

Abstract
Fluorescence lifetime has significant applications in the field of fluorescence microscopy. Effective modulation of fluorescence lifetime can be achieved by controlling the radiative versus nonradiative processes of fluorophores. In this review, we systematically analyze and summarize chemical approaches that achieve fluorescence lifetime modulation for three different types of fluorophores, including small molecules, quantum dots, and metal complexes. In particular, this review is focused on the chemical mechanisms underlying fluorescence lifetime, the structure–function relationship that defines how chemical regulation is achieved, and the chemical principles that can be used to modulate different scaffolds of fluorophores. We aim to provide important resources for gaining a deeper understanding of fluorescence lifetime modulation, through in-depth investigation into the modulation mechanisms of various fluorescence systems. Perspectives are also proposed to enable future investigation on fluorescence lifetime modulation, a field that bears promises to drive the advancement and application of fluorescence imaging technology.
Keywords: fluorescence lifetime, nonradiative process, small molecules, quantum dots, metal complexes
1. Introduction
Over the past few decades, the utilization of fluorescence in the biological sciences has developed significantly. Fluorescence imaging enabled real-time monitoring and quantitative analysis of the morphology and structure of biological samples. However, with the increasing demands on fluorescence technology, fluorescence intensity imaging showed limitations in quantitative detection, multitarget identification, and time-resolved capabilities. Fluorophore lifetime is defined by the average time the molecule spends in the excited state. Accordingly, fluorescence lifetime imaging microscopy (FLIM) is an optimized technique that produced spatially resolved images of fluorophore lifetime, offering an additional dimension of information to visualize fluorophores and an extra source of contrast.1
Fluorescence emission is a complex process, and only a few molecules emitted their photons precisely at t = τ, with the lifetime representing an average value of the time spent in the excited state. Unlike fluorescence intensity imaging, fluorescence lifetime imaging is not affected by the intensity of the excitation light source or sample concentration. This unique advantage made fluorescence lifetime imaging valuable for research in biosensing, molecular interactions, and cell signal transduction. Multiplexed fluorescence microscopy based on fluorescence lifetime is an attractive approach for imaging multiple targets simultaneously. It required only one spectral channel, thus freeing up other channels for further multiplexing.2 Ideally, each probe targeting different components should have exhibited a uniform and narrow distribution of fluorescence lifetimes. Moreover, probes with similar spectra should have demonstrated distinct fluorescence lifetime variations to enable their separation.3 Therefore, the modulation of fluorescence lifetime not only overcomes the limitations of fluorescence lifetime imaging but also enables enhanced detection sensitivity and the implementation of biosensing and molecular probe applications, consequently broadening the applications of fluorescence imaging in diverse research fields.
Both endogenous and exogenous fluorophores are used as fluorescence probes for investigating intracellular environments. In the present review, the modulation of fluorescence lifetime for these exogenous fluorophores is discussed, offering insights and methodologies to optimize fluorescence lifetime imaging. Exogenous fluorophore is a general term concerning small molecules, quantum dots (QDs), and metal complexes, all of which are exogenously loaded in samples. Fluorophores with shorter fluorescence lifetimes are characteristic of weak emitters, and those with longer lifetimes have low photon turnover rates. Lifetime imaging is often hindered when using short-lived fluorophores due to their limited sensitivity and the required long exposition and acquisition time. In fluorescence lifetime modulation studies, the key processes that play a crucial role in determining the fluorescence lifetime are nonradiative processes.4 Nonradiative processes encompass spin–orbit coupling, vibrational relaxation, and energy transfer which competitively deplete the excited energy of fluorophores, thus influencing their fluorescence lifetime.
By precisely controlling these nonradiative processes, effective modulation of fluorescence lifetime can be achieved. The purpose of this review is to comprehensively summarize and analyze the methods of fluorescence lifetime modulation for three different types of fluorophores, namely, small molecules, quantum dots, and metal complexes (Figure 1). This review provides a comprehensive understanding of the field of fluorescence lifetime modulation, driving technological innovations and applications in this area.
Figure 1.

Fluorescence lifetime modulation of small molecules, quantum dots, and metal complexes. Different factors that influence fluorescence lifetime are systematically introduced for three types of fluorophores: small molecules, quantum dots, and metal complexes. For small molecules and metal complexes, mechanisms related to conformational transitions in the excited state, such as MLCT, PET, TICT, and others, have a significant impact on fluorescence lifetimes. The lifetime of quantum dots is primarily influenced by parameters of carrier recombination efficiency.
2. Strategies for Fluorescence Lifetime Control of Small Molecules
2.1. Twisted Intramolecular Charge Transfer (TICT)
In 1973, Grabowski et al. first proposed the TICT mechanism.5 Following this, researchers found the broad applicability of TICT in diverse organic fluorophores, thereby highlighting the critical significance of TICT manipulation in advancing the construction of bright labeling agents and highly responsive molecular probes. The control of TICT formation in a fluorophore hinged on two crucial parameters: the rotational barrier and the driving energy.6 There have been approximately four strategies employed to inhibit TICT: (1) alkyl cyclization; (2) steric hindrance modification; (3) utilization of inductive effects (to reduce the strength of electron-donating groups); and (4) the use of bulky electron-donating groups (to mitigate solvent–solute interactions). The most widely adopted methods involved alkyl cyclization and increasing the ionization potential (IP) of electron-donating groups associated with inductive effects. The primary focus of this section was to modulate the fluorescence lifetime through these two aspects.
How TICT controls fluorescence lifetime (τ) has been extensively studied. The most representative case is provided by rhodamines dyes, wherein two dialkylaniline groups are rigidly bridged by an oxygen linkage. The distinct photophysical properties of rhodamines can be attributed to their nonradiative deactivation by internal conversion7 (Figure 2a). Internal conversion remains inactive, when the nitrogen lacked substitution or when only one nitrogen atom possesses alkyl substitution or the amino group is rigidified. However, in 1 (Figure 2b, 1) with two flexible dialkylamino substituents, internal conversion can be activated, with the activation process being closely linked to the TICT state. In the excited state, the electronic transfer is from the amino group to the xanthene ring in 1, followed by a torsional motion of the bond between the amino group and the xanthene ring. This process underwent nonradiative TICT, thereby resulting in a shorter fluorescence lifetime (2.42 ns in EtOH). For dyes lacking an activated process, such as 2 (Figure 2b, 2), the energy of the TICT state is higher than that of the first excited singlet state. Hence, it is difficult to enter the TICT state, resulting in a reduction of nonradiative pathways and a longer τ of 4.34 ns in EtOH.
Figure 2.

Fluorescence lifetime modulation by TICT. (a) The mechanism of TICT. In the course of the fluorescence to TICT transition, there exists an energy barrier that governs the dynamics of the excited state. (b) Rhodamines with a dialkyl amino group showed the process of TICT. (c) Structures of probes with TICT-modulated fluorescence lifetimes. Fluorescence lifetime of 1–12 was adjusted via alkyl cyclization, and that of 13–16 was adjusted by alkyl cyclization inductive effects.
The impact of the number and rigidity of alkyl groups on the N atoms is an important factor affecting fluorescence lifetime. Among the commercially available rhodamine dyes, the longest lifetime of 4.74 ns in ethanol was found with 2 (R101), whose alkyl carbons are fixed in a six-member ring.8 In addition, 3 (Figure 2b, 3) without alkyl substitution exhibited a relatively high τ of 4 ns. By contrast, 4 (Figure 2b, 4) with two −CH2CH3 on the N atoms showed the shortest τ of 2.92 ns. These observations were attributed to the twisting of the amino group in the excited state, which facilitated the TICT state and promoted nonradiative pathways to reduce the fluorescence lifetime. In the case of 4, the introduction of a second −CH2CH3 group increased the electron-donating ability of the N atom and the mean ground-state twist angle for the amino groups. Because the TICT state needed a perpendicular mutual orientation between the dialkyl amino group and the xanthene ring, these factors facilitated the occurrence of TICT and shortened the fluorescence lifetime of rhodamine.
