

Abstract
Treatment strategies that are efficient against established Alzheimer’s disease (AD) are needed. BRICHOS is a molecular chaperone domain that prevents amyloid fibril formation and associated cellular toxicity. In this study, we treated an AD mouse model seven months after pathology onset, using intravenous administration of recombinant human (rh) Bri2 BRICHOS R221E. Two injections of rh Bri2 BRICHOS R221E per week for three months in AD mice reduced amyloid β (Aβ) burden, and mitigated astro- and microgliosis, as determined by glial fibrillary acidic protein (GFAP) and ionized calcium-binding adaptor molecule 1 (Iba1) immunohistochemistry. Sequencing of RNA from cortical microglia cells showed that BRICHOS treatment normalized the expression of identified plaque-induced genes in mice and humans, including clusterin and GFAP. Rh Bri2 BRICHOS R221E passed the blood–brain barrier (BBB) in age-matched wild-type mice as efficiently as in the AD mice, but then had no effect on measures of AD-like pathology, and mainly affected the expression of genes that affect cellular shape and movement. These results indicate a potential of rh Bri2 BRICHOS against advanced AD and underscore the ability of BRICHOS to target amyloid-induced pathology.
Subject terms: Molecular neuroscience, Diseases
Introduction
Alzheimer’s disease (AD), the most common form of dementia, poses significant medical and socioeconomic problems [1]. The rapidly increasing number of patients afflicted by this ailment underscores the urgent need for the development of efficacious treatments. Recently, antibody-based drugs targeting the amyloid cascade have undergone clinical trials for early symptomatic AD, yielding varied levels of success [2]. Notably, the monoclonal antibodies Aducanumab and Lecanemab, targeting amyloid-β (Aβ) fibrillar assemblies, one hallmark of AD, have been approved by the U.S. Food and Drug Administration (FDA) for treating individuals in the early symptomatic stages of AD. The monoclonal antibody Donanemab, known to bind to amyloid plaques, decelerates memory and cognitive decline in individuals with early-stage AD [3]. While these antibody drugs mitigate memory and cognitive deterioration in early-stage AD, their efficacies are limited, and side effects have been reported [4, 5]. AD is typically diagnosed in its later stages [6], indicating the need for candidate drugs that can affect established AD pathology. AD is characterized by the accumulation of extracellular Aβ plaques and intraneuronal formation of tau neurofibrillary tangles [7, 8]. The Aβ peptide is produced through successive cleavages of amyloid-β precursor protein (APP) by β-secretase 1 (BACE1) and γ-secretase, and AD pathogenesis is coupled to the accumulation of extracellular Aβ, in particular Aβ42, in cortical and subcortical brain regions [9]. AD pathology also includes activation of microglia and astrocytes, and increased levels of proinflammatory mediators [10, 11].
Molecular chaperones play an important role in maintaining cellular proteostasis, and can prevent or resolve protein aggregation [12]. The BRICHOS domain was initially identified in Bri2, chondromodulin-1, and prosurfactant protein C (proSP-C). Each of these proproteins contain a region prone to form amyloid [13, 14], which BRICHOS chaperones during biosynthesis [15]. Furthermore, recombinant human (rh) BRICHOS domains from proSP-C and Bri2 are potent inhibitors of amyloid formation also by non-client proteins such as medin, islet amyloid polypeptide (IAPP), Aβ40 and Aβ42 [16, 17]. Bri2 is highly expressed in neurons of the human hippocampus and cortex, co-localizes with AD senile plaques [18, 19] and releases the BRICHOS domain [20]. Rh Bri2 BRICHOS potently inhibits Aβ42 fibril formation and toxicity in vitro, in hippocampal slice preparations, as well as in vivo in Drosophila melanogaster and AD mouse models [21–24]. The Bri2 BRICHOS monomer is most effective in preventing Aβ42-induced disruption of neuronal network activity [25], and efficiently crosses the blood–brain barrier (BBB) [26]. Therefore, a Bri2 BRICHOS mutant, R221E, which forms long-term stable monomers has been designed [27]. Rh Bri2 BRICHOS R221E administered intravenously (i.v.) to App knock-in mouse models improved recognition and working memory, reduced Aβ plaque deposition and mitigated activation of astrocytes and microglia [24]. However, it remains unclear whether BRICHOS treatment is effective when applied at advanced AD-like pathology stages, and if BRICHOS can normalize Aβ-induced dysregulation of AD-associated genes.
The aim of the current study was to assess the ability of rh Bri2 BRICHOS R221E to affect pathology in an AD mouse model at a stage when pathology is well established. An App knock-in mouse model harbouring the Swedish, Iberian, and Arctic mutations (AppNL-G-F mouse model), which develops AD-like pathology from the second month of age, and reaches near-maximum levels by the seventh month of age [28] was used. Nine-month-old AppNL-G-F mice were treated i.v. with rh Bri2 BRICHOS R221E monomers, or saline, for three months and then sacrificed at thirteen months of age, i.e. one month after the last dose. Age-matched wild-type (WT) mice were treated in the same way. Analysis of amyloid plaque load, reactive gliosis, and bulk microglia RNA sequencing (Fig. 1A), indicate that rh Bri2 BRICHOS treatment is efficient at a late AD-like stage and works by targeting Aβ-induced pathology.
Fig. 1. Overview of rh Bri2 BRICHOS R221E treatment of old AppNL-G-F and WT mice.

A Timeline of treatment with rh Bri2 BRICHOS R221E monomer or administration of vehicle (phosphate-buffered saline, PBS) in AppNL-G-F and age-matched WT mice. B Rh Bri2 BRICHOS R221E monomer analysis by reducing (lane 1) and non-reducing (lane 2) SDS-PAGE. M shows the migration of marker proteins with molecular masses in kDa given to the left. The Bri2 BRICHOS R221E structure at the top was predicted by Alphafold2 [93]. C Body weight of the vehicle (veh)- or rh Bri2 BRICHOS R221E-treated AppNL-G-F (App) and WT mice during the treatment period, presented as mean ± SEM. Two-way ANOVA with Tukey’s correction for multiple comparisons was used to calculate p-values for differences in body weight. ns no significance. D Areas of the hippocampus and cerebral cortex analysed by immunohistochemistry. Auditory auditory cortex, CA Cornu ammunis, DG dentate gyrus, Olfactory olfactory cortex, RSC retrosplenial cortex.
Results
Intravenous BRICHOS R221E administration to aged AppNL-G-F mice
The AppNL-G-F model produces Aβ toxicity by augmenting the overall production of human Aβ, shifting the Aβ42/Aβ40 ratio in favour of the more toxic Aβ42 form, and increasing Aβ42 aggregation propensity [29]. Consequently, robust amyloidosis and pathological changes start to develop at two months of age and reach near-maximal levels by the seventh month [28]. In order to assess the therapeutic potential of rh Bri2 BRICHOS R221E, we started treatment of AppNL-G-F mice at 9 months, and WT mice were treated in the same way, to enable investigation of potential effects not related to the AD-like pathology (Fig. 1A).
Rh Bri2 BRICHOS R221E was produced in E. coli and subsequently purified using immobilized metal ion affinity chromatography and size exclusion chromatography (SEC), as previously established [27]. SDS-PAGE of the SEC-isolated rh Bri2 BRICHOS R221E monomers under reducing or non-reducing conditions, demonstrated high purity (Fig. 1B). Notably, the slightly accelerated migration observed by SDS-PAGE under non-reducing conditions indicates the presence of a conserved intramolecular disulphide bond [27]. AppNL-G-F and WT mice from nine months of age received i.v. administration (in the tail vein) of rh Bri2 BRICHOS R221E monomers at a dose of 10 mg/kg twice a week for a duration of twelve weeks, or vehicle (Veh, i.e. PBS) (Fig. 1A), i.e. the same dosing regimen as previously used in early-stage AppNL-G-F mice [24]. Throughout the treatment period, we monitored the body weight of the mice once a week. Generally, both the AD model mice and their WT counterparts tolerated the rh Bri2 BRICHOS R221E injections. Rh Bri2 BRICHOS-treated AppNL-G-F mice displayed a 5–10% decrease in body weight after 8–9 weeks of treatment compared to those given PBS (p-values 0.5 and 0.6, respectively), followed by a subsequent recovery (Fig. 1C). One month after the completion of the treatment regimen, the mice were sacrificed and immunohistochemical, biochemical and transcriptome analyses on various regions of the hippocampus and cortex, were conducted, i.e. the same interval was employed between sacrifice and analyses as previously used in early-stage AppNL-G-F and late stage AppNL-F mice [24]. The regions analysed included the dentate gyrus (DG), Cornu ammonis (CA) regions 2 and 3 (CA2 and CA3) of the hippocampus, the retrosplenial cortex (RSC), and the auditory and olfactory cortex areas (Fig. 1D).
