

Abstract
Epithelial ovarian carcinoma (EOC) is the most fatal among female reproductive system tumors. The immune tumor microenvironment and ubiquitin-proteasome pathway are closely related to the proliferation, invasion, and response to chemotherapy in EOC. However, their specific roles in EOC have not been fully elucidated. Therefore, we aimed to recognize potential prognostic markers and novel therapeutic targets for EOC. We constructed the ubiquitin-related signature risk model comprising HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, EPB41L2, DNAJB6, VPS18, PPM1G, AKAP12, FRK, and PYGB, specifically for patients with EOC. The high-risk model presented a worse prognosis, primarily associated with the B-cell receptor signaling pathway, ECM receptor interaction, focal adhesion, and actin cytoskeleton regulations. Analysis of the immune landscape revealed a higher abundance of B cells, M2 macrophages, neutrophil CD4 T cells, cancer-associated fibroblasts, macrophage neutrophils, and fibroblasts in the high-risk group. It also exhibited lower tumor mutation burden, mRNAsi, and EREG-mRNAsi and reduced sensitivity to other chemotherapy drugs, except dasatinib. These findings serve as a valuable indicator for personalized treatment strategies and clinical stratification in managing patients with EOC. Additionally, our study will serve as a foundation for future mechanistic research to explore the association between the ubiquitin-proteasome pathway and EOC.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-76945-2.
Subject terms: Cancer, Computational biology and bioinformatics, Genetics, Immunology, Oncology, Risk factors, Biomarkers
Introduction
The mortality rate for ovarian cancer (OC) surpasses those of other female reproductive system tumors. The National Center for Health Statistics (NCHS) reported that OC ranks as the fifth most common female tumor and accounts for 5% of female deaths1. This disease poses a significant threat to the health of women. Epithelial ovarian carcinoma (EOC) is a heterogeneous disease marked by variations in molecular composition and histological features, accounting for approximately 90% of all OC cases2. EOC tends to develop deep within the pelvic cavity and exhibits limited early clinical manifestations, leading to delayed diagnosis in most patients until the disease has advanced significantly. Moreover, patients with EOC exhibit a high rate of postoperative recurrence and frequently show chemotherapy resistance3,4. Consequently, EOC has the highest mortality rate among all gynecological malignancies1. Thus, identifying valuable diagnostic biomarkers, therapeutic targets, and prognostic factors for patients with EOC is crucial.
Protein ubiquitination serves as a signaling mechanism for various cellular processes, including proteasomal degradation, cell cycle regulation, and transcriptional regulation5–9. In EOC, the ubiquitin-proteasome pathway closely interconnects with proliferation, invasion, and chemotherapy responses, and many E3 ubiquitin ligases exhibit abnormal expression patterns. These include the regulation of BRCA1, a gene associated with OC, and p53, a tumor suppressor. Additionally, E3 ubiquitin ligases interfere with the ERK pathway, ERBB2gene expression, and cyclin-dependent cell cycle regulation process10. The ubiquitin-proteasome pathway alters the tumor microenvironment (TME), influencing anti-tumor activity and tumor migration in OC. For instance, UBR5, which has a key role in the ubiquitin proteasome pathway, promotes the recruitment and activation of tumor-associated macrophages through chemokines and cytokines and is critical to the progression of OC11.
The TME encompasses tumor cells and the surrounding fibroblasts, immune and inflammatory cells, interstitial cells, glial cells, microvessels, and infiltrating biomolecules. Three features characterize the TME: hypoxia, chronic inflammation, and immunosuppression. These elements are interconnected, forming a complex network of mechanisms crucial in tumor development. Epithelial ovarian carcinoma is characterized by a highly immunosuppressive TME and distant metastasis. Single-cell RNA sequencing (scRNA-seq) in EOC has revealed its TME map and cell characteristics12. Ovarian cancer cells disrupt the function of dendritic cells (DCs) by disrupting their activation, antigen presentation ability, differentiation, and recruitment; thus, they evade immune control13. In the advanced stages of EOC, macrophages with M2-like phenotypes, which possess a low tumor-killing activity and high potential to promote immune suppression, tumor cell invasion, angiogenesis, and metastasis, are recruited14–16. Cross-talk between OC cells and TME components is related to tumor growth promotion and metastasis and the reprogramming of innate and adaptive immune responses. Despite increasing recognition of the involvement of the ubiquitin pathway and TME in OC, a specific scoring model has not yet been developed to accurately assess this connection. Constructing a risk score model based on ubiquitin-related genes would enable the prediction of overall survival (OS) and immunotherapy response in patients with EOC. Moreover, it would aid oncologists in developing more effective immunotherapy strategies for targeted treatment.
Regarding clinical utility, the prognostic model serves as a convenient tool for assessing OS and recurrence risk, and it holds the potential to contribute to personalized treatment for patients17. Advancements in research have facilitated the identification of patient subgroups with poor prognosis. This has promoted the exploration of alternative treatment strategies tailored for such patients, offering valuable insights into clinical treatment approaches18. Accordingly, this study aimed to determine potential prognostic markers and new therapeutic targets for EOC.
In the present study, we obtained differentially expressed genes (DEGs) by screening out RNAseq data from tissues of patients with EOC, using The Cancer Genome Atlas (TCGA) database, and normal tissues, using the Genotype-Tissue Expression (GTEX) database. These DEGs were intersected with 4299 ubiquitin-related genes of Genecards to establish a risk model for differential expression of ubiquitin-related genes (DEURGs). Clinical features, pathway enrichment, immune status, messenger RNA dry stemness (mRNAsi), epigenetically regulated mRNAsi (EREG-mRNAsi), tumor mutation load, and drug sensitivity were used to analyze the differences between the high- and low-expression groups of risk models. This study verifies the dependability of the risk model and the importance of predicting the prognosis of EOC, addresses that there is no effect of large-scale ubiquitination related genes on the prognosis of EOC, and will help oncologists develop more effective immunotherapy strategies by deepening the understanding of the mechanisms underlying immune infiltration in the TME in patients with EOC.
Results
Prognosis and TME Characteristics in Gene Clusters of a Ubiquitin-Related Risk Model in EOC.
We explored the biological behavior of the molecular patterns in the ubiquitin-related risk model. Subsequently, differential expression analyses were conducted. Using common DEGs, unsupervised clustering was presented. The TCGA-EOC cohort was classified into three gene clusters (Fig. 1A). We further investigated the prognostic implications of ubiquitin-related gene clusters by assessing OS. The results showed that patients in gene cluster A exhibited a longer OS than those of other clusters. However, patients in gene clusters B and C had a pessimistic prognosis (Fig. 1B). Gene cluster A exhibited the lowest risk scores among all the clusters (Fig. 1C).
Fig. 1.

Analyzing Prognosis and TME characteristics using Ubiquitin-Related risk model gene clusters for HGSOC. Consensus clustering matrix when k = 3. (B) OS curve comparison among the three clusters. (C) Distribution of risk score among the three clusters. (D) Estimation of immune, stromal, and estimate scores among the three clusters using the ESTIMATE algorithm. (E) Estimation of immune cell proportions among the three clusters using the CIBERSORT algorithm. (F) Estimation of immune cell proportions among the three clusters using the TIMER algorithm. (G) Estimation of immune cell proportions among the three clusters using the MCP-Counter algorithm.