Based on these results, later studies found that the fluorescence lifetime of rhodamine compounds differed from each other in their substituents on N atoms. In 2015, Lavis and co-workers replaced N,N-dialkylamino groups in the classic dye tetramethylrhodamine (Figure 2b, 5) with differently sized nitrogen-containing rings ranging from aziridine to azepane.9 The relatively short fluorescence lifetime of 5 can be attributed to its tendency to undergo nonradiative transitions into the TICT state upon excitation. In this process, the electron transferred from the nitrogen atom to the xanthene system, accompanied by a distortion of the Caryl-N bond. The introduction of a heterocycle enhances the rigidity of the N atom and prevents rhodamine from undergoing Caryl-N bond twisting to enter the TICT state, thereby increasing the fluorescence lifetime. Among these rhodamine derivatives, the one with azetidine 6 (Figure 2b, 6) exhibited the longest fluorescence lifetime of 3.84 ns. In aziridine derivative 7 (Figure 2b, 7), the ring strain in 7 compelled the rhodamine molecule to adopt a colorless lactone form with no fluorescence. The size of the heterocyclic rings in these rhodamine compounds was found to have no direct correlation with τ. 8 (Figure 2b, 8) had a fluorescence lifetime of 3.6 ns, while compound 6 exhibited a significantly reduced τ of 0.6 ns. However, 9 (Figure 2b, 9) showed a slightly increased lifetime of 1.62 ns compared to compound 6, indicating that the increased flexibility of the larger ring can offset other detrimental structural effects on rhodamine fluorescence. Furthermore, they found an increase in quantum yield (Φ) after introducing azetidine rings as substitutions for the alkyl nitrogen in various fluorophores, including coumarin and naphthalimide, acridines, rhodols, carborhodamines, oxazines, and Si-rhodamines. Therefore, the introduction of azetidine rings as substitutions for alkyl nitrogen provides a universal method to improve fluorescence lifetimes in numerous fluorophores by effectively attenuating nonradiative decay processes.
Studies have also been conducted in fluorophores other than rhodamines. 10 (Figure 2b, 10), 11 (Figure 2b, 11), and 12 (Figure 2b, 12) are structural analogues of rhodamine dyes. The primary distinction between them and the corresponding rhodamine lies in the lack of a carboxyphenyl moiety. It has been demonstrated that the utilization of geometrically constrained azacyclic substituents can elevate the IP of amino groups, thereby suppressing the formation of the TICT state and extending the fluorescence lifetime of rhodamines. Lu and co-workers measured the fluorescence lifetimes of these three rhodamine analogues to investigate how the alkyl groups attached to N on the xanthene core affect fluorescence lifetime.10 They found that 12 exhibited extended the fluorescence lifetime (3.87 ns) and enhanced quantum yield compared to 11 (2.32 ns) and 10 (2.01 ns). These differences are attributed to the influence of alkyl groups on the nitrogen (N) atom, which leads to intensified internal conversion (IC). The increase in the extent of vibrational and rotational motion from the S1 state was accompanied by the increase of the alkyl chain length. The structural variation did not alter the rate constant of the radiation process but changed the nonradiation process significantly.
Replacing the dimethylamino auxochrome with the azetidine ring has been shown to increase the fluorescence lifetime of rhodamine. However, the azetidine substitution strategy appeared to have less pronounced effects on the carboxy rhodamine and silicon rhodamine in the red to near-infrared region. Guo and co-workers suggested that the introduction of electron-withdrawing groups, such as the sulfone group, exerts a significant negative inductive effect (−I effect) on amino groups.11 This modification effectively inhibited the TICT process and increased the fluorescence lifetime (τ) of various fluorophores in aqueous solutions. The geometrically constrained azetidine group had a higher IP value than the diethylamino and piperidine groups. This is presumably due to the significant ring strain of an azetidine group that limits the configuration transformation from the sp3-hybridized N atom to the more planar sp2-hybridized N radical cation upon photoexcitation. The phosphine oxide (P=O) and sulfone (SO2) functionalized piperidine moieties demonstrated higher ionization potential (IP) values than the azetidine group. Hence, their strong −I effect further suppressed the TICT process, resulting in prolonged fluorescence lifetimes of 13 (Figure 2b, 13) and 14 (Figure 2b, 14), measured as 5 and 5.89 ns, respectively. Moreover, the electron-withdrawing group-functionalized piperidine auxochromes can markedly enhance the fluorescence lifetime of all fluorophores suffering from TICT-induced fluorescence quenching in aqueous solution.
Rhodamine compounds differ from each other in their substituents on either xanthene π-skeleton or on N atoms, while their benzoate units are usually the same. In 2010, Hell and co-workers designed a fluorinated rhodamine employing 2,2,2-trifluoroethyl as the amino substituent on rhodamine.12 The strong −I effect of fluorine atoms enhanced the stability of the amino group’s charge, effectively inhibiting the TICT process, thus leading to 15 with significantly higher fluorescence lifetimes (4.2 ns) than those of rhodamines with monoalkylated nitrogen atoms (≤4.0 ns). Rega and co-workers demonstrated that the radiative lifetime of rhodamines can be correlated to the charge transfer from the phenyl group to the xanthene moiety during the S0 ← S1 de-excitation.13 Additionally, the radiative lifetime is influenced by the relative orientation between the xanthene and phenyl groups in the S1 minimum structure, which can vary depending on the phenyl substituents. The S0 ← S1 radiative decay process depends upon the transition energy ΔE and the transition dipole strength |μ10|2. The transition dipole strength is increased by the overlap of the ground and the excited electronic density (transition electronic density) along the x axis. On the contrary, transition electronic density involving the phenyl ring gave a zero contribution to the dipole integral. According to the calculation between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) contributions from the three fragments (xanthene, amino, phenyl fragment), a remarkable linear relationship was found between the fluorescence lifetime and the charge transfer amount involving the phenyl fragment, whereas no dependence is noticed for the electronic density rearrangement on the amino groups. By optimizing the minimum energy conformations of the molecule both in S0 and S1, experimental results indicate that increasing the xanthene–phenyl torsion leads to a larger phenyl charge transfer and a longer lifetime.
Other mechanisms could also regulate the fluorescence lifetime of rhodamine, such as intramolecular charge transfer (ICT), excited-state intramolecular proton transfer (ESIPT), photoinduced electron transfer (PET), and planar intramolecular charge transfer (PICT). Therefore, a comprehensive consideration of these diverse regulatory mechanisms is crucial for understanding and optimizing the performance of rhodamine-based fluorophores. Among these mechanisms, PICT offers an intriguing alternative pathway, wherein the excited-state rigid planar configuration of molecules can substantially reduce nonradiative pathways, leading to an increased fluorescence lifetime of rhodamine. Zhang and co-workers investigated an anilino-substituted rhodamine analogue 16 (Figure 2b, 16).14 They prove that photoexcitation of PICT and TICT occurs simultaneously in 16 with the aid of a potential energy surface. When fluorescence originates from the TICT state, the overlap between two separated and perpendicular orbitals is forbidden, resulting in a reduced transition dipole moment. Consequently, this leads to a shorter fluorescence lifetime.
It was well-established that the prolongation of a molecule’s fluorescence lifetime was often attributed to the presence of relatively rigid excited-state structures with no rotatable bonds, rendering them less susceptible to environmental influences, except in the presence of fluorescence quenchers. The fluorescence lifetimes exceeding 100 ns were only observed in the case of pyrenes and molecules possessing thermally activated delayed fluorescence (TADF) properties. Although the pyrene had a large spectral shift from monomer (400 nm) to the excimer (485 nm), it still cannot extend beyond the blue-green waveband.15 Therefore, pyrenes were mainly applied in fluorescence resonance energy transfer (FRET) imaging related to distance measurements, when the change in fluorescence lifetime indicated mostly a change in the distance between the donor and the acceptor.