Rh Bri2 BRICHOS R221E passes the BBB and reduces Aβ plaque load
To assess the BBB permeability of rh Bri2 BRICHOS R221E following i.v. administration in both WT and AppNL-G-F mice, brain sections were subjected to immunohistochemistry for Bri2 BRICHOS and subsequent quantification. The results revealed higher levels of Bri2 BRICHOS staining in the cortex and in DG, CA2, and CA3 regions of the hippocampus of AppNL-G-F and WT mice after rh Bri2 BRICHOS R221E treatment compared to mice receiving PBS (Fig. 2A–E). Notably, no differences in Bri2 BRICHOS staining were observed between the WT and AppNL-G-F mice, suggesting that the BBB of AppNL-G-F mice has a similar permeability as that of WT mice. These findings strongly suggest that i.v. administered rh Bri2 BRICHOS R221E effectively crosses the BBB and is retained in the brain for at least one month in both WT and AppNL-G-F mice. The same degree of increase in Bri2 BRICHOS staining in AppNL-G-F and WT mice (Fig. 2) was observed, in spite of the fact that rh Bri2 BRICHOS R221E binds to Aβ42 fibrils [30, 31]. This is likely related to that i.v. injected rh Bri2 BRICHOS is internalized into neurons [32] and only a small fraction (ca.10%) of added rh Bri2 BRICHOS R221E binds to Aβ42 fibrils [31].
Fig. 2. Rh Bri2 BRICHOS R221E accumulates in the brains of AppNL-G-F and WT mice after intravenous injections.

Representative images of sections from indicated brain regions (column A, DG and B, CA2 and CA3 hippocampal regions; C, RSC, D, auditory and E, olfactory cortex regions) of rh Bri2 BRICHOS R221E- or vehicle (veh)-treated AppNL-G-F (App) and WT mice stained with anti-Bri2 BRICHOS antibody. The areas marked with boxes are shown in the panels below them. The punctuate pattern of staining indicates that rh Bri2 BRICHOS R221E is present intracellularly, see text for details. The bar charts show the rh Bri2 BRICHOS-positive area (% of total area) in the different brain regions for each treatment group. Scale bars represent 400 μm and apply to all panels in the respective column, except the magnified areas for which the scale bars are 100 μm. Data are shown as mean ± SEM (n = 6–7 mice per group). One-way ANOVA test and Tukey’s test were used to calculate the p-values. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Auditory auditory cortex, CA Cornu ammunis, DG dentate gyrus, Olfactory olfactory cortex, RSC retrosplenial cortex.
Rh Bri2 BRICHOS R221E has been shown to delay Aβ42 amyloid fibril formation in vitro [15], and administration to AppNL-G-F mice starting at three months of age reduced amyloid plaque burden [24]. To investigate the effects of rh Bri2 BRICHOS R221E in older AppNL-G-F mice, brain sections were stained with the Aβ antibody 82E1. The results demonstrated a reduced load of Aβ plaques in the DG, CA2 and CA3 regions of the hippocampus (Fig. 3A, B) and cortex (both auditory and olfactory) (Fig. 3C–E) of mice treated with rh Bri2 BRICHOS R221E compared to PBS. The degree of reduction in the treatment of older AppNL-G-F mice (Fig. 3) is comparable to the effect sizes in early-stage AppNL-G-F mice [24]. These results suggest that the i.v. administration of rh Bri2 BRICHOS R221E reduces amyloid burden in aged AppNL-G-F mice also when amyloid pathology had nearly reached saturation [28] at the start of treatment.
Fig. 3. Plaque load is reduced in AppNL-G-F mice after rh Bri2 BRICHOS R221E treatment.

Representative images of the hippocampus (A, B) and cerebral cortex (C–E) sections from vehicle (veh)- and rh Bri2 BRICHOS R221E-treated AppNL-G-F (App) mice stained with anti-Aβ antibody 82E1. Histograms show plaque load (%) in different hippocampal and cortical regions. The scale bar represents 400 μm and applies to all panels in (A–E). Data are presented as mean ± SEM (n = 6–7 mice/group). An unpaired parametric two-tailed t-test was used for statistical analysis. ∗p < 0.05. Auditory auditory cortex, CA Cornu ammunis, DG dentate gyrus, Olfactory olfactory cortex, RSC retrosplenial cortex.
Rh Bri2 BRICHOS R221E treatment of aged AppNL-G-F mice reduces microgliosis and astrogliosis
To further evaluate the therapeutic effects of rh Bri2 BRICHOS R221E in aged AppNL-G-F mice, we analysed the degree of microgliosis by immunohistochemistry for the ionized calcium-binding adaptor molecule 1 (Iba1), a marker for microglial activation. This clearly showed an overall reduction in Iba1-positive microglia in both the hippocampus and cortex areas after rh Bri2 BRICHOS R221E treatment, compared to the PBS controls, while rh Bri2 BRICHOS R221E treatment of WT mice did not change the levels of Iba1 immunoreactivity (Fig. 4).
Fig. 4. Rh Bri2 BRICHOS R221E treatment mitigates microgliosis in AppNL-G-F mice.

Representative images of hippocampus (A) and cortex (B–D) sections from vehicle (veh)- and rh Bri2 BRICHOS R221E-treated AppNL-G-F (App) and WT mice stained by immunohistochemistry for Iba1. Bar charts at the bottom show the Iba1-positive area (%) in each brain region for the four treatment groups. The areas marked with boxes are magnified in the panels shown below them. All micrographs from the hippocampus are at a lower magnification than the micrographs from the cortex. Scale bars represent 1000 μm (A) and 400 μm (B–D) and apply to all panels in the respective column, except the magnified areas for which scale bars represent 200 μm (A) or 80 μm (B–D). Data are shown as mean ± SEM (n = 6–7 mice/group). One-way ANOVA test and Tukey’s test were used for statistical analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. The entire hippocampus was analysed. Iba1 ionized calcium-binding adaptor molecule 1, RSC retrosplenial cortex.
Next, we analysed the degree of astrogliosis using the astrocyte marker glial fibrillary acidic protein (GFAP). In AppNL-G-F mice treated with rh Bri2 BRICHOS R221E, the levels of GFAP immunoreactivity decreased in the RSC area, while there was no significant change in the entire hippocampus (analysed as whole area) (Fig. 5A, B), nor in the auditory or olfactory cortex areas (not shown) compared to the PBS control group. In order to explore more specifically the effects on astrocytes in the vicinity of Aβ plaques, we double-stained brain sections for GFAP and Aβ and analysed the degree of colocalization of immunoreactivity. The results showed increased colocalization of GFAP and Aβ staining in PBS-treated AppNL-G-F mice compared to WT mice, and significantly less colocalization of Aβ and GFAP staining in the entire hippocampus and RSC of AppNL-G-F mice after rh Bri2 BRICHOS R221E treatment (Fig. 5C, D). Similar to Iba1, GFAP immunoreactivity in WT mice was not affected by rh Bri2 BRICHOS R221E treatment (Fig. 5).
Fig. 5. Rh Bri2 BRICHOS R221E treatment mitigates astrogliosis in AppNL-G-F mice.

