

Abstract
Mucous cell metaplasia (MCM), defined by the appearance of mucous cells in airways where mucous cells were not present, is a consistent pathologic characteristic in the peripheral airways of bronchial asthma. Under mild inflammatory conditions MCM occurs as a result of pre-existing airway epithelial cells (AECs) starting to express mucin genes and differentiating into mucous cells. Under extensive inflammatory responses, AECs proliferate, and the development of MCM involves the differentiation of pre-existing and proliferating cells into mucous cells. Epithelial cell numbers per mm basal lamina are increased by approximately 30%. IL-13 is the central cytokine that is responsible for MCM in asthma through GABA-R- and STAT6-mediated mechanisms involving the calcium-activated chloride channel CLCA. IL-13 is also responsible for the proliferation of AECs by causing cells to produce TGFα that acts on the epidermal growth factor (EGF) receptor. Normally, resolution of MCM involves two distinct mechanisms. 1) Some of the metaplastic mucous cells stop the synthesis of mucus and dedifferentiate into Clara or serous cells to reconstitute the epithelium. 2) When proliferation of epithelial cells had occurred, approximately 30% of metaplastic cells are eliminated during the resolution process. Thus, a safe approach to reducing IL-13-induced MCM would involve blocking mucous synthesis and storage, blocking secretion of stored mucus, and eliminating hyperplastic mucous cells. Understanding the molecular mechanisms of each of these processes is necessary for developing effective therapies for reducing mucous hypersecretion in asthma and leading to a repaired epithelium.
Keywords: mucous cell metaplasia, lung pathogenesis, hyperplasia of airway epithelium, programmed cell death, inflammation, Th2 cytokines, allergy and asthma, proliferation and cell cycle
Introduction
Epithelia lining the skin, the digestive and the reproductive tracts, or the airways have similarities because of their role in maintaining a barrier function. However, upon closer inspection, the functions of the various epithelia are vastly different, because they are exposed to different types of environment. The airway epithelium is primarily exposed to the air we breathe, while the epithelia lining the skin or the digestive tract are constantly exposed to UV light or chemicals contained within foods and other items that we ingest, respectively. Therefore, the airway epithelium has developed various mechanisms to sense bacterial or viral infectious agents and environmental pollutants that are suspended as aerosols and to mount appropriate actions for protection and cleaning. Contact with these environmental agents elicits increased secretion and synthesis of mucus that traps and clears the insult and can cause the epithelium to elicit an inflammatory response that inactivates infectious agents or clears particles.
The airway epithelial surface is covered by mucus that is secreted by mucus-producing goblet cells and by mucous and serous cells in the submucosal glands. The sticky nature of mucus is suited to trap particles and help remove them out of the tracheobronchial tract with the help of ciliated cells. In humans, the site of most mucous hypersecretion is the large airways. Whether the source of the major part of secreted mucus stems from the surface epithelial goblet cells or the submucosal glands is not clear because of limitations in real-time measurement capabilities of synthesized and secreted mucus at rest or under inflammation conditions. However, detailed morphometric analysis of the amount of sustainable stored secretory product in the tracheobronchial airways of the Rhesus monkey reveals at least twice as much mucus in the surface epithelium as in the submucosal glands [1]. In the lower airways of the Rhesus monkey, as in the majority of other animal species, glands are absent, and surface epithelial mucous cells are the sole source of mucus.
In the lungs of mice and humans, the airways are lined by a mixed population of ciliated and nonciliated epithelial cells. In mice, the nonciliated cells comprise 50–70% of the total epithelial population throughout the entire conducting airway network [2]. In naïve mice, the majority of these nonciliated cells are Clara cells, and there are few or no mucous cells in airways distal to the trachea. In healthy humans, there are few Clara cells in the large conducting airways, but their numbers progressively increase in more distal airways [3]. Normally, mucous cells are absent or sparse in airway less than 2 mm in diameter [4].
Mucous cell metaplasia (MCM) is defined by the appearance of mucous cells in airways where mucous cells were not present [5]. Cells that populate the small airways, primarily Clara cells, can proliferate and start to express mucin genes in response to inflammatory responses. AECs that start to store mucins are defined as mucous cells. Following inflammatory responses, mucin gene expression can be regulated by increased transcription [6], [7], [8] or by increased mRNA stabilization [9], [10]. Increased levels of mucin mRNAs translate into increased levels of mucin apoproteins that are glycosylated and constitute the stored mucosubstances.
MCM and Pathological Conditions in Asthma
A history of sputum production is independently associated with an accelerated rate of decline in forced expiratory volume in one second (FEV1) [11]. Furthermore, hypersecretion of mucus plays a central role in the pathogenesis of severe airway obstruction and asphyxiation in fatal attacks of asthma [12]. Asthmatic patients who died suddenly and unexpectedly show luminal occlusion by mucous plugs, plasma exudates, and inflammatory cells in the peripheral airways of autopsied lungs [13]. The blockage of the airway is enhanced by the sticky nature of mucus trapping inflammatory cells that release DNA and further increase its viscosity [14]. The majority of the airways of asphyxiated asthmatic individuals are blocked, not because of airway constriction, but because of widespread, contiguous plugs of a gelatinous substance composed of mucin gene products. However, the mechanisms governing hypersecretion of mucus in asthma are poorly understood. Aikawa et al. [4] were the first to report that in severe asthmatics goblet cell hyperplasia is increased 30–fold when comparing patients with bronchial asthma who died of non-acute asthma attack to patients with bronchial asthma who died of severe acute asthma attack. It is now well established from biopsied specimens [15], and autopsied lungs [12], [13], that mucous cell hyperplasia and MCM in peripheral airways [16] are consistent pathologic characteristics of bronchial asthma.
MCM in Asthma—A Dysregulation of an Innate Protective Response?
MCM is an integral part of the defense system in the airways to help the production of increased levels of mucus to neutralize and remove injurious particles or infections with the help of ciliary action. In rats and mice, MCM can occur as a result of pre-existing Clara or serous cells that begin to express Muc5ac in the absence epithelial cell proliferation [17], [18]. Another study [19] reported that 10% of mucous cells proliferate in conjunction with a compensatory decrease in the number of Clara and ciliated cells. MCM is an integral part of the defense system and helps the necessity of producing increased levels of mucus to neutralize and help remove injurious particles or infection with the help of ciliated cells.