Beyond the role in modulating fluorescence lifetimes, it should be worth noting that the TICT mechanism exerted a profound influence on the quantum yield (Φ) and brightness of fluorescent molecules. First, rigidifying the alkyl groups via cyclization to prevent TICT formation and improve Φ has been applied in many fluorophores where TICT served as a main nonradiative decay channel. By preforming cyclization to rigidify the backbones, Φ increased from 0.055 (17) to 0.66 (18) in coumarin derivatives16 and from 0.15 (19) to 0.69 (20) in Cy5 derivatives (Figure 3a).17 It was noteworthy that Xu et al. conducted an investigation into the impact of azacyclic substituents on the minimization of TICT.18 They performed cyclization with differently sized nitrogen-containing rings (ranging from aziridine to azepane, compounds 21–26; Figure 3b). The TICT-induced conformation changes of them resulted from competing contributions from azacyclic strain energy, steric hindrance, and resonance effect. The resonance effect relied on the electron-donating/withdrawing strength of both the azacyclic rings and the fluorophore scaffolds. Although 21 had the highest Φ, 22 had the highest brightness, which aligned with the conclusions drawn by D. Lavis.9 TICT formation in a dye was primarily governed by two factors: steric hindrance, as shown by Lavis and Xu’s work, and electronic effects. The TICT state arose from full charge separation between the electron donor and acceptor.19 The strong inductive effect achieved by introducing electron-donating groups (EDGs) with high IP proved to be a reliable mechanism for reducing the electron-donating strength and, thereby, effectively suppressing TICT formation. Xiao and co-workers compared the IP of several amino EDGs using density functional theory (DFT) calculations.20 Their discovery indicated that quaternary piperazine possessed a high IP, implying a limited electron-donating capacity resulting from the inductive effect of the positively charged quaternary amino group. Utilizing this quaternary piperazine, they designed a novel rhodamine derivative 27 featuring outstanding quantum yields (Φ = 0.93) and exceptional brightness (ε × Φ = 8.1 × 104 L·mol–1·cm–1) by harnessing the electronic inductive effect to inhibit TICT. This section provided insight into the mechanistic understanding of TICT and its related structure–property relationships. The suppression of TICT can significantly yield bright and long-lasting fluorophores.21
Figure 3.
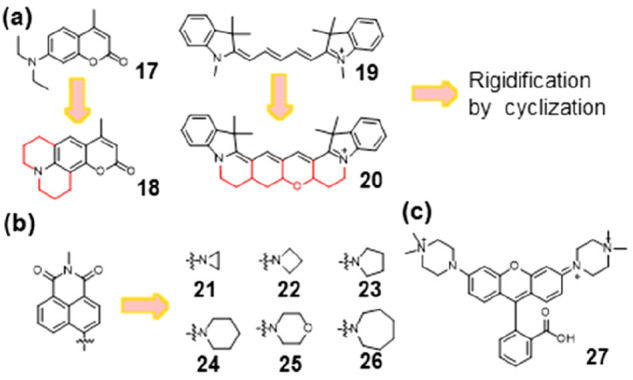
Enhancement of quantum yields and brightness by inhibition of TICT. (a) Alkyl cyclization in coumarin and cyanine dyes. (b) Different sized azacyclic substituents in naphthalimides to inhibit TICT. (c) Quaternary piperazine with a high IP can suppress TICT via inductive effects.
2.2. Effects of Substituents to Fluorophores
Fluorescence lifetime can also be influenced by modulating the electronic nature of the substituents on fluorophores. In the case of boron-dipyrromethene (BODIPY), its fluorescence lifetime can be effectively altered. By increasing or decreasing the electronic density of the substituents, the charge distribution and charge transfer processes within the BODIPY molecule can be controlled, leading to distinct excited states and nonradiative pathways. Through the targeted design and synthesis of BODIPY molecules with diverse substituents, precise modulation of their fluorescence lifetimes can be achieved. Zhang and co-workers found that chlorines on BODIPY can decrease its radiative transition with the reduced fluorescence lifetime.22 According to DFT (Density functional theory) calculations, the electron distribution of the HOMO and the LUMO of 28 (Figure 4a, 28) were localized on the carbazole and BODIPY moieties, respectively (Figure 4b). The introduction of a chlorine atom at the 2,6-positions of 29 (Figure 4a, 29) resulted in reduced LUMO energy and decreased fluorescence lifetime from 4.14 to 3.94 ns.
Figure 4.

Fluorescence lifetime modulation by substituents. (a) Structures of probes with fluorescence lifetime modulation by substituents. (b) The HOMO and LUMO electron distribution of 28. (c) Donor–Acceptor structure of amino rhodamine. The PET process occurred from the amino benzoate unit to the xanthene fluorophore fragment. (d) The HOMO and LUMO electron distribution of 30 and 31. 30 exhibited a relatively limited overlap between HOMO and LUMO orbitals, while 31 demonstrated a significantly higher overlap between HOMO and LUMO orbitals. (b) Reprinted with permission from ref (22). Copyright 2013, American Chemical Society. (d) Reprinted with permission from ref (23). Copyright 2015, American Chemical Society.
Other than BODIPY, Kolehmainen and co-workers investigated the fluorescence properties of molecules containing the NBF2O moiety by introducing the electron-withdrawing or electron-releasing groups in the phenyl ring.23 From strong electron-donating substituents to strong electron-withdrawing substituents, their fluorescence lifetimes gradually decreased from 2.3 ns for 30 (Figure 4a, 30) to 0.1 ns for 31 (Figure 3a, 31). The more electron-rich 30 (compared to 31) with much more significant density change upon excitation may slightly weaken the acceptor ability of the NBF2O moiety, resulting in an increase in the energy of the transition (Figure 4d). As a consequence, this leads to an enhancement of the radiative transition rate. In 30, the dominant structure gave a transition associated with substantial charge reorganization and is the source of the much longer-lived emission (2.5 ns). Thus, the quantum yield and fluorescent lifetime increased with increasing electron-donating properties of the substituent in the phenyl moiety of the BF2-carrying fluorescent dyes.
The electronic nature of substituents significantly influences the photoinduced electron transfer (PET) process. Upon introducing substituents with different electron characteristics into rhodamine, the efficiency and intensity of the PET process may undergo notable changes. Jia and co-workers suggested that the fluorescence lifetime of rhodamine is modulated by the position of substitution on the benzoate.24 The presence of a 5′-COOH substituent on the phenyl moiety in 32 (Figure 4a, 32) resulted in shorter fluorescence lifetime compared to 33 (Figure 4a, 33) with 6′-COOH. The fluorescence lifetime became shorter when the −COOH groups were replaced by −NH2 groups at the 5′ or 6′ position on the benzoate unit. As another example, 34 (Figure 4a, 34) contains the amino benzoate unit as electron donor (D) and the xanthene fragment as electron acceptor (A). For this compound, PET occurred from the amino benzoate unit to the xanthene fluorophore fragment (Figure 4c), leading to decreased radiative transition and reduced fluorescence lifetime.
2.3. Intramolecular and Intermolecular Interactions
The overlap of electronic clouds between adjacent dye molecules leads to strong exciton coupling, thereby altering the fluorescence lifetime of the rhodamine material. Depending on the relative orientation of dye molecules, either H-type or J-type exciton coupling can occur, providing different optical characteristics for solid-state rhodamine dyes. Zhang and co-workers found that solid-state 4 (Figure 5a, 4) showed a longer emission lifetime than that of the corresponding molecular rhodamine, with 4.12 ns in the solid state compared to 1.61 ns in solution.25 Solid-state rhodamines consist of H-type and J-type molecular aggregates as well as individual molecules, among which H-type aggregates appear to be predominant. The steric hindrance of bulky groups in solid-state 4 weakened the intermolecular interactions, leading to an extension of the fluorescence lifetime. On the phenyl of 35 (Figure 5a, 35), the COOR group was replaced by SO3–, and an additional SO3– was present. The negative charges affected the relative orientation or the two negatively charged SO3– groups, which consequently strongly attracted the neighboring positively charged xanthene fluorophores. As a result, the stronger intermolecular interactions quenched the fluorescence, as reflected by the lowest Φ and shortest τ for 35.
Figure 5.

Fluorescence lifetime modulation by intramolecular and intermolecular interactions. (a) 4 with steric repulsion had weak interaction, and 24 with charged substituents had weak interaction. (b) Structures of 39 and 40, and fluorescence decay curves of 39 (blue) and 40 (red) in the solid state. (c) Structures and fluorescence lifetimes of 36, 37, and 38 in THF (black) and solid state (red). (b) Reprinted with permission from ref (30). Copyright 2018, Royal Society of Chemistry. (c) Reprinted with permission from ref (29). Copyright 2023, Multidisciplinary Digital Publishing Institute.