Representative images of brain sections from vehicle (veh)- and rh Bri2 BRICHOS R221E-treated AppNL-G-F (App) and WT mice stained by immunohistochemistry for GFAP (green, rows A, B) and for GFAP and Aβ (green and red, rows C, D). The areas marked with boxes in (C, D) are magnified in the panels shown below them. Scale bars represent 1000 μm (A, C), 400 μm (B, D), and 200 μm (magnified areas) and apply to all panels in the respective row. Bar charts to the right show the GFAP-positive area (%) in the hippocampus (A) and retrosplenial cortex (RSC) (B), or the GFAP and Aβ colocalized (co-loc) area in the hippocampus (C) and RSC (D). Data are shown as mean ± SEM (n = 6–7 mice/group). One-way ANOVA test and Tukey’s test were used for statistical analysis. ∗p < 0.05, ∗∗∗∗p < 0.0001. The entire hippocampus was analysed. GFAP glial fibrillary acidic protein, RSC retrosplenial cortex.
Taken together, these results indicate that i.v. rh Bri2 BRICHOS R221E treatment was able to reduce Aβ-induced astrogliosis and microgliosis in aged AppNL-G-F mice.
Impact of rh Bri2 BRICHOS R221E treatment on gene expression in microglia differs between WT and AppNL-G-F mice
As we observed robust effects of rh Bri2 BRICHOS R221E treatment on microglial activation (Fig. 4), we isolated cortical microglial cells from three mice from each of the four treatment groups, i.e. WT and AppNL-G-F mice treated with either PBS (veh) or rh Bri2 BRICHOS R221E, and performed bulk mRNA sequencing. Comparison of AppNL-G-F with age-matched WT mice, both receiving veh, using a p-value threshold of < 0.05, identified 3470 upregulated genes and 3044 downregulated genes (Fig. 6A). Using the same p-value cut-off we found 288 upregulated genes and 962 downregulated genes between treatment with rh Bri2 BRICHOS R221E or veh in AppNL-G-F mice, and 829 upregulated genes and 370 downregulated genes between treatment with rh Bri2 BRICHOS R221E or veh in WT mice (Fig. 6B, C). There is a 5–7-fold higher number of genes affected in AppNL-G-F vs WT mice compared to the number of genes affected by rh Bri2 BRICHOS R221E vs PBS treatment of AppNL-G-F or WT mice (Fig. 6A–C). This is likely explained by the more profound (genetic) differences between AppNL-G-F and WT mice, compared to the three months of rh Bri2 BRICHOS R221E or PBS treatment. Cluster heatmap analysis of the 100 genes that showed the highest variance between all four treatment groups (Supplementary Fig. 1) confirmed that mouse strain (AppNL-G-F vs WT) had a stronger influence on gene expression levels as compared to treatment (rh Bri2 BRICHOS R221E vs veh). Microglial genes associated with AD, including Trem2, Spp1, Tyrobp, and Cd74, were among the upregulated genes in AppNL-G-F vs WT mice, supporting the activation of microglia in AppNL-G-F mice, consistent with previous findings [33–35].
Fig. 6. Differentially expressed genes (DEGs) in microglia from AppNL-G-F and WT mice.

A–C Volcano plots showing DEGs between A vehicle (veh)-treated AppNL-G-F (App) and WT mice; B rh Bri2 BRICHOS R221E- and veh-treated AppNL-G-F mice, and C rh Bri2 BRICHOS R221E- and veh-treated WT mice. N = 3 mice/group. Different scales are used in panels (A–C) for optimal resolution of gene symbols. Vertical dotted lines indicate | log2fold change | ≥1 border. The ten genes identified in panel (B) are further analysed in Fig. 8. The number of down- or up-regulated genes is indicated for each panel. D Venn diagrams of DEGs in veh-treated WT and AppNL-G-F mice (green circle), veh- and rh Bri2 BRICHOS R221E-treated AppNL-G-F mice (red circle), and veh- and rh Bri2 BRICHOS R221E-treated WT mice (blue circle). The identities of the genes in the different intersections between the circles are given in the Supplementary Excel file 1.
The 1250 differentially expressed genes (DEGs) in AppNL-G-F mice treated with rh Bri2 BRICHOS R221E were mainly downregulated, while the same treatment of WT mice resulted in 1199 DEGs, and most of them were upregulated (Fig. 6B–D). This indicates that rh Bri2 BRICHOS R221E treatment affects AD-relevant genes that are upregulated in the AppNL-G-F mice vs WT mice. The 48/121 DEGs that overlapped between rh Bri2 BRICHOS R221E vs veh treated WT mice and rh Bri2 BRICHOS R221E vs veh treated AppNL-G-F mice and also are differentially regulated in AppNL-G-F vs WT mice (Fig. 6D) represent several different biological processes without known relevance for AD-like pathology (Supplementary Excel file 1). Finally, gene set enrichment analysis [36, 37] of the DEGs between treatment with rh Bri2 BRICHOS R221E or veh in WT mice (Supplementary Fig. 2) showed that many of them were associated with cellular shape and movement processes and membrane channel functions.
Rh Bri2 BRICHOS R221E treatment of AppNL-G-F mice affects the expression of AD-related genes in microglia
To further analyse the DEGs in AppNL-G-F mice as a result of rh Bri2 BRICHOS R221E treatment compared to veh (Fig. 6B, D), we performed a cluster heatmap analysis of the 100 most altered of these genes (Fig. 7). These 100 genes were selected based on the lowest p-values of fold changed expression between AppNL-G-F rh Bri2 BRICHOS R221E and AppNL-G-F veh groups. Inspection of the heatmap in Fig. 7 shows that many of the DEGs in AppNL-G-F mice as a result of rh Bri2 BRICHOS R221E treatment appear to change their expression levels towards normalization, i.e. they approach the levels observed in WT mice treated with either rh Bri2 BRICHOS R221E or veh. Indeed, 57 of the 100 DEGs in Fig. 7 (marked as reverted) fulfilled the criteria that they showed (i) increased expression in veh-treated AppNL-G-F mice compared to veh-treated WT mice, and (ii) altered expression in rh Bri2 BRICHOS R221E-treated AppNL-G-F mice so that the expression levels approached that in veh-treated WT mice. Bar charts of the expression levels of these 57 genes in the four different treatment groups (Supplementary Fig. 3) show that in all cases the expression levels after rh Bri2 BRICHOS R221E treatment of AppNL-G-F mice were fully reverted to the levels in the veh-treated WT mice. Moreover, for essentially all of these 57 genes the expression levels were the same for rh Bri2 BRICHOS R221E- and veh-treated WT mice, corroborating the conclusion from analyses of the DEGs shown in Fig. 6 that rh Bri2 BRICHOS R221E treatment largely affects DEG´s between AppNL-G-F and WT mice. Next, we searched references to these 57 genes within the existing literature on genes relevant for AD or for Aβ aggregation or toxicity. This revealed that 20 of the 57 genes had been concluded to be relevant for the pathogenesis of AD and/or for Aβ aggregation or toxicity (Supplementary Table 1).
Fig. 7. Effects of Rh Bri2 BRICHOS R221E treatment on gene expression.
