronics of the aromatic ring and tends to be most stabilizing for fluorine as compared to other halogens. Although most of the results discussed above detail through-space intramolecular interactions, their applicability to intermolecular interactions is clear. While an individual amino acid may not be polarized, the polarization of an aromatic amino acid residue may be accomplished through adjacent polar amino acid residues in the context of a protein, affecting the π system. Such polarization of aromatic amino acid residues may, in turn, have an attractive interaction with an organofluorine moiety, as noted in the work of Saraogi et al [56].
4. 3rd-generation DFV-taxoids (DFV-fluorotaxoids) and DFV-ortataxel (DFV-OTX) as potent therapeutic agents for anaplastic thyroid and breast cancers
4.1. Potent antitumor activity of DFV-fluorotaxoids in anaplastic thyroid cancer [61]
Thyroid cancer is the fourth most common cancer in China and anaplastic thyroid cancer (ATC) accounts for 1–2% of all thyroid cancer [62,63]. In this study, DFV-fluorotaxoids were evaluated for their therapeutic potential. Table 3 shows the cytotoxicity of five selected DFV-fluorotaxoids against 8505C and 8305 ATC cell lines [61].
Table 3.
Cytotoxicity (IC50 nM) of DFV-fluorotaxoids against 8505C and 8305ATC cell lines [61].
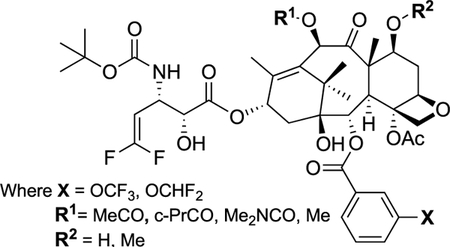
| |||||
|---|---|---|---|---|---|
| X | R1 | R2 | IC50 (Mean ± SD)/nM | ||
| 8505C | 8305C | ||||
| Paclitaxel | 5.86 | 2.51 | |||
| 11a-06 | OCHF2 | MeCO | H | 0.57 | 0.52 |
| 11b-05 | OCF3 | c-PrCO | H | 1.43 | 1.15 |
| 11d-06 | OCHF2 | Me2NCO | H | 1.09 | 0.36 |
| 11e-05 | OCHF2 | Me | H | 1.61 | 0.55 |
| 11f-05 | OCF3 | Me | Me | 1.41 | 0.50 |
The in vitro growth response of DFV-fluorotaxoids and PTX in ATC cells showed significant proliferation inhibition in a dose-dependent manner, and the DFV-fluorotaxoids showed 2–9-fold more effective inhibition than PTX. Moreover, the DFV-fluorotaxoids significantly inhibited cell proliferation than PTX and the colony formation assay demonstrated that the DFV-fluorotaxoids exerted a highly potent inhibitory effect on the colony formation of ATC cells as compared to PTX.
Cell cycle distribution assay of DFV-fluorotaxoids showed a significant mitotic arrest at the G2/M phase. Treatment of ATC cells with taxoid 11d-06 or PTX induced a marked decrease in the expression of cell cycle regulatory components at the G2/M boundary, such as p-Rb, Weel, Cyclin B1, Cyclin E, CDK1, and p21 in ATC cell lines, particularly after 48 h treatment. This indicates that the G2/M arrest induced by 11d-06 is closely associated with an alteration in the expression of cell cycle regulatory proteins.
In vitro tubulin polymerization assay indicated that DFV-fluorotaxoids promoted the polymerization of β-tubulin to microtubules at a much faster rate than PTX. This observation may imply that there is a difference in the structure between the microtubules formed with DFV-fluorotaxoids and those with PTX. As a matter of fact, we observed such structural differences in microtubules formed through rapid polymerization of tubulins promoted by 2nd- and 3rd-generation taxoids [64] and 2nd-generation DFV-taxoids [29].
The in vivo antitumor activity of 11d-06 against the ATC tumor xenografts in mice showed significant tumor growth inhibition (Fig. 12A) and reduction in the mean tumor weight (Fig. 12B) as compared to PTX. Ki-67 is known as a marker that presents in proliferating cells, but not in resting cells [65]. DFV-fluorotaxoid 11d-06 showed a significant decrease in Ki-67 percentage in tumor xenografts compared to PTX (Fig. 12C). Moreover, 11d-06 did not affect the body weight of mice during treatment, indicating a good therapeutic window.
Fig. 12.
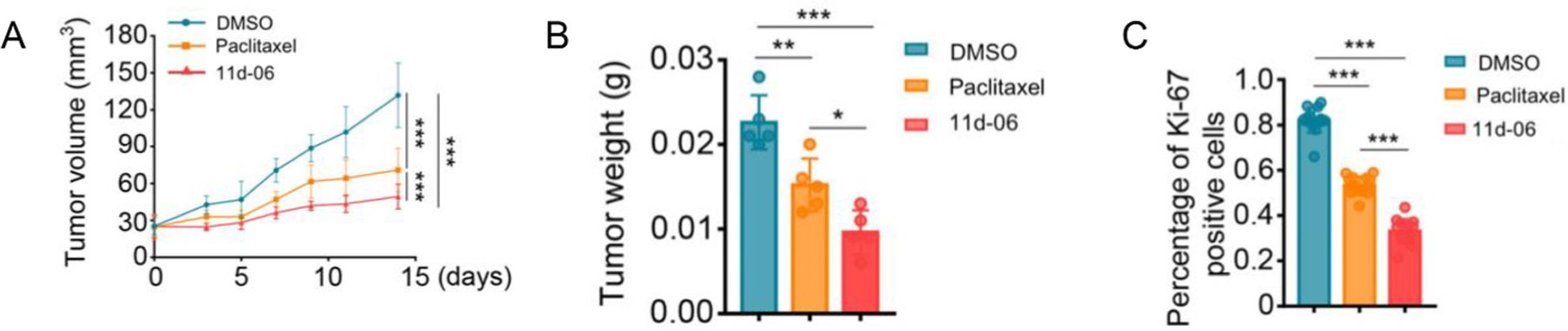
Antitumor effect of 11d-06 and PTX in tumor xenografts. Time course of tumor growth, measured as (A) tumor volume, (B) tumor weight, and (C) the number of Ki-67-positive cells. Adapted from Ref. [61] with permission.