The restriction of intramolecular motion (RIM) mechanism was one of the most important aggregation-induced emission (AIE) mechanisms for designing AIE fluorophores, which can be categorized into the restriction of intramolecular rotation (RIR) mechanism and the restriction of intramolecular vibration (RIV) mechanism.26 Many AIEgens, such as hexaphenylsilole (HPS), tetraphenylethene (TPE), and tetraphenylpyrazine (TPP), were predominantly designed based on the RIR mechanism, significantly advancing AIE materials research.27 Therefore, inhibiting the RIR to reduce energy loss through nonradiative pathways was one of the effective strategies for extending fluorescence lifetimes. Triphenylamines possessed a characteristic propeller-like structure and were frequently employed in the construction of AIE fluorophores.28 Lu and co-workers designed a series of phenylmethylene pyridineacetonitrile derivatives bearing a triphenylamine (TPA) structure and pyridine ring with the nitrogen atom at the ortho (36), meta (37), and para (38) position in Figure 5c.29 In tetrahydrofuran (THF), their fluorescence lifetimes were only approximately 1 ns, but in the solid state, these lifetimes were significantly extended. Specifically, the fluorescence lifetime of 37 was prolonged to 3.32 ns, and that of 38 was extended to 5.94 ns (Figure 5c). Remarkably, 36 exhibited the longest fluorescence lifetime in the solid state, reaching 7.96 ns, which represented an over 7-fold increase compared to its fluorescence lifetime in THF. The longest fluorescence lifetime was likely a result of a more planar molecular configuration, which was driven by the interaction between the ortho-nitrogen and the vinyl hydrogen atom (−CH=C−). Based on triphenylamine and pyridinium salt moieties, Tang et al. designed 39 and 40 with green and near-infrared emission properties, respectively (Figure 5b).30 In aqueous solutions in their single-molecule state, these compounds were nearly nonemissive. This can be primarily attributed to the rotational motions of molecular rotors, including the phenyl rings within the triphenylamine moiety and carbon–carbon double bonds as well as the pyridinium and thiophene units. These rotations dissipate exciton energy and elevate nonradiative decay rates, consequently preventing emission. In the solid state, the fluorescence decay curves of 39 and 40 revealed that their lifetimes were 5.75 and 0.92 ns. The increased emissions observed in aggregates can be attributed to the restriction of rotor motions, which facilitates radiative decay. The twisted conformation of the triphenylamine segment can extend the intermolecular distance and prevent emission quenching by the reduction of the intermolecular π–π interaction, thus switching on the luminescence process in the aggregation state.
2.4. Fluorescence Lifetime Affected by Geometric Symmetry
In the excited state, conformational changes of fluorescent molecules occurred primarily due to the higher energy level of excitation, leading to increased vibrational amplitudes of the atoms and bond angles. This enhanced vibrational motion facilitated the exploration of more stable conformations, thereby reducing the energy of the excited-state molecules. The influence of molecular geometric symmetry on the conformational changes in the excited state of a molecule arose from the determination of the arrangement of internal atoms and bonds. This, in turn, affected the spatial distribution and vibrational modes of the molecule in the excited state. Due to the molecular geometric symmetry, certain vibrational modes may be forbidden or more active in the excited state. As the excited-state molecule undergoes vibrations, specific symmetry elements may restrict or facilitate conformational adjustments. Consequently, the molecular symmetry impacted the conformational transitions in the excited state, thus influencing the pathways of radiative transitions.
The structural symmetry of rhodamine had a significant impact on its fluorescence lifetime. Jockusch and co-workers showed that 41 (Figure 6, 41) and 42 (Figure 6, 42) with one and three methyl groups on the nitrogen atoms, respectively, exhibit extended lifetimes compared to both the unmethylated 43 (Figure 6, 43) and 44 (Figure 6, 44) containing four methyl groups.31 Thus, rhodamines with a tendency toward Cs symmetry in an excited state are more susceptible to relaxation through intramolecular PET, resulting in shorter lifetimes compared to those that adopt more asymmetrical conformations.
Figure 6.

Structures of probes with fluorescence lifetime modulation by geometric symmetry. The configurations of 41 and 42 exhibited lower levels of symmetry, whereas those of 43 and 44 were comparatively more symmetric.
2.5. Fluorescence Lifetime Affected by π-Expansion
The S1 energy profile is believed to share some connection with the fluorescence lifetime, although there is limited substantial empirical evidence to confirm this connection.32 Conventionally, alterations in the S1 energy profile have been achieved through various methods, including the introduction of electron-donating/withdrawing substituents, replacement of heteroatoms, and imposition of internal steric constraints. Additionally, the manipulation of π-systems has frequently been employed to facilitate the transition of the S1 profile from a bent to a planar conformation. Accurate control of π-expansion enables the fine-tuning of excited-state energy levels and conformational changes in the excited state, effectively governing the related nonradiative process in these organic fluorescent π-systems. Consequently, this control reduced energy losses arising from conformational transitions, leading to an enhancement in fluorescence lifetime.
The form and structure of the S1 energy profile play a crucial role in the optimization of photofunctional molecule design. Subtle variations within the S1 energy profiles determined the properties and functions of photochromic compounds, molecular rotors, and ESIPT dyes. Saito and co-worker explored the impact of aromaticity on the energy variation of the S1 state during the transition of fluorescence molecules from a bent conformation to a planar conformation by adjusting the aromaticity of oxepins.33 Modifying the aromaticity of oxepins would influence the energy distribution of the S1 state, rather than solely affecting the energy level of the S1 state. In these 3 π-expanded oxepins, further π-expansion was shown to lead to weaker S1 aromaticity and produce a flatter S1 downhill planarization energy profile. These flatter S1 downhill led to a shorter fluorescence lifetime. Computational analysis revealed that the radiative decay constants kr of 45–47 (Figure 7a, 45–47) exhibited negligible changes, whereas the nonradiative decay constants knr increased with π-expansion. In summary, the π-expansion decreased the aromaticity of the S1 state, resulting in a flatter energy distribution corresponding to the molecular conformational changes. This, in turn, led to higher nonradiative decay constants and consequently decreased fluorescence lifetimes.
Figure 7.

Fluorescence lifetime modulation by π-expansion. (a) Structures of probes with fluorescence lifetime modulation by π-expansion. The degree of π-conjugation progressively increased from 45 to 47. Phenyl groups at the third and ninth positions extended the π-conjugation of DPND in 48. Different P-heteropines in π-systems had influences on fluorescence lifetime of 49 and 50. π-Expansion affected the conformation changes in the excited state of terpyridines (51) and triarylborane derivatives (52–54). (b) Excited-state aromaticity made the conformation tend to be planar. (c) Aromatic stabilization in the excited state. (b) Reprinted with permission from ref (36). Copyright 2023, American Chemical Society. (c) Reprinted with permission from ref (33). Copyright 2020, American Chemical Society.
A number of studies have reported that highly planar dipyrrolonaphthyridinedione (DPND) molecules undergo nonradiative transitions through π–π stacking, a phenomenon commonly widely recognized as aggregation-caused quenching (ACQ). Tang introduced the concept of aggregation-induced emission (AIE), wherein molecules with limited degrees of freedom in the solid state exhibit luminescence due to a reduction in vibrational deexcitation pathways.34 Gryko and co-workers obtained π-extended 48 (Figure 7a, 48) by introducing phenyl groups at the third and ninth positions of DPND.35 The substitution of DPND with a benzene ring at positions 3 and 9 mainly affected the HOMO energy level, increasing the oscillator strength of the S0–S1 transition. In the solid state, 48 differs from DPND in terms of π–π stacking interactions. The presence of multiple interactions between aromatic H and O resulted in varying distances between adjacent DPND core planes, thereby alleviating π–π stacking and increasing the fluorescence lifetime.
The π-conjugation also exerts an influence on the fluorescence properties of P-heteropines. Ren and co-workers explored the photophysical properties of a series of ionic P-heteropines with various aryl backbones.36 Since excited-state aromaticity-induced planarity (EAIP) was hardly applicable to the P-heteropines due to the high-energy barrier of the planarization for the pyramidal P-center, ionic P-heteropines with various aryl backbones were limited to fine-tune the excited-state characteristics (Figure 7b). 49–50 (Figure 7a, 49–50) with maleimide (MI)-conjugated P-heteropines with NPN-type P-environment exhibited a significantly longer fluorescence lifetime of approximately 9 ns compared to various π-conjugated P-heteropines in other P-environments.