Subsequently, we explored whether these three gene clusters exhibited distinct TME features. The results showed that cluster B exhibited a low immunological score (Fig. 1D). CIBERSORT analysis revealed that cluster B had the highest proportion of resting memory CD4 T cells and monocytes among all the clusters (Fig. 1E). The TIMER algorithm revealed that CD4 T cell and macrophage levels were highest in cluster C (Fig. 1F). The MCP-Counter algorithm indicated that in cluster B, the expression levels of the B lineage, neutrophils, and endothelial cells were higher than those in the others. However, fibroblasts had the highest expression levels in cluster C (Fig. 1G). Overall, the consistency observed between prognostic and TME features across the three gene clusters suggested the reliability and validity of this classification.
Construction of Ubiquitin-Related Signature for Patients with EOC.
We employed the limma package in R software to detect DEGs between normal and EOC tissues, with the aim of identifying genes implicated in TME regulation. The volcano plot depicted 6123 DEGs, with 3,073 and 3,050 upregulated and downregulated genes, respectively (Fig. 2A). A Venn plot was constructed depicting the number of DEGs and their intersection with ubiquitin-related genes in EOC (Fig. 2B). These overlapping genes were subsequently utilized as inputs for LASSO-Cox regression analysis. The LASSO and multivariate Cox analyses were performed on 1,530 ubiquitin-related genes in the TCGA cohort. This process was used for identifying the best risk genes to evaluate the prognoses of EOC patients. Figure 2C and D show the resulting variation trajectories of the respective variables in which LASSO regression analysis was conducted. Overall, twelve cancer-ubiquitin-related genes were selected: HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, EPB41L2, DNAJB6, VPS18, PPM1G, AKAP12, FRK, and PYGB. Analysis of the GEPIA website showed that there was no high probability of multicollinearity among genes (supplementary Table 3). Univariate Cox regression analysis of EOC patients revealed that seven factors (HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, DNAJB6, and PPM1G) exhibited a hazard ratio (HR) < 1, indicating a protective effect. Conversely, EPB41L2, VPS18, AKAP12, FRK, and PYGB were observed as risk factors, with HR > 1 (Fig. 2E). This was based on the median of the risk score derived from the risk formula that categorized patients into low- and high-risk groups. In the GEO database, there is no probe that covers all genes of the risk model, which cannot be externally verified. Therefore, in the TCGA database, the EOC patients were randomly divided into test and training groups at a ratio of 6:4 to validate the accuracy of the model. The Kaplan-Meier survival curves in the training and test cohorts indicated that patients categorized in the high-risk group exhibited a lower survival rate than those in the low-risk group (Fig. 2F–H). The ROC analysis revealed that the training group exhibited potential in predicting OS among patients in the TCGA cohort (AUC values of 0.737, 0.762, and 0.793 for 1-, 3-, and 5-year OS, respectively; [Fig. 2I]). Among the risk features in the test group, the 1-year OS had a high AUC value (Fig. 2J). The 3- and 5-year OS rates in the entire group showed a high AUC value, suggesting that the modified risk model exhibited superior early prediction accuracy (Fig. 2K). IHC staining of DEURGs was performed using the HPA database (Fig. 3A–K). Additionally, no protein FBXO9 expression was observed in the HPA database. The expression of DEURGs in high grade serous ovarian cacinoma (HGSOC) and normal tubal epithelial tissues was analyzed using qRT-PCR in 54 tissues (Fig. 4A–L, supplementary Table 4).
Fig. 2.

Construction of the Ubiquitin-Related Signature for Patients with OC. (A) Volcano map illustrating the differential expression of 6,123 genes in normal and OC tumor tissues. Red and blue dots denote upregulated and downregulated genes in tumor tissue, respectively. (B) Correlations between the signature and the five genes. The Venn diagram illustrates overlapping genes for two screening datasets. Overall, 1,530 genes were identified as ubiquitin-related genes. (C, D) LASSO regression analysis in the TCGA database. The determination of ‘lambda’ for selecting optimal gene signature. (E) Forest plot illustrating the prognostic ability of the 12 ubiquitin-related genes included in the risk signature for OC. (F–H) Kaplan–Meier survival analysis of patients with OC in different groups. (I–K) ROC curves evaluate the specificity and sensitivity of 1-, 3-, and 5-year OS based on the risk score from TCGA datasets.
Fig. 3.

Construction of the Ubiquitin-Related Signature for Patients with OC. Immunohistochemical staining of DEURGs in HPA. The expression of (A) HSP90AB1, (B) SIGMAR1, (C) STAT1, (D) SH3KBP1, (E) EPB41L2, (F) DNAJB6, (G) VPS18, (H) PPM1G, (I) AKAP12, (J) FRK, and (K) PYGB in the HPA.
Fig. 4.

Construction of the Ubiquitin-Related Signature for Patients with OC. The RNA expression of DEURGs through qRT-PCR. The expression of (A) HSP90AB1, (B) FBXO9,(C)SIGMAR1, (D) STAT1, (E) SH3KBP1, (F) EPB41L2, (G) DNAJB6, (H) VPS18, (I) PPM1G, (J) AKAP12, (K) FRK, and (L) PYGB, p < 0.05 indicates differential expression.
Establishment and Validation of a Nomogram Combined with Clinical Characteristics.
Based on the median of the risk score derived from the risk formula that categorized patients into low- and high-risk groups, the high-risk group exhibited more deaths among the training, test, and whole sets, suggesting that the clinical outcomes of low-risk patients were superior to those of high-risk patients, meanwhile, differences in the expression levels of the same gene within the risk model between high-risk and low-risk groups were also shown. (Fig. 5A–C). We considered the risk score as an independent protective factor (p < 0.001, 95% CI HR: 2.003–3.033) (Fig. 5D). Moreover, even after adjusting for age, stage, and grade, this association remained statistically significant (p < 0.001, 95% CI HR: 1.883–2.897) (Fig. 5E). To quantitatively predict the probability of survival, we constructed a nomogram that integrated the risk score and clinical characteristics (Fig. 5F). The nomogram achieved a C-index of 0.632. The calibration plots and ROC curves indicated the satisfactory performance of the derived nomogram. Similarly, the nomogram model indicated that the AUC values for the 1-year, 3-year, and 5-year OS predictive ability were 0.927, 0.928, and 0.912, respectively (Fig. 5G, H). By employing the ROC curve to compare the predictive value of various factors (including nomogram, risk score, age, grade, and stage) the nomogram showed excellent predictive capacity (Fig. 5I). Combining risk scores with clinical characteristics in the nomogram showed high specificity and sensitivity in predicting survival. These nomograms suggest the strong potential of the constructed risk model for prognostic prediction in patients with EOC.
Fig. 5.

Establishment and Validation of a Nomogram Combining Clinical Characteristics. (A–C) Prognosis value of the 12 ubiquitin-related genes model in the training, test, and entire sets. This includes displaying the predictive model, survival time, survival status, and heatmap illustrating the 12 genes. (D–E) Univariate and multivariate Cox regression analyses evaluating the prognostic value of the risk model score and clinical features. (F) Development of a clinical prognostic nomogram for predicting 1-, 3-, and 5-year survival. (G) Calibration curves illustrate the performance of the nomogram predictions for 1-, 3-, and 5-year survival. (H–I) Time-dependent ROC curve analyses assessing the ability to predict OS using risk scores and clinical features.
Analyzing Biological Pathways and Functional Enrichment in the Ubiquitin-Related Signature.