4.2. DFV-OTX effectively overcomes PTX resistance in breast cancer in vitro and in vivo [34]
Breast cancer is the second leading cause of cancer-related death in women [66] and metastatic breast cancer, especially triple-negative breast cancer (TNBC), is considered incurable, with an expected survival of only 2–3 years [67]. Even though the fact that ortataxel (OTX) derived from 14-OH-DAB has advanced to the phase II human clinical trials [68], no fluorine-containing analogs of OTX had been synthesized and evaluated until we synthesized the first fluorine-containing analog of OTX, 3′-difluorovinylortataxel (DFV-OTX) and investigated its potency and profile as a new anticancer agent [34]. The synthesis of DFV-OTX is shown in Fig. 13, wherein the same protocols as those used for the synthesis of 2nd- and 3rd-generation DFV-taxoids described above.
Fig. 13.
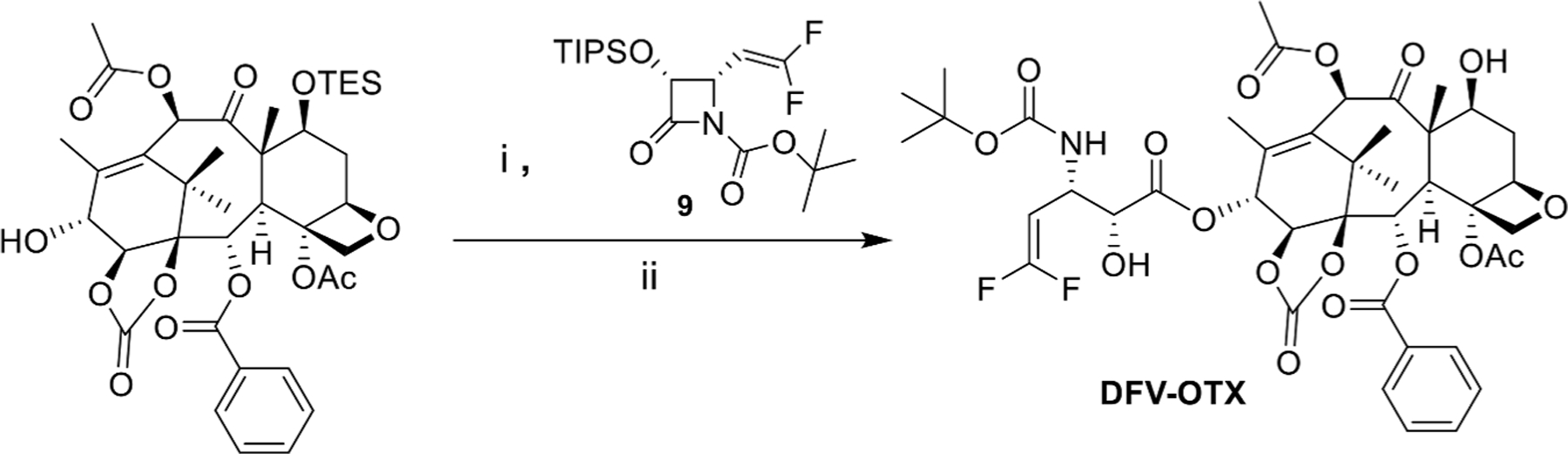
Synthesis of DFV-OTX. Reagents and conditions: (i) LiHMDS, THF, −40°C; ii) HF/pyridine, pyridine/MeCN, 0°C-r.t) [34].
DFV-OTX exhibited higher potency than PTX against most of the breast cancer cell lines assayed, especially drug-resistant cell lines including TNBC cell lines, as shown in Table 4. In addition to the cytotoxicity assay, the colony formation inhibitory assays were also performed, which disclosed the high inhibitory activity of DFV-OTX on the tumorigenesis of MCF-7R and MDA-MB-231R cells, as well as the parental MCF-7 and MDA-MB-231 cells. The results indicate a highly promising anticancer activity of DFV-OTX against PTX-resistant breast cancer cells, including TNBC cells (e.g., BT549) which lack effective chemotherapeutics agents.
Table 4.
Cytotoxicity (IC50 nM) of DFV-OTX in various drug-sensitive and drug-resistant breast cancer cells.
| Cell lines | PTX IC50 (nM) | DFV-OTX IC50 (nM) |
|---|---|---|
| MCF-7R | 17.37 | 8.15 |
| MDA-MB-231R | 550.80 | 138.50 |
| MCF-7 | 2.94 | 3.11 |
| MDA-MB-231 | 5.31 | 3.97 |
| BT474 | 243.60 | 31.44 |
| BT549 | 143.60 | 79.24 |
| MDA-MB-453 | 3.39 | 3.07 |
| 293T | 5.82 | 6.32 |
| MCF-10A | 3.40 | 2.68 |
MCF-7R: drug-resistant human breast cancer cell line.
MCF-7: human breast cancer cell line.