The effect of π-conjugation substituent length, namely, torsional hindrance between the aryl and terpy units, played key roles in the fluorescence properties of terpyridines. Machura and co-workers introduced different π-extended aromatic groups onto terpyridines to investigate the impact of π-extension aromatic groups on the characteristics of electronic transitions.37 These terpyridines displayed twisted ground-state conformations, with notable structural changes observed in the dihedral angle between the central pyridine ring and the plane of the polyaromatic group. These modifications were primarily influenced by steric hindrance arising from both inter-ring H···H interactions and hydrogen−π-ring steric repulsions. Among these terpyridines, 51 (Figure 7a, 51) with 9-phenanthryl-substituted terpyridines had the highest fluorescence lifetime, which was approximately 11 ns.
In triarylboranes, the presence of multiple aromatic rings allowed for the formation of a conjugated structure, enabling the molecule to adopt a stable π–π* excited state upon excitation. This conjugated structure facilitated efficient electron transfer within the molecule, leading to reduced nonradiative energy loss and, consequently, longer fluorescence lifetimes. Halet and co-workers performed DFT and time-dependent (TD) DFT calculations to investigate the photophysical properties of a series of triarylborane derivatives 52–54 (Figure 7a, 52–54).3852, bearing tert-butyl substitution, exhibited a longer fluorescence lifetime compared to 53 and 54 with diphenylamine substitution.
2.6. Fluorescence Lifetime Affected by ESIPT
The phenomenon of excited-state intramolecular proton transfer (ESIPT) has garnered significant interest due to its distinctive four-level photophysical mechanism and long Stokes shift (Figure 7a). 2-(2-Hydroxyphenyl) benzothiazole (HBT) is a prototypical ESIPT chromophore with enol form (E) changed to keto form (K) upon photoexcitation. Zhao and co-workers coupled rhodamine with the HBT to investigate the ESIPT characteristics of the HBT-rhodamine dyad as well as the fluorescence switching under acidic and alkaline conditions.39 According to their investigation, 55 (Figure 8b, 55) exhibited an emission band at 404 nm due to the emission of the respective enol-form. Upon the addition of trifluoroacetic acid (TFA), 55 exhibited pronounced fluorescence radiation at 579 nm due to the open-ring amide form. 55 displayed a fluorescence lifetime of approximately 4–7 ns in polar aprotic solvents.
Figure 8.
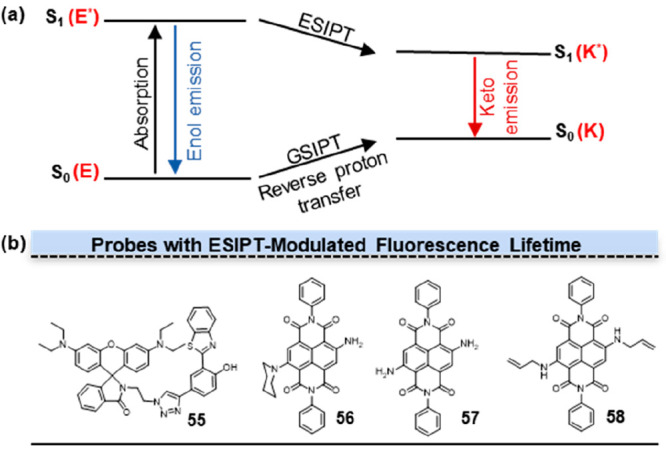
Fluorescence lifetime modulation by ESIPT. (a) The mechanism of ESIPT. ESIPT chromophore with enol form (E) changes to keto form (K) upon photoexcitation. The keto form exhibits additional fluorescence emission at longer wavelengths. (b) Structures of probes with fluorescence lifetime modulation by ESIPT.
By introducing a series of alkylamine substituents on the core or NDI-shoulder positions of naphthalenediimide (NDI), fluorescent moieties with excited-state intramolecular proton transfer (ESIPT) properties can be obtained. Mukhopadhyay and co-workers reported a new class of symmetrically and asymmetrically-NH2-substituted core-NDIs.40 Previous investigations have suggested that the introduction of free amine (−NH2) groups to the NDI core can serve as a promising strategy to promote hydrogen bonding and facilitate the ESIPT process. Furthermore, this approach was expected to promote an increase in fluorescence lifetime. The short fluorescence lifetime of 56 (Figure 8b, 56) can be attributed to the occurrence of photoinduced electron transfer (PET) from the donor piperidine-substituted nitrogen atom to the acceptor NDI scaffold. The PET process competed with the ESIPT process, resulting in a decrease of fluorescence lifetime. Through the comparison of 57 (Figure 8b, 57) and 58 (Figure 8b, 58), it was found that an increase in the number of hydrogen atoms in the amino groups at the NDI core can effectively facilitate the ESIPT process and notably extend the lifetime.
2.7. Fluorescence Lifetime Affected by Thermally Activated Delayed Fluorescence (TADF)
Typically, the fluorescence lifetimes of the majority of organic small molecules range from 0 to 20 ns.41 Exceptionally long fluorescence lifetimes exceeding 100 ns, however, were only observed in the case of pyrenes and molecules exhibiting TADF properties. These final emissive singlet states of TADF emitted in the microsecond to millisecond range due to the involvement of the longer-lived triplet states. When TADF molecules were excited, they underwent activation from the S0 to S1 state.42 On one hand, the excited TADF molecules underwent rapid radiative decay back to S0, emitting prompt fluorescence with a short lifetime (typically less than 10 ns) for conventional fluorescence imaging. Alternatively, the excited TADF molecules underwent an intersystem crossing (ISC) process from the S1 state to the T1 state. Subsequently, the small singlet–triplet energy gap (ΔEST) facilitated a thermally activated up-conversion from nonemissive triplet excitons into spin-allowed singlet excitons through an efficient reverse intersystem crossing (RISC) process.43 This led to delayed fluorescence (DF) emission, which constituted the core of the TADF mechanism. The most common strategy in the design of TADF emitters, aiming to achieve a sufficiently low ΔEST to facilitate efficient RISC, involved the minimization of electron exchange energy in the excited state.44
In general, the TADF structure was a donor–acceptor-type structure in order to have a small ΔEST value by minimizing the orbital overlap between the LUMO and HOMO for effective RISC.45 The weak electronic communication that occurred through an intensely twisted conjugated linker46 or a single sigma (σ) bond47 still resulted in some overlap between the HOMO and LUMO (Figure 9a). Another design strategy involved physically separating the D and A moieties, resulting in minimal overlap between the HOMO and LUMO. In this design, electronic communication between the D and A within the molecule predominantly occurred through spatial charge transfer interactions, leading to a significantly reduced ΔEST and enhanced radiative decay rate. In 59, triphenylamine functions as the donor and dicyanoquinoxaline as the acceptor, which were placed on the different fins of a triptycene scaffold (Figure 9a).48 TD-DFT calculations reveal that the HOMO was located on the triphenylanime moiety and the LUMO was localized on quinoxaline. In Figure 9c, TD-DFT calculations revealed that the HOMO resided on the triphenylamine moiety, while the LUMO was localized on the quinoxaline portion. There existed only a minimal overlap between HOMO and LUMO, which facilitated singlet-state emissions. With a ΔEST value of 111 meV, 59 exhibited TADF potential and displayed a phosphorescence lifetime of 2.4 μs when tested under deoxygenated conditions (Figure 9b). M. Swager and co-workers designed the D–A structure 60 with a 9,9-dimethylxanthene bridge connected (Figure 9d).49 Crystallographic analysis revealed a donor–acceptor distance of 3.423 Å within 60, accompanied by coplanar alignment fostering π–π intramolecular interactions. Additionally, intermolecular C–H···π interactions were observed in a unit cell at the distance of 2.7–2.9 Å that stabilized the crystal packing and restricted the intramolecular motion giving rise to AIE. As illustrated in Figure 9d, a negligible overlap between HOMO and LUMO was observed with ΔEST as low as 1 meV. Under nitrogen, 60 displayed distinctive delayed 2.3 μs relaxation in addition to a prompt 2.8 ns relaxation. This provided a viable strategy for designing TADF probes with extended lifetimes.
Figure 9.

Fluorescence lifetime modulation TADF. (a) Donor (red) and acceptor (blue) geometry of TADF molecules and 59. (b) Photoluminescence lifetime of 59 measurement in cyclohexane at room temperature. (c) HOMO (left) and LUMO (right) of 59. (d) HOMO and LUMO orbital distributions and calculated bandgaps, S1 and T1 energy levels, and oscillator strengths (f) for 60. (e) Structure of 61. (f) Structure of 62. (g) HOMO (left) and LUMO (right) of 61. (h) HOMO (left) and LUMO (right) of 62. (a–c) Reprinted with permission from ref (48). Copyright 2015, American Chemical Society. (d) Reprinted with permission from ref (49). Copyright 2017, American Chemical Society. (g, h) Reprinted with permission from ref (51). Copyright 2019, Wiley-VCH.