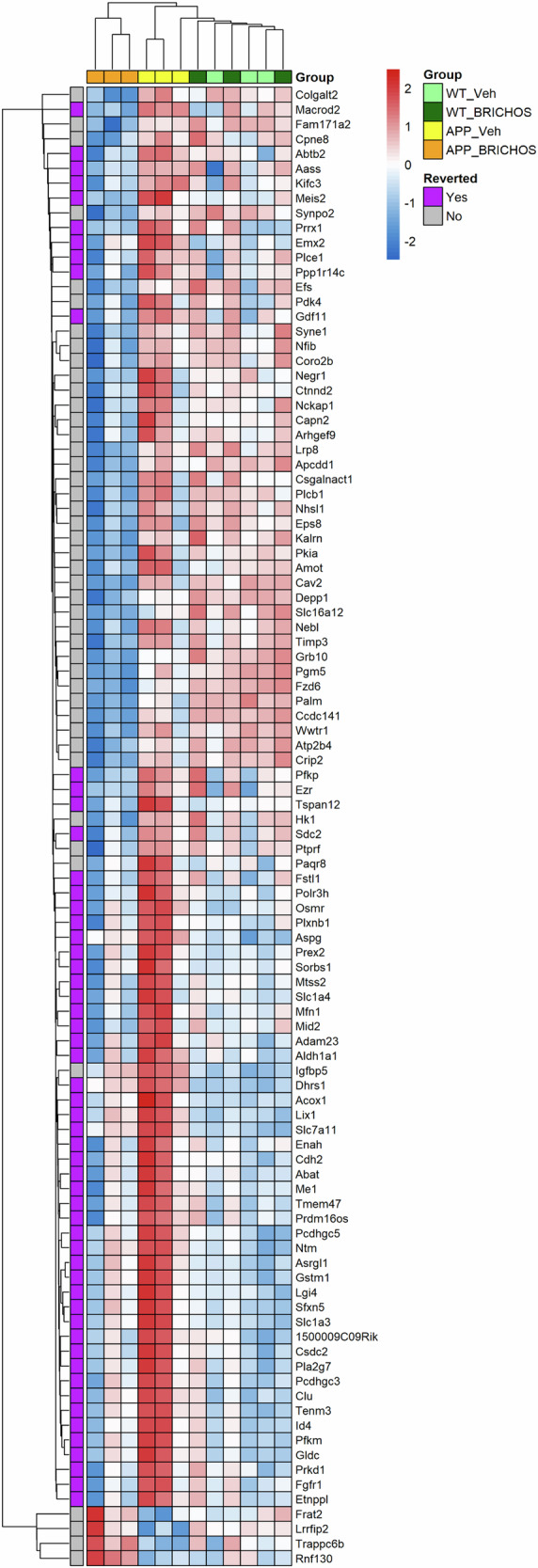
Heatmap for all four treatment groups for the 100 top differentially expressed genes (DEGs) between rh Bri2 BRICHOS R221E- and vehicle (veh)-treated AppNL-G-F (App) and WT mice. The genes were selected based on p-values for changed gene expression between AppNL-G-F mice given veh or BRICHOS, with the gene order determined by the clustering outcome. The genes reverted by rh Bri2 BRICHOS R221E treatment are further analysed in Supplementary Fig. 3 and discussed in the text. A row-based pseudo-scale has been used for the heatmap colouring, with the highest expression in red and the lowest in blue. n = 3 mice/group. Heat colouring is shown as | log2fold change | ≥1 and p < 0.05.
Comparison of genes affected by rh Bri2 BRICHOS R221E treatment with plaque-induced genes and genes specific for subpopulations of microglia
Next, we compared the 1250 microglial DEGs identified between treatment with rh Bri2 BRICHOS R221E or veh in AppNL-G-F mice (Fig. 6B, D) with those identified in previously published studies of genes and microglial populations found to be relevant in AD and AD model mice (Fig. 8). Chen et al. [33] identified so-called plaque-induced genes (PIGs) that were found to be relevant both for AppNL-G-F mice and human AD. Four of the 53 PIGs [33] were found among the 1250 DEGs in AppNL-G-F mice treated with rh Bri2 BRICHOS R221E vs veh as well as in the AppNL-G-F vs WT mice treated with veh (Fig. 8A). Bar charts of the expression levels of these four genes (Clu, Gfap, Igfbp5, and Prdx6) in the four different treatment groups (Fig. 8A) show that they are upregulated in AppNL-G-F vs WT mice, and, except for Prdx6, significantly downregulated by treatment with rh Bri2 BRICHOS R221E towards the levels seen in veh-treated WT mice, while the treatment of WT mice caused no change in the expression levels.
Fig. 8. Genes affected by rh Bri BRICHOS R221E treatment in relation to plaque-induced genes and genes in microglia subpopulations.

Venn diagrams illustrating differentially expressed genes (DEGs) in the vehicle (veh)-treated WT and AppNL-G-F (App) mice (green circle), veh- and rh Bri2 BRICHOS R221E-treated AppNL-G-F mice (red circle), and A plaque-induced genes (PIGs, blue circle); B activated response microglia genes (ARMs, blue circle); and C disease-associated microglia genes (DAMs, blue circle). Histograms of normalized (NOR) counts for the four treatment groups of genes in the indicated intersections for panels (A, B) are shown to the right. For panel C, the genes in the indicated intersection are identified in Supplementary Excel file 2, together with genes in intersections from panels (A, B). See main text for details. n = 3 mice/group, one-way ANOVA test and Tukey’s test were used for statistical analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, 0.05 < #p < 0.1.
The expression patterns of the four PIGs affected by rh Bri2 BRICHOS R221E in Fig. 8A may indicate that rh Bri2 BRICHOS treatment affects the plaques so that the release of compounds that can activate microglia is reduced, which would be in accordance with the ability of rh Bri2 BRICHOS R221E to block fibril surface catalysed generation of toxic Aβ42 species [38, 39]. The expression levels of the PIGs Trem2 and ApoE, both known to be associated with risk of developing AD, were increased in AppNL-G-F mice compared to WT mice, and treatment with rh Bri2 BRICHOS R221E resulted in a further increase, ca. 20% (Supplementary Fig. 4). The effects of rh Bri2 BRICHOS R221E on Trem2 and Apoe, as on Clu, Gfap, Igfbp5 and Prdx6 are likely related to the AD-like pathology in the AppNL-G-F mice, since rh Bri2 BRICHOS R221E treatment of WT mice caused no changes in Clu, Gfap, Igfbp5 and Prdx6, Trem2 or Apoe expression levels (Fig. 8A and Supplementary Fig. 4). Analysis of rh Bri2 BRICHOS R221E treatment effects on ligand-receptor pathways [40] (Supplementary Fig. 5) showed that receptor-mediated endocytosis and innate immunity pathways were affected in AppNL-G-F mice, while in WT mice the effects were linked to several different processes. Taken together, these results suggest that rh Bri2 BRICHOS R221E treatment may enhance the microglial response to amyloid deposits, promoting a more effective clearance, while at the same time mitigating overall microglial activation as seen by reduced staining for Iba1 (Fig. 4).
Activated response microglia (ARMs) [35] have been defined as a subpopulation that expresses known AD risk genes. Of the 1250 DEGs in AppNL-G-F mice treated with rh Bri2 BRICHOS vs veh, seven overlapped with ARMs (Fig. 8B). Clu is found among these genes, but three of the remaining six genes were not up- or down-regulated in AppNL-G-F vs WT mice, and none of them was normalized in AppNL-G-F mice after treatment with rh Bri2 BRICHOS R221E (Fig. 8B).
Finally, disease-associated microglia (DAMs) represent a population that can restrict neurodegeneration in AD and AD model mice [41]. Of the 1250 DEGs in AppNL-G-F mice treated with rh Bri2 BRICHOS vs vehicle, 212 (17%) overlap with DAMs (Fig. 8C and Supplementary Excel file 2). Clu, but none of the other PIGs affected by rh Bri2 BRICHOS R221E are found among these 212 genes (Supplementary Excel file 2).
Discussion
Early diagnosis of AD presents a challenge due to the subtle nature of symptoms, often initially attributed to normal ageing. Consequently, AD is typically identified in its later stages, when individuals begin to undergo noticeable cognitive decline and memory impairment [42]. In the current study, we explored intravenous administration of rh Bri2 BRICHOS R221E in a late-stage AD mouse model. The results revealed improvements across various AD-related parameters, encompassing reduction in Aβ plaque deposition, and mitigated overall astro- and microgliosis, as well as modulation of AD-relevant microglial gene expression. The BRICHOS domain, in particular rh Bri2 BRICHOS R221E, thus emerges as a compelling candidate for the treatment of AD, holding promises also against the challenges posed by late-stage pathology.
