

Abstract
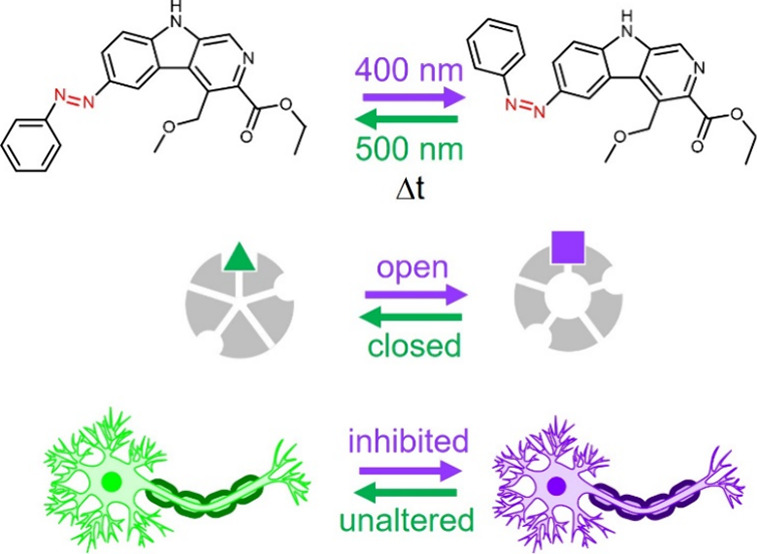
Gamma aminobutyric acid type A receptors (GABAARs) play a key role in the mammalian central nervous system (CNS) as drivers of neuroinhibitory circuits, which are commonly targeted for therapeutic purposes with potentiator drugs. However, due to their widespread expression and strong inhibitory action, systemic pharmaceutical potentiation of GABAARs inevitably causes adverse effects regardless of the drug selectivity. Therefore, therapeutic guidelines must often limit or exclude clinically available GABAAR potentiators, despite their high efficacy, good biodistribution, and favorable molecular properties. One solution to this problem is to use drugs with light-dependent activity (photopharmacology) in combination with on-demand, localized illumination. However, a suitable light-activated potentiator of GABAARs has been elusive so far for use in wildtype mammals. We have met this need by developing azocarnil, a diffusible GABAergic agonist-potentiator based on the anxiolytic drug abecarnil that is inactive in the dark and activated by visible violet light. Azocarnil can be rapidly deactivated with green light and by thermal relaxation in the dark. We demonstrate that it selectively inhibits neuronal currents in hippocampal neurons in vitro and in the dorsal horns of the spinal cord of mice, decreasing the mechanical sensitivity as a function of illumination without displaying systemic adverse effects. Azocarnil expands the in vivo photopharmacological toolkit with a novel chemical scaffold and achieves a milestone toward future phototherapeutic applications to safely treat muscle spasms, pain, anxiety, sleep disorders, and epilepsy.
Introduction
Gamma aminobutyric acid type A receptors (GABAARs) are the main inhibitory synaptic receptors in the mammalian central nervous system (CNS) and are important targets in pharmaceutical research.1 They form heteropentameric transmembrane assemblies with a central chloride-ion-permeable pore that opens upon GABA binding, hyperpolarizing the cell membrane and inhibiting neuronal activity (Figure 1A). GABAAR agonists and potentiators (positive allosteric modulators, PAM) like barbiturates and especially benzodiazepines (Figure 1B), are widely used to treat muscle spasms, alcohol withdrawal, insomnia, anxiety, delirium, and seizures, as well as to induce sedation and anesthesia.2 Propofol is a GABAAR PAM exclusively used for general anesthesia and binds to a different receptor site than benzodiazepines. GABAAR antagonists like bicuculline and gabazine are useful as research tools to functionally dissect neuronal GABAAR current components and behavioral phenotypes, but their stimulant and convulsant effects in vivo offer limited clinical applications, like treating benzodiazepine withdrawal syndrome and overdose.3
Figure 1.
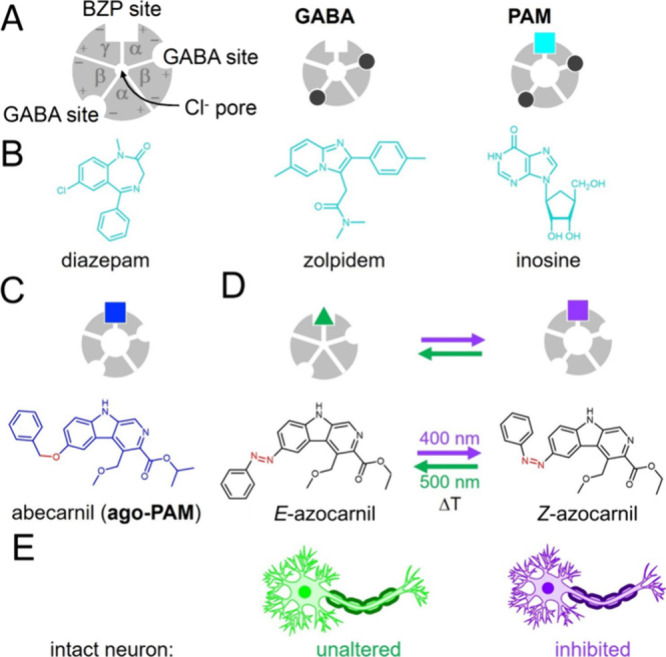
Design of a neuroinhibitory photoswitchable drug azocarnil. (A) Diagram of the GABAAR, as seen from the extracellular side, showing the subunit (α, β, and γ) arrangement and the principal (+) and complementary (−) subunit interfaces, as well as the central Cl− ion pore, the orthosteric GABA ligand sites, and the primary allosteric ligand site of benzodiazepines (BZPs) and β-carbolines. Other allosteric potentiator sites (propofol, barbiturate, ethanol) are not shown for simplicity. Binding of the endogenous orthosteric ligand GABA (black dots) opens the Cl– permeable pore, thus hyperpolarizing (i.e., inhibiting the activity of) the neuron. Potentiators or positive allosteric modulators (PAMs, cyan square) enlarge Cl– currents, thus enhancing the inhibitory effect of GABA binding. (B) Examples of PAMs binding at the canonical high-affinity benzodiazepine binding site: diazepam, zolpidem,30 and the endogenous GABAAR allosteric ligand inosine.31,32 (C) A special class of PAMs (termed ago-PAMs, blue square) can open the ion channel in the absence of GABA, like abecarnil25,33 below. (D) Design of a photoswitchable potentiator based on the structure of abecarnil by replacing the ether bond by an aza bond (azolog) and the isopropyl ester group by an ethyl ester group for synthetic accessibility reasons. Azobenzene photoisomerization between the E and Z configurations (represented as a green triangle and a violet square, respectively) is intended to change the agonist-potentiator activity and control GABAAR channel opening with light. ΔT indicates the thermal relaxation from Z to E in the dark. (E) The activity of intact neurons can be modulated with light by means of their endogenous GABAARs and azocarnil applied in the medium. Neuronal activity should not be altered in the dark or under green light, corresponding to safe conditions regardless of the presence of the drug (e.g., systemically). Neuroinhibition should be elicited on demand and in localized regions using violet illumination.