MCF-10A: non-malignant breast epithelial cells.
MDA-MB-231R: drug-resistant human breast cancer cell line.
MDA-MB-231: human breast cancer cell line.
MDA-MB-453: human metastatic breast cancer cell line.
BT474: human invasive breast cancer cell line.
BT549: human invasive and metastasized breast cancer cell line.
293T: human embryonic kidney cells.
The immunofluorescence assay was performed to examine whether DFV-OTX could polymerize the tubulin/microtubules in PTX-resistant breast cancer cells. The results clearly indicated that DFV-OTX effectively induced tubulin polymerization in MCF-7R and MDA-MB-231R cells, while PTX only showed a weak effect on tubulin polymerization at the same drug concentration.
The molecular docking analysis of DFV-OTX in comparison to PTX and OTX displayed significant overlapping of binding orientations (Fig. 14A). DFV-OTX conserved the H-bonding interactions with Gly262 and His229, as well as the favorable van der Waals interactions with His229 as PTX (Fig. 14B). The observed strong H-bonding between the NH of the imidazole moiety of His229 and the carbonyl oxygen of the 1,14-carbonate moiety is unique to DFV-OTX, which does not exist in PTX. Furthermore, the two fluorine atoms of the DFV group make attractive interactions with Ala233, Ser236, and Phe272 (Fig. 14C).
Fig. 14.
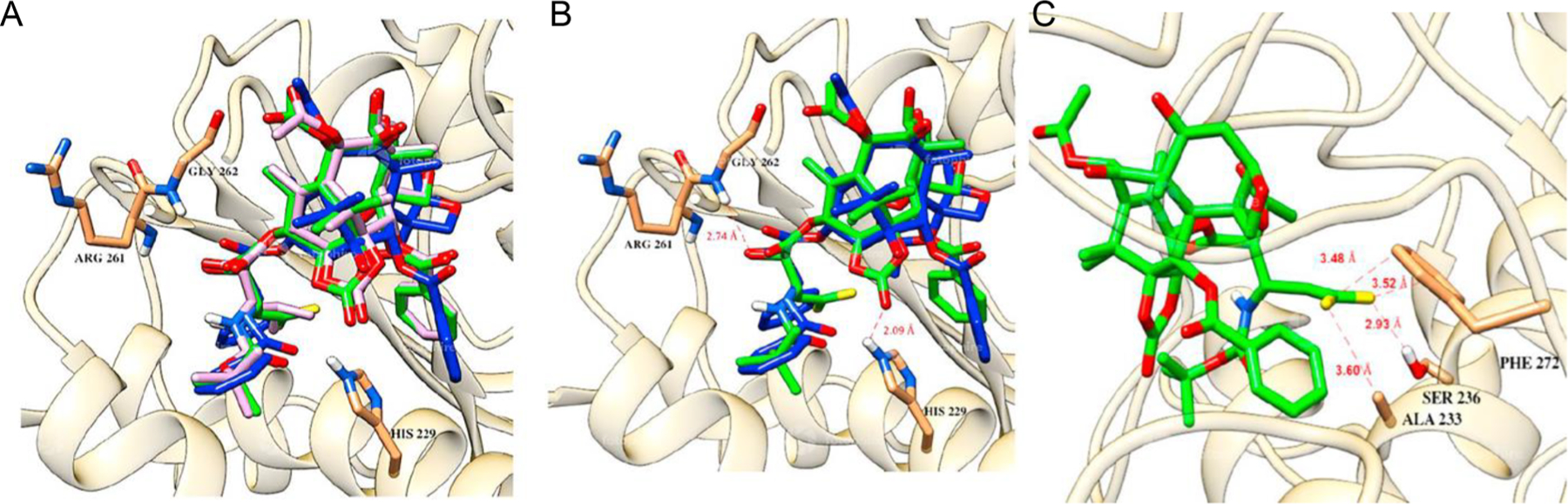
(A) Overlay of PTX (blue), OTX (pink), and DFV-OTX (green) in β-tubulin. (B) Overlay of PTX (blue) and DFV-OTX (green), with important H-bonding and van der Waals interactions. (C) The critical H-bonding and van der Waals interactions of the DFV group with nearby amino acid residues; Ala233, Ser236, and Phe272. Adapted from Ref. [34] with permission.
DFV-OTX induced cell cycle arrest at the G2/M phase in PTX-resistant MCF-7R and MDA-MB-231R cells. Cell cycle-related protein expression analysis showed that DFV-OTX caused an increase in cyclin B1 and a decrease in cyclin D1 in a dose-dependent manner in MCF-7R and MCF-7 cells as well as MDA-MB-231R and MDA-MB-231 cells. Interestingly, p53 and p21 were upregulated in a dose-dependent manner in MCF-7R and MCF-7 cells, while p21 was slightly downregulated in MDA-MB-231 cells. However, p21 was slightly upregulated after PTX treatment, while it was slightly downregulated after DFV-OTX treatment in MDA-MB-231R cells. The observed difference in the p21 expression pattern may suggest a difference in the MOA between DFV-OTX and PTX.
Cell apoptosis induced by DFV-OTX was significantly higher than that by PTX in MCF-7R and MDA-MB-231R cells in a dose-dependent manner. DFV-OTX induced the upregulation of p-eIF2α and p-p38, which are the markers of endoplasmic reticulum (ER) stress response, which suggests a similar cell death induction mechanism between DFV-OTX and PTX.