While MCM can arise by non-mucous cells starting to differentiate into a mucin-storing and -secreting cells, an extensive inflammatory response can also cause epithelial cells to proliferate [20], [18]. Epithelial cells undergo proliferation in remodeling airways [21], [22], [23], [24], and the extent of proliferation appears to be dependent on the extent of injury and inflammatory response associated with the length of allergen exposure. Proliferation of epithelial cell is also relevant for human asthma because an increased cellular proliferation in the airway epithelia is found in subjects with severe asthma, as compared with subjects with mild asthma or normal non-diseased control subjects [25].
Under conditions where extensive inflammation occurs the epithelial layer may be disrupted and these responses may initiate the proliferation of epithelial cells to close the injury-induced gap in the epithelium [26], [16], [27], [28]. Therefore, MCM can occur as a result of pre-existing and proliferating cells differentiating into mucous cells [18], [29], [30]. The number of epithelial cells per mm basal lamina increases by approximately 30–40% following repeated allergen exposure over 4–5 d [30]. The increase in epithelial cells essentially constitutes the increase in the number of mucous cells, suggesting that newly formed cells are mucus-secreting cells. Several studies have shown that secretory cells are the cells that proliferate following injury to the epithelium [31], [32], [33], [34], [18], suggesting that AECs capable synthesizing and secreting mucus may also be the first to proliferate and enhance protection to the epithelium. This response may be explained by the effort of the epithelium to produce sufficient mucus for protection from further injury.
IL-13 is necessary for the development of MCM in asthma [35] by activating the IL-13 receptor alpha 1 together with the IL-4 receptor alpha [36]. The role of IL-9 in independently inducing MCM or in conjunction with IL-13 has been debated for a long time [37], [6], [38]. However, several studies now support the idea that IL-13 either produced by epithelial cells or by Th2 cells is the primary regulator of MCM in vivo. The blockade of IL-13 in IL-9-overexpressing mice completely prevents the development of MCM [39]. All other Th cytokines depend on IL-13 to induce MCM and IL-13 acts, not through intermediate inflammatory cells, but on structural cells within the lung, likely the airway epithelium itself [40]. The potency of IL-13 is shown, requiring its complete blockade for a significant reduction in mucus production [41], [42].
Ligation of the IL-13α1/IL-4R receptor complex by IL-13 results in activation of JAK1 and Tyk-2 kinases, and monomers of STAT6 bind through their Src homology 2 domains to phosphorylated tyrosine residues of IL-4Rα. After phosphorylation, STAT6 dimerizes and translocates to the nucleus to activate promoter regions regulated by IL-13. Studies with STAT6−/− mice have shown that STAT6 is necessary for IL-13 to induce MCM [43]. The direct effect of IL-13 on epithelial cells was confirmed when reconstitution of STAT6 only in epithelial cells was sufficient for IL-13 to induce MCM in the absence of inflammation [40]. Despite the importance of IL-13 in inducing MCM the precise mechanisms by which it causes MCM remain unclear. However, a recent study found that IL-13 enhances expression of both the γ-aminobutyric acid (GABA) receptor and the GABA synthetic enzyme signaling pathway in the airway epithelium [44], [45]. Blockade of the GABA pathway inhibited the development of IL-13 or allergen-induced MCM. Therefore, it appears that GABA has a role in stimulating the metaplastic transition from the non-secretory cell type to a secretory one. Whether this pathway is independent from the IL-13 receptor and STAT6-mediated pathway remains to be determined but these findings are starting to unravel the complex mechanistic pathways by which IL-13 may be acting to induce MCM.
In murine models of asthma, this MCM is associated with the induction of Muc5ac mRNA expression [8]. In the mouse models, MUC1, MUC2 and 3 mRNA levels are not affected, but IL-13-induced MCM is primarily driven by the expression of Muc5ac. However, the events connecting IL-13R activation to Muc5ac gene expression are incompletely defined, but preliminary work with inhibitors indicates requirements for MEK/ERK, p38MAPK, and PI3K activation in cultured cells [46]. Whether these signaling proteins are mediators for the expression and activation of the calcium-activated chloride channel (CLCA) protein family, which are also key molecules for the development of MCM in asthma is unclear. The murine CLCA3, the mouse homologue of human CLCA1, is induced by IL-13 and suppressing mCLCA3 inhibits the development of MCM in the mouse model of asthma while overexpression of this gene induces the expression of MUC5AC in human airway cell lines [47]. In addition, mCLCA3 gene transfer to mouse airway epithelium was sufficient to induce MCM, and expression of hCLCA1 is significantly upregulated in patients with bronchial asthma compared with control subjects as determined by in situ hybridization and immunohistochemical analysis [48]. In light of all this supporting evidence for mCLCA3 to be crucial for the development of IL-13-induced MCM, it was an unexpected surprise that allergic challenge with ovalbumin or intranasal administration of IL-13 produced a robust allergic response in both mCLCA3+/+ and mCLCA3−/− mice [49]. These unexpected findings suggested that other compensatory mechanisms may be present and other CLCA family members may also mediate IL-13-induced MCM [50]. Despite all these studies to unravel the role of the CLCA family, how hCLCA1 fits within the signaling pathway of the IL-13-induced mucin gene expression is still not understood. However, NF-κB, important in pseudomonas-induced MCM [51] was excluded from being involved in the development of IL-13-induced MCM [42].
IL-13 also directly induces proliferation of normal human airway epithelial cells (NHBE) cells, as reflected by tritiated thymidine uptake and cell counts in differentiated normal human bronchial epithelial cells in culture [52]. The proliferative effects of IL-13 are mediated through the epidermal growth factor receptor (EGFR), as proliferation was attenuated by AG1478, an EGFR tyrosine kinase inhibitor. IL-13 induces release of TGFα from the epithelial cells that, in turn, binds via an autocrine/paracrine-type action to the EGFR, initiating proliferation [52]. However, which pathways and cell cycle regulatory proteins are activated or induced to drive cell proliferation in NHBEs and other AECs is largely unknown. In addition, whether proliferating cells in airway epithelia are always programmed to immediately produce and store mucus or whether the pathways that induce cell proliferation or mucus gene expression are distinct pathways is not well-understood.