Neurons expressing GABAARs in the dorsal horns of the spinal cord are key elements in regulation of sensory information and pain sensation. The complex local inhibitory circuitry in the dorsal horns is also known as the “gate” of peripheral sensory information.4 Hence, downregulation of inhibitory transmission in this spinal cord region leads to the development of chronic pain.5
For these reasons, GABAARs in the spinal cord and other CNS regions have a privileged position as drivers of neuroinhibitory circuits that can be targeted for therapeutic purposes with potentiator drugs. However, due to their widespread expression and strong inhibitory action, systemic and indiscriminate potentiation of GABAARs with drugs gives rise to adverse effects regardless of the drug selectivity for these receptors. Therefore, despite the efficacy of clinically available GABAAR potentiators like phenobarbital and clonazepam, they are not recommended in modern therapeutic guidelines to treat neuropathic pain and to restore inhibitory drive in the spinal cord because they lead to intolerable systemic neuroinhibition in supraspinal CNS centers.6,7
One possibility to avoid undesired GABAAR potentiation at the systemic level while locally keeping it at the intended neuronal circuits (e.g., at the spinal cord for neuropathic pain) is to use drugs with light-dependent activity (photopharmacology) in combination with localized illumination. Drugs modified with chemical groups that change their configuration under different wavelengths of light have been shown to control protein activity, cell and tissue function,8 and animal behavior.9 These groups are dubbed photoswitches. One of them is azobenzene, which reversibly photoisomerizes between E and Z configurations of its N=N double bond and changes its length and dipole moment.10 These photoswitchable groups can be coupled by design to bioactive molecules with suitable pharmacological properties like agonism, antagonism, and modulation, thus producing powerful molecular tools to investigate physiological processes and to develop new therapeutic modalities.11 A successful photopharmacological design strategy involves the direct replacement of biaryl amides or biaryl ethers by azobenzene (azobenzene analogs or “azologs”12,13) in a drug of interest, which endows the parent compound with photocontrolled activity while largely retaining all other valuable molecular properties.14
The ideal GABAAR photoswitchable drug profile for therapeutic purposes is the following: (1) It should have agonist-potentiator activity to provide robust neuroinhibition regardless of the presence of endogenous GABA, which may be reduced in certain conditions. (2) It should target the benzodiazepine site to avoid the anesthetic effects and the lack of analgesic action associated with the propofol site. (3) It should remain inactive in the dark to eliminate adverse effects at unintended locations in case that they are exposed to the drug (e.g., by systemic administration or diffusion from the local administration site). (4) It should be activated under illumination, preferably by visible light to avoid the cellular toxicity of UV light (<380 nm). (5) It should be a freely diffusible and drug-like small molecule to facilitate administration, biodistribution, and drug development.
Although considerable efforts have been invested to develop GABAAR photopharmacology (recently reviewed by Kramer and Rajappa15), the quest remains for a GABAergic photoswitchable agonist-potentiator that is inactive in the dark and demonstrates therapeutic effects in wildtype mammals under illumination. The only GABAergic compounds tested in mice so far are photoswitchable antagonists that must be covalently tethered to cysteine residues introduced in a GABAAR subunit by site-directed mutagenesis (optogenetic pharmacology).16,17 Thus, they require using knock-in mice, which is advantageous for fundamental studies of inhibitory neurotransmission but not useful to develop therapeutic applications in wildtype animals, including humans. The tethered approach has also been applied to propofol agonists in vitro, including both dark-inactive18 and dark-active19 tethering sites. Besides, all freely diffusible GABAAR photoswitches that have been reported are dark-active (either targeting the channel pore,20 the GABA site,21 or the propofol site22) except for fulgazepam, a benzodiazepine modified with a fulgimide photoswitch that is dark-inactive and acts as a GABAAR potentiator under UV (365 nm) light.23 Azopropofol displays anesthetic activity in tadpoles that is reduced under 360 nm light,22 and fulgazepam allows photocontrolling zebrafish locomotion;23 however, none of these ligands has been tested in rodents.
Thus, there is an unmet need for dark-inactive photoswitchable agonist potentiators of GABAARs to produce neuroinhibition with light in mammals. To achieve the photopharmacological profile outlined above, we focused on diffusible ligands targeting the benzodiazepine site, like fulgazepam,23 but having a nonbenzodiazepine scaffold. We searched among β-carbolines,24 which bind to the same site and produce similar pharmacological effects as benzodiazepines, and selected abecarnil25 due to its drug-likeness and azologization potential (Figure 1C). Abecarnil acts as a partial agonist and PAM of GABAARs with high binding affinity.26 It shows anxiolytic and anticonvulsant effects, but in contrast to benzodiazepines, it exerts weak or no sedative and ataxic actions.27−29
We designed an azobenzene analogue of abecarnil (termed azocarnil) by replacing the ether bond by an azo group (Figure 1C). In the absence of the receptor-bound atomic structure of abecarnil, we used molecular docking calculations to test whether such modification is compatible with receptor binding and whether there are differences in binding between the Z and E isomers. Our computational results suggested that Z-azocarnil, but not E, can bind in the benzodiazepine site similar to abecarnil and diazepam. Such a prediction is confirmed by our experimental observation that azocarnil potentiates GABAergic currents in hippocampal neurons under 400 nm illumination (Z configuration). Upon intrathecal injection in wildtype mice, azocarnil decreases the withdrawal responses to cutaneous mechanical stimulation as a function of spinal cord illumination and does not display systemic adverse effects. Thus, photoswitchable analogs of abecarnil significantly expand the GABAAR photopharmacological toolkit based on a novel chemical scaffold and achieve photoactivated neuroinhibition without adverse effects in mammals, a milestone toward future therapeutic applications to safely treat muscle spasms, pain, anxiety and sleep disorders, and epilepsy.