The inherent and acquired resistance to PTX in tumors is mainly attributed to the facts that (i) high expression of efflux pumps, such as ABCB1, ABCG2, and/or ABCC1, (ii) mutations at the PTX binding site in β-tubulin, (iii) the overexpression of β-tubulin subtypes, especially βIII-tubulin (TUBB3), and (iv) the overexpression of microtubule regulatory proteins (MAP4) and/or γ-actin [50,69]. It was found that ABCB1 was drastically overexpressed in MDA-MB-231R and MCF-7R cells compared to the corresponding PTX-sensitive cancer cells which promotes drug efflux, which eventually causes major resistance to PTX. The combination of PTX and Elacridar (Ela) induced substantial tubulin/microtubule polymerization, compared to PTX alone, whereas DFV-OTX alone induced extensive tubulin/microtubule polymerization, indicating the capability of DFV-OTX to overcome PTX-resistance caused by the ABC-efflux pumps. Further, the blocking of ABC-efflux pumps with Ela can sufficiently restore the cytotoxicity of PTX in MDA-MB-231R cells, but not in MCF-7R cells. This suggests the involvement of other PTX-resistance mechanisms, such as point mutations in the PTX binding site of β-tubulin and the overexpression of β-III-tubulin subtype, in this MCF-7R cell line. Importantly, DFV-OTX can overcome the drug-resistance in MCF-7R cells.
It was found that DFV-OTX treatment minimally lowered the mRNA expression levels of ABCB1 in MCF-7R and MDA-MB-231R cells. It was also observed that the intracellular level of DFV-OTX is lower than PTX at the same extracellular concentrations in both MCF-7R and MDA-MB-231R cells. Furthermore, the efflux rate of DFV-OTX showed no difference as compared to that of PTX in MDA-MB-231R cells, which would indicate that DFV-OTX and PTX possess a similar affinity to the ABC-efflux pumps. These results may suggest that the remarkable activity of DFV-OTX against PTX-resistant breast cancer cells can be attributed to its exceptionally strong interaction with β-tubulin to promote tubulin polymerization and stabilization of the formed microtubules, thereby enhancing cell cycle arrest at G2/M and triggering apoptosis.
The antineoplastic activity of DFV-OTX in vivo using the female BALB/c nude mice model showed that DFV-OTX significantly inhibited the growth of MCF-7R and MDA-MB-231R tumor xenografts, which indicates that DFV-OTX can effectively block the growth of subcutaneous PTX-resistant breast cancer tumor xenografts in vivo (Fig. 15). The sections of the harvested xenografted tumors were examined for tumor cell proliferation and apoptotic status by immunohistochemical staining of the Ki 67 cell proliferation marker. The results clearly showed that DFV-OTX blocked cell proliferation in both MCF-7R and MDA-MB-231R tumors, which indicates that DFV-OTX possesses a highly promising therapeutic potential for the treatment of PTX-resistant breast tumors.
Fig. 15.
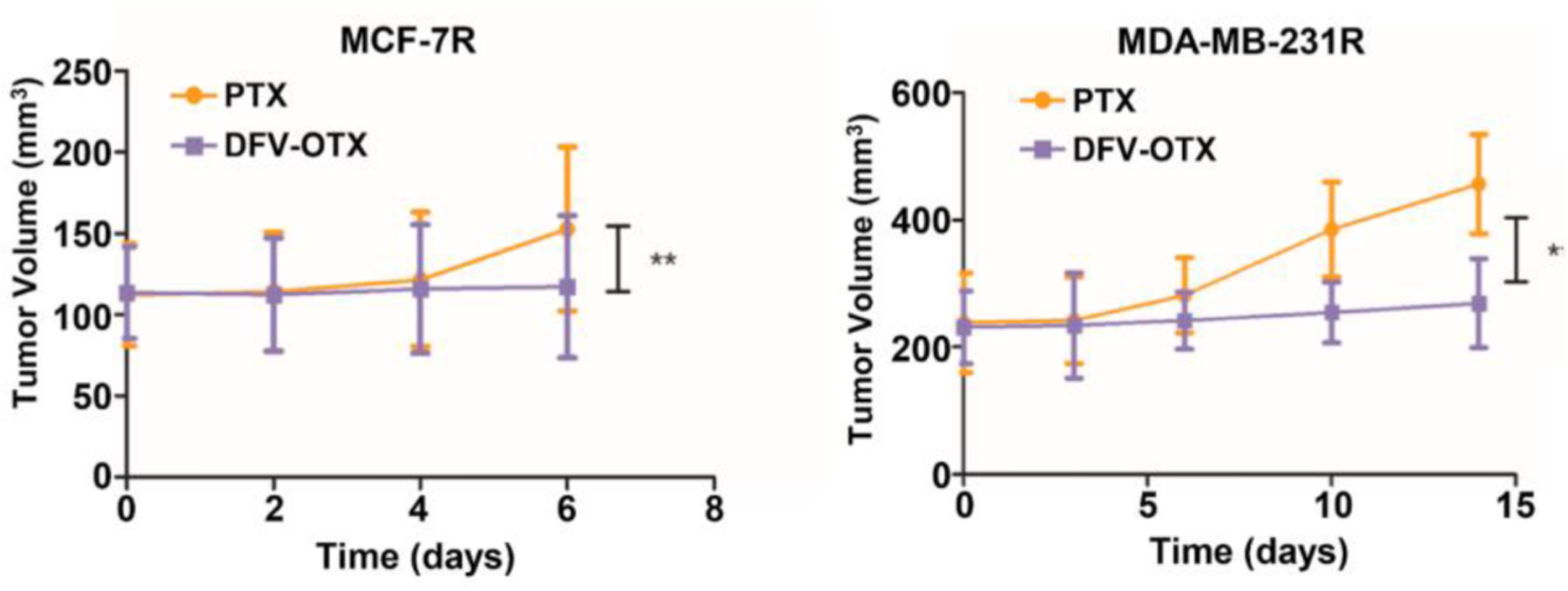
DFV-OTX represses the subcutaneous PTX-resistant tumor growth. Tumor growth of MCF-7R and MDA-MB-231R xenografts in mice upon treatment with 10 mg/ kg PTX or DFV-OTX. * p<0.05; ** p<0.01. Adapted from Ref. [34] with permission.