The recent positive treatment effects in phase III clinical trials using monoclonal antibodies [2] suggest that the approach to target Aβ represents a valid strategy for treating AD, particularly in its early stages. The BRICHOS domain has been demonstrated to inhibit Aβ from forming amyloid fibrils by binding to the fibril surface, affecting secondary nucleation, a major pathway for generating neurotoxic amyloid oligomers [27, 30, 31, 43]. Reactive astrocytes and microglia are frequently observed in close vicinity to Aβ plaques in cases of AD [44–47]. The intrinsic pro-inflammatory properties of Aβ, coupled with its neurotoxic effects, lead to persistent activation of microglia, astrocyte proliferation, and a continuous release of inflammatory factors. This, in turn, contributes to further Aβ production [11]. Rh Bri2 BRICHOS R221E was shown to reduce plaque load and neuroinflammation, and to improve memory and cognition when starting the treatment at 3 months of age in AppNL-G-F mice [24]. Importantly, our current study markedly expanded the potential treatment window of rh Bri2 BRICHOS R221E to advanced AD stages.
Microglia participate in brain inflammation and are instrumental in the clearance of cellular debris [48, 49]. Genome-wide association studies (GWAS) revealed that a majority of AD risk genes are predominantly or exclusively expressed in microglia [50], suggesting that microglia play a significant role in AD development. Despite the advantageous functions of microglia, such as phagocytosis of Aβ, their excessive or unmanaged activation and proliferation can lead to the release of pro-inflammatory mediators, thereby contributing to neuronal cell death [11] and induction of Aβ production [51]. Here we show that treatment with rh Bri2 BRICHOS R221E resulted in an overall reduction in microglial activation, as evidenced by Iba1 immunohistochemistry, as well as altered expression levels of several PIGs.
The normalisation of expression of Clu, Gfap, Igfbp5 and Prdx6 by treatment with rh Bri2 BRICHOS R221E is noteworthy. The risk of developing AD is associated with common polymorphisms within the human clusterin gene (CLU) [52, 53]. Clusterin, also known as apolipoprotein J (ApoJ), can bind Aβ and affect its solubility, thereby preventing the formation of amyloid fibrils [54–56], suggesting that it is protective against amyloid-mediated neurotoxicity [57]. CLU upregulation may play a protective function [52, 58–60], but a correlation between increased CLU and brain atrophy and rapid clinical progression in AD patients [61] has also been observed. GFAP, suggested as a blood-based biomarker of AD [62], is upregulated in reactive astrocytosis, and a component of the neuropathological alterations seen in AD [63]. Plasma GFAP concentrations are elevated in individuals with AD or at risk of developing AD, and the levels apparently correlate with cognitive impairment [64–68]. Moreover, a close association between the concentration of GFAP in plasma and the decline in cognitive function in patients with AD has been observed [64, 65, 69, 70]. Further studies are needed of the gene expression patterns in other cells and brain areas than the cortical microglia now analysed. It will also be important to find out whether any of the gene expression changes detected here, in particular down-regulation of Gfap, is associated with altered GFAP levels in plasma or CSF. Insulin-like growth factor binding protein 5 (IGFBP-5) has been found to be upregulated in the hippocampus and cortex of an AD mouse model and in the cerebrospinal fluid of AD cases [71, 72]. This could contribute to neurodegeneration through oxidative stress, Aβ toxicity, and tauopathy [73, 74], and IGFBP-5 may influence AD pathology via direct interactions with Aβ [75]. Finally, peroxiredoxin-6 (PRDX6) has antioxidative effects and its expression is upregulated in AD [76]. The expression levels of Prdx6 in mouse models were observed to correlate with the recruitment of astrocytes and microglia to Aβ plaques, the infiltration of these plaques by activated astrocytes, and the phagocytic activation of periplaque microglia [77].
In addition to Clu, Gfap, Igfbp5, and Prdx6, rh Bri2 BRICHOS R221E treatment also affected the expression of Trem2 and ApoE. It is interesting to note that activation of the TREM2-ApoE pathway was found to shift microglia towards a phagocytic state with activity against neuritic amyloid plaques and apoptotic neurons [78], and that the ApoE3 isoform was more efficient in increasing TREM2 expression and in mounting an Aβ-induced microglial response compared to the ApoE4 isoform, which is associated with increased risk of developing AD [79]. Moreover, it was recently shown that Bri2 BRICHOS and TREM2 proteins interact in transfected cells and that deletion of Bri2 (ITM2B) expression reduces phagocytosis similarly to a pathogenic TREM2 variant [80]. The activities of phagocytosis clearance of the tissue and downregulating inflammation are generally considered part of the resolution of inflammation, see [81, 82]. Failure in this process leads to chronic inflammation, which is known as part of AD pathology [83, 84], and there are indeed experiments indicating that the resolution of inflammation is disturbed in AD [85–89]. The data presented herein suggest that that rh Bri2 BRICHOS R221E treatment enhances the microglial response to amyloid deposits, and at the same time mitigates overall microglial activation. The basis and implications of these observations require further studies.
Materials and methods
Animals
AppNL-G-F mice [28] and their background strain C57BL/6J (Jackson Laboratory, Bar Harbour, ME, USA) were maintained in temperature-controlled environments with a regular 12-h light/12-h dark cycle and with free access to food and water. The experiment was performed on male mice.
Treatments
Forty male AppNL-G-F and forty WT mice were randomly selected and equally divided into four groups. The mice were treated for 12 weeks with i.v. injections of PBS (vehicle) or rh Bri2 BRICHOS R221E (10 mg/kg, n = 10/group) twice a week (total of 24 injections) starting from the age of 9 months. The mice were anaesthetized with 2% isoflurane by inhalation in a plastic induction chamber for a short period of time necessary to administer the i.v. injection.
After twelve weeks of treatment, and another four weeks without treatment as used in previous studies [24] the mice were anaesthetized and perfused intracardially with physiological saline. The brains were dissected out and divided into hemispheres. The left hemispheres were further dissected into the olfactory bulb, hippocampus, cerebral cortex, brain stem and cerebellum, frozen in dry ice and stored at −80 °C for biochemical analyses. The right hemispheres were fixed in 4% paraformaldehyde (PF) in 0.1 M phosphate buffer and stored at +4 °C for immunohistochemical analysis. The experimenters performing the analyses were blinded to the nature of the intervention group the sample originated from.
Rh Bri2 BRICHOS R221E protein expression and purification
A gene coding for human Bri2, residues 113–231, with Arg at position 221 replaced with Glu, referred to as Bri2 BRICHOS R221E, was fused to a gene fragment coding for the NT* solubility tag followed by a thrombin cleavage site, and expressed in Shuffle T7 Escherichia coli cells as described previously [27]. Shuffle T7-competent E. coli cells were cultured at +30 °C in lysogeny broth (LB) medium supplemented with 15 μg/mL kanamycin. The temperature was lowered to +20 °C after the optical density at 600 nm (OD600) reached 0.8–1.0, and after adding 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) the cells were incubated overnight. The cells were collected and resuspended in 20 mM Tris-HCl (pH 8.0). The cells were lysed with a 5-min sonication on ice (2 s on, 2 s off, 65% power), followed by centrifugation at +4° (24,000×g) for 30 min. The resulting supernatant with target proteins was purified with an immobilized metal affinity chromatography (IMAC) column (Ni Sepharose 6 Fast Flow; GE Healthcare, UK) equilibrated with 20 mM Tris-HCl (pH 8.0). The fusion protein was eluted with 300 mM imidazole in 20 mM Tris-HCl (pH 8.0) and dialysed against 20 mM Tris-HCl (pH 8.0) overnight in a cold room. Subsequently, the NT*-Bri2 BRICHOS R221E fusion proteins were incubated with thrombin (1:600 enzyme-to-substrate weight ratio, Merck) for 72 h at +4 °C and applied onto a second IMAC column to remove the His6-NT∗ tag. The monomeric rh Bri2 BRICHOS R221E fractions were isolated using an ÄKTA system (GE Healthcare, UK) and a Superdex 75 PG column (GE Healthcare, UK). The resulting monomeric R221E proteins were dialysed against filtered and autoclaved PBS (pH 7.4), concentrated to the required concentration (~2 mg/mL), and filtered through a 0.2 μm Millex-GV filter (Merck Millipore, Ireland). Coomassie staining after SDS-PAGE was used to assess the final purity of rh monomeric Bri2 BRICHOS R221E.