Results
Molecular Design of a Photoactivated Agonist-Potentiator of GABAARs
We started by asking whether azocarnil retains the binding properties of abecarnil and whether the properties can be controlled by Z–E photoisomerization. Since the GABAAR structure bound to abecarnil is not available, we looked for other compounds that bind to the same receptor site and whose structure in complex with GABAAR has been determined experimentally, with the aim of using molecular docking calculations to compare them. We first validated our docking protocol by redocking methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (DMCM, a negative allosteric modulator of the same β-carboline family as abecarnil, Figure 2A) and diazepam (another potentiator binding to the same site but from the benzodiazepine family, Figure 2B) in their respective cryo-electron microscopy structures in complex with the human GABAAR α1β2γ2 subtype (PDB codes 8DD3(34) and 6X3X,35 respectively). The representative docking poses obtained are almost identical with the experimentally determined binding poses for both ligands (Figure S6), confirming the reliability of the docking protocol used.
Figure 2.
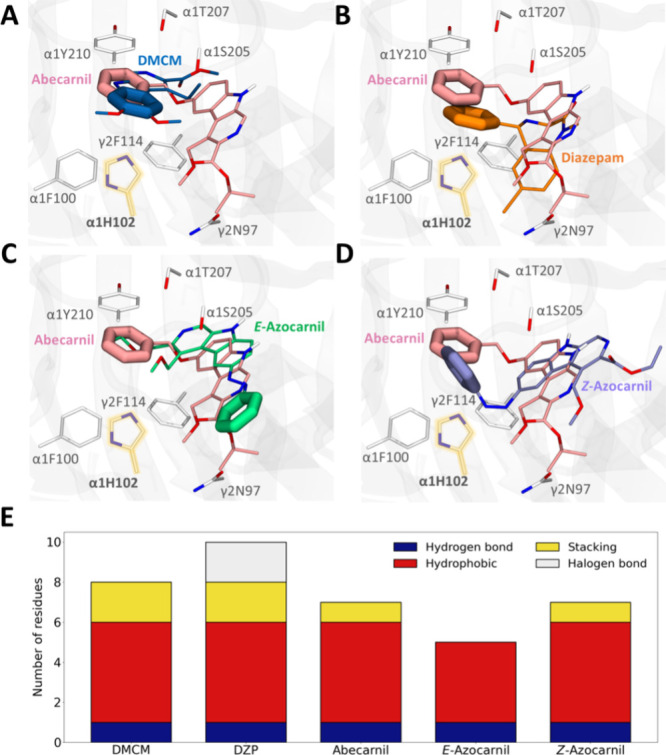
Molecular docking calculations of abecarnil and E- and Z-azocarnil in the benzodiazepine binding site of the GABAA receptor. Comparison of the representative docking pose of the most populated cluster between abecarnil (pink) and (A) DMCM (blue, experimental structure with PDB code 8DD3), (B) diazepam (orange, experimental structure with PDB code 6X3X), (C) E-azocarnil (green), and (D) Z-azocarnil (violet). Only the GABAAR residues (white sticks) for which mutagenesis data is available are shown.34,36−40,42 The α1 His102 residue (Table S1 and Figures S3–S5), which is essential for β-carboline activity, is highlighted in yellow.37 The phenyl rings of the compounds are shown as thicker lines to emphasize their positions in the benzodiazepine site. (E) Number of GABAAR residues for which mutagenesis data are available that are in contact with the compounds, quantifying separately hydrogen bond (dark blue), hydrophobic (red), stacking (yellow), and halogen bond (gray) interactions.
We then computed the binding mode of the parent compound abecarnil (potentiator), focusing on the benzodiazepine binding site between the principal (+) side of the α1 subunit and the complementary (−) side of the γ2 subunit, as this is the known primary, high-affinity site for both β-carbolines and benzodiazepines in GABAARs.24,26 Most of the experimentally observed interactions for diazepam and DMCM are retained for abecarnil (Table S1). We also observe that the phenyl pendant ring of abecarnil is placed in the same region as the phenyl pendant ring of diazepam and the phenyl ring of DMCM. This region contains most of the residues for which mutagenesis data is available, demonstrating their importance for β-carboline and benzodiazepine binding.34,36−40
To evaluate the feasibility that azocarnil maintains the same binding mode as the parent compound abecarnil in the benzodiazepine site, we then performed molecular docking calculations of both E and Z isomers, as previously done for other photoswitchable ligands targeting GABAARs.41 The orientation of the docked E-azocarnil does not resemble abecarnil, diazepam, or DMCM, as it is turned around 180° (Figure 2C). Moreover, the E form is missing a key interaction with the α1 His102 residue (Table S1 and Figure S4B), which is essential for β-carboline activity according to mutagenesis data.37 In contrast, the preferred binding pose of docked Z-azocarnil resembles that of docked abecarnil (Figure 2D), as well as that of diazepam and DMCM. The azobenzene moiety of Z-azocarnil is superimposed with the pendant phenyl ring of the diazepam molecule, thus conserving the strong stabilization by the surrounding aromatic residues35 (Figures 2E and S4A). Altogether, the binding modes predicted by docking in the benzodiazepine binding site suggest that Z-azocarnil could act as a potentiator. To test this prediction experimentally, we proceeded with the synthesis of azocarnil and the characterization of its photopharmacological properties.
Synthesis of Azocarnil
To design a photoswitchable analogue of abecarnil (Figure 1C), the ether bond between the carboline scaffold and the pendant phenyl ring was replaced by an azo group, and the isopropyl ester group had to be substituted by an ethyl ester group for synthetic accessibility reasons. The synthetic approach (Scheme 1) to the resulting compound (azocarnil, (1)) started from 1,1,2-trimethoxyethane (2) which was hydrolyzed to afford methoxyacetaldehyde (3) which was, due to its thermal instability and volatility, directly used in the reaction with isopropylamine (4) to form 5. Intermediate 5 was, without further purification, reacted with indole (6) to give compound 7. In a reaction executed in parallel, the reaction of 4-methoxybenzaldehyde (8) and glycine ethyl ester (9) gave imine 10. The three-step synthesis toward β-carboline 14 starting from 7 and 10, including the cyclization with paraformaldehyde (12) in a Pictet-Spengler reaction and oxidation with trichloroisocyanuric acid (TCCA) to obtain the fully aromatic scaffold, was done in one pot without isolation of the intermediates 11 and 13. The next step involved the nitration of 14 with a mixture of concentrated and fumic nitric acid to get nitro compound 15. Subsequent reduction of the nitro group led to amine 16. The photochromic moiety of 1 was introduced in the last step by performing a Mills reaction using nitrosobenzene (17) and the amine 16 in acetic acid.
Scheme 1. Synthetic Route toward Azocarnil (1).