In summary, this study identified DFV-OTX as a highly promising new taxane anticancer agent, which effectively overcame PTX-resistance in both MCF-7R and MDA-MB-231R cancer cell lines and exhibited efficacy in tumor xenografts in mouse models. Moreover, DFV-OTX has a potentially lower risk of promoting tumor metastasis compared to PTX. Molecular docking analysis showed unique H-bonding and van der Waals interactions between DFV-OTX and β-tubulin, involving the 1,14-cyclic carbonate and DFV moieties contributing to the remarkable activity against PTX-resistant breast cancer cell lines, MCF-7R and MDA-MB-231R, with different drug resistance mechanism.
5. Concluding remarks
To cover the recent progress in the exploration of next-generation taxane anticancer agents, novel 3rd-generation taxoids bearing 3-OCF3 or 3-OCF2H at the C2 benzoate moiety were reviewed first. These fluorotaxoids exhibited up to 7 times better potency against drug-sensitive cancer cell lines (MCF7 and LCC6-WT breast) and are 2–3 orders of magnitude more potent against drug-resistant cancer cell lines (NCI/ADR, LCC6-MDR, and LDL-1) than PTX. The SAR study on these fluorotaxoids disclosed that OCF3 and OCHF2 groups are well tolerated at the C2-benzoate position with enhanced potency, especially against MDR-cancer cell lines. Molecular modeling analysis of the 3rd-generation fluorotaxoids, 1a-05 and 1a-06, identified considerably favorable van der Waals interactions of OCF3 and OCF2H groups with hydrophobic amino acid residues in the binding site, as compared to CH3 and OCH3 groups. It has also been shown that the inclusion of OCF3 and OCHF2 groups in drug leads often improve their pharmacological properties, especially metabolic stability, membrane permeability, and PK profile. The unique non-spherical structure of the OCHF2 group can provide interesting characteristics, and the 1–06 series of 3rd-generation fluorotaxoids exhibited the highest potencies against MDR-cancer cell lines and CSC-enriched cancer cell lines.
Next, a series of 3rd-generation taxoids bearing a DFV group at C3′ and a CF3O/CHF2O group at C2 benzoate moiety was reviewed. These novel DFV-fluorotaxoids exhibited 2–4 orders of magnitude higher potency against extremely drug-resistant cancer cell lines (LCC6-MDR and DLD-1) and better dose–response than PTX and SBT-1214 (1c). The results indicate that these DFV-fluorotaxoids can effectively overcome MDR caused by the overexpression of Pgp and other ABC cassette transporters. A DFV-mimic of cabazitaxel, 11f-06, exhibited remarkably high potency against MCF7, PANC-1, PC3, and LCC6-MDR cancer cell lines. The molecular docking analysis revealed that DFV-fluorotaxoids occupy the PTX binding site in β-tubulin, forming a critical H-bonding interaction of C2′-OH and Gly362. Furthermore, the 3′-DFV moiety induces a little more compact structure, allowing unique attractive interactions between the gem-difluoro group and Phe272, as well as an H-boding with Ser236. In addition, the two fluorine atoms of the OCF3/OCHF2 group have interactions with Phe272 and H-bonding (OF2C-F—HN or OF2C-H—N) with His229, which enhances the binding affinity.
Extensive in vitro and in vivo evaluations of five novel DFV-fluorotaxoids (11a-06, 11b-05, 11d-06, 11e-06 and 11f-05), disclosed exceptionally high potency of these DFV-fluorotaxoids against ATC cells. These DFV-fluorotaxoids were found to induce GTP-independent tubulin polymerization much faster than PTX. Furthermore, these DFV-fluorotaxoids promoted effective inhibition of cell proliferation, colony formation, and tumorigenic potential in nude mice. Importantly, no serious toxic side effects were observed in the treated mice, indicating a possibly safe and effective treatment for ATC.
Finally, a novel DFV-analog of ortataxel (OTX), DFV-OTX, was reviewed. DFV-OTX effectively overcame PTX-resistance in MCF-7R and MDA-MB-231R breast cancer cell lines and exhibited efficacy in tumor xenografts in mouse models. It is suggested that DFV-OTX is a promising new-generation taxane anticancer agent for the treatment of drugresistant breast tumors, including TNBCs.
Overall, the case studies covered in this account suggest that these next-generation fluorotaxoids are highly promising candidates as chemotherapeutic agents, as well as payloads for tumor-targeting drug conjugates such as antibody-drug conjugates (ADCs).
Acknowledgment
This work has been supported by grants from the National Institutes of Health (CA103314 and CA237154).
Footnotes
Dedicated to Professor Dr. Beate Koksh of Free University Berlin for her outstanding achievements of exceptional originality and creativity in fluorine chemistry at the interface of chemistry, biology and protein science on the occasion of her ACS Award for Creative Work in Fluorine Chemistry 2021.
Declaration of Competing Interest
Authors declare no conflict of interest.
Data availability
No data was used for the research described in the article.