We conducted KEGG pathway and GO analyses by using GSEA to explore the underlying mechanisms contributing to the varied outcomes stratified by the risk model. The GO analysis, encompassing19–21 biological processes, molecular functions, and cellular components, contend that numerous functions and processes in low-risk patients were chiefly associated with metabolic-related biological processes and protein synthesis (Fig. 6A–C). The KEGG pathway enrichment analysis revealed that the top five considerably pathway terms in the low-risk group included DNA replication, oxidative phosphorylation, proteasomes, and spliceosomes (Fig. 6D). However, patients in the high-risk group were primarily concentrated in B cell receptor signaling pathway, ECM receptor interaction, focal adhesion, and regulation of the actin cytoskeleton (Fig. 6D). Furthermore, the hallmark pathway analysis revealed that E2F targets, MYC targets, V1, the G2M checkpoint, and oxidative phosphorylation were predominantly concentrated among low-risk patients. Conversely, high-risk patients exhibited dominance in signaling pathways that promote tumor progression, including epithelial-mesenchymal transition, TGF-β signaling, TNFα signaling via NFκB, and UV response DN (Fig. 6E). These findings characterized the ubiquitin-associated risk model, affirming its potential mechanism for assessing the prognosis of patients with EOC.
Fig. 6.

Biological Pathways and Functional Enrichment Analysis of the Ubiquitin-Related Signatures. (A–C) GO enrichment analysis, (D) KEGG enrichment analysis, and (E) hallmark pathway analysis.
Different TME and Immune Status in the Risk Score.
The correlation between the risk model and tumor-infiltrating immune cells was investigated to elucidate its correlation with immune-related biological pathways. The TCGA cohort was quantified using the ESTIMATE algorithm for immune, stromal, and infiltrating immune cells. The results showed a lower immune score in the low-risk group, suggesting infiltration of TME immune cells increased (Fig. 7A).
Fig. 7.

Varied TIME and immune status based on Risk Scores. (A) Comparison of immune scores and ESTIMATE scores between low- and high-risk patients using the ESTIMATE algorithm. (B) Estimation of immune cell proportions in low- and high-risk patients employing the CIBERSORT algorithm. (C) Estimation of immune cell proportions in low- and high-risk patients using the TIMER algorithm. (D) Estimation of immune cell proportions in low- and high-risk patients using the MCP-Counter algorithm. (E) Estimation of immune cell proportions in low- and high-risk patients utilizing the quantiseq algorithm. (F) Estimation of immune cell proportions in low- and high-risk patients using the EPIC algorithm. *p < 0.05; **p < 0.01; ***p < 0.001.
The CIBERSORT algorithm revealed a lower proportion of monocytes and macrophage M2 in the low-risk group (Fig. 7B). The study revealed a significant association between a low monocyte count and favorable survival outcomes, specifically regarding OS and progression-free survival (PFS) in patients with EOC. We further analyzed the risk model in the two subgroups to elucidate specificity differences among invasive immune cells. The CIBERSORT algorithm revealed that the high-risk group exhibited substantially higher numbers of B cells and macrophages (Fig. 7C). The MCP-Counter algorithm indicated that the high-risk group exhibited higher expression levels in neutrophils and fibroblasts than those of the low-risk group (Fig. 7D). The quantiSeq algorithm showed that the high-risk group had a greater percentage of B cells, M2 macrophages, and neutrophils (Fig. 7E). Moreover, the EPIC algorithm indicated a higher abundance of CD4 + T cells and cancer-associated fibroblasts (CAFs) in the high-risk group compared to those in the low-risk group (Fig. 7F).
Correlation Between mRNAsi, EREG-mRNAsi, Tumor Mutation Burden, and Risk Model.
We evaluated the relationship between stemness index (mRNA expression based-index, mRNAsi and epigenetically regulated mRNAsi, and EREG-mRNAsi) and the risk models. Gene expression is reflected by mRNAsi. The findings revealed that patients in the low-risk group exhibited higher mRNAsi and EREG mRNAsi expression levels than those of the high-risk group (Fig. 8A). Using TIDE to assess tumor immune dysfunction and rejection, the results showed elevated scores within the high-risk group. A high TIDE score proposes a greater potentiality of evading immune surveillance and a lower success rate for immunotherapy (Fig. 8B).
Fig. 8.

Relationships between risk model, mRNAsi, EREG-mRNAsi, and tumor mutation burden. (A) Differences of the stemness index in distinct risk groups. (B) Variations in the dysfunction index among distinct risk groups. (C) Differences in TMB among distinct risk groups. (D) Survival curve comparison between high and low TMB groups. (E) Survival curve based on risk score and TMB. (F) Waterfall plots illustrating the somatic mutations in high- and low-risk groups. *p < 0.05; **p < 0.01; ***p < 0.001.
The KM analysis of the TMB subtype revealed that the low-risk group exhibited a higher tumor mutation burden (TMB) score than that of the high-risk group. Patients with higher TMB scores universally experienced a better prognosis than those with lower TMB scores (Fig. 8C, D). Generally, the higher the TMB in most tumor cells, the more neoantigens that may be produced, leading to increased tumor immunogenicity. This suggests greater potential benefits from PD-1/PD-L1 immune checkpoint inhibitor (ICI) treatments. Patients in the low-risk group demonstrated a survival benefit regarding the TMB score despite having significantly lower performance (Fig. 8E). Consequently, we examined mutations in different risk subgroups. Somatic mutation distribution among low- and high-risk patients from the TCGA cohort was specifically evaluated. Additionally, we ranked the top 20 genes associated with each mutation. Figure 8F shows mutations frequency was higher in patients with low-risk scores (99.21%) than in those with high-risk scores (98.33%). Although the TMB was similar, the frequency of the top five mutated genes differed among various groups. In the high-risk group, they were TP53 (93%), TTN (25%), CSMD3 (13%), MUC16 (11%), and USH2A (11%). Conversely, the low-risk group showed TP53 (94%), TTN (28%), RYR2 (11%), CSMD3 (10%), and FAT3 (10%).
Correlation Between the Ubiquitin-Related Signature and Drug Sensitivity.
To assess the practicality of the risk score in clinical therapy, we analyzed the sensitivity to chemotherapeutic drugs in the subtype groups using the GDSC(genomics of drug sensitivity in cancer). The results showed that patients in the low-risk group demonstrated lower IC50 values for 5-fluorouracil, cisplatin, cyclophosphamide, docetaxel, oxaliplatin 1089, paclitaxel, sorafenib, and topotecan (Fig. 9A–C, E–I). Conversely, patients in the high-risk group exhibited a lower IC50 value for dasatinib than that of the low-risk group (Fig. 9D). These findings have significant implications; for instance, the low-risk group may exhibit more sensitivity to molecular-targeted drugs and common chemotherapeutic agents. However, dasatinib showed a therapeutic significance in high-risk groups. These findings indicate that the risk model can predict drug sensitivity in patients with EOC to some extent.
Fig. 9.

Analysis of the Correlation Between the Ubiquitin-Related Signature and Drug Sensitivity. Association between the Ubiquitin-Related signature risk model and drug sensitivity, encompassing chemotherapeutics and molecular targeting drug. (A) 5-Fluorouracil, (B) Cisplatin, (C) Cyclophosphamide, (D) Dasatinib, (E) Docetaxel, (F) Oxaliplatin, (G) Paclitaxel, (H) Sorafenib, (I), and Topotecan. IC50: half-maximal inhibitory concentration.