Recent studies have started to address whether the proliferative responses may be driven by pathways that are different from those driving mucous cell differentiation. In rats, IL-13-induced MCM required signaling through EGFR, because selective EGFR tyrosine kinase inhibitors prevented IL-13-induced MCM in a dose-dependent manner [53]. In addition, how EGFR signaling is involved in the IL-13 signaling through IL-13 and IL-4Rα was clarified by a mouse model of viral infection that showed ciliated cells proliferate and persistent activation of EGFR signaling prevents apoptosis of ciliated cells, presumably through the PI3/Akt pathway [54]. In that setting, ciliated cells transdifferentiate into goblet cells establishing MCM in the presence of IL-13. Under those conditions, blockage of the EGFR signaling prevents virus-induced increases in ciliated and goblet cells because ciliated cells undergo apoptosis [54]. Inhibition of IL-13 signaling reduces synthesis and storage of mucus and increases the number of ciliated cells, suggesting that it does not affect the cell death of ciliated cells. These findings suggest that EGFR sustains ciliated cell hyperplasia while IL-13 causes the transdifferentiation of ciliated cells to mucous cells. Future studies are needed to define the interaction of these pathways in various model systems representing allergen-induced MCM.
Resolution of MCM
Normally, MCM is transient and spontaneous resolution of MCM has been observed after cessation of allergen exposure. In BALB/c mice, when MCM develops as a result of a single allergen challenge in the absence of cell proliferation, the mucous cell phenotype resolves after 3-4 weeks [17]. Resolution occurs over a course of 10 d when the same mouse strain is repeatedly exposed to allergen to increase MCM and the epithelial cell numbers per mm basal lamina [55]. For C57Bl/6 mice repeatedly exposed to allergen for 5 d, approximately 10-11 d are necessary for the resolution of MCM in the absence of further allergen challenge [56].
Depending on the frequency and concentration of allergen AECs proliferate in C57Bl/6 mice [19]. This defensive response is normally transient and resolved in the absence of further challenge so that the original epithelium is restored in which none or very few mucous cells remain [56].
When the number of mucous cells/mm basal lamina is subtracted from the total epithelial cell number at various time points during the resolution of epithelial cell hyperplasia the number of mucous cells is reduced, while the number of non-mucous cells remains largely unaffected indicating that the increase in total epithelial cell numbers is entirely composed of hyperplastic mucous cells [29]. The resolution of MCM involves various mechanisms. First, some of the mucous cells appear to transdifferentiate into non-mucus cells. This change must involve reducing mucus synthesis and possibly differentiating into ciliated [54] or serous cells (personal unpublished observation). This process of transdifferentiation could be due to the decline in cytokines and other inflammatory mediators responsible for mucin gene expression and the presence of a combination of inflammatory mediators stimulating the differentiation of these cells into another epithelial cell phenotype. Second, the resolution of MCM involves the reduction of approximately 30% of AECs being removed from the epithelium. Because all of these cells represent mucus-producing cells, this mechanism may account for the reduction of mucus production by at least one-third. This resolution is in part orchestrated by the Bcl-2 family of proteins such that anti-apoptotic proteins are downregulated allowing the pro-apoptotic members to elicit cell death and reduce the number of hyperplastic epithelial cells [57]. Whether the decline in specific cytokine levels causes the downregulation of anti-apoptotic proteins or an increase in pro-apoptotic regulators, and thereby the cell death of hyperplastic epithelial cells is being investigated.
Resolution of MCM also occurs when mice are exposed to the allergen continuously for prolonged periods [56]. In rodents, chronic exposure to an allergen initially induces inflammation, which decreases over time [58], [59]; while in humans with allergic asthma, inflammation persists and is chronic during their lifetime [60]. This transition from antigen sensitization to immunological tolerance is accompanied by a shift in the lymphocyte content in the lung tissue and bronchial lavage fluid (BALF) [61], [58]. Antigen-specific regulatory T cells are believed to produce IL-10 transiently and inhibit the asthma phenotype during the development of tolerance [62], [63]. Prolonged exposure of mice to an allergen causes the cytokine IL-13 which is secreted by T helper cell type 2 (Th2) to decrease and IFNγ, which is secreted by Th1 cells, to increase in the BALF [64]. IFNγ mediates the reduction of IL-13-induced MCM by inducing programmed cell death [56].
Resolution of MCM during prolonged exposure to allergen involves an active process that requires cell death-inducing conditions. The experimental role of IFNγ signaling in the resolution process of MCM was identified when instillation of IFNγ enhanced resolution of allergen-induced MCM by causing cell death in airway mucous cells. The importance of IFNγ in resolving MCM was confirmed by the finding that mice deficient in IFNγ (unpublished personal observation) and STAT1 [56], [65], a protein that plays an obligated and dedicated role in mediating IFNγ-dependent responses, were unable to resolve MCM during prolonged exposure to allergen. The main cell type that undergoes cell death in allergic mice is the mucous cell. However, our studies demonstrate that growing NHBE cells are susceptible to IFNγ-induced cell death regardless of whether they express mucin genes or not. Interestingly, non-proliferating NHBE cells are generally unaffected by IFNγ [56]. The mechanism for the selective susceptibility of proliferating cells to IFNγ is not clear. However, the reason why mucous cells are reduced during the resolution process may be due to their hyperplastic nature that makes them a target of IFNγ. Overall, these findings suggest that IFNγ causes a reduction of MCM through the apoptosis of hyperplastic cells, which restores the normal number of cells in the epithelium.
Classic apoptotic epithelial cells are not obvious during the recovery of the airway epithelium. Whether some of the metaplastic cells slough off during this process is still under investigation. It is possible that selected cells are removed by extrusion and involve detachment from the basement membrane, which is also called anoikis [66], thereby being sloughed off before the classic apoptotic morphology appears.
IFNγ-Induced Cell Death and Check Points Controlling this Pathway in AECs
Many studies have shown that T lymphocytes, neutrophils, and eosinophils undergo cell death in massive numbers within 3–6 h after cytokine deprivation or stimulation with death agonists [67]. However, it is widely recognized that epithelial cells in general and AECs in particular are resistant to cell death [57].
