

Abstract
Introduction:
Predicting Self-interacting proteins (SIPs) is a crucial area of research in predicting protein functions, as well as in understanding gene-disease and disease-drug associations. These interactions are integral to numerous cellular processes and play pivotal roles within cells. However, traditional methods for identifying SIPs through biological experiments are often expensive, time-consuming, and have long cycles. Therefore, the development of effective computational methods for accurately predicting SIPs is not only necessary but also presents a significant challenge.
Results:
In this research, we introduce a novel computational prediction technique, VGGNGLCM, which leverages protein sequence data. This method integrates the VGGNet deep convolutional neural network (VGGN) with the Gray-Level Co-occurrence Matrix (GLCM) to detect Self-interacting proteins associations. Specifically, we initially utilized Position Specific Scoring Matrix (PSSM) to capture protein evolutionary information and integrated key features from PSSM using GLCM. We then employed VGGNet as a predictive classifier, leveraging its capabilities for powerful learning and classification prediction. Subsequently, the extracted features were input into the VGGNet deep convolutional neural network to identify Self-interacting proteins. To evaluate the performance of the VGGNGLCM model, we conducted experiments using yeast and human datasets, achieving average accuracies of 95.68% and 97.72% respectively. Additionally, we compared the prediction performance of the VGGNet classifier with that of the Convolutional Neural Network (CNN) and the state-of-the-art Support Vector Machine (SVM) using the same feature extraction method. We also compared the prediction ability of VGGNGLCM with other existing approaches. The comparison results further demonstrate the superior performance of VGGNGLCM over other prediction models in this domain.
Conclusion:
The experimental verification further strengthens the evidence that VGGNGLCM is effective and robust compared to existing methods. It also highlights the high accuracy and robustness of the VGGNGLCM model in predicting Self-interacting proteins (SIPs). Consequently, we believe that the VGGNGLCM method serves as a valuable computational tool and can catalyze extensive bioinformatics research related to SIPs prediction.
Keywords: SIPs, VGGNet, GLCM PSSM
Introduction
A considerable body of research has unveiled the significance of Protein-protein interactions (PPIs) across numerous biological activities. However, a fascinating research inquiry lies in the potential for proteins to interact with themselves. Self-interacting proteins (SIPs) represent a distinct category within PPIs, where multiple copies of a protein can interact with each other, originating from the same gene. This phenomenon may lead to the emergence of homo-oligomerization issues. Recent studies have underscored the pivotal role of SIPs in diverse cellular physiological functions and the evolutionary dynamics of protein-protein interaction networks (PPINs).1 -4 Therefore, understanding whether a protein can self-interact is paramount for deciphering its functions. Investigations into SIPs can significantly enhance our comprehension of protein function regulation, molecular mechanisms underlying biological activities, and the fundamental cellular and genetic disease mechanisms. Numerous studies have delved into homo-oligomerization, a crucial biological function essential for various processes including signal transduction, gene expression regulation, enzyme activation, and immune response.5 -9 Moreover, SIPs have been shown to increase the functional diversity of proteins without increasing genome length. Furthermore, SIPs can also improve protein stability and prevent denaturation by decreasing surface area exposure10 -12 Consequently, there is a growing need to develop reliable and highly effective computational approaches based on protein sequences for predicting SIPs.