Discussion
Epithelial ovarian carcinoma is a highly aggressive tumor of the female reproductive system. Its incidence rises annually, and it ranks as the leading cause of mortality among malignant tumors of the female reproductive tract, being typically diagnosed at an advanced stage and exhibiting a high recurrence rate1,3.
The ubiquitin-proteasome system serves as one of the crucial mechanisms for degrading intracellular oncoproteins and tumor suppressor proteins. Moreover, it is the primary regulator of protein expression9,22. Recent findings highlight the significance of ubiquitin-related genes as potential therapeutic targets in cancer management. Recently, prognostic signatures derived from ubiquitin-related targets have been discovered in clear cell renal cell carcinoma, triple-negative breast cancer, hepatocellular carcinoma, and prostate cancer23–26. In this study, unsupervised clustering was conducted based on DEGs. A TCGA-EOC cohort was divided into three gene clusters. The results showed that cluster A exhibited longer OS time; cluster B showed low immune scores, the highest B lineage levels, and more neutrophils and endothelial cells. Furthermore, cluster C exhibited the highest T cell CD4 and macrophage levels. A comprehensive analysis was conducted to examine the clinical significance and characteristics of the TME associated with ubiquitin-related genes in EOC. Furthermore, a risk model of ubiquitin-related diseases was introduced to assess the proteasome pathway in individuals. This model may enhance our understanding of immune infiltration in the TME and aid oncologists in developing more effective and targeted immunotherapeutic strategies.
By analyzing the mRNA expression profiles of ubiquitin-related genes, we established a molecular pattern risk model to predict the prognosis of patients with EOC. Firstly, we utilized the limma package to identify differentially expressed genes (DEGs) between epithelial ovarian carcinoma and normal ovarian tissues, revealing 3,073 upregulated and 3,050 downregulated genes, respectively, that potentially contribute to EOC progression. Subsequently, we aimed to identify differential genes related to the regulation of ubiquitination modification. Leveraging the GeneCards database, we procured a list of 4,299 ubiquitin-associated genes. We employed a Venn diagram to intersect these ubiquitin-related genes with our differentially expressed ones, yielding 1,530 genes that were uniquely differentially expressed between cancer and normal tissues and are linked to ubiquitination. These genes may have a direct role in the progression of EOC. Given the absence of an efficacious prognostic model for ovarian cancer, we aspire to devise a ubiquitination-based risk prognostic model that can forecast EOC progression and patient outcomes with heightened accuracy and efficacy. To this end, univariate cox regression analysis was used to search for differentially expressed ubiquitin-related genes associated with prognosis, which may play a role in both tumor progression and tumor prognosis.
Further through the LASSO and multivariate Cox analyses, we established a prognostic signature for EOC that incorporates several ubiquitin-related genes: HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, EPB41L2, DNAJB6, VPS18, PPM1G, AKAP12, FRK, and PYGB. The risk model demonstrated strong performance in predicting survival and employed to observe the TME landscape and anti-tumor drug sensitivity. The heat shock protein 90-kDa alpha, class B member 1 (HSP90AB1), belongs to the HSP family and functions as a molecular chaperone27. In native and vitrified-thawed human ovarian cortical tissues, HSP90AB1maintains stable expression at the protein and RNA levels28. F-box-only protein 9 (FBXO9) exhibits varying expressions and functions across different human cancer types, regulating the stability and activity of oncogenes and tumor suppressor genes29. Previous studies have demonstrated the association of FBXO9 with advanced tumor type, histological severity, tumor stage progression, and advanced tumor metastasis in OC. These findings suggest that FBXO9functions as a potential tumor suppressor in OC30. In HepG2 cells, Sigmar1 overexpression has demonstrated the capacity to inhibit cell proliferation, enhance cell apoptosis, and reduce NF-κB levels, indicating that it exerts protective effects31. Our IHC analysis revealed that elevated STAT1levels correlated positively with longer OS and PFS in patients with OC32–35. Tumor cells employ various mechanisms to evade immune destruction, such as alterations in antigen, reduced immunogenicity, changes in TME, and diminished immune response36. STAT1has been associated with enhancing immune responses by upregulating cytotoxic immune cells such as cytotoxic T lymphocytes and natural killer (NK) cells37,38. SH3KBP1is involved in the complex responsible for regulating EGFR endocytosis. In EOC, the loss of this gene is detected in 70% of resistant tumors39. EPB41L2 consistently showed overexpression in the initial tumor samples of subsequent non-responders across various PFS cut-off times in OC. Persistently high expression of EPB41L2 is associated with a marked deterioration in PFS, suggesting that elevated initial EPB41L2expression correlates with shorter subsequent PFS40. According to the UniProtKB database (www.uniprot.org/uniprot/O75190), two types of DNAJB6 transcript variants exist: 1 and 2. These variants possess distinct DNAJB6 mRNA 3’ untranslated regions. circPLEKHM3 can function as a ceRNA for miR-9, leading to the downregulation of DNAJB6expression and subsequently suppressing the proliferation and migration abilities of OC cells41. VPS18is recognized for its pivotal role in vesicle-mediated protein transport to lysosomes, encompassing endocytic membrane transport and autophagy pathways42,43. Moreover, VPS18 is markedly linked to treatment response in OC44. PPM1Gbelongs to the metal-dependent protein phosphatase (PPM) family, which is implicated in various diseases, such as dysfunction, tumors, and metabolic diseases45. PPM1Gfunctions by dephosphorylating pre-mRNA splicing factors, thereby affecting protein diversity46. Consequently, impairing PPM1Gfunction might stimulate cancer progression through its effect on pre-mRNA splicing45. Additionally, AKAP12belongs to the kinase scaffolding protein family, anchoring protein kinases A and C to the plasma membrane47. AKAP12 expression positively correlated with the IC50 value of sorafenib in EOC. This finding suggests that patients with high AKAP12levels may exhibit resistance to anti-VEGF inhibitor therapy48. Fyn-related kinase (FRK) belongs to the BRK family, originally known as RAK49,50, and it functions as a tumor promoter or suppressor in various cancers49. The overexpression of FRK overexpression was reported to enhance the proliferation, migration, and invasion abilities of OC cells51. Additionally, PYGBcan regulate the biological characteristics of a variety of cancer cells, including their proliferation and invasion abilities, apoptosis, and metastasis phenotypes52–57. PYGBexpression is upregulated in OC tissues, and a high level of its expression is significantly associated with poor prognosis in patients with OC58.