However, the two primary types of D–A (TADF) molecular architectures developed had limitations in their design tunability (in fixed-constrained systems) or exhibit unpredictable spatial arrangement (when grafted on flexible backbones),50 leading to a restricted comprehension of through-space interactions between D and A. Adachi and co-workers explored a novel TADF probe design, employing a dithia[3.3]paracyclophane building block as a versatile three-dimensional (3D) platform to enhance through-space interactions.51 Such a 3D platform allowed them to bring together the D and A units into close proximity and to probe the effect of their orientation, contact site, and distance on their TADF emission properties. They employed the dithiacyclophane to control the distance and orientation between D and A, resulting in the design of two isomers, 61 and 62 (Figure 9e,f). These isomers featured a core composed of a dithiacyclophane, with a donor ditolylphenylamine unit grafted onto the upper layer in an eclipsed and staggered configuration, while two acceptor nitrile groups were attached to the lower layer. The effective separation of electron densities induced by the cyclophane bridge significantly reduced the overlap between the HOMO and LUMO (Figure 8g-h). This resulted in very low calculated ΔEST values of 20 meV for 61 and 62. Compared to 62, which possessed a low quantum yield, 61 demonstrated a significantly enhanced quantum yield and an extended delayed lifetime of up to 1.9 μs. This work demonstrated that the development of new highly efficient TADF molecules based on this molecular 3D platform should be possible by a careful selection of the type, orientation, and contact site of the D and A units.
3. Strategies for Fluorescence Lifetime Control of Quantum Dots
3.1. CdS and CdSe Quantum Dots
Auger nonradiative recombination, as the predominant pathway for nonradiative recombination of multicarrier states, refers to the process in which an electron–hole pair annihilates, transferring its energy to a third carrier. Peng and co-workers developed a range of CdSe-based core/shell quantum dots with optimized optical properties for single excitonic states and employed them to study the Auger nonradiative recombination rate.52 Currently, two strategies have been employed for controlling the Auger nonradiative process: (1) tuning the core size and/or shell thickness/shape to manipulate wave function overlap, conduction- and valence-band mixing, and dielectric screening; and (2) refining the core/shell interface to regulate the intensity of the intraband transition. For single-exciton emission, the photoluminescence (PL) lifetime of 63 (Figure 10a) (20.3 ns) exhibited a notable decrease compared to that of 64 (Figure 10b) (24.5 ns). This could be attributed to the increased overlap of electron–hole wave functions within 63. As the excitation power was elevated, higher-order excitons would be generated within the QDs. The presence of additional radiative recombination pathways and the occurrence of Auger nonradiative recombination contributed to the shorter PL lifetime observed in these higher-order excitonic states compared to their corresponding single exciton counterparts. Experimental results indicated that the influence of interfacial potential smoothness on the dynamics of Auger recombination was limited. In contrast, substantial modulation of the Auger nonradiative recombination rate and the PL lifetime could be achieved by adjusting the wave function delocalization of the hole or electron states within the QDs.
Figure 10.
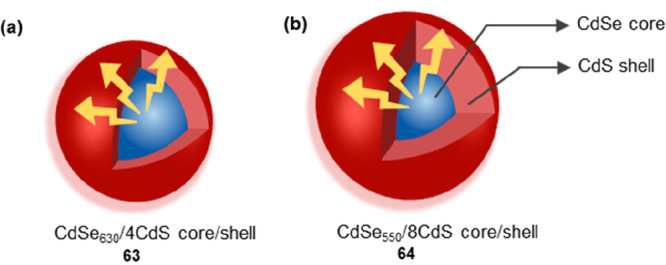
Scheme of 63 and 64. The CdS shell (bule) is distributed within the interior, while the CdSe core (red) is distributed on the exterior. (a) 63 with increased overlap of electron–hole wave functions. (b) 64 with decreased overlap of electron–hole wave functions.
When studying electrochemically stable electroluminescent quantum dots, Peng and co-workers observed that the fluorescence lifetimes of 65 and 66 (Figure 11a, 65–66) functionalized with cadmium-carboxylate ligands and fatty amine ligands showed no significant difference.53 This observation indicated that the positive or negative charges of the ligands have negligible impact on the fluorescence lifetime of CdSe/CdS core/shell QDs (Figure 11b,c). Later, they successfully engineered trap-insensitive QDs with long PL lifetimes in aqueous environments by the epitaxy of ZnS shells into an inorganic structure (CdSe/CdS/ZnS core/shell/shell structure) (Figure 11d).54 Upon photoexcitation, electron–hole pairs underwent both radiative and nonradiative recombination processes within QDs. The radiative process referred to the recombination of electrons and holes with the emission of photons, resulting in fluorescence emission. However, holes can be trapped in defects or localized energy levels within QDs, forming “hole traps”, leading to the formation of stable energy states. These hole traps can influence the recombination process of electrons and holes, causing an increase in the nonradiative process and a reduction in the fluorescence lifetime. The epitaxial ZnS shells with significantly wide bandgap play a crucial role in effectively isolating surface traps from the photogenerated excitons within QDs. As a result, 67 (Figure 11e–g) with three monolayers of ZnS shell exhibited longer lifetimes (36.3 ns) in water.
Figure 11.

PL lifetime modulation of CdSe/Cds-type QDs by charged ligands and traps. (a) Scheme of 65 and 66. Ligand of 65 with cadmium-carboxylates (with a small amount of negatively charged carboxylates). Ligand of probe 45 with primary amines. (b) Absorption and PL spectra of 65 and 66. (c) PL lifetime of 65 and 66. (d) Scheme of three types of QDs with three monolayers of ZnS outer shells. (e–g) PL spectra of three types of QDs before (black) and after (red) transfer from toluene to water. (a–c) Reprinted with permission from ref (53). Copyright 2020, Nature. (d–g) Reprinted with permission from ref (54). Copyright 2020, American Chemical Society.
Because of the finite physical size of QDs, if surface defects resided energetically within or near the bandgap, they function as electronic traps to effectively compete with the radiative recombination of excitons (electron–hole pairs) generated by either photoexcitation or electroexcitation within a nanocrystal. Peng and co-workers investigated the impact of surface defects on the fluorescence lifetime of QDs with different chemical terminations.55 Two types of chemically terminated CdSe/CdS QDs were prepared: 68 (Figure 12a) terminated with cadmium ions coordinated with carboxylate ligands and 69 (Figure 12a) terminated with sulfur. For 68 terminated with cadmium ions coordinated with carboxylate ligands, the fluorescence lifetime (28 ns) was shorter compared to sulfur-terminated 69. This phenomenon was attributed to the presence of more surface electron traps in 68, leading to increased nonradiative transitions and consequently shorter fluorescence lifetimes. Conversely, 69 exhibited a relatively longer fluorescence lifetime (>78 ns) due to its more intact surface and fewer defects. This enhancement can be ascribed to the formation of delocalized surface electronic bands originating from all sulfur sites located on the basal planes of the CdSe/CdS QDs, instead of the discrete anionic surface sites acting as deep hole traps within the bandgap.
Figure 12.

PL lifetime modulation by surface defects and confining of QDs. (a) Scheme of 68 and 69. Delocalized surface electronic states of 69 enhanced its PL lifetime. (b) Scheme of 70. Strongly confined CdSe/CdS with decreased thickness of CdS can have shortened lifetime. (a) Reprinted with permission from ref (55). Copyright 2020, American Chemical Society.
The wave function of a QD was a quantum mechanical concept that described the spatial and momentum-related information on electrons confined within the quantum dot. When the wave function was spatially localized (compact), it resulted in a reduced energy gap between the excited and ground states, leading to enhanced probabilities for the electron to decay from the excited to the ground state as well as an increased possibility of nonradiative transitions, both of which contributed to the shortened fluorescence lifetime. Conversely, when the wave function was spatially delocalized (expanded), the energy difference between the ground and excited states in quantum dots becomes less well-defined, resulting in reduced transition probabilities. In such cases, the fluorescence lifetime of QDs was typically longer, as electrons tended to remain in the excited state for a more extended period due to the decreased likelihood of transitions back to the ground state. Peng and co-workers investigated the processes of weakly confined and cubic-shaped cadmium (CdSe, CdS, and CdSe/CdS core/shell) nanocrystals.56 It was observed that with an increasing thickness of CdS shells in 70 (Figure 12b), the delocalization of electron wave functions gradually extended into the CdS shell, resulting in an increase in the PL lifetime of approximately 200 ns.