Based on the aforementioned analysis, an increasing number of studies have concentrated on computational methodologies grounded in protein sequences. This has led to the development of numerous computational techniques aimed at predicting Protein-Protein Interactions (PPIs). Wong et al 13 proposed a sequence-based method for predicting PPIs by combining Rotation Forest classifier with a novel feature extraction method called Local Phase Quantization (LPQ). The method obtained the high prediction accuracy. Wei et al 14 proposed a novel method called weighted Position Specific Scoring Matrix (PSSM) histogram for extracting features and adopted a random forests classifier for predicting PPIs. Wang et al 15 presented an effective computational method for predicting SIPs,which combined with histogram of oriented gradients (HOG),synthetic minority oversampling technique (SMOTE) and rotation forest (RF) classifier. Li et al 16 proposed a new sequence-based computational method, which used Position Specific Scoring Matrix to capture features and employed an ensemble classifier for identifying PPIs. Liu et al 17 introduced a novel computational method, RF-PSSM, which integrates rotation forest and PSSM to predict protein interactions. The PSSM was utilized to characterize each protein, while the RF was employed for classification. This approach yielded superior prediction results. Wang et al 18 presented a new computational approach for detecting PPIs using Rotation Forest and matrix-based protein sequence. The method can capture biological evolution information from PSSM matrix and created feature vectors by using the 2-dimensional Principal Component Analysis (2DPCA) algorithm. Li et al 19 present a method to predict PPIs based on protein sequences, which combined Position Weight Matrix (PWM) with Scale-Invariant Feature Transform (SIFT) for extracting features. The method used Weighted Extreme Learning Machine (WELM) classifier for predicting PPIs at last and obtained higher prediction accuracy. Jia et al 20 designed a novel method called NLPEI for detecting PPIs by using evolutionary information of protein and natural language understanding theory. A number of key features can be integrated by using serial multi-feature Fusion. The above methods can explore the correlational information between protein pairs, such as, coevolution, co-localization and co-expression.21 -25 The existing methods, while effective for predicting general protein-protein interactions (PPIs), may not be directly applicable to predicting self-interacting proteins (SIPs) due to several reasons. Firstly, these methods often rely on correlational information between protein pairs, such as coevolution, co-localization, and co-expression, which may not adequately capture the specific characteristics of SIPs. SIPs involve proteins interacting with identical copies of themselves, a phenomenon not fully addressed by traditional PPI prediction methods. Moreover, the datasets used to train these prediction models do not contain interactions between the same protein partners, which are important for SIP identification. Without this information, the models cannot learn how to distinguish SIPs from other types of interactions. In response to these challenges, Liu et al 26 proposed a method named SLIPPER, which integrates multiple known properties specifically tailored for predicting SIPs. This approach represents a step forward in addressing the unique characteristics of SIPs and demonstrates the importance of developing specialized computational methods for this purpose. However, the accuracy of the existing methods has still room for improvement. Therefore, it is meaningful to develop more efficient computational methods to improve the prediction accuracy of identifying SIPs, which will be critical for advancing our understanding of protein-protein interactions and their role in biological processes.
In this research, we introduce a novel computational prediction technique, VGGNGLCM, which leverages protein sequence data. This method integrates the VGGNet deep convolutional neural network (VGGN) with the Gray-Level Co-occurrence Matrix (GLCM) to detect Self-interacting proteins associations. Specifically, we initially utilized Position Specific Scoring Matrix (PSSM) to capture protein evolutionary information and integrated key features from PSSM using GLCM. We then employed VGGNet as a predictive classifier, leveraging its capabilities for powerful learning and classification prediction. Subsequently, the extracted features were input into the VGGNet deep convolutional neural network to identify Self-interacting proteins. To evaluate the performance of the VGGNGLCM model, we conducted experiments using yeast and human datasets, achieving average accuracies of 95.68% and 97.72% respectively. Additionally, we compared the prediction performance of the VGGNet classifier with that of the Convolutional Neural Network (CNN) and the state-of-the-art Support Vector Machine (SVM) using the same feature extraction method. We conducted a comparative analysis of the predictive capabilities of VGGNGLCM against other existing methodologies. The outcomes of this comparison further underscore the superior performance of VGGNGLCM in relation to other predictive models within this particular domain.
Method
Datasets
The UniProt database hosts 20 199 curated human protein sequences. 27 Previous research has utilized PPI datasets from various sources, including DIP, 28 BioGRID, 29 IntAct, 30 InnateDB, 31 and MatrixDB. 32 In the study, the SIP datasets were constructed to only include interactions between the same 2 protein sequences, defined as “direct interaction” in the relevant databases. To assess the performance of VGGNGLCM, a total of 2994 human Self-interactions protein sequences were identified to construct the experimental datasets, following the steps outlined below 33 : (1) Exclusion of protein sequences shorter than 50 residues and longer than 5000 residues from the entire human proteome; (2) Creation of positive samples based on the following conditions: (a) Self-interaction proteins are detected by at least 2 large-scale experiments or 1 small-scale experiment; (b) Self-interaction proteins are defined as a homooligomer (including homodimer and homotrimers) by the UniProt; (c) it has been reported by at least 2 publications for its Self-interactions; (3) Construction of negative samples by removing all types of SIPs from the entire human proteome (including proteins annotated as ‘direct interaction’ and more extensive “physical association”) and the UniProt database. This resulted in 15 938 non-SIPs as negative samples and 1441 SIPs as positive samples for the human dataset. The similar strategy was employed to create the yeast dataset, comprising 5511 negative and 710 positive samples. Notably, the yeast dataset exhibits approximately 8 times as many positive samples as negative samples, while the human dataset contains roughly 11 times as many positive samples as negative samples.