Two major characteristics define IFNγ-induced cell death in AECs: 1) Only proliferating AECs are sensitive and die, while confluent AECs are resistant to IFNγ-induced cell death [56]. The sensitivity of only proliferating cells may ensure the selective removal of hyperplastic cells during the resolution of MCM and that the majority of AECs, which are quiescent and in the resting G0 phase of the cell cycle, are protected. This selective cell killing provides a mechanism to restore the epithelial structure without affecting the rest of the airway epithelium. 2) Cell death induced by IFNγ treatment is not evident until after 3 d of treatment. The role of this slow execution of the cell death process may be to allow neighboring cells time to close the resulting gap and maintain an intact barrier function.
The apparent requirement of 2–3 d for IFNγ-induced cell death suggests that apoptosis in AECs is mediated by an indirect pathway, i.e., the cells must produce the factor that induces cell death. Alternatively, the delayed cell death could stem from checkpoints that are in place to abrogate direct activation of the death signals. AECs may have developed signaling pathways that allow activation of pro- and anti-apoptotic pathways in response to an injury stimulus, making the cell very refractory to cell death unless the anti-apoptotic factors are overwhelmed by a sustained death stimulus. Such complex activation of checkpoints allows the airway epithelium to resist massive cell death and fulfill its ancient role as a crucial innate protector from environmental insults.
In the context of apoptosis to restore homeostasis in hyperplastic epithelium, it remains unclear how the epithelium determines the number of cells that must be eliminated to restore the original condition. Is it possible that the epithelial cells that populate the epithelium signal through gauging the intercellular pressure that there is not enough room. Furthermore, it is unknown whether the discarded cells represent abnormal or damaged epithelial cells and whether there are signals that determine which cells should be discarded from the airway epithelium to restore the normal proportions of epithelial cell types so that the remaining cells differentiate into serous, Clara, ciliated, and mucous cells to reconstitute the proportions found in normal epithelia.
One of the regulators of cell death during the resolution of MCM is Bax that translocates to the ER and causes calcium release [64], [65]. The percentage of Bax-immunopositive mucous cells increases from approximately 3 to 25%, while the number of metaplastic mucous cells decreases [64]. Overall, approximately 25–35% of mucus cells express Bax after repeated exposure to allergen for 15 d. Trifilieff et al. [27] found that by 3 d post allergen exposure, approximately 30% of epithelial cell nuclei are BrdU-positive, a marker for cells that undergo DNA synthesis during the cell cycle. Taken together, the observed Bax positivity in approximately 25–35% of mucus cells during the resolution of allergen-induced GCM suggests that the Bax-positive mucous cells may represent cells that must be eliminated to reconstitute the original cell number of the repaired epithelium.
Sustained MCM in Asthma – Possible Therapies to Reduce Mucous Hypersecretion
It is generally believed that MCM in asthma does not resolve because of persistent inflammation in diseased airways that ensures the continuous presence of IL-13, the central regulator of MCM. Several studies have reported that IL-13 is present in the lungs of asthmatics at concentrations that are sufficient to induce cell proliferation and MCM. In patients with asthma, IL-13 levels are increased only upon allergen challenge to concentrations ranging from 0.4–3 ng/ml [68], sufficient to cause proliferation of NHBE cells [52]. IL-13 levels are found to be higher even in the sputum of mild asthmatics than in non-asthmatic controls [69]. Therefore, the presence of this cytokine may be the main reason for sustained MCM in asthma.
Given the importance of IL-13 in the development of MCM, efficient therapies for reducing mucous hypersecretion need to be based at least in part on the understanding of how the overabundance of IL-13 in the lungs of asthmatics is sustained. Dysregulation of IL-13 production in asthma may occur due to polymorphisms in the IL-13 gene that have been shown to be associated with the asthmatic phenotype [70], [71]. Several studies have demonstrated that blocking IL-13 and its signaling proteins reduce or eliminate the development of MCM in animal models of asthma [35], [54]. Clinical trials with IL-13 inhibitors will define the importance of this approach in reducing MCM in human asthma. Because CLCA proteins are critical for IL-13-induced MCM, while CLCA1 expression may not be a selective determinant of MCM so that shared homologies between CLCA family members may still represent a useful target for focused therapeutic intervention in hypersecretory airway disease.
The biosynthesis of mucus can be blocked by inhibiting the signaling proteins for mucin gene expression or blocking the glycosylation process. Inhibition of MUC mRNA biosynthesis using MAP kinase inhibitors has been proposed to inhibit mucin biosynthesis [51]. However, for developing effective therapies directed toward reducing the biosynthesis of mucins understanding the molecular mechanisms of the IL-13-mediated increase in MUC5AC gene expression and/or mRNA stabilization needs to be expanded. In addition to the biosynthesis of mucin glycoproteins, increased mucous hypersecretion requires proteins such as myristolated, alanine-rich C-kinase substrate (MARCKS) that is a target for phosphorylation by protein kinase C [72]. A MARCKS-related peptide was successfully used to block mucus hypersecretion in a mouse model of asthma [73], and human studies are under way to determine whether this approach will be clinically relevant.
N-acetyl cysteine has been used to reduce the viscosity of mucins so that the airways are not blocked by this viscous substrate [74]. However, in asthma it is the acute secretion of mucus in areas where MCM is prevalent that causes blockade of the airways and airway obstruction. This approach may only be suitable for the acute condition and not a treatment for the chronic presence of MCM.
Because surface epithelial mucous cells represent a great potential for secretion of mucosubstances [16], it is important to investigate how their numbers are regulated in airway epithelia. Therefore, reducing MCM based on the understanding of AEC apoptosis is regulated may also be highly effective in reducing mucous hypersecretion in asthma.
There is the possibility that the normal resolution of MCM is disrupted in patients with asthma. One could also speculate that humans with allergic asthma are deficient in developing such immune deviation to specific compounds or in IFNγ signaling to reduce MCM. In some instances, serum levels of IFNγ can be increased in severe asthma cases [75], and IFNγ levels in BALF are sometimes found even in mild asthma [76], [77]. Although these data seem paradoxical, IFNγ levels may not be high enough to increase the pro-apoptotic Bcl-2 family members. Supporting this hypothesis, the instillation of 100 ng IFNγ causes the reduction of MCM, while 50 ng does not [56]. Furthermore, polymorphisms in genes encoding for IFNγ and IFN regulatory factor-1 (IRF-1) confer genetic susceptibility to allergic asthma in Japanese children [78] and Stat 1 is constitutively activated in epithelial cells of asthmatics [79]. Therefore, it is also possible that in a subpopulation of asthmatics a deficiency in the IFNγ-signaling pathway may render IFNγ incapable of inducing cell death in epithelial cells and in sustaining MCM. In that context, initiating cell death of hyperplastic AECs using pro-apoptotic Bcl-2 family members as part of a general therapeutic effort to reduce inflammation may be highly effective. Chronic inflammation in animal models needs to be established to adequately address these complex issues of MCM in asthma.