Immunohistochemistry
Following dissection, the right brain hemispheres were placed in 4% PF and kept at +4 °C overnight. Next, the tissues were soaked in 30% sucrose for 24 h and then moved to a cryoprotectant solution and stored at −20°C. The brain hemispheres were cut in the coronal plane using a Leica cryostat to obtain 20 μm thick sections, which were stored free-floating in the cryoprotectant at −20 °C until further processing. The sections were transferred into 24-well plates with 0.01 M PBS and after three washes they were incubated for 20 min with PBS containing 0.3% Triton X-100 at room temperature (RT). Following another PBS wash, the sections were blocked for 30 min in a solution containing 0.1% Triton X-100 in PBS (PBST) and 5% normal donkey or goat serum. The sections were then incubated overnight at +4 °C with primary antibodies. Normal donkey serum in the absence of primary antibodies was used as a negative control. After a PBS wash, the sections were incubated with secondary antibodies for one h at RT. After three washes in PBST, the sections were mounted with an anti-fading mounting medium (ProLong™ Glass Antifade Mountant, Catalogue No. P36984). The primary and secondary antibodies used are listed in Supplementary Tables 2 and 3.
Image analysis
Images in four regions were analysed: hippocampus (approximately 1000–3800 µm posterior to bregma in the mouse brain), retrosplenial cortex (1000–2500 µm posterior to bregma), auditory cortex (around 2500–3800 µm posterior to bregma) and olfactory cortex hippocampal level (around 2800–4500 µm anterior to bregma) in coronal sections.
Images of the entire hippocampus were captured at 4× magnification and analysed for Iba1 and GFAP staining. The cortex regions, DG, and the CA2 and CA3 regions were captured at 10× and analysed for Bri2 and Aβ staining. A Nikon Eclipse microscope (Eclipse E800, Nikon, Japan) was used to capture the images. Three consecutive sections per animal were stained and subjected to quantitative analysis using Image J software (NIH, MD, USA). Background fluorescence values from negative controls (only stained with secondary antibodies) were subtracted from all images. The lowest threshold for detection of fluorescence signal from samples stained with primary and secondary antibodies was then manually determined for 20 images, including all treatment groups, and averaged. The area (in per cent of the total image area) that showed fluorescence above this threshold was averaged for three consecutive sections per animal. For each group, 6–7 animals were analysed.
Colocalization of GFAP and Aβ staining was quantitatively assessed using Image J software. Image adjustments for brightness, contrast and threshold were uniformly applied to sections double-stained with antibodies specific to GFAP and Aβ. These images were of identical dimensions and represented the same anatomical region. Subsequently, the GFAP-stained image was pseudo-coloured green and the Aβ-stained image red. Merging of these images resulted in a composite wherein areas of GFAP and Aβ colocalization appeared yellow. The percentage of the total area that showed yellow colour was then determined as for the single stainings described above, and plotted as per cent of colocalization.
Protein extraction
Soluble and insoluble protein fractions were extracted from the hippocampus and cerebral cortex. Fresh-frozen brain samples were mechanically homogenised by a 3-step extraction protocol. TRIS-buffered saline (TBS) (20 mM Tris-HCl, 150 mM NaCl, pH 7.4) containing cocktails of phosphatase and protease inhibitors (Chemical Co., Stockholm, Sweden, and Thermo Fisher Scientific, Stockholm, Sweden, respectively) was used for homogenization of the tissues. The supernatant was collected as a TBS fraction after centrifugation (56,000 rpm, +4 °C, 20 min). The remaining pellet was homogenised in TBST (TBS containing 1% Triton X-100) and centrifuged (56,000 rpm, +4 °C, 20 min). The supernatant was collected as a TBST fraction. The remaining pellet was sonicated (15 s on, 15 s off, 15 s on, 15 s off, 20% power) in 80% formic acid and centrifuged (56,000 rpm, +4 °C, 20 min), and the resulting supernatant collected as a formic acid fraction.
Microglia and RNA isolation
Papain dissociation kits (Worthington, Catalogue. No. LK003178) along with magnetic cell isolation were used to isolate CD11b-positive microglia from cerebral cortex samples [90] according to the manufacturer’s instructions. The cortical tissue was minced and incubated in papain enzyme solution at +37 °C for 40 min to digest the connective tissue. A single-cell pellet was collected by density gradient centrifugation. The cells were filtered through 70 μm cell strainers, incubated with magnetic beads conjugated with anti-CD11b antibodies and the CD11b-positive microglia were isolated. The microglial cells were suspended in RNAlater solution (Thermo Fisher, Catalogue No. AM7020) and stored at −80 °C until used.
The RNeasy micro kit (Qiagen, Germany) was used to isolate and purify RNA from the isolated microglia. Procedures were performed according to the manufacturer’s specifications. The quality and purity of the RNA was measured photometrically.
RNA-sequencing
Adaptor and read quality trimming were performed by Cutadapt (v3.5). Using MultiQC (v1.14) and FastQC (v0.11.8), the quality of the samples was evaluated. STAR (v2.7.9d) was used to align reads to the Ensembl GRCm38 reference genome. Gene counts were acquired by employing featureCounts (v1.5.1). For library size normalization of raw counts and sample group comparisons with Wald tests, the Bioconductor package DESeq2 (v1.36.0) was utilised. p-values adjusted for multiple testing (padj) were estimated using the Benjamini-Hochberg method. For differential gene expression analysis throughout the study, we applied specific cutoffs: log2FC > 1 (indicating upregulation, FC = fold change, 2-fold), log2FC < −1 (indicating downregulation, 2-fold), and p < 0.05.
Statistical analysis
All statistical analyses were performed in GraphPad Prism 10 (GraphPad, CA, USA) or Excel. The results were expressed as bar graphs as mean ± SEM. p < 0.05 was considered statistically significant, if not stated otherwise. Normal distribution was checked by the Shapiro–Wilk test. To analyse differences between treatments, Tukey’s test and one-way ANOVA were used to test for group differences. An unpaired parametric two-tailed t-test was used to test the difference between the two groups. Figure legends describe the type of statistical test, the definition of significance for various p-values, and the number of biological replicates (n) for each group. The DESeq2 comparisons and enrichment testing were done by R 4.3.1. The DESeq2 comparisons (Wald test statistics) are used for the selection of regulated genes (described in the Results section), volcano plots and enrichment analysis, while the bar chart p-values are based on ANOVA/t-tests of normalized counts. Enrichment analysis of functional groups was performed using the Bioconductor package fgsea together with records from the Molecular Signatures Database (MSigDB), accessible via Bioconductor package msigdbr [36, 91, 92]. Log2 fold changes from the DESeq2 comparisons were used as input data.
Supplementary information
Acknowledgements
We thank Drs. Per Nilsson and Simone Tambaro from Karolinska Institutet for providing AppNL-G-F mice, and Sofia Fredin and Zoë Tuinman for assisting with sampling of mouse tissues and immunohistoichemistry. This study was supported by the Swedish Research Council, Swedish Brain Foundation, U.S. Alzheimer’s Association, Olle Engkvists Stiftelse, the Petrus and Augusta Hedlunds Stiftelse, Åke Wibergs Stiftelse, the Swedish Alzheimer Foundation, Åhlén Stiftelsen, Karolinska Institutet Research Foundation Grant, Stiftelsen för Gamla Tjänarinnor, Stiftelsen Sigurd och Elsa Goljes Minne, Loo and Hans Osterman Foundation, Geriatric Diseases Foundation at Karolinska Institutet, Gun and Bertil Stohne’s Foundation, Magnus Bergvall foundation, and the China Scholarship Council. We also would like to thank the core facility at NEO, BEA, Bioinformatics and Expression Analysis, which is supported by the board of research at the Karolinska Institutet and the research committee at the Karolinska Hospital.