Feature extraction method
Position Specific Scoring Matrix (PSSM)
Proteins exhibit functional conservation, making the utilization of evolutionary information from protein sequences crucial for enhancing prediction accuracy. The Position-Specific Scoring Matrix (PSSM) encapsulates both the positional and evolutionary information of protein sequences, reflecting their conservation. To represent the characteristics of protein sequences using PSSM, we employ the Position-Specific Iterative BLAST (PSI-BLAST) tool. 34 This tool converts protein sequences into an L × 20 PSSM matrix, where L represents the length of different protein sequences. The schematic diagram of the PSSM matrix is illustrated in Figure 1.
Figure 1.
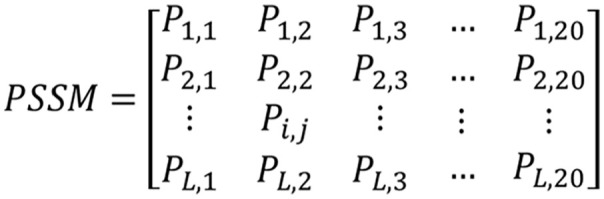
The diagram of PSSM.
In the PSSM matrix, the number 20 corresponds to the 20 amino acids, with Pi,j denoting the probability of the ith type amino acid mutating to the jth type amino acid during biological evolution. The value of Pi,j can be positive, negative, or zero. A positive value indicates a higher probability of mutation, signifying a less conservative region, while a negative value suggests a lower probability of mutation, indicating a more conserved region. By employing the PSI-BLAST tool with parameters such as an e-value of 0.001 and 3 iterations, we can convert each protein sequence into a PSSM matrix, thereby extracting crucial feature information embedded in the evolutionary history of protein sequences. This approach enables us to capture essential evolutionary patterns and improve the prediction accuracy of SIPs.
Gray level co-occurrence matrix (GLCM)
In this study, the efficacy of the prediction model is directly influenced by the quality of the feature extraction method. Consequently, we utilized an advanced feature technique known as the Gray Level Co-occurrence Matrix (GLCM) to extract the evolutionary information from protein sequences in PSSMs. This process resulted in the generation of feature vectors of identical dimensions. The GLCM is a classic texture-based feature extraction algorithm introduced by Haralick et al. 35 While commonly used in various applications, particularly in image processing, to obtain spatial variation features of matrices, GLCM can also be applied to capture essential characteristics of protein sequences. The GLCM features are created by calculating the pixel brightness value (gray level) that contains a specific spatial relationship and a specific value in a given image. This spatial relationship is defined by parameters (Ā, D), where Ā represents the direction of 2 pixels and D defines the spatial distance between the 2 pixels, typically representing the pixel of interest and its horizontally adjacent counterpart. The mathematical expression of the GLCM is as follows: Let f(x,y) be a 2-dimensional digital image with a size of M × N and N gray levels. Then, the Gray-Level Co-occurrence Matrix that meets a certain spatial relationship is derived as follows:
In practical use, it is essential to represent pairs of parameter sets (Ā, D) and combine these parameters with the GLCM matrix to define the rotation invariance of GLCM by setting up rotation parameters. Typically, the parameter is set to 8 directions with an interval of π/4 radians. The grayscale value Ng represents the number of unique brightness values presented in the image. The image is scaled from [0, 255] to [0, Ng] before calculating the GLCM. Ng represents both the gray level and determines the size of the GLCM matrix. 36 In our experiment, the GLCM algorithm was used to extract texture features from the PSSM, including correlation, contrast, homogeneity, and energy. 34 The characteristic expression of GLCM is as follows, where Mij of each expression defines the value at the (i,j) position in the gray co-occurrence matrix. 36
| (1) |
| (2) |
| (3) |
Where represent the averages and the variances of the column and row. Their mathematical description is as follows:
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
Through the aforementioned processing, we generated a 60-dimensional feature vector for each protein sequence by employing the GLCM feature extraction method. Figure 2 illustrates the technology roadmap of the feature extraction method.
Figure 2.

The technology roadmap of the feature extraction method.