In summary, the amount of mucus in the airways of asthmatics may be controlled at various levels. First, the biosynthesis of mucins including the transcription of mucin genes followed by translation and posttranslational glycosylation, can be crucial stages for controlling the amount of mucin synthesized. Second, the secretion of the synthesized mucins may be suppressed by affecting the mechanisms underlying the transport and release of vesicles containing the mucins. Third, the number of mucus-producing cells can be reduced by initiating cell death by driving the proapoptotic pathways that are involved in the normal resolution of airway epithelial hyperplasia and thereby help reconstitute the normal epithelium.
Fig. 1 (A):
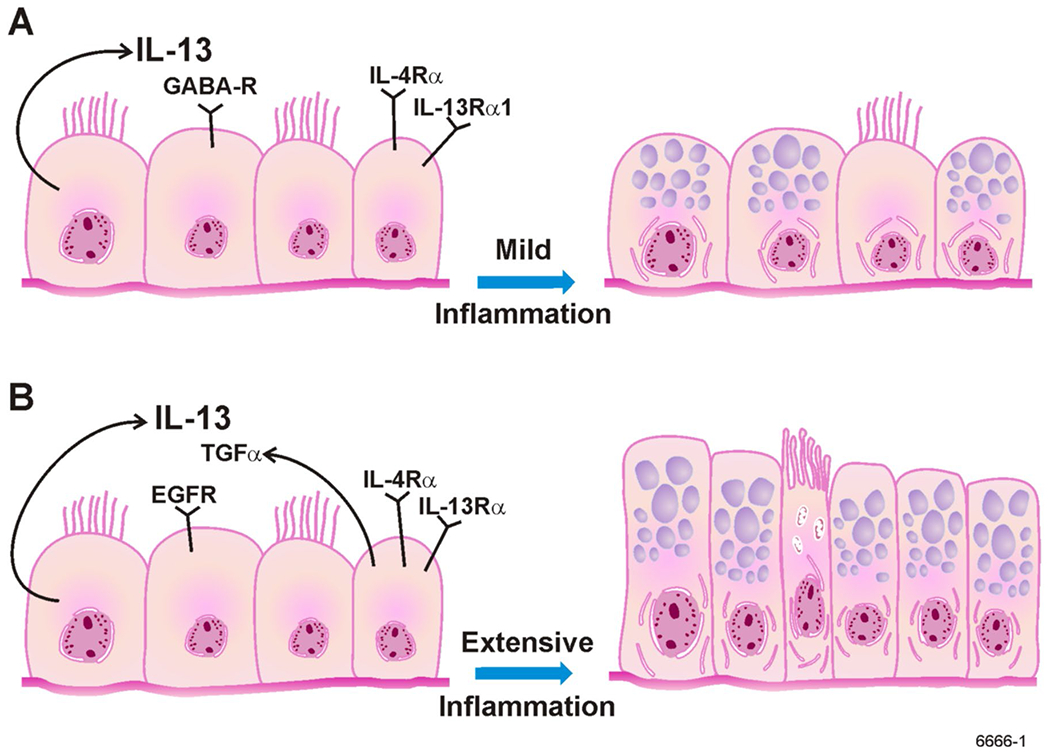
Under conditions of mild inflammation, MCM is induced when AECs start to produce and store mucous proteins. IL-13 that is produced by AECs acts through IL-4α and IL-13α1 and on GABA-R to increase mucin gene expression and protein synthesis. (B): Under conditions of extensive inflammation, MCM is induced by AECs undergoing proliferation and storing MCM. IL-13 causes proliferation of AECs through EGF-R/TGFα-dependent mechanisms.
Acknowledgments
This research was supported by grants from the National Heart Lung and Blood Institute (HL068111).
Abbreviations:
- AECs
airway epithelial cells
- BALF
bronchial lavage fluid
- CLCA
calcium-activated chloride channel
- FEV
forced expiratory volume
- GABA
γ-aminobutyric acid
- MARCKS
myristolated, alanine-rich C-kinase substrate
- MCM
mucous cell metaplasia
- NHBEs
normal human airway epithelial cells
References
- [1].Plopper CG, Heidsiek JG, Weir AJ, George JA, Hyde DM, Tracheobronchial epithelium in the adult rhesus monkey: a quantitative histochemical and ultrastructural study, Am J Anat 184 (1989) 31–40. [DOI] [PubMed] [Google Scholar]
- [2].Pack RJ, Al-Ugaily LH, Morris G, The cells of the tracheobronchial epithelium of the mouse: a quantitative light and electron microscope study, J Anat 132 (1981) 71–84. [PMC free article] [PubMed] [Google Scholar]
- [3].Boers JE, Ambergen AW, Thunnissen FB, Number and proliferation of clara cells in normal human airway epithelium, American journal of respiratory and critical care medicine 159 (1999) 1585–1591. [DOI] [PubMed] [Google Scholar]
- [4].Aikawa T, Shimura S, Sasaki H, Ebina M, Takishima T, Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack, Chest 101 (1992) 916–921. [DOI] [PubMed] [Google Scholar]
- [5].Robbins, Cellular Growth and Differentiation: Normal Regulation and Adaptations., in: J. SF (Ed.), Pathologic Basis of Disease, W.B. Saunders Company, Philadelphia, 1994, pp. 35–50. [Google Scholar]
- [6].Longphre M, Li D, Gallup M, Drori E, Ordonez CL, Redman T, Wenzel S, Bice DE, Fahy JV, Basbaum C, Allergen-induced IL-9 directly stimulates mucin transcription in respiratory epithelial cells, The Journal of clinical investigation 104 (1999) 1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Temann UA, Prasad B, Gallup MW, Basbaum C, Ho SB, Flavell RA, Rankin JA, A novel role for murine IL-4 in vivo: induction of MUC5AC gene expression and mucin hypersecretion, American journal of respiratory cell and molecular biology 16 (1997) 471–478. [DOI] [PubMed] [Google Scholar]
- [8].Zuhdi Alimam M, Piazza FM, Selby DM, Letwin N, Huang L, Rose MC, Muc5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways, American journal of respiratory cell and molecular biology 22 (2000) 253–260. [DOI] [PubMed] [Google Scholar]
- [9].Borchers MT, Carty MP, Leikauf GD, Regulation of human airway mucins by acrolein and inflammatory mediators, The American journal of physiology 276 (1999) L549–555. [DOI] [PubMed] [Google Scholar]
- [10].Fischer BM, Voynow JA, Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species, American journal of respiratory cell and molecular biology 26 (2002) 447–452. [DOI] [PubMed] [Google Scholar]
- [11].Lange P, Parner J, Vestbo J, Schnohr P, Jensen G, A 15-year follow-up study of ventilatory function in adults with asthma, The New England journal of medicine 339 (1998) 1194–1200. [DOI] [PubMed] [Google Scholar]
- [12].Shimura S, Andoh Y, Haraguchi M, Shirato K, Continuity of airway goblet cells and intraluminal mucus in the airways of patients with bronchial asthma, Eur Respir J 9 (1996) 1395–1401. [DOI] [PubMed] [Google Scholar]
- [13].Saetta M, Di Stefano A, Rosina C, Thiene G, Fabbri LM, Quantitative structural analysis of peripheral airways and arteries in sudden fatal asthma, Am Rev Respir Dis 143 (1991) 138–143. [DOI] [PubMed] [Google Scholar]
- [14].Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Pare PD, The nature of small-airway obstruction in chronic obstructive pulmonary disease, The New England journal of medicine 350 (2004) 2645–2653. [DOI] [PubMed] [Google Scholar]
- [15].Dunnell MS, The pathology of asthma, with special references to changes in the bronchial mucosa, The Journal of clinical investigation 13 (1960) 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rogers DF, Airway goblet cells: responsive and adaptable front-line defenders, Eur Respir J 7 (1994) 1690–1706. [PubMed] [Google Scholar]