Author contributions
RZ, MO, AM, and YW performed experiments. RZ, MO, DB, AM, MS, GC, and JJ analysed data. JJ and MS conceptualised the study. RZ, GC, MS, and JJ wrote the manuscript. All authors commented on the manuscript.
Funding
Open access funding provided by Karolinska Institute.
Data availability
The RNA sequencing data are publically available under the GEO ID GSE263166. All data and materials related to this paper are available upon request.
Competing interests
The authors declare no competing financial interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. Procedures for handling and conducting animal experiments were carried out in compliance with the comparative medicine guidelines (KM-B, Karolinska Institutet, Sweden) and the experiments were approved by the Stockholm Ethical Committee for Animal Experiments (15949-2020). No human participants were included in any of the studies.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Marianne Schultzberg, Email: Marianne.Schultzberg@ki.se.
Gefei Chen, Email: Gefei.Chen@ki.se.
Jan Johansson, Email: Janne.Johansson@ki.se.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-024-03161-x.
References
- 1.Winblad B, Amouyel P, Andrieu S, Ballard C, Brayne C, Brodaty H, et al. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol. 2016;15:455–532. [DOI] [PubMed] [Google Scholar]
- 2.Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022;21:306–18. [DOI] [PubMed] [Google Scholar]
- 3.Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330:512–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388:9–21. [DOI] [PubMed] [Google Scholar]
- 5.Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–6. [DOI] [PubMed] [Google Scholar]
- 6.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- 8.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–31. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJHJ. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–72. [DOI] [PubMed] [Google Scholar]
- 11.Emre C, Arroyo-García LE, Do KV, Jun B, Ohshima M, Alcalde SG, et al. Intranasal delivery of pro-resolving lipid mediators rescues memory and gamma oscillation impairment in AppNL-G-F/NL-G-F mice. Commun Biol. 2022;5:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson MR, Satapathy S, Vendruscolo M. Extracellular protein homeostasis in neurodegenerative diseases. Nat Rev Neurol. 2023;19:235–45. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci. 2002;27:329–32. [DOI] [PubMed] [Google Scholar]
- 14.Hedlund J, Johansson J, Persson B. BRICHOS—a superfamily of multidomain proteins with diverse functions. BMC Res Notes. 2009;2:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leppert A, Poska H, Landreh M, Abelein A, Chen G, Johansson J. A new kid in the folding funnel: molecular chaperone activities of the BRICHOS domain. Protein Sci. 2023;32:e4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oskarsson ME, Hermansson E, Wang Y, Welsh N, Presto J, Johansson J, et al. BRICHOS domain of Bri2 inhibits islet amyloid polypeptide (IAPP) fibril formation and toxicity in human beta cells. Proc Natl Acad Sci USA. 2018;115:E2752–E2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willander H, Presto J, Askarieh G, Biverstal H, Frohm B, Knight SD, et al. BRICHOS domains efficiently delay fibrillation of amyloid beta-peptide. J Biol Chem. 2012;287:31608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martins F, Santos I, Da Cruz OAB, Tambaro S, Rebelo S. The role of the integral type II transmembrane protein BRI2 in health and disease. Cell Mol Life Sci. 2021;78:6807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Campo M, Hoozemans JJ, Dekkers LL, Rozemuller AJ, Korth C, Müller-Schiffmann A, et al. BRI2-BRICHOS is increased in human amyloid plaques in early stages of Alzheimer’s disease. Neurobiol Aging. 2014;35:1596–604. [DOI] [PubMed] [Google Scholar]
- 20.Del Campo M, Teunissen CE. Role of BRI2 in dementia. J Alzheimers Dis. 2014;40:481–94. [DOI] [PubMed] [Google Scholar]
- 21.Hermansson E, Schultz S, Crowther D, Linse S, Winblad B, Westermark G, et al. The chaperone domain BRICHOS prevents CNS toxicity of amyloid-beta peptide in Drosophila melanogaster. Dis Model Mech. 2014;7:659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poska H, Haslbeck M, Kurudenkandy FR, Hermansson E, Chen G, Kostallas G, et al. Dementia-related Bri2 BRICHOS is a versatile molecular chaperone that efficiently inhibits Aβ42 toxicity in Drosophila. Biochemical Journal. 2016;473:3683–704. [DOI] [PubMed] [Google Scholar]
- 23.Dolfe L, Tambaro S, Tigro H, Del Campo M, Hoozemans JJM, Wiehager B, et al. The Bri2 and Bri3 BRICHOS domains interact differently with abeta(42) and Alzheimer amyloid plaques. J Alzheimers Dis Rep. 2018;2:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manchanda S, Galan-Acosta L, Abelein A, Tambaro S, Chen G, Nilsson P, et al. Intravenous treatment with a molecular chaperone designed against beta-amyloid toxicity improves Alzheimer’s disease pathology in mouse models. Mol Ther. 2023;31:487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Abelein A, Nilsson HE, Leppert A, Andrade-Talavera Y, Tambaro S, et al. Bri2 BRICHOS client specificity and chaperone activity are governed by assembly state. Nat Commun. 2017;8:2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tambaro S, Galan-Acosta L, Leppert A, Chen G, Biverstal H, Presto J, et al. Blood–brain and blood-cerebrospinal fluid passage of BRICHOS domains from two molecular chaperones in mice. J Biol Chem. 2019;294:2606–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen G, Andrade-Talavera Y, Tambaro S, Leppert A, Nilsson HE, Zhong X, et al. Augmentation of Bri2 molecular chaperone activity against amyloid-beta reduces neurotoxicity in mouse hippocampus in vitro. Commun Biol. 2020;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17:661–3. [DOI] [PubMed] [Google Scholar]
- 29.Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, et al. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017;36:2473–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen SIA, Arosio P, Presto J, Kurudenkandy FR, Biverstål H, Dolfe L, et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nature Structural & Molecular Biology. 2015;22:207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R, Le Marchand T, Adam L, Bobrovs R, Chen G, Fridmanis J, et al. Identification of potential aggregation hotspots on Abeta42 fibrils blocked by the anti-amyloid chaperone-like BRICHOS domain. Nat Commun. 2024;15:965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galan-Acosta L, Sierra C, Leppert A, Pouliopoulos AN, Kwon N, Noel RL, et al. Recombinant BRICHOS chaperone domains delivered to mouse brain parenchyma by focused ultrasound and microbubbles are internalized by hippocampal and cortical neurons. Mol Cell Neurosci. 2020;105:103498. [DOI] [PubMed] [Google Scholar]
- 33.Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 2020;182:976–91.e919. [DOI] [PubMed] [Google Scholar]
- 34.Gedam M, Comerota MM, Propson NE, Chen T, Jin F, Wang MC, et al. Complement C3aR depletion reverses HIF-1alpha-induced metabolic impairment and enhances microglial response to Abeta pathology. J Clin Invest. 2023;133:e167501. [DOI] [PMC free article] [PubMed]
- 35.Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to abeta plaques. Cell Rep. 2019;27:1293–306.e1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abelein A, Johansson J. Amyloid inhibition by molecular chaperones in vitro can be translated to Alzheimer’s pathology in vivo. RSC Med Chem. 2023;14:848–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, Johansson J. Potential of molecular chaperones for treating Alzheimer’s disease. Neural Regeneration Research. 2024;19:2343–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badolati I, van der Heiden M, Brodin D, Zuurveld M, Szilagyi S, Bjorkander S, et al. Staphylococcus aureus-derived factors promote human Th9 cell polarization and enhance a transcriptional program associated with allergic inflammation. Eur J Immunol. 2023;53:e2250083. [DOI] [PubMed] [Google Scholar]
- 41.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–90.e1217. [DOI] [PubMed] [Google Scholar]
- 42.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson J. Treatment with BRICHOS domain helps to clarify issues with Alzheimer mouse models. EMBO Mol Med. 2024;16:715–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15. [DOI] [PubMed] [Google Scholar]