VGGNet convolutional neural network
Convolutional neural network is a multi-layer neural network inspired by the hierarchical processing mechanism of information in the biological visual cortex channel. 37 Convolutional neural network is mainly composed of convolutional layer, activation function, pooling layer and fully-connected layer. 38
The VGGNet Convolutional Neural Network was pioneered by a team of researchers from the Visual Geometry Group at Oxford University and Google DeepMind. This network comprises 6 distinct models, each with varying depths that range between 11 and 19 layers. 39 Among them, the 16 and 19-layer models are considered the best for classification and location tasks. The overall structure of VGGNet includes 5 convolutional layers, where the convolution kernel size is 3 × 3, the stride length is 1, and the padding is 1. After each convolutional layer, a maximum pooling layer with a size of 2 × 2 and a stride length of 2 is applied. Following the last maximum pooling layer, 3 fully-connected layers are connected to integrate the features from the image feature map. The final layer of the network is the SoftMax layer, which is used for classification and normalization. Compared to traditional convolutional neural networks, VGGNet makes several improvements, such as reducing the size of the convolution and pooling kernels, increasing the number of convolutional layers, and using pre-trained data to initialize parameters. 40 Additionally, during the test phase, VGGNet transforms the fully-connected layers into convolutional layers. The technology roadmap of the VGGNGLCM prediction model is shown in Figure 3.
Figure 3.

The technology roadmap of the VGGNGLCM prediction model.
Performance evaluation
To assess the performance of the proposed computational model, we used the following metrics: accuracy (AC), specificity (TPR), precision (PPV), and Matthews’s correlation coefficient (MCC):
| (10) |
| (11) |
| (12) |
| (13) |
In the provided formulas, TP and TN represent the number of true positive and true negative interaction sequence pairs accurately predicted, respectively. FP and FN denote the count of falsely predicted non-interaction and interaction protein sequence pairs, respectively. Additionally, we utilize the Receiver Operating Characteristic (ROC) curve to further evaluate the prediction ability of VGGNGLCM in the experiment.
Results and Discussion
Performance of the proposed VGGNGLCM model
The prediction performance of VGGNGLCM on yeast and human datasets was assessed by fivefold cross-validation. To avoid the influence of overfitting, we split the whole datasets into training and independent test sets. In particular, the human dataset was randomly divided into 5 parts, 4 of which were used as the training set and the rest as the independent test dataset. The same strategy was applied to process the yeast dataset. To ensure fairness in comparison, we use a 19 to 1ayer VGGNet network model, in which the convolutional layer is divided into 5 segments, each segment contains 2 to 4 convolutional layers, and each segment is connected with a max- pooling layer at the end, while leaving other parameters at default values. 41 The prediction results of the VGGNGLCM model are presented in Tables 1 and 2.
Table 1.
Fivefold cross validation results shown using VGGNGLCM model on yeast.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 95.31 | 93.59 | 92.24 | 90.50 |
| 2 | 96.49 | 94.56 | 90.66 | 91.43 |
| 3 | 95.25 | 93.67 | 91.20 | 90.74 |
| 4 | 95.78 | 93.81 | 91.63 | 90.35 |
| 5 | 95.58 | 93.37 | 92.29 | 91.16 |
| (Average of 5 tests) | 95.68 ± 0.38 | 93.80 ± 0.44 | 92.01 ± 0.66 | 90.84 ± 0.49 |
Table 2.
Fivefold cross validation results shown using VGGNGLCM model on human.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 97.20 | 96.76 | 97.22 | 92.67 |
| 2 | 97.51 | 96.21 | 97.11 | 92.81 |
| 3 | 97.74 | 95.61 | 97.27 | 91.72 |
| 4 | 98.61 | 97.38 | 96.43 | 91.80 |
| 5 | 97.55 | 97.22 | 96.78 | 92.28 |
| (Average of 5 tests) | 97.72 ± 0.65 | 96.64 ± 0.66 | 96.96 ± 0.48 | 92.30 ± 0.51 |
As shown in Table 1, the proposed VGGNGLCM achieved an average accuracy of 95.68%, an average True Positive Rate (TPR) of 93.80%, an average Positive Predictive Value (PPV) of 92.01%, and an average Matthews Correlation Coefficient (MCC) of 90.84%. Similarly, from Table 2, the VGGNGLCM also demonstrated improved prediction results on the human dataset, with average accuracy, TPR, PPV, and MCC of 97.72%, 96.64%, 96.96%, and 92.30% respectively. In addition, the ROC curves in Figures 4 and 5 also displayed the fivefold cross-validation results of the VGGNGLCM model on yeast and human. These experimental results indicate that the VGGNGLCM model exhibits strong prediction performance for SIPs.
Figure 4.

The fivefold cross-validation ROC curve of VGGNGLCM was performed on yeast.
Figure 5.

The fivefold cross-validation ROC curve of VGGNGLCM was performed on human.