- [17].Evans CM, Williams OW, Tuvim MJ, Nigam R, Mixides GP, Blackburn MR, DeMayo FJ, Burns AR, Smith C, Reynolds SD, Stripp BR, Dickey BF, Mucin is produced by clara cells in the proximal airways of antigen-challenged mice, American journal of respiratory cell and molecular biology 31 (2004) 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tesfaigzi Y, Harris JF, Hotchkiss JA, Harkema JR, DNA synthesis and Bcl-2 expression during the development of mucous cell metaplasia in airway epithelium of rats exposed to LPS., American journal of physiology 286 (2004) L268–274. [DOI] [PubMed] [Google Scholar]
- [19].Reader JR, Tepper JS, Schelegle ES, Aldrich MC, Putney LF, Pfeiffer JW, Hyde DM, Pathogenesis of mucous cell metaplasia in a murine asthma model, Am J Pathol 162 (2003) 2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Harris JF, Aden J, Lyons R, Tesfaigzi Y, Resolution of LPS-induced airway inflammation and goblet cell hyperplasia is independent of IL-18., Respir Res 8 (2007) 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Druilhe A, Wallaert B, Tsicopoulos A, Lapa e Silva JR, Tillie-Leblond I, Tonnel AB, Pretolani M, Apoptosis, proliferation, and expression of Bcl-2, Fas, and Fas ligand in bronchial biopsies from asthmatics, American journal of respiratory cell and molecular biology 19 (1998) 747–757. [DOI] [PubMed] [Google Scholar]
- [22].Harkema J, Hotchkiss J, Griffith W, Mucous cell metaplasia in rat nasal epithelium after a 20-month exposure to ozone: a morphometric study of epithelial differentiation., American journal of respiratory cell and molecular biology 16 (1997) 521–530. [DOI] [PubMed] [Google Scholar]
- [23].Longphre M, Zhang L, Harkema JR, Kleeberger SR, Ozone-induced pulmonary inflammation and epithelial proliferation are partially mediated by PAF, J Appl Physiol 86 (1999) 341–349. [DOI] [PubMed] [Google Scholar]
- [24].Harkema JR, Hotchkiss JA, Barr EB, Bennett CB, Gallup M, Lee JK, Basbaum C, Long-lasting effects of chronic ozone exposure on rat nasal epithelium, American journal of respiratory cell and molecular biology 20 (1999) 517–529. [DOI] [PubMed] [Google Scholar]
- [25].Cohen L, E X, Tarsi J, Ramkumar T, Horiuchi TK, Cochran R, Demartino S, Schechtman KB, Hussain I, Holtzman MJ, Castro M, Epithelial cell proliferation contributes to airway remodeling in severe asthma, American journal of respiratory and critical care medicine 176 (2007) 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Erjefalt JS, Erjefalt I, Sundler F, Persson CG, In vivo restitution of airway epithelium, Cell Tissue Res 281 (1995) 305–316. [DOI] [PubMed] [Google Scholar]
- [27].Trifilieff A, El-Hashim A, Bertrand C, Time course of inflammatory and remodeling events in a murine model of asthma: effect of steroid treatment, American journal of physiology 279 (2000) L1120–1128. [DOI] [PubMed] [Google Scholar]
- [28].Keenan KP, Combs JW, McDowell EM, Regeneration of hamster tracheal epithelium after mechanical injury. I. Focal lesions: quantitative morphologic study of cell proliferation, Virchows Arch B Cell Pathol Incl Mol Pathol 41 (1982) 193–214. [DOI] [PubMed] [Google Scholar]
- [29].Harris JF, Fischer MJ, Hotchkiss JA, Monia BP, Randell SH, Harkema JR, Tesfaigzi Y, Bcl-2 sustains increased mucous and epithelial cell numbers in metaplastic airway epithelium., American journal of respiratory and critical care medicine 171 (2005) 764–772. [DOI] [PubMed] [Google Scholar]
- [30].Pierce J, Rir-Sima-Ah J, Estrada I, Wilder JA, Strasser A, Tesfaigzi Y, Loss of pro-apoptotic Bim promotes accumulation of pulmonary T lymphocytes and enhances allergen-induced goblet cell metaplasia, American journal of physiology 291 (2006) L862–870. [DOI] [PubMed] [Google Scholar]
- [31].Keenan KP, Combs JW, E.M. M, Regeneration of hamster tracheal epithelium after mechanical injury. I. Focal lesions: Quantitative morphologic study of cell proliferarion., Virchows Arch. B 41 (1982) 193–214. [DOI] [PubMed] [Google Scholar]
- [32].Keenan KP, Combs JW, McDowell EM, Regeneration of hamster tracheal epithelium after mechanical injury. II. Multifocal lesions: stathmokinetic and autoradiographic studies of cell proliferation, Virchows Arch B Cell Pathol Incl Mol Pathol 41 (1982) 215–229. [DOI] [PubMed] [Google Scholar]
- [33].Jeffery PK, Reid L, New observations of rat airway epithelium: a quantitative and electron microscopic study, J Anat 120 (1975) 295–320. [PMC free article] [PubMed] [Google Scholar]
- [34].Harkema JR, Hotchkiss JA, In vivo effects of endotoxin on intraepithelial mucosubstances in rat pulmonary airways. Quantitative histochemistry, Am J Pathol 141 (1992) 307–317. [PMC free article] [PubMed] [Google Scholar]
- [35].Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD, Interleukin-13: central mediator of allergic asthma., Science 282 (1998) 2258–2261. [DOI] [PubMed] [Google Scholar]
- [36].Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, Corry DB, Requirement for IL-13 independently of IL-4 in experimental asthma, Science 282 (1998) 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vermeer PD, Harson R, Einwalter LA, Moninger T, Zabner J, Interleukin-9 induces goblet cell hyperplasia during repair of human airway epithelia, American journal of respiratory cell and molecular biology 28 (2003) 286–295. [DOI] [PubMed] [Google Scholar]