Photochemical Properties of Azocarnil
The photochemical properties of azocarnil were characterized by using absorption spectroscopy in DMSO and aqueous buffer. It can be toggled reversibly between its Z- and E-isomer using LEDs emitting 400 nm violet light and 505 nm green light, respectively (Figure S8A). The E-isomer absorption plot is colored green and shows the typical azobenzene absorption maximum at 360 nm. Upon switching under 400 nm light during 1 min, a new maximum at 450 nm appears corresponding to the Z-isomer (violet in Figure S8A). Azocarnil displays a high fatigue resistance in DMSO and water (Figure 3B). The thermal relaxation half-life in DMSO is 10.4 h (lifetime 15 h, rate 0.07 h–1), which allows for investigating the photostationary states (PSS) by HPLC (Figure S8C). It exhibits a PSS of 87% Z for the E- to Z-isomerization after 400 nm light and 25% Z for the Z- to E-isomerization after 505 nm light. The thermal relaxation half-life of azocarnil in aqueous buffer is 1.5 min (lifetime 2.1 min, rate 0.47 min–1, Figure S8C). Hence, the PSS could not be determined by HPLC or NMR. In the next sections, Z-azocarnil refers to the maximum Z isomer fraction at the PSS under 400 nm illumination, and E-azocarnil refers to the maximum E isomer fraction at the PSS under 505 nm illumination. After thermal relaxation in the dark, azocarnil solutions are 100% E.
Figure 3.

Effect of azocarnil on endogenous GABAergic currents in cultured primary hippocampal neurons. (A) Voltage-clamp recordings of GABAergic currents (represented in pA units, see scale bar at the bottom of the panel) in control conditions (TTX 1 μM to block voltage-dependent sodium channels, CNQX 10 μM, and AP5 40 μM to block glutamatergic currents), after application of azocarnil (10 μM), and after addition of bicuculline (25 μM, competitive antagonist of the GABA site in GABAARs). Illumination with 400 and 500 nm light is depicted by violet and green boxes, respectively. Vhold = −70 mV. (B) GABAegic currents in the control, after addition of azocarnil (25 μM) and after addition of the GABAAR pore blocker picrotoxin (PTX, 100 μM). (C) Cumulative graph demonstrating the relative mean amplitude of macroscopic GABAergic currents recorded during simultaneous application of different concentrations of azocarnil and 400 nm illumination. Here and elsewhere the values represent mean ± SD. Comparison was done using a paired t test, n = 5–7 cells for each concentration; ns, not significant; **, P < 0.01; ***, P < 0.001. (D) Cumulative graph showing the mean amplitude of macroscopic GABAergic currents in two different sets of experiments: when current induced by application of azocarnil (25 μM) and 400 nm light was inhibited by bicuculine (25 μM; orange column) and by PTX (100 μM; red column). (E) Azocarnil (25 μM) under 400 nm light increases the decay time of the mIPSCs. Recording of mIPSCs at 400 nm illumination in control conditions (gray trace) and mIPSCs at 400 nm illumination combined with the application of azocarnil (25 μM, black trace). (F) Cumulative graph demonstrating the effect of azocarnil (25 μM) combined with 400 nm light on the decay time (ms) of mIPSCs in cultured hippocampal neurons. Comparison was done using an unpaired t test, n = 78 events from 4 independent experiments; **, P < 0.001. TTX: tetrodotoxin (voltage-gated sodium channel blocker). CNQX: 6-cyano-7-nitroquinoxaline-2,3-dione (AMPA receptor antagonist). AP5: (2R)-amino-5-phosphonovaleric acid (NMDA receptor antagonist).
Azocarnil is a Light-Activated GABAergic Agonist-Potentiator in Vitro
We next evaluated the photopharmacological properties of the compound in vitro using whole cell patch clamp electrophysiology in hippocampal neurons under different illumination conditions. We chose primary neurons because they express a representative diversity of physiological GABAAR subunits and heteropentameric subtypes, in contrast to overexpressed receptors in cell lines or oocytes, which provide specific receptor responses but involve longer screening processes and can lead to nonphysiologically relevant results.43,44 Thus, we tested the effect of E- and Z-azocarnil on endogenous GABAARs in cultured neurons by measuring whole-cell macroscopic currents and transient miniature inhibitory postsynaptic currents (mIPSCs). When voltage clamped neurons are illuminated at 400 nm in the absence of the compound (control conditions), a small but noticeable deflection of the baseline is observed (Figure 3A,B, upper panels). To take into account this baseline photocurrent in the quantifications for every cell, we recorded it under 400 and 500 nm before the application of azocarnil, and all currents measured at the peak of the response were normalized to this baseline.
Upon perfusion of 500 nM to 25 μM azocarnil in the absence of illumination, the current did not change significantly. When cells were illuminated with 400 nm light during azocarnil perfusion, the magnitude of the photocurrent exceeded the baseline photocurrent. At 500 nM to 1 μM azocarnil, the relative amplitude of GABAergic currents under 400 nm light increased nonsignificantly by a factor of 1.1 ± 0.4 (n = 7 cells) and 1.4 ± 0.6 (n = 6 cells), respectively (paired t test). When azocarnil concentration was raised to 10 μM and 25 μM, the macroscopic GABAergic currents increased significantly by a factor of 3.4 ± 1.2 (n = 7 cells, P < 0.01, 10 μM) and 3.1 ± 0.5 from control (n = 5 cells, P < 0.001, 25 μM) as shown in Figure 3A,C. Upon application of the vehicle (DMSO 0.5%) and 400 nm illumination, macroscopic currents were of the same amplitude as baseline photocurrents, and the kinetics of synaptic GABAergic currents was not altered (Figure S11D), confirming the specificity of the azocarnil effect.
To confirm that the recorded currents have GABAergic origin, after every experiment, we applied bicuculline, a GABAAR inhibitor antagonizing the GABA site (Figure 3A,D). The application of bicuculline completely suppressed GABAergic mIPSCs and partially but significantly inhibited the macroscopic photocurrents to a factor of 1.3 ± 0.4 from the control (n = 4; Figure 3A,D). Note that, in contrast to the photocurrent baseline in the absence of azocarnil, the characteristic time course of the GABAergic macroscopic photocurrent with a desensitizing component is retained in the presence of azocarnil and bicuculine (Figure 3A). In a separate set of experiments after recording GABAergic currents, we applied the GABAAR pore blocker picrotoxin (PTX, 100 μM) and observed a complete suppression of both the mIPSCs and the macroscopic GABAergic photocurrents induced by Z azocarnil (Figure 3B,D). The partial inhibition of photocurrents by bicuculline (which displaces GABA from its binding sites, thus suppressing any potentiator effects) reveals an agonist-like activity of azocarnil. In contrast, PTX abolishes both the agonist and potentiator effects of azocarnil, which corresponds to a pore blocker effect. Note that the different effects of 25 μM azocarnil between the experiments to separately test bicuculline and PTX on the photoresponses (Figure 3D) are due to differences across individual neurons and neuronal cultures (we used 7 different animals and hippocampal dissections, which include heterogeneous neuronal populations and protein expression profiles like GABAARs).