The promising prediction results of the VGGNGLCM model can be attributed to the effective feature extraction methods of the Gray-Level Co-occurrence Matrix and the high-performance classifier of the VGGNet Convolutional Neural Network. The main advantages can be summarized as follows:
First, the Position Specific Scoring Matrix (PSSM) encompasses not only the positional data of protein sequences but also incorporates evolutionary and prior information that mirrors the conserved functions of proteins. This feature facilitates the efficient extraction of evolutionary and prior information from protein sequences via the PSSM matrix. Second, the Gray-Level Co-occurrence Matrix (GLCM) feature extraction method calculates the grayscale spatial correlation between protein sequences at certain distances in the PSSM matrix, extracting hidden key features and generating high-quality protein sequence feature vectors. Finally, compared to traditional convolutional neural networks, VGGNet brings several key improvements: (1) VGGNet reduces the size of the convolutional and pooling kernels to 3 × 3 and 2 × 2 respectively, which allows for more detailed feature extraction and reduces the number of parameters. (2) VGGNet significantly increases the depth of the network by using more convolutional layers. This deeper architecture allows for the extraction of more complex features from the input data. (3) VGGNet utilizes pre-trained data to initialize parameters. This pre-training process helps the network converge faster and can lead to better generalization on a wide range of image recognition tasks. This makes it a valuable tool for improving prediction accuracy, as evidenced in Tables 1 and 2.
In summary, the experimental findings corroborate the assertion that the VGGNGLCM model markedly improves prediction accuracy and is adept at forecasting Self-Interacting Proteins.
Comparison with the method of CNN-based and SVM-based
In order to demonstrate the prediction performance of the VGGNGLCM model, we compared the VGGNet classifier with the CNN and SVM classifiers using the same GLCM approach on the yeast and human datasets. To ensure a fair comparison, we optimized several parameter settings of the CNN using a grid search approach. Specifically, we set the CNN’s epochs (training time), eta (learning rate), batch size per training, and weight values to 98, 0.3, 0.6, and 0.82, respectively. Similarly, using a similar strategy, we optimized the RBF kernel parameters of the SVM, setting c to 0.6 and g to 5.31, while leaving other parameters at their default values. Furthermore, the SVM classifier utilized the LIBSVM tool 42 for classification.
The results of the fivefold cross-validation of CNNGLCM and SVMGLCM on the yeast and human datasets are presented in Tables 3 to 6, while the comparison of ROC curves on these datasets between VGGNet, CNN, and SVM is illustrated in Figures 6 and 7. As shown in Tables 3 and 4, the CNNGLCM model achieved an average accuracy of 92.07%, and the SVMGLCM model obtained an average accuracy of 89.69% on the yeast dataset. Similarly, from Tables 5 and 6 the CNNGLCM and SVMGLCM models achieved average accuracies of 94.13% and 92.15% on the human dataset, respectively. When comparing these results with those of the CNNGLCM and SVMGLCM models, it is evident that the performance of the VGGNet classifier is significantly better than the other 2 classifiers. This is further supported by the ROC curves in Figures 6 and 7, which demonstrate the superior performance of the VGGNet classifier.
Table 3.
Fivefold cross validation results shown by using CNNGLCM model on yeast.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 92.88 | 87.88 | 87.63 | 81.44 |
| 2 | 91.79 | 93.43 | 86.42 | 83.95 |
| 3 | 91.21 | 85.78 | 87.58 | 78.38 |
| 4 | 92.38 | 87.79 | 86.71 | 80.27 |
| 5 | 92.12 | 85.59 | 86.28 | 77.51 |
| (Average of 5 tests) | 92.07 ± 0.62 | 88.09 ± 3.23 | 86.92 ± 0.71 | 80.31 ± 2.51 |
Table 6.
Fivefold cross validation results shown by using SVMGLCM model on human.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 92.67 | 37.91 | 82.67 | 58.58 |
| 2 | 91.60 | 35.43 | 87.82 | 53.52 |
| 3 | 91.58 | 30.21 | 86.17 | 46.87 |
| 4 | 92.45 | 33.67 | 87.33 | 52.62 |
| 5 | 92.47 | 36.12 | 88.13 | 57.11 |
| (Average of 5 tests) | 92.15 ± 0.76 | 34.67 ± 3.82 | 86.42 ± 2.11 | 53.74 ± 3.96 |
Figure 6.

Comparison of ROC curves between RNN, BPNN and SVM on human dataset.
Figure 7.

Comparison of ROC curves between RNN, BPNN and SVM on yeast dataset.
Table 4.
Fivefold cross validation results shown by using SVMGLCM model on yeast.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 89.67 | 31.53 | 81.57 | 49.58 |
| 2 | 90.15 | 35.41 | 85.29 | 55.38 |
| 3 | 89.23 | 30.42 | 89.56 | 48.91 |
| 4 | 90.13 | 33.56 | 87.22 | 52.72 |
| 5 | 89.31 | 30.43 | 81.41 | 46.48 |
| (Average of 5 tests) | 89.69 ± 0.42 | 32.27 ± 2.36 | 85.01 ± 6.02 | 50.61 ± 3.35 |
Table 5.