3.2. CdTeSe Type Multi-Component Quantum Dots
The fluorescence lifetimes of single QDs generally ranged from 10 to 40 ns.57 CdTeSe/ZnS QDs exhibited a shorter fluorescence lifetime of 31 ns.58 CdSeTe QDs synthesized in an oil phase demonstrated a slightly longer fluorescence lifetime of 30–40 ns.59 In 2020, Elibol synthesized CdSeTe alloy QD by the organometallic method.60 The QDs were subsequently treated with chloride solutions of varying mass% (m/m) to enhance their PL lifetime. The treatment of CdSeTe (71) alloy QDs with CdCl2 solutions (72) resulted in an increase in the PL lifetime from 28 to 33 ns (Figure 13a,b). Chloride passivation balanced the charge of cation-rich nanocrystals and was compact enough to fill cation spers. This enhancement can be attributed to the reduction of surface traps induced by long ligands and steric barriers on the QD surfaces, leading to a decrease in nonradiative recombination processes.
Figure 13.

Effects of ion exchange and alloying on the fluorescence lifetime of QDs. (a) Scheme of 71 and 72. The treatment of CdSeTe alloy QDs (71) with CdCl2 solutions (72) resulted in an increase in the PL lifetime. (b) Lifetime measurement of 71 and 72. (c) Scheme of 73 and 74. (d) Lifetime measurement of 73 and 74. (b) Reprinted with permission from ref (60). Copyright 2020, Elsevier. (d) Reprinted with permission from ref (61). Copyright 2016, Royal Society of Chemistry.
Ternary AgInS2 QDs exhibited favorable properties as cadmium-free luminescent bioprobes, offering red-shifted emission, tunability, and efficient Stokes and anti-Stokes excitations as well as long luminescence lifetimes. Strek and co-workers developed ternary AgInS2 QDs with a longer fluorescence lifetime greater than 100 ns.61 The fluorescence lifetime of AgInS2 QDs was significantly influenced by the Auger effect in nonradiative recombination due to its strong correlation of electrons and large enough charge imbalance. Alloying the QDs with the Zn2+ ions can alter the electronic structure and band structure of the quantum dots, consequently influencing the fluorescence lifetime. The maximum average luminescence lifetime (τave) for 73 (AgInS2) and 74 (AgInS2/ZnS) were estimated to be approximately 120 and 550 ns, respectively (Figure 13c,d).
The core/shell quantum dots have demonstrated an extended fluorescence lifetime due to the tunable wave function delocalization.62 Pham and co-workers investigated the 75 (CdTe/CdTe0.5Se0.5/CdSe core/alloyed/shell) (C/A/S) type-II structure and compared it with CdTe core and CdTe/CdSe core/shell (C/S) QDs (Figure 14a).63 The modulation of the core/shell composition and structure held the potential to enhance the fluorescence lifetime. They made adjustments to the conventional C/S structure by confining the holes within the CdTe core while concentrating the electrons in the CdSe shell, separated by an alloy CdTeSe. With the excessive growth of the shell on the CdTe core, the electrons began to delocalize between the shell and the alloy, attributed to the size effect. The enhanced wave function delocalization resulted in an increase in fluorescence lifetime of the QDs with the C/A/S structure, reaching 51.59 ns, more than twice the fluorescence lifetime of the C/S sample (24.22 ns) (Figure 14b).
Figure 14.

Fluorescence lifetime modulation by alloying of QDs. (a) Scheme of 75. (b) Lifetime measurement of three types of alloyed. The C/A/S structure was regulated by a medium amount of Te. (c) Fluorescence lifetime of CdTe1–xSex QDs with Se content (x) changed. (b) Reprinted with permission from ref (63). Copyright 2019, Elsevier. (c) Reprinted with permission from ref (64). Copyright 2017, Elsevier.
Alloy quantum dots (QDs) provide additional degrees of freedom for tailoring the desired properties of quantum dots due to their dependence on size and composition. The physical and optical characteristics of these QDs can be optimized accordingly. Water-soluble ternary-alloyed CdTe1–xSex QDs with longer lifetime between 80 and 174.64 ns were synthesized in the aqueous phase by Chen’s group.64 The fluorescence lifetime of alloyed QDs in CdTe1–xSex with different compositions was influenced by the reactant components. Increasing the Se content led to a reduction in the fluorescence lifetime of the alloyed QDs (Figure 14c). This can be attributed to the substitution of larger Te atoms with smaller Se atoms, resulting in an enhanced plasmonic coupling effect at smaller interlattice fluorophore distance.
3.3. Other Alloyed Quantum Dots
To overcome the challenges in controlling the size of binary nanocrystals, alloyed nanocrystals have been developed to achieve an intended size control by adjusting the Se/Te ratio. Compared to undoped QDs, doped alloyed QDs exhibited several exceptional properties, including photothermal stability, tunable dual-emissive behavior, and extended fluorescence lifetime. Transition-metal-doped quantum dots (TM-doped QDs) have received significant interest as important semiconductor nanomaterials in recent years. The introduction of transition-metal ions, such as Mn, Mg, and Cu, into the core or shell allowed for the tuning of photoluminescence lifetime of QDs. Four types of C/S heterostructures have been classified based on the features of band alignment and carrier localization, including type-I, reverse type-I, type-II, and quasi-type-II. Among them, type-II C/S heterostructures exhibit superior optical properties due to the staggered band alignment. This property resulted in spatially separated charge carriers between the core and shell regions, leading to the formation of indirect excitons.65
Doped C/S heterostructures exhibited dual emission, wherein one emission originates from the near band-edge excitonic transition, and the other arose from impurities and/or defects. The peak position was strongly dependent on dopant concentration and the size of core and shell parts. Doped CdS/ZnSe type-II core/shell QDs can be classified into two categories: one involved Cu doping in the CdS core, while the other involved Cu doping in the ZnSe shell. In 2015, Yang and co-workers synthesized CdS core-ZnSe shell QDs with a Cu-doped CdS core (76).66 This core–shell structure showed dual emission consistent with doped photoluminescence (D-PL) and type-II photoluminescence (T-PL); the former was attributed to Cu:CdS cores (d-dots), and the latter resulted from the CdS–ZnSe type-II core–shell structure (Figure 15a). The lifetime of the Cu dopant was associated with the Cu energy level and should be less coupled with lattice vibrations in comparison with the excitonic emission of undoped QDs. Due to the doping of Cu, the lifetime of D-PL (τ = 317.3 ns) was significantly longer than that of T-PL (τ = 180 ns). Ca and co-workers fabricated Cu-doped CdS/ZnSe core/shell type-II QDs, in which Cu was doped in the CdS core (77) or ZnSe shell (78) part (Figure 15b).67 The Cu2+-related emission was dominant with the average lifetimes longer than those of the undoped QDs. The Cu doping in both the CdS core and ZnSe shell of the C/S QDs significantly increased the PL lifetime, reaching a maximum value of τ = 295.62 ns (Figure 15c). The doping of Cu resulted in an increased PL lifetime, attributed to the combined effects of surface states, lattice strain, and spatial overlap between the conduction band electron wave functions and localized Cu states. Hien and co-workers found that, compared to the CdTe1–xSex host, Cu-doped CdTe1–xSex QDs have much longer PL decay times, up to ∼1.8 μs.68 The average lifetime showed a significant enhancement ranging from 9- to 18-fold when the concentration of Cu is increased from 1% to 10% (Figure 15d). The increase in the lifetime can be attributed to the localization of deeper states, which resulted in weaker overlap between the hole and electron wave functions.
Figure 15.