We also studied the decay time of the mIPSCs under 400 nm illumination before and after azocarnil application and observed a statistically significant increase at 25 μM, from 32 ± 11 ms to 43 ± 19 ms (Figure 3 EF; P < 0.01, n = 78 events from 4 independent experiments).
In addition, we investigated the effect of azocarnil on glutamatergic miniature excitatory postsynaptic currents (mEPSCs). Application of azocarnil in combination with 400 or 500 nm light did not induce changes in either the amplitude of the macroscopic current (Figure S11A) or the decay time of the mEPSCs (Figure S11B,C).
Together, electrophysiological and pharmacological experiments in hippocampal neurons demonstrate that azocarnil is inactive in the dark and that it acts as a micromolar agonist-potentiator of GABAARs under 400 nm light, producing macroscopic GABAergic currents and prolonging the decay kinetics of synaptic mIPSCs.
Azocarnil Can Be Safely Injected and Photocontrols Mechanical Sensitivity in Vivo
We next tested the effect of photoactivated azocarnil on spinal nociception in vivo. The spinal cord dorsal horn is the first site of synaptic processing in pain pathways. At this site, sensory information conveyed by peripheral nerve fibers is processed by a local network consisting of excitatory glutamatergic and inhibitory GABAergic (and glycinergic) neurons before it is relayed to higher central nervous system areas. The magnitude of aversive responses triggered by cutaneous mechanical stimulation strongly depends on the activity of inhibitory (GABAergic and glycinergic) interneurons of the spinal cord.45
The sensitivity of mice to mechanical stimulation of the plantar side of the left hind paw was assessed with von Frey filaments (for a schematic presentation of the experimental design, see Figure 4A). The first baseline measurement was taken 7 days after cannula implantation. On the following day, a second baseline was taken 20 min after E-azocarnil was injected intrathecally (i.e., into the cerebrospinal fluid). E-Azocarnil did not cause any significant change relative to the baseline sensitivity (Figure 4B). Photoactivation of E-azocarnil was started immediately after, by delivering violet light (405 nm, 10–20 μW) on the dorsal side of the lumbar spinal cord at levels L4/L5 through an optical fiber. At this stage, the von Frey filament test was performed during continuous light stimulation, showing a significant decrease in the mechanical sensitivity (Figure 4B). Illumination was then discontinued, and motor coordination was subsequently assessed. No change in the rotarod performance was observed (Figure 4C). The von Frey filament test was performed again 1 h after the illumination was stopped, showing a recovery to baseline mechanical sensitivity (Figure 4B).
Figure 4.

Azocarnil exerts photoactivated neuroinhibition in mice. (A) Experimental setup to measure changes in mechanical sensitivity using the von Frey filament test. Photoactivation of E-azocarnil was done with UV light of 405 nm (10–20 μW) delivered by an optical fiber connected to the lumbar spine. (B) Withdrawal thresholds measured at baseline (7 days after cannula implantation, 20 min after intrathecal injection of azocarnil (300 μM; 10 μL), during violet illumination (405 nm; 10–20 μW), and 1 h after violet light was turned off). (C) Motor coordination assessed in the rotarod test. Each data point represents one mouse. Red lines show mean and SEM *, P < 0.05, ** P < 0.01, *** P < 0.001. One-way ANOVA followed by Bonferroni posthoc test. Figure 4A was created using the free version of BioRender (https://www.biorender.com).
Overall, azocarnil achieves a qualitative advancement in the photopharmacology of GABAARs due to its unique properties. Its agonist-potentiator activity provides robust neuroinhibition independently of GABA. Our molecular modeling results predict that azocarnil retains binding to the benzodiazepine site and other favorable properties of its precursor abecarnil and that it establishes stronger receptor-drug interactions in the illuminated Z form than in the dark-relaxed E form. Accordingly, azocarnil has lower GABAAR activity in the dark, it is photoactivated by visible violet light in vitro and in vivo, and it does not produce systemic adverse effects when injected intrathecally in mice. Thus, azocarnil is a freely diffusible and drug-like small molecule photoswitch that is amenable to further development for therapeutic purposes in which on-demand and localized neuroinhibition is required.
We have demonstrated that under 400 nm illumination azocarnil induces macroscopic GABAergic current and prolongs the decay time of synaptic currents in cultured hippocampal neurons (Figure 3). These effects are specific to GABAARs and do not alter glutamatergic currents (Figure S11ABC). Visible light of 400 nm can alter retinal and cortical neurons,46 but unlike UV light, it does not damage nucleic acids or produce reactive oxygen species.47 Azocarnil is about 100-fold less potent than abecarnil,48 as it is sometimes found in photopharmacology.23 Considering the excellent performance of azocarnil in vitro, we tested it to control the transmission of sensory information in the spinal cord in vivo. We observed that 405 nm light delivered to the lumbar spine after intrathecal injection of azocarnil significantly decreases the hind paw sensitivity in the von Frey test without affecting motor coordination (Figure 4). Thus, it is a photoactivated potentiator of GABAergic neurotransmission that efficiently controls mechanical sensitivity without adverse effects.
Compared to other reported diffusible photoswitches, azocarnil is less potent than the antagonist azogabazine but ten times more potent than other photoswitchable GABAAR potentiators like MPC088, AP-2, and fulgazepam.21−23 In contrast to the propofol derivatives MPC088 and AP-2, it is inactive in the dark. Azocarnil is also more synthetically accessible than fulgazepam23 and constitutes the first GABAAR photoswitch activated by visible light, allowing the use of compact 405 nm laser diodes.
Azocarnil does not induce anesthesia at the doses studied here (Figure 4C), in contrast to the action of propofol. This result agrees with the properties of benzodiazepine site ligands. However, the observed changes in the receptor decay kinetics (Figure 3D,E) diverge from the canonical benzodiazepine mechanism49 and may involve secondary binding sites50,51 that require further studies.