Fivefold cross validation results shown by using CNNGLCM model on human.
| Testing set | AC (%) | TPR (%) | PPV (%) | MCC (%) |
|---|---|---|---|---|
| 1 | 94.05 | 89.16 | 90.69 | 85.79 |
| 2 | 95.17 | 90.82 | 90.72 | 84.27 |
| 3 | 93.22 | 86.22 | 91.29 | 84.88 |
| 4 | 93.86 | 87.33 | 90.32 | 82.98 |
| 5 | 94.37 | 88.28 | 91.46 | 84.47 |
| (Average of 5 tests) | 94.13 ± 0.72 | 88.36 ± 1.85 | 90.89 ± 0.36 | 84.48 ± 1.26 |
The favorable comparison results obtained may be attributed to the following reasons: compared with the traditional convolution neural network, the most obvious improvement of VGGNet is to reduce the size of convolution kernel and pool kernel, increase the number of convolution layers, use the pre-trained data to initialize parameters, and adopts a way to transform the fully-connected layers into the convolutional layers in the test phase. The experimental findings further underscore the potential of the VGGNGLCM prediction model as a valuable instrument for forecasting SIPs, demonstrating high predictive performance.
Comparison with other methods
To further validate the prediction ability of the VGGNGLCM model, we compared its performance with previous methods using the yeast and human datasets, as displayed in Tables 7 and 8. As shown in Table 7, the average accuracy of the VGGNGLCM model is notably higher than that of the other 6 approaches on the yeast dataset. Similarly, Table 8 depicts that the prediction accuracy achieved by the VGGNGLCM model is significantly better than that of the other 6 methods on the human dataset.
Table 7.
Comparison results between VGGNGLCM and other methods on yeast dataset.
Table 8.
Comparison results between VGGNGLCM and other methods on human dataset.
Based on the comparison results from Tables 7 and 8, it can be concluded that the proposed VGGNGLCM model exhibits high accuracy and robustness, and is capable of better predicting Self-Interacting Proteins. These findings indicate that the VGGNGLCM prediction model can serve as a valuable computational tool for predicting SIPs. The positive experimental outcomes can be ascribed to the application of high-performance VGGNet classifiers in the VGGNGLCM method, coupled with the use of the GLCM feature extraction technique. This combination effectively captures both essential and concealed feature information from protein sequences.
Conclusion
In our research, we have introduced a novel computational prediction technique, VGGNGLCM, which leverages protein sequence data. This method integrates the VGGNet deep convolutional neural network (VGGN) with the Gray-Level Co-occurrence Matrix (GLCM) to detect Self-interacting proteins associations. The VGGNGLCM model obtained average accuracies of 95.68% and 97.72% respectively on yeast and human datasets. The exceptional performance of VGGNGLCM can be attributed to the following key factors:(1) The Position Specific Scoring Matrix (PSSM) captures both the positional and evolutionary information of protein sequences, enabling the extraction of sequence evolutionary information while retaining a wealth of prior knowledge. This aspect contributes to the extraction of critical sequence evolutionary information. (2) The Gray-Level Co-occurrence Matrix (GLCM) feature extraction technique computes grayscale spatial correlation attributes between protein sequences at designated distances within the PSSM matrix. This process facilitates the identification of concealed key features from the PSSM matrix, leading to the creation of superior-quality protein sequence feature vectors. (3) Compared to traditional convolutional neural networks, VGGNet brings several key improvements: (a) VGGNet reduces the size of the convolutional and pooling kernels to 3 × 3 and 2 × 2 respectively, which allows for more detailed feature extraction and reduces the number of parameters. (b) VGGNet significantly increases the depth of the network by using more convolutional layers. This deeper architecture allows for the extraction of more complex features from the input data. (c) VGGNet utilizes pre-trained data to initialize parameters. This pre-training process helps the network converge faster and can lead to better generalization on a wide range of image recognition tasks. Through rigorous experimental validation, we have substantiated the efficacy and robustness of VGGNGLCM. This has been evidenced by its superior accuracy and ability to predict SIPs more accurately than existing methodologies. The experimental validation further underscores the effectiveness and robustness of VGGNGLCM in comparison to current methods. It also reveals that the VGGNGLCM model exhibits high accuracy and robustness, surpassing other methods in the prediction of SIPs. These findings underscore the potential of VGGNGLCM as a valuable computational tool for advancing bioinformatics research related to SIPs prediction.