- [38].Louahed J, Toda M, Jen J, Hamid Q, Renauld JC, Levitt RC, Nicolaides NC, Interleukin-9 upregulates mucus expression in the airways., American journal of respiratory cell and molecular biology 22 (2000) 649–656. [DOI] [PubMed] [Google Scholar]
- [39].Temann UA, Ray P, Flavell RA, Pulmonary overexpression of IL-9 induces Th2 cytokine expression, leading to immune pathology, The Journal of clinical investigation 109 (2002) 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ, Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma, Nature medicine 8 (2002) 885–889. [DOI] [PubMed] [Google Scholar]
- [41].Temann UA, Laouar Y, Eynon EE, Homer R, Flavell RA, IL9 leads to airway inflammation by inducing IL13 expression in airway epithelial cells, International immunology 19 (2007) 1–10. [DOI] [PubMed] [Google Scholar]
- [42].Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A, Homer RJ, Cohn L, Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9, American journal of respiratory cell and molecular biology 27 (2002) 593–602. [DOI] [PubMed] [Google Scholar]
- [43].Kuperman D, Schofield B, Wills-Karp M, Grusby MJ, Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production, J Exp Med 187 (1998) 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xiang YY, Wang S, Liu M, Hirota JA, Li J, Ju W, Fan Y, Kelly MM, Ye B, Orser B, O’Byrne P M, Inman MD, Yang X, Lu WY, A GABAergic system in airway epithelium is essential for mucus overproduction in asthma, Nature medicine 13 (2007) 862–867. [DOI] [PubMed] [Google Scholar]
- [45].Corry DB, Kheradmand F, A new link to airway obstruction in asthma, Nature medicine 13 (2007) 777–778. [DOI] [PubMed] [Google Scholar]
- [46].Atherton HC, Jones G, Danahay H, IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation, American journal of physiology 285 (2003) L730–739. [DOI] [PubMed] [Google Scholar]
- [47].Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, Fujisawa Y, Nishimura O, Fujino M, Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma, Proceedings of the National Academy of Sciences of the United States of America 98 (2001) 5175–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, Nakanishi A, Ashida Y, Nishimura O, Fujisawa Y, Fujino M, Increased expression of the human Ca2+-activated Cl- channel 1 (CaCC1) gene in the asthmatic airway, American journal of respiratory and critical care medicine 165 (2002) 1132–1136. [DOI] [PubMed] [Google Scholar]
- [49].Robichaud A, Tuck SA, Kargman S, Tam J, Wong E, Abramovitz M, Mortimer JR, Burston HE, Masson P, Hirota J, Slipetz D, Kennedy B, O’Neill G, Xanthoudakis S, Gob-5 is not essential for mucus overproduction in preclinical murine models of allergic asthma, American journal of respiratory cell and molecular biology 33 (2005) 303–314. [DOI] [PubMed] [Google Scholar]
- [50].Patel AC, Morton JD, Kim EY, Alevy Y, Swanson S, Tucker J, Huang G, Agapov E, Phillips TE, Fuentes ME, Iglesias A, Aud D, Allard JD, Dabbagh K, Peltz G, Holtzman MJ, Genetic segregation of airway disease traits despite redundancy of calcium-activated chloride channel family members, Physiological genomics 25 (2006) 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li JD, Feng W, Gallup M, Kim JH, Gum J, Kim Y, Basbaum C, Activation of NF-kappaB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells, Proceedings of the National Academy of Sciences of the United States of America 95 (1998) 5718–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Booth BW, Adler KB, Bonner JC, Tournier F, Martin LD, Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-alpha, American journal of respiratory cell and molecular biology 25 (2001) 739–743. [DOI] [PubMed] [Google Scholar]
- [53].Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA, IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils, American journal of physiology 280 (2001) L134–140. [DOI] [PubMed] [Google Scholar]
- [54].Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, Battaile JT, Patel AC, Patterson GA, Castro M, Spoor MS, You Y, Brody SL, Holtzman MJ, Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals, The Journal of clinical investigation 116 (2006) 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Blyth DI, Pedrick MS, Savage TJ, Bright H, Beesley JE, Sanjar S, Induction, duration, and resolution of airway goblet cell hyperplasia in a murine model of atopic asthma: effect of concurrent infection with respiratory syncytial virus and response to dexamethasone, American journal of respiratory cell and molecular biology 19 (1998) 38–54. [DOI] [PubMed] [Google Scholar]
- [56].Shi ZQ, Fischer MJ, De Sanctis GT, Schuyler M, Tesfaigzi Y, IFNγ but not Fas mediates reduction of allergen-induced mucous cell metaplasia by inducing apoptosis, J Immunol 168 (2002) 4764–4771. [DOI] [PubMed] [Google Scholar]