The photoactivation of azocarnil with visible light and its prominent effect on mechanical sensitivity make it eligible for selective, on-demand, and localized inhibition of pathologic pain signaling at the dorsal horn of the spinal cord. Peripheral neuropathic pain involves the release of inflammatory molecules that alter the activity of a wide variety of targets on dorsal root ganglion neurons, including voltage gated ion channels (NaV, KV, CaV) and GABAARs, among others.52 However, drugs targeting the latter are not used clinically to treat pain due to their systemic adverse effects. Several studies have been published to control pain in vivo with photopharmacology.53−58 A photoswitchable adenosine receptor agonist decreases the inflammatory pain when injected locally in the affected paw. However, it is active in the dark and deactivated under illumination, which limits its usability.53 Several photoswitchable modulators of metabotropic glutamate receptors can be used to treat inflammatory and neuropathic pain in vivo, but they require highly invasive injections in the brain and are also dark-active.54−57 A photoswitchable carbamazepine (carbadiazocine) for neuropathic pain is also active in the dark.58 None of them can take advantage of the GABAergic gate of sensory information in the dorsal horn like azocarnil, which is poised as a promising photopharmacological drug candidate against pathological pain.
For that purpose, the molecular properties of azocarnil that can be improved include the activation wavelength, which has a low penetration in tissue. Photoactivation with orange-red light would be convenient, and azobenzene substitutions to achieve it have been described;59 however, they involve bulky substituents that may affect azocarnil potency, drug-likeness, and the prospects of oral bioavailability and CNS exposure derived from abecarnil. The lack of adverse systemic effects of azocarnil after intrathecal injection and localized illumination (Figure 4) prompts future pharmacokinetic studies by other routes of administration that give good tolerability and efficacy in humans with abecarnil.60,61
Conclusions
To address the need for photoswitchable GABAergic drugs for therapies with high efficacy and without adverse effects, we developed azocarnil, a small molecule photoswitchable analogue of the drug abecarnil. This compound overcomes several limitations of previously reported photoswitchable GABAergic ligands. It is an agonist-potentiator of GABAARs that is inactive in the dark and can be activated with visible light and with precise spatiotemporal selectivity, both in hippocampal neurons and in the dorsal horns of the spinal cord of mice. Azocarnil is the first diffusible GABAergic photoswitch that is demonstrated in wildtype mammals, achieving a prominent decrease in their sensitivity to touch without concomitant motor effects.
Acknowledgments
This research was funded by the EU Horizon 2020 Framework Programme for Research and Innovation, including the European Innovation Council Pathfinder (Phototheraport, 101130883), Human Brain Project (WaveScalES, SGA3, 945539), ERA SynBIO Grant MODULIGHTOR (PCIN-2015-163-C02-01), and Information and Communication Technologies (Deeper, ICT-36-2020-101016787). It was also supported by the Government of Catalonia (CERCA Programme; AGAUR 2021-SGR-01410 to P.G. and 2021-SGR-00680 to C.R.); by the Spanish Ministry of Science and Innovation (DEEP RED, grant PID2019-111493RB-I00; EPILLUM, grant AEI/10.13039/501100011033; Research Network in Biomedicine eBrains-Spain, RED2022-134823-E; and MICINN/AEI/FEDER, UE, PID2020-118893GB-100, to C.R.); the Spanish Structures of Excellence María de Maeztu (CEX2021-001202-M, to C.R. and A.N.-H.); and in part by the DFG Research Unit FOR2518 “Functional Dynamics of Ion Channels and Transporters – DynIon” (project P6, AL 2511/1-2 to M.A.-P). G.M. was supported by a Ramón y Cajal Investigator grant (RYC2021-033056-I financed by MCIN/AEI/10.13039/501100011033 and by the European Union NextGeneration EU/PRTR). This study has also benefitted from the postdoctoral fellowship Margarita Salas (A.N.-H.) funded by the Spanish Ministry of Science, Innovation and Universities (MICIU). Part of the work presented here has been adapted from the PhD dissertation of U.W. (“Photochromic Molecules for Biological Applications”, University of Regensburg, Germany, 2024), under a CC BY 4.0 license. Permission for all animal experiments was obtained from the Kanton of Zurich (license 097/2021). All animal experiments complied with the relevant ethical regulations.
Glossary
Abbreviations
- AcOH
acetic acid
- AMPA receptor
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- AP5
(2R)-amino-5-phosphonovaleric acid
- ATP
adenosine triphosphate
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DIV
days in vitro
- DMCM
methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- DMSO
dimethyl sulfoxide
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- GABA
γ-aminobutyric acid
- GTP
guanosine-5′-triphosphate
- HPLC
high performance liquid chromatography
- mEPSCs
miniature excitatory postsynaptic currents
- mIPSCs
miniature inhibitory postsynaptic currents
- ms
millisecond
- NMDA receptor
N-methyl-d-aspartate receptor
- NMR
nuclear magnetic resonance
- pA
picoampere
- PAM
positive allosteric modulator
- PDB
protein data bank
- PE
petroleum ether
- P
postnatal
- PSS
photostationary state
- PTX
picrotoxin
- RP
reverse phase
- s
second
- TFA
trifluoroacetic acid
- TCCA
trichloroisocyanuric acid
- TTX
tetrodotoxin
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c08446.