Analysis of the KEGG pathway and GSEA revealed that the low-risk group exhibited enrichment in DNA replication, oxidative phosphorylation, proteasomes, and spliceosomes. The high-risk subgroup mainly showed enrichment for cytoskeletal regulation. In approximately 50% of high grade serous ovarian carcinoma (HGSOC) cases, alterations in homologous recombination genes, which play crucial roles in DNA repair pathways, are observed. Therapeutic strategies for OC, combining paclitaxel and carboplatin with DNA-damaging agents tailored to specific gene alterations, could benefit patients with recurrence in OC59. Findings from a previous study have indicated a close relationship between the reliance of ovarian tumor cells on oxidative phosphorylation (OXPHOS) and the survival and proliferation of cancer-derived stem cells60. Therefore, OXPHOS plays a key role in OC tumorigenesis; thus, targeting OXPHOS may serve as a promising therapeutic strategy for OC treatment. The ubiquitin-proteasome system serves as a crucial mechanism for degrading intracellular oncoproteins and tumor suppressor proteins. Moreover, it functions as the primary regulator of protein expression9,22, which is involved in controlling tumor-related cellular processes, such as cell proliferation, apoptosis, gene transcription, and receptor downregulation5–8. Several proteasome system inhibitors have demonstrated an effect on the proliferation, migration, and invasion abilities of OC cells. For example, YSY01A, a proteasome inhibitor, enhanced cisplatin cytotoxicity in cisplatin-resistant human OC cells61. Moreover, the proteasome inhibitor PS-341 decelerates the growth of ES-2 OC xenografts in immunodeficient mice62. Alternative splicing (AS) is essential for normal development, and the regulated expression of splicing factors holds significance in cancer development and progression. Numerous AS markers have been identified in OC, including the aberrant expression of the splicing factors SRSF3 and SFPQ63. The cytoskeleton provides structural support for cells, serving as a physical link between the extracellular biochemical, physical, and physiological environments and the cell surface. This linkage extends to intracellular signaling pathways and nuclear events that regulate processes such as apoptosis, contact inhibition, proliferation, and anchorage-independent growth64,65. During cancer development, alterations in cellular architecture bolster cell plasticity, modulating cell-cell interactions and dynamic adhesion. These changes enhance the motility and invasion abilities of cancer cells64,66. Consequently, in the high-risk group, the progression of EOC may be promoted via the regulation of this pathway, potentially worsening the prognosis.
The findings from the chemotherapeutic drugs in the GDSC database suggest that the low-risk group may exhibit higher sensitivity to standard chemotherapeutic agents and molecular-targeted drugs. FBXO9 may regulate the degradation of proteins involved in chemoresistance67. In EOC, STAT1 activation can influence the tumor microenvironment, potentially modulating responses to immunotherapy combined with chemotherapy. Chemotherapy-induced inflammation and immune cell infiltration may be altered by STAT1 status, impacting drug efficacy33. SH3KBP1 may regulate endocytosis and degradation of membrane receptors crucial for drug transport and chemosensitivity-modulating signaling pathways. A high-resolution CGH study of 40 stage III ovarian cancer samples treated with paclitaxel/carboplatin found that DNA alterations in SH3KBP1 and SH3GL2 impacted their activity, with losses in either locus occurring in 70% (14/20) of resistant cases39. EPB41L2 through cell adhesion and migration, potentially influencing metastasis and treatment resistance40. PYGB is involved in glycogen metabolism, which may impact the energy available for cells to respond to stress caused by chemotherapy. However, dasatinib holds a therapeutic significance for high-risk patients. Recent in vitro experiments demonstrated that knockdown PYGB in LUSC (lung squamous cell carcinoma) cells enhances their sensitivity to chemotherapy drugs, including dasatinib68. Also proposes a new regimen for chemotherapeutic drug treatment in high-risk groups. These genes, through their diverse functions in signal transduction, protein homeostasis, and metabolism, can modulate the drugs sensitivity of EOC.
In our study, the CIBERSORT algorithm revealed that the low-risk group showed a lower M2 ratio of monocytes to macrophages, warranting further discussion. The TME constitutes a complex ecosystem comprising tumor, invasive immune, and stromal cells intertwined with non-cellular components. Various cell and functional phenotypes, coupled with dynamic interactions within and between these components, influence tumor biology and potentially result in different immunotherapy responses12. Monocytes exert an influence on other immune cells in the TME. They regulate the recruitment and function of lymphocytes within TME through paracrine signaling, interacting with the adaptive immune system, and they serve as antigen-presenting cells69. Studies have demonstrated that monocytes isolated from the peripheral blood or peritoneal fluid of patients with OC, upon in vitro activation, diminished the ability of antibody-dependent cells to phagocytose and engulf tumor cells70. This helps to explain the correlation between monocyte count and OC outcomes in this study.
Tumor-associated macrophages (TAMs), including types 1 (M1) and 2 (M2) macrophages, have distinct anti- and pro-tumor functions, respectively. These macrophages are pivotal in innate and adaptive immune responses influencing cancer cell growth. TAMs exhibiting the M2 phenotype are associated with a poorer prognosis in patients with cancer than those with the M1 phenotype.
Notably, TAMs facilitate OC progression across various stages of disease development. They play roles in immune escape by tumor cells, migration, invasion, and metastasis of cancer cells, along with angiogenesis71. M2-like TAMs contribute to OC progression by exerting significant immunosuppressive effects on immune cells within the TME, consequently enabling cancer cells to evade attacks. The immunosuppressive function of M2-like TAMs strongly correlates with STAT3protein overactivation. This activation subsequently upregulates IL-10 and IL-6 production71, aligning with our observation of elevated M2-like TAMs in the high-risk group.
In the present study, the low-risk group displayed a higher TMB; the TMB denotes counts of mutations per megabase (mut/Mb) of DNA sequence in a specific cancer72. As the number of detected mutations and new epitopes increase, one or more of these neoantigens are likely to become immunogenic, potentially triggering a T cell response. We analyzed 20 cancer types in 6,035 patients from the TCGA database. The findings revealed that TMB significantly affected the OS in 14 cases, based on the effect of TMB on OS. They were categorized into TMB-worse (eight cancer types), TMB-better (six cancer types), and TMB-similar (six cancer types) groups, with higher TMB being associated with poorer, better, or similar OS, respectively. In the context of TMB, EOC belongs to the TMB-better group, signifying that a higher TMB indicates a better prognosis73. Substantial evidence suggests a strong association between somatic mutations in solid tumors are strongly and immunotherapy. The survival effects observed in the high-low expression TMB group remain generally consistent across the two FDA-approved panels [68], which was consistent with our findings. Despite the recent accelerated approval by the US Food and FDA for the anti-PD-1 drug pembrolizumab in treating TMB solid tumors74and advancement in blood tests for evaluating TMB, numerous challenges persist in the further development of TMB as a clinical biomarker. We anticipate the use of this therapy in patients with EOC by incorporating multiple variables into the composite biomarker. This integration will enhance the accuracy of predicting ICI outcomes and fully harness the potential benefits of immunotherapy75.
Methods.
Study aim
This study aimed to recognize potential prognostic markers and novel therapeutic targets for EOC.
Data collection
Transcriptomic information, clinical data, and somatic mutation data of EOC (n = 375) and normal ovarian tissues (n = 88) were obtained from TCGA and the GTEx databases. Patients with incomplete clinical information were removed. Data were downloaded from the public database Xena website (https://xena.ucsc.edu/). Quantile normalization was used to eliminate systematic differences in large scale RNA-Seq samples from GTEx and TCGA. Overall, 4299 ubiquitin-related genes with a relevance score > 2 were obtained from GeneCards (https://www.genecards.org/).(supplementary Table 1). Immunohistochemical (IHC) data obtained from the Human Protein Atlas (HPA) website (http://www.proteinatlas.org/) was included for validation purposes.
Identifying differentially expressed ubiquitination-related genes
The limma R package, which was obtained from the Bioconductor (https://www.bioconductor.org/), was employed to screen DEGs. The DEGs were then filtered based on a filtering threshold of absolute Log2(Fold change) > 1 and false discovery rate (FDR) < 0.05.(supplementary Table 2).
The differentially expressed ubiquitin-related genes (DEURGs) were achieved by determining the intersections using Venn diagrams.