- [57].Tesfaigzi Y, Roles of Apoptosis in Airway Epithelia, American journal of respiratory cell and molecular biology 34 (2006) 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yiamouyiannis CA, Schramm CM, Puddington L, Stengel P, Baradaran-Hosseini E, Wolyniec WW, Whiteley HE, Thrall RS, Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model, Am J Pathol 154 (1999) 1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Haczku A, Moqbel R, Elwood W, Sun J, Kay AB, Barnes PJ, Chung KF, Effects of prolonged repeated exposure to ovalbumin in sensitized Brown Norway rats, American journal of respiratory and critical care medicine 150 (1994) 23–27. [DOI] [PubMed] [Google Scholar]
- [60].Howarth PH, The airway inflammatory response in allergic asthma and its relationship to clinical disease, Allergy 50 (1995) 13–21. [DOI] [PubMed] [Google Scholar]
- [61].Cui ZH, Joetham A, Aydintug MK, Hahn YS, Born WK, Gelfand EW, Reversal of allergic airway hyperreactivity after long-term allergen challenge depends on gammadelta T cells, American journal of respiratory and critical care medicine 168 (2003) 1324–1332. [DOI] [PubMed] [Google Scholar]
- [62].Stock P, Akbari O, Berry G, Freeman GJ, Dekruyff RH, Umetsu DT, Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity, Nat Immunol 5 (2004) 1149–1156. [DOI] [PubMed] [Google Scholar]
- [63].Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, Berry G, DeKruyff RH, Umetsu DT, Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity, Nature medicine 8 (2002) 1024–1032. [DOI] [PubMed] [Google Scholar]
- [64].Tesfaigzi Y, Fischer MJ, Green FHY, De Sanctis GT, Wilder JA, Bax is crucial for IFNγ-induced resolution of allergen-induced mucous cell metaplasia., J Immunol 169 (2002) 5919–5925. [DOI] [PubMed] [Google Scholar]
- [65].Stout B, Melendez K, Seagrave J, Holtzman MJ, Wilson B, Xiang J, Tesfaigzi Y, STAT1 activation causes translocation of Bax to the endoplasmic reticulum during the resolution of airway mucous cell hyperplasia by IFNγ, J Immunol 178 (2007) 8107–8116. [DOI] [PubMed] [Google Scholar]
- [66].Grossmann J, Molecular mechanisms of “detachment-induced apoptosis--Anoikis”, Apoptosis 7 (2002) 247–260. [DOI] [PubMed] [Google Scholar]
- [67].Teague TK, Marrack P, Kappler JW, Vella AT, IL-6 rescues resting mouse T cells from apoptosis, J Immunol 158 (1997) 5791–5796. [PubMed] [Google Scholar]
- [68].Huang SK, Xiao HQ, Kleine-Tebbe J, Paciotti G, Marsh DG, Lichtenstein LM, Liu MC, IL-13 expression at the sites of allergen challenge in patients with asthma, J Immunol 155 (1995) 2688–2694. [PubMed] [Google Scholar]
- [69].Berry MA, Parker D, Neale N, Woodman L, Morgan A, Monk P, Bradding P, Wardlaw AJ, Pavord ID, Brightling CE, Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis, The Journal of allergy and clinical immunology 114 (2004) 1106–1109. [DOI] [PubMed] [Google Scholar]
- [70].Howard TD, Meyers DA, Bleecker ER, Mapping susceptibility genes for allergic diseases, Chest 123 (2003) 363S–368S. [DOI] [PubMed] [Google Scholar]
- [71].Hunninghake GM, Soto-Quiros ME, Avila L, Su J, Murphy A, Demeo DL, Ly NP, Liang C, Sylvia JS, Klanderman BJ, Lange C, Raby BA, Silverman EK, Celedon JC, Polymorphisms in IL13, total IgE, eosinophilia, and asthma exacerbations in childhood, The Journal of allergy and clinical immunology 120 (2007) 84–90. [DOI] [PubMed] [Google Scholar]
- [72].Li Y, Martin LD, Spizz G, Adler KB, MARCKS protein is a key molecule regulating mucin secretion by human airway epithelial cells in vitro, The Journal of biological chemistry 276 (2001) 40982–40990. [DOI] [PubMed] [Google Scholar]
- [73].Singer M, Martin LD, Vargaftig BB, Park J, Gruber AD, Li Y, Adler KB, A MARCKS-related peptide blocks mucus hypersecretion in a mouse model of asthma, Nature medicine 10 (2004) 193–196. [DOI] [PubMed] [Google Scholar]
- [74].Decramer M, Rutten-van Molken M, Dekhuijzen PN, Troosters T, van Herwaarden C, Pellegrino R, van Schayck CP, Olivieri D, Del Donno M, De Backer W, Lankhorst I, Ardia A, Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial, Lancet 365 (2005) 1552–1560. [DOI] [PubMed] [Google Scholar]
- [75].Corrigan CJ, Kay AB, CD4 T-lymphocyte activation in acute severe asthma. Relationship to disease severity and atopic status, Am Rev Respir Dis 141 (1990) 970–977. [DOI] [PubMed] [Google Scholar]
- [76].Cembrzynska-Nowak M, Szklarz E, Inglot AD, Teodorczyk-Injeyan JA, Elevated release of tumor necrosis factor-alpha and interferon-gamma by bronchoalveolar leukocytes from patients with bronchial asthma, Am Rev Respir Dis 147 (1993) 291–295. [DOI] [PubMed] [Google Scholar]
- [77].Holtzman MJ, Sampath D, Castro M, Look DC, Jayaraman S, The one-two of T helper cells: does interferon-gamma knock out the Th2 hypothesis for asthma?, American journal of respiratory cell and molecular biology 14 (1996) 316–318. [DOI] [PubMed] [Google Scholar]
- [78].Nakao F, Ihara K, Kusuhara K, Sasaki Y, Kinukawa N, Takabayashi A, Nishima S, Hara T, Association of IFN-gamma and IFN regulatory factor 1 polymorphisms with childhood atopic asthma, The Journal of allergy and clinical immunology 107 (2001) 499–504. [DOI] [PubMed] [Google Scholar]
- [79].Sampath D, Castro M, Look DC, Holtzman MJ, Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma, The Journal of clinical investigation 103 (1999) 1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]