Further details on the experimental and computational procedures used in this study, spanning molecular modeling, synthesis and analytical characterization of azocarnil, and electrophysiological characterization in neuronal cell cultures and mice experiments. Additional figures and tables supplementing the results presented in the main text, including validation of the docking protocol, photophysical and NMR characterization and electrophysiological control recordings (PDF)
Author Present Address
□ Toulouse Biotechnology Institute, TBI, Université de Toulouse, CNRS, INRAE, INSA, F-31077 Toulouse Cedex, France
Author Present Address
○ Iris Biotech GmbH, Marktredwitz, 95615 Germany
Author Present Address
△ Institute of Neuroscience, Kazan State Medical University, Kazan, 420012 Russia
Author Contributions
+ G.M. and A.N.-H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Korpi E. R.; Sinkkonen S. T. GABAA receptor subtypes as targets for neuropsychiatric drug development. Pharmacol Ther 2006, 109, 12–32. 10.1016/j.pharmthera.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Edwards Z.; Preuss C. V.. GABA receptor positive allosteric modulators. In StatPearls; StatPearls Publishing, 2020. [PubMed] [Google Scholar]
- George K.; Preuss C. V.; Sadiq N. M.. GABA inhibitors. In StatPearls; StatPearls Publishing, 2023. [PubMed] [Google Scholar]
- Melzack R.; Wall P. D. Pain Mechanisms: A New Theory: A gate control system modulates sensory input from the skin before it evokes pain perception and response. Science 1965, 150, 971–979. 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- Zeilhofer H. U.; Wildner H.; Yévenes G. E. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev. 2012, 92, 193. 10.1152/physrev.00043.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor A. B.; Dworkin R. H. Treatment of neuropathic pain: an overview of recent guidelines. Am. J. Med. 2009, 122, S22–S32. 10.1016/j.amjmed.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Cruccu G.; Truini A. A review of neuropathic pain: from guidelines to clinical practice. Pain Ther 2017, 6, 35–42. 10.1007/s40122-017-0087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobauri P.; Dekker F. J.; Szymanski W.; Feringa B. L. Rational design in photopharmacology with molecular photoswitches. Angew. Chem. 2023, 135, e202300681 10.1002/ange.202300681. [DOI] [PubMed] [Google Scholar]
- Gomila A. M. J.; Gorostiza P. In Vivo Applications of Photoswitchable Bioactive Compounds. Molecular Photoswitches: Chemistry, Properties, and Applications, 2 Volume Set 2022, 811–842. 10.1002/9783527827626.ch34. [DOI] [Google Scholar]
- Mukherjee A.; Seyfried M. D.; Ravoo B. J. Azoheteroarene and Diazocine Molecular Photoswitches: Self-Assembly, Responsive Materials and Photopharmacology. Angew. Chem., Int. Ed. 2023, 62, e202304437 10.1002/anie.202304437. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: beyond proof of principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Schoenberger M.; Damijonaitis A.; Zhang Z.; Nagel D.; Trauner D. Development of a new photochromic ion channel blocker via azologization of fomocaine. ACS Chem. Neurosci. 2014, 5, 514–518. 10.1021/cn500070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittolo S.; et al. An allosteric modulator to control endogenous G protein-coupled receptors with light. Nat. Chem. Biol. 2014, 10, 813–815. 10.1038/nchembio.1612. [DOI] [PubMed] [Google Scholar]
- Matera C.; et al. Photoswitchable antimetabolite for targeted photoactivated chemotherapy. J. Am. Chem. Soc. 2018, 140, 15764–15773. 10.1021/jacs.8b08249. [DOI] [PubMed] [Google Scholar]
- Kramer R. H.; Rajappa R. Interrogating the function of GABAA receptors in the brain with optogenetic pharmacology. Curr. Opin Pharmacol 2022, 63, 102198 10.1016/j.coph.2022.102198. [DOI] [PubMed] [Google Scholar]
- Lin W.-C.; et al. A comprehensive optogenetic pharmacology toolkit for in vivo control of GABAA receptors and synaptic inhibition. Neuron 2015, 88, 879–891. 10.1016/j.neuron.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport C. M.; et al. Relocation of an extrasynaptic GABAA receptor to inhibitory synapses freezes excitatory synaptic strength and preserves memory. Neuron 2021, 109, 123–134. 10.1016/j.neuron.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L.; et al. Robust photoregulation of GABAA receptors by allosteric modulation with a propofol analogue. Nat. Commun. 2012, 3, 1095. 10.1038/ncomms2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghese C. M.; et al. Modulation of α1β3γ2 GABAA receptors expressed in X. laevis oocytes using a propofol photoswitch tethered to the transmembrane helix. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2008178118 10.1073/pnas.2008178118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleeva G.; et al. A photoswitchable GABA receptor channel blocker. Br. J. Pharmacol. 2019, 176, 2661–2677. 10.1111/bph.14689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M.; Huckvale R.; Pandurangan A. P.; Baker J. R.; Smart T. G. Optopharmacology reveals a differential contribution of native GABAA receptors to dendritic and somatic inhibition using azogabazine. Neuropharmacology 2020, 176, 108135 10.1016/j.neuropharm.2020.108135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M.; et al. Azo-propofols: photochromic potentiators of GABAA receptors. Angew. Chem., Int. Ed. 2012, 51, 10500–10504. 10.1002/anie.201205475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustler K.; et al. Optical Control of GABAA Receptors with a Fulgimide-Based Potentiator. Chemistry A European Journal 2020, 26, 12722–12727. 10.1002/chem.202000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens D. N.Anxiolytic β-Carbolines: From Molecular Biology to the Clinic; Springer Science & Business Media, 2013; Vol. 11. [Google Scholar]
- Stephens D. N.; et al. Abecarnil, a metabolically stable, anxioselective beta-carboline acting at benzodiazepine receptors. J Pharmacol Exp Ther. 1990, 253, 334–343. [PubMed] [Google Scholar]
- Pribilla I.et al. Abecarnil is a full agonist at some, and a partial agonist at other recombinant GABAA receptor subtypes. Anxiolytic β-carbolines: from molecular biology to the clinic; Stephens D. N., Eds.; Psychopharmacology Series; Springer: Berlin, Heidelberg, 1993; Vol 11, pp 50–61. [DOI] [PubMed] [Google Scholar]
- Ozawa M.; et al. Pharmacological characterization of the novel anxiolytic β-carboline abecarnil in rodents and primates. Japanese Journal of Pharmacology 1994, 64, 179–187. 10.1254/jjp.64.179. [DOI] [PubMed] [Google Scholar]
- Barbaccia M. L.; et al. Stress-induced increase in brain neuroactive steroids: antagonism by abecarnil. Pharmacol., Biochem. Behav. 1996, 54, 205–210. 10.1016/0091-3057(95)02133-7. [DOI] [PubMed] [Google Scholar]
- Aufdembrinke B. Abecarnil, a new beta-carboline, in the treatment of anxiety disorders. British Journal of Psychiatry 1998, 173, 55–63. 10.1192/S0007125000293537. [DOI] [PubMed] [Google Scholar]