Signature construction
The prognostic DEURGs were screened using univariate Cox regression analysis. The performance of the patients in the TCGA database were evaluated by randomly dividing the data set into training and test sets, and training and test were performed iteratively. For further gene selection, minimum absolute contraction and selection operator (LASSO) regression was utilized. The selected genes served as inputs for stepwise regression. Subsequently, a ubiquitin-associated risk score (UBrisk) was constructed using multivariate Cox regression analysis. UBrisk was developed based on HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, EPB41L2, DNAJB6, VPS18, PPM1G, AKAP12, FRK, and PYGB. Following that, the risk score of each patient was used in the following formula to calculate the risk:
The risk score =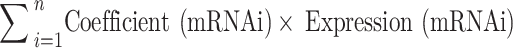
According to the median risk score the patients were divided into low-risk and high-risk groups. Kaplan–Meier curves were plotted to compare the OS between the training, test, and entire sets by using the R package named ‘survival’. Additionally, time-dependent receiver operating characteristics (ROC) curve analyses were constructed, including 1-, 3-, and 5-year survival groups, to use the R package named ‘survivalROC’, which indicated the specificity and sensitivity of risk characteristics. The area under the curve (AUC) was counted. Subsequently, it was used to determine the ROC effects.
RNA extraction and quantitative reverse transcription polymerase chain reaction analyses
Total RNA was extracted from the cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Extracted RNA was converted to cDNA using a miScript II RT kit (QIAGEN, Hilden, Germany), and quantitative polymerase chain reaction (qPCR) was performed using a miScript SYBR Green PCR kit (QIAGEN). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using a QuantiFast SYBR Green PCR kit (QIAGEN) in accordance with the manufacturer’s instructions. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the endogenous control and RNA expression was quantified using the 2-ΔΔCt method. Primers were purchased from Sangon Biotech (Shanghai, China). The primers were as follows:
GAPDH forward, 5′-CCTTCATTGACCTCAACTACATGG-3′; GAPDH reverse, 5′-CTCGCTCCTGGAAGATGGTG-3′.
HSP90AB1 forward, 5′-TGGAGAGGAGGAGGTGGAGAC-3′; HSP90AB1 reverse, 5′-AGGCATCAGAAGCATTAGAGATCAAC-3′.
FBXO9 forward, 5′-CTGGACAACATAGCAAGACCTCATC-3′; FBXO9 reverse, 5′-AAGTGATCCTCCTGCCTTAGCC-3′.
SIGMAR1 forward, 5′-CTCTTCTATACTCTTCGCTCCTATGC-3′; SIGMAR1 reverse, 5′-CTGCCCGCTCCTGTCTATCC-3′.
STAT1 forward, 5′-TCTCTGCCCGTTGTGGTGATC-3′; STAT1 reverse, 5′-ACATGGTGGAGTCAGGAAGAAGG-3′.
SH3KBP1 forward, 5′-CATCGACGTAGGCTGGTGG-3′; SH3KBP1 reverse, 5′-CCTTCCTTTTCAAAGTCCGGTG-3′.
EPB41L2 forward, 5′-GCAGCAGCAGCAGCAGTG-3′; EPB41L2 reverse, 5′-GTTCTTCCTCCACCTCTTCTTCATAC-3′.
DNAJB6 forward, 5′-TGCCTCGCTGCTGAGACAC-3′; DNAJB6 reverse, 5′-CTCTGCTTCTGCTTCTTCCTCTTG-3′.
VPS18 forward, 5′-GGTGAGGTCCAGCAGGTGAG-3′; VPS18 reverse, 5′-GCATACAGTGACAGCAGGTAGTTG-3′.
PPM1G forward, 5′-CACCGAGGAGGCTGAAGAGG-3′; PPM1G reverse, 5′-CACCGCTGTTGTACCACTGTC-3′.
AKAP12 forward, 5′-AGTCAGAGTCAACCGCAGTGG-3′; AKAP12 reverse, 5′-AGTGGAACCTTCTACCTCAACAGTC-3′.
FRK forward, 5′-TAGCACCTCCAGCCACAGAAAG-3′; FRK reverse, 5′-GTCAGCACCAACTCACCATACTTC-3′.
PYGB forward, 5′-GCACGCAGCAGCACTACTAC-3′; PYGB reverse, 5′-ATGGCTTCATCGCAGGCATTC-3′.
Nomogram construction
Variables, including UB risk score, age, grade, and stage, were matched with clinical outcomes to proceed univariate and multivariate Cox regression analyses. Multivariate Cox regression analysis was performed on prognostic related variables to explore independent prognostic variables. A nomogram was constructed using the variables above to prognosticate the probability of 3-, 5-, and 7-year survival. Calibration and ROC curves were used to assess clinical efficacy and discriminative accuracy of the nomogram model.
Enrichment analysis
According to the median risk score, the patients were divided into low-risk and high- risk groups. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and hallmark pathway analyses were conducted using GSEA software (version 4.3.2). Gene sets, including c5.go.bp, c5.go.cc, c5.go.mf, c2.cp.kegg, and hallmarks were collected from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea). The filtering criteria included a normalized enrichment score |>1, a P-value < 0.05, and an FDR < 0.25.
Immune Infiltration analysis
The expression data (ESTIMATE) algorithm was employed to estimate stromal and immune cell proportions within malignant tumors. Subsequently, three distinct immune-related scores—stromal, immune, and estimate —were calculated. Variations in immune cell infiltration levels, depicting differences in the immune landscape, were compared between the high- and low-risk groups utilizing CIBERSORT, TIMER, MCP-Counter, quantiseq, and EPIC through the IBOR R-package.
Stemness Index and Tumor Mutational Burden (TMB) analysis
RNA-Seq data from pluripotent stem cell samples were downloaded from the Progenitor Cell Biology Consortium database. Subsequently, one-class logistic regression algorithms were utilized to calculate the mRNAsi and EREG mRNAsi for each patient with OC.
Drug sensitivity analysis
The Genomics of Drug Sensitivity in Cancer (GDSC) website (https://www.cancerrxgene.org/) was used to identify the molecular features of cancers that predict the response to anticancer drugs. Connectivity scores for the half-maximal inhibitory concentration (IC50) were calculated to determine drug sensitivity. A lower score (0 < IC50 < 1) indicated higher drug effectiveness for the high- and low-risk groups.
Cluster analysis
Ubiquitin-related molecule subtypes were recognized by conducting cluster analysis using the ‘Non-negative matrix factorization’ package. This analysis was based on prognosis-related DEURG expression. Following this, survival analysis was performed to compare the prognoses among the three clusters.
Statistical analysis
Statistical analyses were performed using the R software (4.3.1). The Wilcoxon test was employed to analyze the differences in median values between the two groups. To screen independent prognostic factors, univariate and multivariate Cox regression analyses were utilized. The correlation Coefficient between variables was analyzed by Pearson analysis using the GEPIA website (http://gepia.cancer-pku.cn/index.html) to detect multicollinearity. The significance of the KM analysis was assessed using the log-rank test. The Spearman’s method was utilized to evaluate the correlation between the expression levels of the two genes. The prediction accuracies of the risk and nomogram models were evaluated using calibration, C-index, and ROC curves. Statistical significance was set at p < 0.05. *p < 0.05; **p < 0.01; ***p < 0.001.