Acknowledgments
The authors would like to thank all the guest editors and anonymous reviewers for their constructive advices.
Footnotes
Author Contributions: CDH and AJY conceived the algorithm, carried out analyses, prepared the data sets, carried out experiments, and wrote the manuscript; NXM designed, performed and analyzed experiments and wrote the manuscript; all authors read and approved the final manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by “the Fundamental Research Funds for the Central Universities (2019XKQYMS88).”
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval and Consent to Participate: Not applicable.
Consent for Publication: Not applicable.
ORCID iD: Ji-Yong An  https://orcid.org/0000-0001-9546-3654
https://orcid.org/0000-0001-9546-3654
References
- 1. Gottschalk M, Nilsson H, Roos H, Halle B. Protein self-association in solution: the bovine beta -lactoglobulin dimer and octamer. Protein Sci. 2003;12:2404-2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Le Brun V, Friess W, Schultz-Fademrecht T, Muehlau S, Garidel P. Lysozyme-lysozyme self-interactions as assessed by the osmotic second virial coefficient: impact for physical protein stabilization. Biotechnol J. 2009;4:1305-1319. [DOI] [PubMed] [Google Scholar]
- 3. Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG[J]. Nucleic Acids Res. 2006;34(Database issue):D354-D357. doi:10.1093/nar/gkj102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Palamà IE, Maiorano G, Barbarella G, Gigli G. Small thiophene fluorophores in live cells promote protein self-assembly into nanostructured fluorescent and electroactive microfibers. Nano Sel. 2023;4:463-485. [Google Scholar]
- 5. Baisamy L, Jurisch N, Diviani D. Leucine zipper-mediated homo-oligomerization regulates the Rho-GEF activity of AKAP-Lbc. J Biol Chem. 2005;280:15405-15412. [DOI] [PubMed] [Google Scholar]
- 6. Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K. C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene. 2003;22:1273-1280. [DOI] [PubMed] [Google Scholar]
- 7. Katsamba P, Carroll K, Ahlsen G, et al. Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci. 2009;106:11594-11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koike R, Kidera A, Ota M. Alteration of oligomeric state and domain architecture is essential for functional transformation between transferase and hydrolase with the same scaffold. Protein Sci. 2009;18:2060-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woodcock JM, Murphy J, Stomski FC, Berndt MC, Lopez AF. The dimeric versus monomeric status of 14-3-3zeta is controlled by phosphorylation of Ser58 at the dimer interface. J Biol Chem. 2003;278:36323-36327. [DOI] [PubMed] [Google Scholar]
- 10. Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618-625. [DOI] [PubMed] [Google Scholar]
- 11. Bacot-Davis VR, Bassenden AV, Berghuis AM. Drug-target networks in aminoglycoside resistance: hierarchy of priority in structural drug design. MedChemComm. 2016;7:103-113. [Google Scholar]
- 12. Wei W, Yue D. CoGSPro-net:A graph neural network based on protein-protein interaction for classifying lung cancer-relatrd proteins. Comput Biol Med. 2024;172:108251. [DOI] [PubMed] [Google Scholar]
- 13. Wong Y, Huang Z, Liu Y. Detection of protein-protein interactions from amino acid sequences using a rotation forest model with a novel PR-LPQ descriptor. Lect Notes Artif Int. 2015. [Google Scholar]
- 14. Wei Z, Yang J, Yu D. Predicting protein-protein interactions with weighted PSSM histogram and random forests. In: International conference on intelligence science and big data engineering, 2015. [Google Scholar]
- 15. Wang Z, Li Y, Li LP, You ZH, Huang WZ. Self-interacting proteins prediction from PSSM based on evolutionary information. Sci Program. 2021;2021:1-10. [Google Scholar]
- 16. Li JQ, You ZH, Li X, Ming Z, Chen X. PSPEL: in silico prediction of self-interacting proteins from amino acids sequences using ensemble learning. IEEE/ACM Trans Comput Biol Bioinform. 2017;14:1165-1172. [DOI] [PubMed] [Google Scholar]
- 17. Liu X, Lu Y, Wang L, et al. RF-PSSM: A combination of rotation forest algorithm and position-specific scoring matrix for improved prediction of protein-protein interactions between hepatitis C virus and Human. Big Data Min Anal. 2023;6:21-31. [Google Scholar]
- 18. Wang L, You ZH, Yan X, et al. Using two-dimensional principal component analysis and rotation forest for prediction of protein-protein interactions. Sci Rep. 2018;8:12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Shi X, You ZH, et al. Using weighted extreme learning machine combined with scale-invariant feature transform to predict protein-protein interactions from protein evolutionary information. IEEE/ACM Trans Comput Biol Bioinform. 2020;17:1546-1554. [DOI] [PubMed] [Google Scholar]