- 45.Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–21. [DOI] [PubMed] [Google Scholar]
- 46.Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: an integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2016;144:121–41. [DOI] [PubMed] [Google Scholar]
- 47.Olsen M, Aguilar X, Sehlin D, Fang XT, Antoni G, Erlandsson A, et al. Astroglial responses to amyloid-beta progression in a mouse model of Alzheimer’s disease. Mol Imaging Biol. 2018;20:605–14. [DOI] [PubMed] [Google Scholar]
- 48.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. [DOI] [PubMed] [Google Scholar]
- 49.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94. [DOI] [PubMed] [Google Scholar]
- 50.Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emre C, Do KV, Jun B, Hjorth E, Alcalde SG, Kautzmann MI, et al. Age-related changes in brain phospholipids and bioactive lipids in the APP knock-in mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2021;9:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9. [DOI] [PubMed] [Google Scholar]
- 54.Ghiso J, Matsubara E, Koudinov A, Choi-Miura NH, Tomita M, Wisniewski T, et al. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J. 1993;293:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsubara E, Soto C, Governale S, Frangione B, Ghiso J. Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem J. 1996;316:671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA, et al. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1–40) peptide. Nat Struct Mol Biol. 2011;19:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boggs LN, Fuson KS, Baez M, Churgay L, McClure D, Becker G, et al. Clusterin (Apo J) protects against in vitro amyloid-beta (1–40) neurotoxicity. J Neurochem. 1996;67:1324–7. [DOI] [PubMed] [Google Scholar]
- 58.May PC, Johnson SA, Poirier J, Lampert-Etchells M, Finch CE. Altered gene expression in Alzheimer’s disease brain tissue. Can J Neurol Sci. 1989;16:473–6. [DOI] [PubMed] [Google Scholar]
- 59.Koudstaal SchrijversEM, Hofman PJ, Breteler AMM. Plasma clusterin and the risk of Alzheimer disease. JAMA. 2011;305:1322–6. [DOI] [PubMed] [Google Scholar]
- 60.Schurmann B, Wiese B, Bickel H, Weyerer S, Riedel-Heller SG, Pentzek M, et al. Association of the Alzheimer’s disease clusterin risk allele with plasma clusterin concentration. J Alzheimers Dis. 2011;25:421–4. [DOI] [PubMed] [Google Scholar]
- 61.Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. 2010;67:739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benedet AL, Mila-Aloma M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78:1471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garwood CJ, Ratcliffe LE, Simpson JE, Heath PR, Ince PG, Wharton SB. Review: astrocytes in Alzheimer’s disease and other age-associated dementias: a supporting player with a central role. Neuropathol Appl Neurobiol. 2017;43:281–98. [DOI] [PubMed] [Google Scholar]
- 64.Verberk IMW, Thijssen E, Koelewijn J, Mauroo K, Vanbrabant J, de Wilde A, et al. Combination of plasma amyloid beta((1–42/1–40)) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res Ther. 2020;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chatterjee P, Pedrini S, Stoops E, Goozee K, Villemagne VL, Asih PR, et al. Plasma glial fibrillary acidic protein is elevated in cognitively normal older adults at risk of Alzheimer’s disease. Transl Psychiatry. 2021;11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simren J, Leuzy A, Karikari TK, Hye A, Benedet AL, Lantero-Rodriguez J, et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimers Dement. 2021;17:1145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elahi FM, Casaletto KB, La Joie R, Walters SM, Harvey D, Wolf A, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early- and late-onset Alzheimer’s disease. Alzheimers Dement. 2020;16:681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oeckl P, Halbgebauer S, Anderl-Straub S, Steinacker P, Huss AM, Neugebauer H, et al. Glial fibrillary acidic protein in serum is increased in Alzheimer’s disease and correlates with cognitive impairment. J Alzheimers Dis. 2019;67:481–8. [DOI] [PubMed] [Google Scholar]
- 69.Cicognola C, Janelidze S, Hertze J, Zetterberg H, Blennow K, Mattsson-Carlgren N, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. 2021;13:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asken BM, Elahi FM, La Joie R, Strom A, Staffaroni AM, Lindbergh CA, et al. Plasma glial fibrillary acidic protein levels differ along the spectra of amyloid burden and clinical disease stage. J Alzheimers Dis. 2020;78:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barucker C, Sommer A, Beckmann G, Eravci M, Harmeier A, Schipke CG, et al. Alzheimer amyloid peptide abeta42 regulates gene expression of transcription and growth factors. J Alzheimers Dis. 2015;44:613–24. [DOI] [PubMed] [Google Scholar]
- 72.Salehi Z, Mashayekhi F, Naji M. Insulin like growth factor-1 and insulin like growth factor binding proteins in the cerebrospinal fluid and serum from patients with Alzheimer’s disease. Biofactors. 2008;33:99–106. [DOI] [PubMed] [Google Scholar]
- 73.Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Freude S, Hettich MM, Schumann C, Stohr O, Koch L, Kohler C, et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009;23:3315–24. [DOI] [PubMed] [Google Scholar]
- 75.Rauskolb S, Andreska T, Fries S, von Collenberg CR, Blum R, Monoranu CM, et al. Insulin-like growth factor 5 associates with human Ass plaques and promotes cognitive impairment. Acta Neuropathol Commun. 2022;10:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Power JH, Asad S, Chataway TK, Chegini F, Manavis J, Temlett JA, et al. Peroxiredoxin 6 in human brain: molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008;115:611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pankiewicz JE, Diaz JR, Marta-Ariza M, Lizinczyk AM, Franco LA, Sadowski MJ. Peroxiredoxin 6 mediates protective function of astrocytes in Abeta proteostasis. Mol Neurodegener. 2020;15:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–81.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fitz NF, Nam KN, Wolfe CM, Letronne F, Playso BE, Iordanova BE, et al. Phospholipids of APOE lipoproteins activate microglia in an isoform-specific manner in preclinical models of Alzheimer’s disease. Nat Commun. 2021;12:3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yin T, Yesiltepe M, D’Adamio L. Functional BRI2-TREM2 interactions in microglia: implications for Alzheimer’s and related dementias. EMBO Rep. 2024;25:1326–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dyall SC, Balas L, Bazan NG, Brenna JT, Chiang N, da Costa, et al. Polyunsaturated fatty acids and fatty acid-derived lipid mediators: Recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res. 2022;86:101165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Panigrahy D, Gilligan MM, Serhan CN, Kashfi K. Resolution of inflammation: an organizing principle in biology and medicine. Pharmacol Ther. 2021;227:107879. [DOI] [PubMed] [Google Scholar]
- 83.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016;388:505–17. [DOI] [PubMed] [Google Scholar]
- 85.Do KV, Hjorth E, Wang Y, Jun B, Kautzmann MI, Ohshima M, et al. Cerebrospinal fluid profile of lipid mediators in Alzheimer’s disease. Cell Mol Neurobiol. 2023;43:797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Emre C, Hjorth E, Bharani K, Carroll S, Granholm AC, Schultzberg M. Receptors for pro-resolving mediators are increased in Alzheimer’s disease brain. Brain Pathol. 2020;30:614–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, et al. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, Zhu M, Hjorth E, Cortés-Toro V, Eyjolfsdottir H, Graff C, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer’s & Dementia. 2015;11:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC, et al. Pro-resolving lipid mediators improve neuronal survival and increase Aβ42 phagocytosis. Mol Neurobiol. 2016;53:2733–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Holt LM, Stoyanof ST, Olsen ML. Magnetic cell sorting for in vivo and in vitro astrocyte, neuron, and microglia analysis. Curr Protoc Neurosci. 2019;88:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data are publically available under the GEO ID GSE263166. All data and materials related to this paper are available upon request.