Conclusions
In the present study, we drastically analyzed the clinical significance and features of TME features associated with ubiquitin-related genes in EOC. We established the ubiquitin-related signature risk model, comprising HSP90AB1, FBXO9, SIGMAR1, STAT1, SH3KBP1, EPB41L2, DNAJB6, VPS18, PPM1G, AKAP12, FRK, and PYGB, specifically for patients with EOC. We conducted an analysis combining clinical characteristics to verify and enrich the pathways, immune status, mRNAsi, EREG-mRNAsi, tumor mutation burden, and drug sensitivity. The risk model used in this study was based on the GTEx database, GeneCards website, and TCGA data, with a large sample size and relatively complete clinical information. Our model relies on specific genes that are more cost-effective and clinically practical, assisting clinicians in determining the prognosis of individual patients. However, owing to insufficient verification samples, the verification work cannot be carried out, and the reliability of the verification results is difficult to be guaranteed owing to the lack of multi-centers and large sample population for evaluation. Furthermore, a risk model of ubiquitin-related diseases was introduced to assess the proteasome pathway in individuals. This model may aid in improving the understanding of TME immune infiltrations and help oncologists in determining more effective immunotherapeutic strategies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the patients as well as their Family members for their consent and participation. Additionally, we thank and appreciate the Pathological Diagnosis Clinical Medical Research Center of Xinjiang Production and Construction Corps for their valuable assistance.
Author contributions
WJ and SYL designed the study; LDL, LW, YKY, YENEH, and LT prepared the figures; LDL and XNL wrote the main manuscript text . All authors read and approved the final manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos.82360494); International Science and Technology Cooperation promotion program of Shihezi university (GJHZ202301); and Strong Youth Science and Technology Leading Talents in Science and Technology Innovation project of Corps (2023CB008-02). No funding or other benefits related to the subject of this article were received from any commercial entity.
Data availability
The datasets used in this study is available from the corresponding author upon reasonable requests.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Donglin Luo and Xiaoning Li contributed equally to this work.
Contributor Information
Siyuan Li, Email: 503654536@qq.com.
Wei Jia, Email: jiawei@shzu.edu.cn.
References
- 1.Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Gadducci, A. et al. Current strategies for the targeted treatment of high-grade serous epithelial ovarian cancer and relevance of BRCA mutational status. J. Ovarian Res. 12 (1), 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munoz-Galvan, S. et al. Downregulation of MYPT1 increases tumor resistance in ovarian cancer by targeting the Hippo pathway and increasing the stemness. Mol. Cancer. 19 (1), 7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asare-Werehene, M. et al. The exosome-mediated autocrine and paracrine actions of plasma gelsolin in ovarian cancer chemoresistance. Oncogene. 39 (7), 1600–1616 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz, A. L. & Ciechanover, A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 49, 73–96 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Devoy, A., Soane, T., Welchman, R. & Mayer, R. J. The ubiquitin-proteasome system and cancer. Essays Biochem. 41, 187–203 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Hickey, C. M., Xie, Y. & Hochstrasser, M. DNA binding by the MATalpha2 transcription factor controls its access to alternative ubiquitin-modification pathways. Mol. Biol. Cell. 29 (5), 542–556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumeister, W., Walz, J., Zuhl, F. & Seemuller, E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 92 (3), 367–380 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Nath, D. & Shadan, S. The ubiquitin system. Nature. 458 (7237), 421 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Rao, Z. & Ding, Y. Ubiquitin pathway and ovarian cancer. Curr. Oncol. 19 (6), 324–328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song, M. et al. Tumor derived UBR5 promotes ovarian cancer growth and metastasis through inducing immunosuppressive macrophages. Nat. Commun. 11 (1), 6298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hornburg, M. et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell. 39 (7), 928–944e926 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Jiang, Y., Wang, C. & Zhou, S. Targeting tumor microenvironment in ovarian cancer: Premise and promise. Biochim. Biophys. Acta Rev. Cancer. 1873 (2), 188361 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Kawamura, K., Komohara, Y., Takaishi, K., Katabuchi, H. & Takeya, M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol. Int. 59 (5), 300–305 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Xu, J. et al. Single-cell RNA sequencing reveals the tissue Architecture in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 28 (16), 3590–3602 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang, T. et al. Lymphocyte and macrophage infiltration in omental metastases indicates poor prognosis in advance stage epithelial ovarian cancer. J. Int. Med. Res. 49 (12), 3000605211066245 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahner Hendrickson, A. E. et al. Assessment of published models and prognostic variables in epithelial ovarian cancer at Mayo Clinic. Gynecol. Oncol. 137 (1), 77–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodelon, C. et al. Molecular classification of epithelial ovarian Cancer based on methylation profiling: evidence for Survival Heterogeneity. Clin. Cancer Res. 25 (19), 5937–5946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51 (D1), D587–d592 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zwickl, P., Voges, D. & Baumeister, W. The proteasome: a macromolecular assembly designed for controlled proteolysis. Philos. Trans. R Soc. Lond. B Biol. Sci. 354 (1389), 1501–1511 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu, Y. et al. Development of an Individualized Ubiquitin Prognostic signature for Clear Cell Renal Cell Carcinoma. Front. Cell. Dev. Biol. 9, 684643 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, X. et al. Development of an ubiquitin-proteasome system signature for predicting prognosis and providing therapeutic guidance for patients with triple-negative breast cancer. J. Gene Med. 26(1):e3584. (2023). [DOI] [PubMed]
- 25.Ni, W. et al. Identification and validation of Ubiquitin-Specific proteases as a Novel Prognostic signature for Hepatocellular Carcinoma. Front. Oncol. 11, 629327 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song, G. et al. Identification of a Ubiquitin related genes signature for Predicting prognosis of prostate Cancer. Front. Genet. 12, 778503 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haase, M. & Fitze, G. HSP90AB1: helping the good and the bad. Gene. 575 (2 Pt 1), 171–186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikishin, D. A. et al. Selection of stable expressed reference genes in native and vitrified/thawed human ovarian tissue for analysis by qRT-PCR and Western blot. J. Assist. Reprod. Genet. 35 (10), 1851–1860 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hussain, S. et al. F-box only protein 9 and its role in cancer. Mol. Biol. Rep. 49 (2), 1537–1544 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Foulkes, W. D., Ragoussis, J., Stamp, G. W., Allan, G. J. & Trowsdale, J. Frequent loss of heterozygosity on chromosome 6 in human ovarian carcinoma. Br. J. Cancer. 67 (3), 551–559 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu, Q. et al. Sigma-1 receptor (σ1R) is downregulated in hepatic malignant tumors and regulates HepG2 cell proliferation, migration and apoptosis. Oncol. Rep. 39 (3), 1405–1413 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Koti, M. et al. A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br. J. Cancer. 112 (7), 1215–1222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Au, K. K. et al. STAT1-associated intratumoural T(H)1 immunity predicts chemotherapy resistance in high-grade serous ovarian cancer. J. Pathol. Clin. Res. 2 (4), 259–270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Josahkian, J. A. et al. Increased STAT1 expression in high Grade Serous Ovarian Cancer is Associated with a better outcome. Int. J. Gynecol. Cancer. 