- 20. Jia LN, Yan X, You ZH, et al. NLPEI: a novel self-interacting protein prediction model based on natural language processing and evolutionary information. Evol Bioinform. 2020;16:9848-133323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia J, Liu Z, Xiao X, Liu B, Chou KC. iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J Theor Biol. 2015;377:47-56. [DOI] [PubMed] [Google Scholar]
- 22. Jia J, Liu Z, Xiao X, Liu B, Chou KC. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition. J Biomol Struct Dyn. 2016;34:1946-1961. [DOI] [PubMed] [Google Scholar]
- 23. Jia J, Liu Z, Xiao X, Liu B, Chou KC. iPPBS-Opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules. 2016;21:E95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Solomonov A, Kozell A, Shimanovich U. Designing multifunctional biomaterials via protein self-assembly. Angew Chem. 2024;63:63. [DOI] [PubMed] [Google Scholar]
- 25. Sana B, Ding K, Siau JW, et al. Thermostability enhancement of polyethylene terephthalate degrading PETase using self- and nonself-ligating protein scaffolding approaches. Biotechnol Bioeng. 2023;120:3200-3209. [DOI] [PubMed] [Google Scholar]
- 26. Liu Z, Guo F, Zhang J, et al. Proteome-wide prediction of self-interacting proteins based on multiple properties. Mol Cell Proteomics. 2013;12:1689-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gane P, Bateman A, Martin M, Zhang J. UniProt: A hub for protein information. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salwinski L, Miller CS, Smith AJ, et al. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32(Database issue):D449-D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stark C, Breitkreutz B-J, Chatr-Aryamontri A, et al. The BioGRID interaction database: 2011 update. Nucleic Acids Res. 2011;39:D698-D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sandra O, Mais A, Bruno A, et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;D1(2014):358-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Breuer K, Foroushani AK, Laird MR, et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228-D1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Launay G, Salza R, Multedo D, Thierry-Mieg N, Ricard-Blum S. MatrixDB, the extracellular matrix interaction database: updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2014;43:D321-D327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu X, Yang S, Li C, Zhang Z, Song J. SPAR: a random forest-based predictor for self-interacting proteins with fine-grained domain information. Amino Acids. 2016;48:1655-1665. [DOI] [PubMed] [Google Scholar]
- 34. Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: detection of distantly related proteins. Proc Natl Acad Sci. 1987;84:4355-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haralick RM, Shanmugam K, Dinstein I. Textural features for image classification. Stud Media Commun. 1973;SMC-3:610-621. [Google Scholar]
- 36. Lohithashva B, Aradhya V, Guru D. Violent video event detection based on integrated LBP and GLCM texture features. 2020. [Google Scholar]
- 37. Meng Z, Liu P, Cai J, Han S, Tong Y. Identity-aware convolutional neural network for facial expression recognition. In: IEEE international conference on automatic face & gesture recognition, 2017. [Google Scholar]
- 38. Wang M, Tan P, Zhang X, Kang Y. Facial expression recognition based on CNN. J Phys Conf Ser. 2020;1601(5):052027(5pp). [Google Scholar]
- 39. Simonyan K, Zisserman A. Very deep convolutional networks for large-scale image recognition. Computer Science, 2014. [Google Scholar]
- 40. Saxe A, Pang W, Koh Z, et al. Ng: on random weights and unsupervised feature learning. In: International conference on machine learning, 2011. [Google Scholar]
- 41. Jun H, Shuai L, Jinming S, Yue L, Peng J. Facial expression recognition based on VGGNet convolutional neural network. In: 2018 Chinese automation congress (CAC), 2018. [Google Scholar]
- 42. Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol. 2007;2(3);Article 27. [Google Scholar]
- 43. Zahiri J, Yaghoubi O, Mohammad-Noori M, Ebrahimpour R, Masoudi-Nejad A. PPIevo: protein-protein interaction prediction from PSSM based evolutionary information. Genomics. 2013;102:237-242. [DOI] [PubMed] [Google Scholar]
- 44. Zahiri J, Mohammad-Noori M, Ebrahimpour R, et al. LocFuse: human protein-protein interaction prediction via classifier fusion using protein localization information. Genomics. 2014;104:496-503. [DOI] [PubMed] [Google Scholar]