- Sigel E.; Ernst M. The benzodiazepine binding sites of GABAA receptors. Trends Pharmacol. Sci. 2018, 39, 659–671. 10.1016/j.tips.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Yarom M.; et al. Identification of inosine as an endogenous modulator for the benzodiazepine binding site of the GABAA receptors. J. Biomed Sci. 1998, 5, 274–280. 10.1159/000025340. [DOI] [PubMed] [Google Scholar]
- MacDonald J. F.; Barker J. L.; Paul S. M.; Marangos P. J.; Skolnick P. Inosine may be an endogenous ligand for benzodiazepine receptors on cultured spinal neurons. Science (1979) 1979, 205, 715–717. 10.1126/science.37602. [DOI] [PubMed] [Google Scholar]
- Stephens D. N.; Turski L.; Jones G. H.; Steppuhn K. G.; Schneider H. H. Abecarnil: a novel anxiolytic with mixed full agonist/partial agonist properties in animal models of anxiety and sedation. Anxiolytic β. Carbolines: From Molecular Biology to the Clinic 1993, 79–95. 10.1007/978-3-642-78451-4_7. [DOI] [PubMed] [Google Scholar]
- Zhu S.; et al. Structural and dynamic mechanisms of GABAA receptor modulators with opposing activities. Nat. Commun. 2022, 13, 4582. 10.1038/s41467-022-32212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J.; et al. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. 10.1038/s41586-020-2654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M. D.; et al. Role of the histidine residue at position 105 in the human α5 containing GABAA receptor on the affinity and efficacy of benzodiazepine site ligands. Br. J. Pharmacol. 2002, 135, 248–256. 10.1038/sj.bjp.0704459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiulis S.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. 10.1038/s41586-018-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F.; Assandri R.; Täuber M.; Martin J. R.; Rudolph U. Contribution of the α1-GABAA receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists. Neuropharmacology 2002, 43, 679–684. 10.1016/S0028-3908(02)00159-4. [DOI] [PubMed] [Google Scholar]
- Kasaragod V. B.; et al. The molecular basis of drug selectivity for α5 subunit-containing GABAA receptors. Nat. Struct Mol. Biol. 2023, 30, 1936–1946. 10.1038/s41594-023-01133-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramerstorfer J.; Furtmüller R.; Vogel E.; Huck S.; Sieghart W. The point mutation γ2F77I changes the potency and efficacy of benzodiazepine site ligands in different GABAA receptor subtypes. Eur. J. Pharmacol. 2010, 636, 18–27. 10.1016/j.ejphar.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin-Hill A.; Mueller N. P. F.; Molteni C.; Rovira C.; Alfonso-Prieto M. Photopharmacology of Ion channels through the light of the computational microscope. Int. J. Mol. Sci. 2021, 22, 12072. 10.3390/ijms222112072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr A.; Baur R.; Sigel E. Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 1997, 272, 11799–11804. 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- Whiting P. J. GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery?. Drug Discov Today 2003, 8, 445–450. 10.1016/S1359-6446(03)02703-X. [DOI] [PubMed] [Google Scholar]
- Qian X.; et al. Current status of GABA receptor subtypes in analgesia. Biomedicine & Pharmacotherapy 2023, 168, 115800 10.1016/j.biopha.2023.115800. [DOI] [PubMed] [Google Scholar]
- Foster E.; et al. Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron 2015, 85, 1289–1304. 10.1016/j.neuron.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theruveethi N. Impact of light-emitting diodes on visual cortex layer 5 pyramidal neurons (V1-L5PNs)—A rodent study. Mol. Vis 2024, 30, 67. [PMC free article] [PubMed] [Google Scholar]
- Rodrigues R. B.; et al. Dithiothreitol reduces oxidative stress and necrosis caused by ultraviolet A radiation in L929 fibroblasts. Photochemical & Photobiological Sciences 2024, 23, 271–284. 10.1007/s43630-023-00516-z. [DOI] [PubMed] [Google Scholar]
- Vorobjev V. S.; Sharonova I. N.; Skrebitsky V. G.; Schneider H. H.; Stephens D. N. Abecarnil enhances GABA-induced currents in acutely isolated cerebellar Purkinje cells. Neuropharmacology 1995, 34, 157–163. 10.1016/0028-3908(94)00139-J. [DOI] [PubMed] [Google Scholar]
- McAdam L. C.Propofol and benzodiazepine modulation of GABA (A) R function. Thesis, 1998. [Google Scholar]
- Thomet U.; Baur R.; Scholze P.; Sieghart W.; Sigel E. Dual mode of stimulation by the β-carboline ZK 91085 of recombinant GABAA receptor currents: molecular determinants affecting its action. Br. J. Pharmacol. 1999, 127, 1231–1239. 10.1038/sj.bjp.0702639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio M. T.; et al. GABAA receptor ligands often interact with binding sites in the transmembrane domain and in the extracellular domain—can the promiscuity code be cracked?. Int. J. Mol. Sci. 2020, 21, 334. 10.3390/ijms21010334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez A. L.; et al. In Vitro Models for Neuropathic Pain Phenotypic Screening in Brain Therapeutics. Pharmacol. Res. 2024, 202, 107111 10.1016/j.phrs.2024.107111. [DOI] [PubMed] [Google Scholar]
- Hüll K.; et al. Optical control of adenosine-mediated pain modulation. Bioconjug Chem. 2021, 32, 1979–1983. 10.1021/acs.bioconjchem.1c00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V.; Arias J. A.; Llebaria A.; Goudet C. Photopharmacological manipulation of amygdala metabotropic glutamate receptor mGlu4 alleviates neuropathic pain. Pharmacol. Res. 2023, 187, 106602 10.1016/j.phrs.2022.106602. [DOI] [PubMed] [Google Scholar]
- Zussy C.; et al. Dynamic modulation of inflammatory pain-related affective and sensory symptoms by optical control of amygdala metabotropic glutamate receptor 4. Mol. Psychiatry 2018, 23, 509–520. 10.1038/mp.2016.223. [DOI] [PubMed] [Google Scholar]
- Ricart-Ortega M.; et al. Mechanistic insights into light-driven allosteric control of GPCR biological activity. ACS Pharmacol Transl Sci. 2020, 3, 883–895. 10.1021/acsptsci.0c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Santacana X.; et al. Illuminating phenylazopyridines to photoswitch metabotropic glutamate receptors: from the flask to the animals. ACS Cent Sci. 2017, 3, 81–91. 10.1021/acscentsci.6b00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerin L.; et al. Photoswitchable Carbamazepine Analogs for Non-Invasive Neuroinhibition In Vivo. Angew. Chem. 2024, e202403636 10.1002/anie.202403636. [DOI] [PubMed] [Google Scholar]
- Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Red-shifting azobenzene photoswitches for in vivo use. Acc. Chem. Res. 2015, 48, 2662–2670. 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]
- Krause W.; Schütt B.; Duka T. Pharmacokinetics and acute toleration of the beta-carboline derivative abecarnil in man. Arzneimittelforschung 1990, 40, 529–532. [PubMed] [Google Scholar]
- Trenité D. G. A. K.-N.; Groenwold R. H. H.; Schmidt B.; Löscher W. Single dose efficacy evaluation of two partial benzodiazepine receptor agonists in photosensitive epilepsy patients: a placebo-controlled pilot study. Epilepsy Res. 2016, 122, 30–36. 10.1016/j.eplepsyres.2016.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable request.