28 (3), 459–465 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Li, X., Wang, F., Xu, X., Zhang, J. & Xu, G. The dual role of STAT1 in Ovarian Cancer: insight into Molecular mechanisms and Application potentials. Front. Cell. Dev. Biol. 9, 636595 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beatty, G. L. & Gladney, W. L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21 (4), 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meissl, K., Macho-Maschler, S., Müller, M. & Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 89, 12–20 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Jin, Y. et al. IL-21 reinvigorates exhausted natural killer cells in patients with HBV-associated hepatocellular carcinoma in STAT1-depedent pathway. Int. Immunopharmacol. 70, 1–8 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Osterberg, L. et al. Potential predictive markers of chemotherapy resistance in stage III ovarian serous carcinomas. BMC Cancer. 9, 368 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menyhárt, O., Fekete, J. T. & Győrffy, B. Gene expression indicates altered Immune Modulation and Signaling Pathway Activation in Ovarian Cancer patients resistant to Topotecan. Int. J. Mol. Sci. 20(11), 2750 (2019). [DOI] [PMC free article] [PubMed]
- 41.Zhang, L. et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol. Cancer. 18 (1), 144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang, T. et al. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 23 (5–6), 247–267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segala, G. et al. Vps11 and Vps18 of Vps-C membrane traffic complexes are E3 ubiquitin ligases and fine-tune signalling. Nat. Commun. 10 (1), 1833 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Arca, D. et al. Serum Mass Spectrometry Proteomics and Protein Set Identification in response to FOLFOX-4 in drug-resistant ovarian carcinoma. Cancers (Basel) 15(2), 412 (2023). [DOI] [PMC free article] [PubMed]
- 45.Lin, Y. R., Yang, W. J. & Yang, G. W. Prognostic and immunological potential of PPM1G in hepatocellular carcinoma. Aging (Albany NY). 13 (9), 12929–12954 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di, C. et al. Function, clinical application, and strategies of Pre-mRNA splicing in cancer. Cell. Death Differ. 26 (7), 1181–1194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radeva, M. Y., Kugelmann, D., Spindler, V. & Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS One. 9 (9), e106733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang, Q. et al. Pan-cancer analysis of the prognosis and immunological role of AKAP12: a potential biomarker for resistance to anti-VEGF inhibitors. Front. Genet. 13, 943006 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goel, R. K. & Lukong, K. E. Understanding the cellular roles of fyn-related kinase (FRK): implications in cancer biology. Cancer Metastasis Rev. 35 (2), 179–199 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Brauer, P. M. & Tyner, A. L. RAKing in AKT: a tumor suppressor function for the intracellular tyrosine kinase FRK. Cell. Cycle. 8 (17), 2728–2732 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu, X. Z. et al. TRIM44 facilitates ovarian cancer proliferation, migration, and invasion by inhibiting FRK. Neoplasma. 68 (4), 751–759 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Altemus, M. A. et al. Breast cancers utilize hypoxic glycogen stores via PYGB, the brain isoform of glycogen phosphorylase, to promote metastatic phenotypes. PLoS One. 14 (9), e0220973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, Z., Han, G., Liu, Q., Zhang, W. & Wang, J. Silencing of PYGB suppresses growth and promotes the apoptosis of prostate cancer cells via the NF–κB/Nrf2 signaling pathway. Mol. Med. Rep. 18 (4), 3800–3808 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimada, S., Shiomori, K., Tashima, S., Tsuruta, J. & Ogawa, M. Frequent p53 mutation in brain (fetal)-type glycogen phosphorylase positive foci adjacent to human ‘de novo’ colorectal carcinomas. Br. J. Cancer. 84 (11), 1497–1504 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tashima, S., Shimada, S., Yamaguchi, K., Tsuruta, J. & Ogawa, M. Expression of brain-type glycogen phosphorylase is a potentially novel early biomarker in the carcinogenesis of human colorectal carcinomas. Am. J. Gastroenterol. 95 (1), 255–263 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Takashi, M., Koshikawa, T., Kurobe, N. & Kato, K. Elevated concentrations of brain-type glycogen phosphorylase in renal cell carcinoma. Jpn J. Cancer Res. 80 (10), 975–980 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shimada, S., Tashima, S., Yamaguchi, K., Matsuzaki, H. & Ogawa, M. Carcinogenesis of intestinal-type gastric cancer and colorectal cancer is commonly accompanied by expression of brain (fetal)-type glycogen phosphorylase. J. Exp. Clin. Cancer Res. 18 (1), 111–118 (1999). [PubMed] [Google Scholar]
- 58.Zhou, Y., Jin, Z. & Wang, C. Glycogen phosphorylase B promotes ovarian cancer progression via Wnt/β-catenin signaling and is regulated by miR-133a-3p. Biomed. Pharmacother. 120, 109449 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Murai, J. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int. J. Clin. Oncol. 22 (4), 619–628 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Nayak, A. P., Kapur, A., Barroilhet, L. & Patankar, M. S. Oxidative phosphorylation: a target for Novel therapeutic strategies against ovarian Cancer. Cancers (Basel) 10(9), 337 (2018). [DOI] [PMC free article] [PubMed]
- 61.Huang, W. et al. Proteasome inhibitor YSY01A enhances cisplatin cytotoxicity in cisplatin-resistant human ovarian Cancer cells. J. Cancer. 7 (9), 1133–1141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bazzaro, M. et al. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 66 (7), 3754–3763 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Klinck, R. et al. Multiple alternative splicing markers for ovarian cancer. Cancer Res. 68 (3), 657–663 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Pawlak, G. & Helfman, D. M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 11 (1), 41–47 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Provenzano, P. P. & Keely, P. J. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and rho GTPase signaling. J. Cell. Sci. 124 (Pt 8), 1195–1205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ayollo, D. V., Zhitnyak, I. Y., Vasiliev, J. M. & Gloushankova, N. A. Rearrangements of the actin cytoskeleton and e-cadherin-based adherens junctions caused by neoplasic transformation change cell-cell interactions. PLoS One. 4 (11), e8027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo, X. et al. Identification of a Prognostic Signature for Ovarian Cancer Based on Ubiquitin-Related Genes Suggesting a Potential Role for FBXO9. Biomolecules ; 13(12). (2023). [DOI] [PMC free article] [PubMed]
- 68.Song, G. Q. et al. The necroptosis signature and molecular mechanism of lung squamous cell carcinoma. Aging (Albany NY). 15 (22), 12907–12926 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olingy, C. E., Dinh, H. Q. & Hedrick, C. C. Monocyte heterogeneity and functions in cancer. J. Leukoc. Biol. 106 (2), 309–322 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Almeida-Nunes, D. L., Mendes-Frias, A., Silvestre, R., Dinis-Oliveira, R. J. & Ricardo, S. Immune Tumor Microenvironment in Ovarian Cancer Ascites. Int J Mol Sci ; 23(18). (2022). [DOI] [PMC free article] [PubMed]
- 71.Nowak, M. & Klink, M. The role of Tumor-Associated macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 9(5), 1299 (2020). [DOI] [PMC free article] [PubMed]
- 72.Addeo, A., Friedlaender, A., Banna, G. L. & Weiss, G. J. TMB or not TMB as a biomarker: that is the question. Crit. Rev. Oncol. Hematol. 163, 103374 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Wu, H. X. et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann. Transl Med. 7 (22), 640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marcus, L. et al. FDA approval Summary: Pembrolizumab for the treatment of Tumor Mutational Burden-High Solid tumors. Clin. Cancer Res. 27 (17), 4685–4689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jardim, D. L., Goodman, A., de Melo Gagliato, D. & Kurzrock, R. The challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 39 (2), 154–173 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used in this study is available from the corresponding author upon reasonable requests.