References
- [1].Ojima I, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, 2009. [Google Scholar]
- [2].Müller K, Faeh C, Diederich F, Fluorine in pharmaceuticals: looking beyond intuition, Science 317 (2007) 1881–1886. [DOI] [PubMed] [Google Scholar]
- [3].Bégué JP, Bonnet-Delpon D, Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural products, J. Fluor. Chem 127 (2006) 992–1012. [Google Scholar]
- [4].Isanbor C, O’Hagan D, Fluorine in medicinal chemistry: a review of anti-cancer agents, J. Fluor. Chem 127 (2006) 303–319. [Google Scholar]
- [5].Wang J, Sánchez-Roselló M, Aceña JL, Del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H, Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011), Chem. Rev 114 (2014) 2432–2506. [DOI] [PubMed] [Google Scholar]
- [6].Cottet F, Marull M, Lefebvre O, Schlosser M, Recommendable routes to trifluoromethyl-substituted pyridine-and quinolinecarboxylic acids (2003), Eur. J. Org. Chem (8) (2003) 1559–1568. [Google Scholar]
- [7].Kirk KL, Fluorine in medicinal chemistry: recent therapeutic applications of fluorinated small molecules, J. Fluor. Chem 127 (2006) 1013–1029. [Google Scholar]
- [8].Yamazaki T, Taguchi T, Ojima I, Unique properties of fluorine and their relevance to medicinal chemistry and chemical biology, Fluor. Med. Chem. Chem. Biol (2009) 1. [Google Scholar]
- [9].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, 2021, CA: Cancer J. Clin 71 (2021) 7–33. [DOI] [PubMed] [Google Scholar]
- [10].Dalerba P, Cho RW, Clarke MF, Cancer stem cells: models and concepts, Annu. Rev. Med 58 (2007) 267–284. [DOI] [PubMed] [Google Scholar]
- [11].Ojima I, Use of fluorine in the medicinal chemistry and chemical biology of bioactive compounds—a case study on fluorinated taxane anticancer agents, ChemBioChem 5 (2004) 628–635. [DOI] [PubMed] [Google Scholar]
- [12].Botchkina GI, Zuniga ES, Das M, Wang Y, Wang H, Zhu S, Savitt AG, Rowehl RA, Leyfman Y, Ju J, New-generation taxoid SB-T-1214 inhibits stem cell-related gene expression in 3D cancer spheroids induced by purified colon tumorinitiating cells, Mol. Cancer 9 (2010) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Botchkina GI, Zuniga ES, Rowehl RH, Park R, Bhalla R, Bialkowska AB, Johnson F, Golub LM, Zhang Y, Ojima I, Shroyer KR, Prostate cancer stem cell-targeted efficacy of a new-generation taxoid, SBT-1214 and novel polyenolic zinc-binding curcuminoid, CMC2.24, PLoS One 8 (2013) e69884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bissery MC, Nohynek G, Sanderink GJ, Lavelle F, Docetaxel (Taxotere): a review of preclinical and clinical experience. Part I and Part II: preclinical experience, Anticancer Drugs 6 (1995) 339–355, and 356–368. [DOI] [PubMed] [Google Scholar]
- [15].Wang C, Aguilar A, Ojima I, Strategies for the drug discovery and development of taxane anticancer therapeutics, Expert Opin. Drug Discov 17 (2022) 1193–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ojima I, Wang X, Jing YR, Wang CW, Quest for efficacious next-generation taxoid anticancer agents and their tumor-targeted delivery, J. Nat. Prod 81 (2018) 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bono JS, Oudard S, Ozguroglu M, Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial, Lancet 376 (2010) 1147–1154. [DOI] [PubMed] [Google Scholar]
- [18].Rowinsky M, Eric K, The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents, Annu. Rev. Med 48 (1997) 353–374. [DOI] [PubMed] [Google Scholar]
- [19].De Bono J, Oudard S, Ozguroglu M, Hansen S, Machiels J, Shen L, Matthews P, Sartor A, Investigators T, Cabazitaxel or mitoxantrone with prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC) previously treated with docetaxel: Final results of a multinational phase III trial (TROPIC), J. Clin. Oncol 28 (2010) 4508. -4508. [Google Scholar]
- [20].Ojima I, Lichtenthal B, Lee S, Wang C, Wang X, Taxane anticancer agents: a patent perspective, Expert. Opin. Ther. Pat 26 (2016) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ojima I, Kamath A, Seitz JD, Natural Products in Medicinal Chemistry, Wiley-VCH, Weinheim, 2013, pp. 127–180. S. Hanessian. [Google Scholar]
- [22].Bissery MC, Nohynek G, Sanderink GJ, Lavelie F, Docetaxel (Taxotere®) a review of preclinical and clinical experience. Part I: preclinical experience, Anticancer Drugs 6 (3) (1995) 339–355. [DOI] [PubMed] [Google Scholar]
- [23].Fojo AT, Ueda K, Slamon DJ, Poplack D, Gottesman M, Pastan I, Expression of a multidrug-resistance gene in human tumors and tissues, Proc. Natl. Acad. Sci 84 (1987) 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gottesman MM, Pastan I, Biochemistry of multidrug resistance mediated by the multidrug transporter, Annu. Rev. Biochem 62 (1993) 385–427. [DOI] [PubMed] [Google Scholar]
- [25].Haranahalli K, Honda T, Ojima I, Recent progress in the strategic incorporation of fluorine into medicinally active compounds, J. Fluor. Chem 217 (2019) 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mohelnikova-Duchonova B, Kocik M, Duchonova B, Brynychova V, Oliverius M, Hlavsa J, Honsova E, Mazanec J, Kala Z, Ojima I, Hedgehog pathway overexpression in pancreatic cancer is abrogated by new-generation taxoid SB-T-1216, Pharmacogenomics J 17 (2017) 452–460. [DOI] [PubMed] [Google Scholar]
- [27].Seitz JD, Wang T, Vineberg JG, Honda T, Ojima I, Synthesis of a next-generation taxoid by rapid methylation amenable for 11C-labeling, J. Org. Chem 83 (2018) 2847–2857. [DOI] [PubMed] [Google Scholar]
- [28].Kuznetsova LV, Pepe A, Ungureanu IM, Pera P, Bernacki RJ, Ojima I, Syntheses and structure–activity relationships of novel 3′-difluoromethyl and 3′-trifluoromethyl-taxoids, J. Fluor. Chem 129 (2008) 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kuznetsova L, Sun L, Chen J, Zhao X, Seitz J, Das M, Li Y, Veith JM, Pera P, Bernacki RJ, Synthesis and biological evaluation of novel 3′-difluorovinyl taxoids, J. Fluor. Chem 143 (2012) 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ojima I, Slater JC, Kuduk SD, Takeuchi CS, Gimi RH, Sun CM, Park YH, Pera P, Veith JM, Bernacki RJ, Syntheses and structure activity relationships of taxoids derived from 14β-hydroxy-10-deacetylbaccatin III, J. Med. Chem 40 (1997) 267–278. [DOI] [PubMed] [Google Scholar]
- [31].Ojima I, Wang C, Wang X, Third Generation Taxoids and Methods of Using Same, Google Patents, 2020. [Google Scholar]
- [32].Ojima I, Strategic incorporation of fluorine into taxoid anticancer agents for medicinal chemistry and chemical biology studies, J. Fluor. Chem 198 (2017) 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang C, Chen L, Sun Y, Guo W, Taouil AK, Ojima I, Design, synthesis and SAR study of Fluorine-containing 3rd-generation taxoids, Bioorg. Chem 119 (2022), 105578. [DOI] [PubMed] [Google Scholar]
- [34].Rong D, Wang C, Zhang X, Wei Y, Zhang M, Liu D, Farhan H, Ali SAM, Liu Y, Taouil A, Guo W, Wang Y, Ojima I, Yang S, Wang H, A novel taxane, difluorovinyl-ortataxel, effectively overcomes paclitaxel-resistance in breast cancer cells, Cancer Lett 491 (2020) 36–49. [DOI] [PubMed] [Google Scholar]
- [35].Ojima I, Slater JC, Michaud E, Kuduk SD, Bounaud PY, Vrignaud P, Bissery MC, Veith JM, Pera P, Bernacki RJ, Syntheses and structure activity relationships of the second-generation antitumor taxoids: exceptional activity against drug-resistant cancer cells, J. Med. Chem 39 (1996) 3889–3896. [DOI] [PubMed] [Google Scholar]
- [36].Beer M, Lenaz L, Amadori D, Group OS, Phase II study of ortataxel in taxane-resistant breast cancer, J. Clin. Oncol 26 (2008) 1066. -1066.18212337 [Google Scholar]
- [37].Sonnichsen DS, Liu Q, Schuetz EG, Schuetz JD, Pappo A, Relling MV, Variability in human cytochrome P450 paclitaxel metabolism, J. Pharmacol. Exp. Ther 275 (1995) 566–575. [PubMed] [Google Scholar]
- [38].Chaudhary AG, Gharpure MM, Rimoldi JM, Chordia MD, Kingston DG, Grover S, Lin CM, Hamel E, Gunatilaka AL, Unexpectedly facile hydrolysis of the 2-benzoate group of taxol and syntheses of analogs with increased activities, J. Am. Chem. Soc 116 (1994) 4097–4098. [Google Scholar]
- [39].Kingston DG, Chaudhary AG, Chordia MD, Gharpure M, Gunatilaka AL, Higgs PI, Rimoldi JM, Samala L, Jagtap PG, Giannakakou P, Synthesis and biological evaluation of 2-acyl analogues of paclitaxel (Taxol), J. Med. Chem 41 (1998) 3715–3726. [DOI] [PubMed] [Google Scholar]
- [40].Guengerich FP, Waterman MR, Egli M, Recent structural insights into cytochrome P450 function, Trends Pharmacol. Sci 37 (2016) 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang C, Wang X, Sun Y, Taouil AK, Yan S, Botchkina GI, Ojima I, Design, synthesis and SAR study of 3rd-generation taxoids bearing 3-CH3, 3-CF3O and 3-CHF2O groups at the C2-benzoate position, Bioorg. Chem 95 (2020), 103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Besset T, Jubault P, Pannecoucke X, Poisson T, New entries toward the synthesis of OCF 3-containing molecules, Org. Chem. Front 3 (2016) 1004–1010. [Google Scholar]
- [43].Xing L, Blakemore DC, Narayanan A, Unwalla R, Lovering F, Denny RA, Zhou H, Bunnage ME, Fluorine in drug design: a case study with fluoroanisoles, ChemMedChem 10 (2015) 715–726. [DOI] [PubMed] [Google Scholar]
- [44].Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M, Fluorine in medicinal chemistry, ChemBioChem 5 (2004) 637–643. [DOI] [PubMed] [Google Scholar]
- [45].Leroux F, Jeschke P, Schlosser M, α-Fluorinated ethers, thioethers, and amines: anomerically biased species, Chem. Rev 105 (2005) 827–856. [DOI] [PubMed] [Google Scholar]
- [46].Müller K, Simple vector considerations to assess the polarity of partially fluorinated alkyl and alkoxy groups, Chimia 68 (2014) 356–362 (Aarau). [DOI] [PubMed] [Google Scholar]
- [47].Xing L, Keefer C, Brown MF, Fluorine multipolar interaction: Toward elucidating its energetics in binding recognition, J. Fluor. Chem 198 (2017) 47–53. [Google Scholar]
- [48].Zheng X, Wang C, Xing Y, Chen S, Meng T, You H, Ojima I, Dong Y, SB-T-121205, a next-generation taxane, enhances apoptosis and inhibits migration/invasion in MCF-7/PTX cells, Int. J. Oncol 50 (2017) 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [49].Ojima I, Habus I, Zhao M, Zucco M, Park YH, Sun CM, Brigaud T, New and efficient approaches to the semisynthesis of taxol and its C-13 side chain analogs by means of β-lactam synthon method, Tetrahedron 48 (1992) 6985–7012. [Google Scholar]
- [50].Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB, Mechanisms of Taxol resistance related to microtubules, Oncogene 22 (2003) 7280–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gut I, Ojima I, Vaclavikova R, Simek P, Horsky S, Linhart I, Soucek P, Kondrova E, Kuznetsova L, Chen J, Metabolism of new-generation taxanes in human, pig, minipig and rat liver microsomes, Xenobiotica 36 (2006) 772–792. [DOI] [PubMed] [Google Scholar]
- [52].Taylor R, The hydrogen bond between N—H or O—H and organic fluorine: favourable yes, competitive no, Acta. Crystallogr. B Struct. Sci. Cryst. Eng. Mater 73 (2017) 474–488. [DOI] [PubMed] [Google Scholar]
- [53].Holl MG, Struble MD, Singal P, Siegler MA, Lectka T, Positioning a carbon–fluorine bond over the π cloud of an aromatic ring: a different type of Arene activation, Angew. Chem. Int. Ed 55 (2016) 8266–8269. [DOI] [PubMed] [Google Scholar]
- [54].Zafrani Y, Yeffet D, Sod-Moriah G, Berliner A, Amir D, Marciano D, Gershonov E, Saphier S, Difluoromethyl bioisostere: examining the “lipophilic hydrogen bond donor” concept, J. Med. Chem 60 (2017) 797–804. [DOI] [PubMed] [Google Scholar]
- [55].Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W, Revealing noncovalent interactions, J. Am. Chem. Soc 132 (2010) 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Saraogi I, Vijay V, Das S, Sekar K, Row TG, C–halogen… π interactions in proteins: a database study, Cryst. Eng 6 (2003) 69–77. [Google Scholar]
- [57].Esrafili MD, Mahdavinia G, Javaheri M, Sobhi HR, A theoretical study of substitution effects on halogen–π interactions, Mol. Phys 112 (2014) 1160–1166. [Google Scholar]
- [58].Kawahara S, Tsuzuki S, Uchimaru T, Theoretical study of the C F/π interaction: attractive interaction between fluorinated alkane and an electron-deficient π-system, J. Phys. Chem. A 108 (2004) 6744–6749. [Google Scholar]
- [59].Li P, Maier JM, Vik EC, Yehl CJ, Dial BE, Rickher AE, Smith MD, Pellechia PJ, Shimizu KD, Stabilizing fluorine–π interactions, Angew. Chem 129 (2017) 7315–7318. [DOI] [PubMed] [Google Scholar]
- [60].Jian J, Poater J, White PB, McKenzie CJ, Bickelhaupt FM, Mecinovic J, Probing halogen-π versus CH-π interactions in molecular balance, Org. Lett 22 (2020) 7870–7873. [DOI] [PubMed] [Google Scholar]
- [61].Wang M, Wang C, Feng C, Guo W, Chen H, Liu B, Li E, Liu W, Taouil A, Ojima I, Potent antitumor activity of novel taxoids in anaplastic thyroid cancer, Endocrine 75 (2022) 465–477. [DOI] [PubMed] [Google Scholar]
- [62].Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J, Cancer statistics in China, 2015, CA: Cancer J. Clin 66 (2016) 115–132. [DOI] [PubMed] [Google Scholar]
- [63].Molinaro E, Romei C, Biagini A, Sabini E, Agate L, Mazzeo S, Materazzi G, Sellari-Franceschini S, Ribechini A, Torregrossa L, Anaplastic thyroid carcinoma: from clinicopathology to genetics and advanced therapies, Nat. Rev. Endocrinol 13 (2017) 644–660. [DOI] [PubMed] [Google Scholar]
- [64].Ojima I, Chen J, Sun L, Borella CP, Wang T, Miller ML, Lin S, Geng X, Kuznetsova L, Qu C, Design, synthesis, and biological evaluation of new-generation taxoids, J. Med. Chem 51 (2008) 3203–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gerdes J, Schwab U, Lemke H, Stein H, Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation, Int. J. Cancer 31 (1983) 13–20. [DOI] [PubMed] [Google Scholar]
- [66].Liang Y, Zhang H, Song X, Yang Q, Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets, Semin. Cancer Biol (2020) 14–27. [DOI] [PubMed] [Google Scholar]
- [67].Hu X, Huang W, Fan M, Emerging therapies for breast cancer, J. Hematol. Oncol (2017) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ojima I, Kamath A, Seitz JD, Taxol, Taxoids and Related Taxanes, Natural Products in Medicinal Chemistry, 60, Wiley-VCH, Weinheim, 2014, pp. 127–180. [Google Scholar]
- [69].Kavallaris M, Microtubules and resistance to tubulin-binding agents, Nat. Rev. Cancer 10 (2010) 194–204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.