PL lifetime modulation by metal doping. (a) (left) Schematic of band alignments of bulk Cu-doped CdS, ZnSe, and ZnS. (right) Cu-doped CdS core–ZnSe, shell–ZnS, and shell QDs in the form of d-core–shell–shell structure (76) with dual emissions under excitation. (b) Band alignment of bulk CdS and ZnSe together with the excitonic and Cu-related emissions for 77 and 78. (c, d) Normalized decay curves of some typical Cu-doped QDs. (e) PL decay curves of Mn:ZnSe d-dots with different amounts of Mn2+ ions. (f) The relationship between PL lifetime and Mn2+ concentration. (a) Reprinted with permission from ref (66). Copyright 2015, Royal Society of Chemistry. (b, c) Reprinted with permission from ref (67). Copyright 2019, Elsevier. (d) Reprinted with permission from ref (68). Copyright 2019, Elsevier. (e, f) Reprinted with permission from ref (69). Copyright 2015, American Chemical Society.
Dopant emission typically arose from the d-shell orbitals of intentionally incorporated ions within the lattice of a host semiconductor nanocrystal. This phenomenon was observed in Mn2+-doped ZnSe nanocrystals (Mn: ZnSe d-dots), where the emission mechanism was intricately governed by the intricate interplay of the 3d orbitals of Mn2+ ions. Peng and co-workers synthesized stable Mn: ZnSe QDs with single-exponential PL decay and large tunable PL lifetime.69 By adjusting the quantity of Mn2+ ions per dot in Mn2+-doped ZnSe nanocrystals (referred to as Mn: ZnSe d-dots) from 500 to 20, the decay lifetime of the PL exhibited a continuous variation from around 50 to 1000 μs (Figure 15e,f). In the event of the presence of MnSe cluster(s) in a QD, it would lead to the emergence of a considerably short component of PL decay lifetime, coupled with a complicated PL decay.
The photoluminescence lifetime was a critical parameter that describes the excited-state lifetime of QDs and directly impacted their performance in optical devices and bioimaging applications. The presence of oxygen can significantly influence the excited-state lifetime of individual QDs. The impact of oxygen on the PL behavior can be regarded as a chemical reaction between the QDs and oxygen. The Peng group further investigated the influence of oxygen on the PL of conventional CdSe/CdS core/shell QDs. QDs in an electrically neutral state emit photons continuously creating a bright state in the PL emission.70 However, when the electrons or holes of excitons interacted with the surrounding environment and were lost, the QDs become charged. The excitation of charged QDs led to the formation of a “trion” state, consisting of two electrons and one hole or one electron and two holes. The efficient Auger nonradiative recombination in the trion state (dim state) significantly reduced the fluorescence lifetime. The analysis of Sionnest and co-workers suggested that the rapid decay component was primarily attributed to the PL of charged QDs in the trion state, while the slower decay component was predominantly associated with the PL of neutral QDs in the single-exciton state.71 When the charged quantum dots neutralize again, they re-emit photons and return to the single-exciton state (bright state). In the case of a photoreduced QD, the absorption of a photon resulted in the formation of a negative trion state comprising two electrons and one hole, instead of a single-exciton state. In an oxygen environment, CdSe/CdS core/shell quantum dots primarily existed in the single exciton state after optical excitation, with a photoluminescence lifetime of 22 ns. However, in an inert gas environment, the QDs exhibited a shorter photoluminescence lifetime of 2 ns due to the formation of a trion state (dark state) upon illumination. Oxygen, functioning as a strong electron acceptor, reacted with the negatively charged QDs, removing the extra electron and restoring the QD to a neutral state. This process facilitated the recovery of PL properties and enhanced the lifetime.
4. Strategies for Fluorescence Lifetime Control of a Metal Complex
4.1. Fluorescence Lifetime Control of Cu Complexes
The presence of a fully filled d10 electronic configuration in Cu(I) results in a symmetrical distribution of electron density around the Cu center. This electronic configuration strongly favors the adoption of a flexible pseudotetrahedral geometry by the Cu complexes.72 Cu(I) diimine complexes in the metal-to-ligand-charge-transfer (MLCT) excited state exhibit a structural reorganization phenomenon (Figure 16a). This transformation involves a change from the pseudotetrahedral D2d symmetry in the ground state to a flattened D2 symmetry in the MLCT state. This structural change enables the coordination of a solvent molecule, leading to the formation of an exciplex intermediate. Generally, in the process of MLCT, the excited state induced by the metal–ligand charge transfer exhibits a lower spin multiplicity, thereby reducing the likelihood of nonradiative decay and resulting in a comparatively longer fluorescence lifetime.
Figure 16.

Excitation lifetime modulation by ligands. (a) The mechanism of MLCT. The exceptionally long radiative lifetimes in metal complexes arise from intricate and extended processes of excited-state energy-level transitions: complexes: internal conversion (IC), intersystem crossing (ISC), Franck–Condon (FC), metal-to-ligand charge transfer (MLCT), nonradiative deactivation (NRD), pseudo-Jahn–Teller distortion (PJT distortion), reverse intersystem crossing (RISC), absorption (abs), fluorescence (FL), flattened geometry (flat), phosphorescence (PH). (b) Structures of Cu complexes.
In 2021, Chen et al. investigated the possibility of manipulating the excited-state properties by constraining the distortion of the angle between the two ligand planes from the orthogonal to flattened conformation, preventing the solvent accessibility to the Cu center.73 In Cu(I) bis-phenanthroline complexes, which are a subset of Cu(I) diimine complexes, the introduction of substituents at the 2 and 9 positions of the phenanthroline ligands exerted a significant impact on the structural characteristics of the MLCT state. The energy gap between the singlet and triplet states was inversely proportional to the decrease in the angle φ between the two ligand planes. Consequently, a reduction in the angle φ leads to a decrease in the energy gap between the singlet and triplet states. The decrease in the energy gap was associated with a corresponding increase in the fluorescence lifetime. In 79 (Figure 16b, 79), the presence of methyl groups at the 2 and 9 positions offered a degree of shielding against ligand interactions with the Cu center, resulting in a longer excited-state lifetime of approximately 100 ns. Due to the relatively smaller size of the methyl groups compared to tert-butyl groups (80), they induce only minor van der Waals steric repulsions (Figure 16b, 80). Consequently, this molecule underwent significant flattening distortions upon excitation, leading to a notable singlet–triplet energy gap of 2950 cm–1.
Cu complexes with MLCT properties exhibit structural distortions in the excited state. Viscosity-responsive probes are molecular sensors that undergo twisting or rotation around specific chemical bonds to adapt to changes in environmental viscosity. These probes display distinct photophysical properties in different viscosity media. Hence, there was a hypothesis that Cu complexes in the excited state may also be influenced by the viscosity of the surrounding environment, thereby affecting their fluorescence lifetimes. In 2017, Ostrowski and co-workers investigated the correlation between the excited-state lifetime of 79 in various supramolecular hydrogen-bonded polymer systems and their mechanical properties within a certain viscosity range.74 They explored the behavior of these polymer systems under different viscosity conditions and analyzed the relationship between the excited-state lifetime and the mechanical performance of the systems. The lifetimes of 79 were measured in H-bonding polymer P2 materials, yielding an average value of 210 ± 4 ns. This lifetime was approximately 3.5 times longer than that observed in an aerated dichloromethane (DCM) solution. They further observed that polymers with higher viscosity resulted in restricted Jahn-Teller (J-T) distortion in the excited state, leading to an extended MLCT lifetime of 79. Consequently, a linear correlation was found between the excited-state lifetime of 79 and the logarithm of the viscosity of the surrounding polymer matrix. To enhance the fluorescence imaging performance of Cu(I) complexes, one can employ Cu(I) complexes with increased emission quantum yield and greater steric hindrance in a more crowded environment.
Cu(I) complexes that possess an appropriate ligand environment have the potential to demonstrate prolonged excited states and vibrant luminescence. In 2018, by selecting two phenanthroline ligands without substituents in the 2,9 positions (phen, Bphen), Armaroli et al. systematically investigated the electronic and structural properties of ten highly stable and luminescent [Cu(NN)(PP)]+ complexes, where PP indicates five different bis-phosphine ligands.75 Homoleptic and heteroleptic Cu(I) compounds lacking substituents in the 2,9 positions of phenanthroline exhibit relatively short excited-state lifetimes in solution. This was attributed to the inability of these ligands to effectively prevent the photoinduced distortion of the Cu(I) tetrahedral complexes, leading to square planar excited-state geometries that promote nonradiative deactivations. Theoretical calculations have demonstrated the crucial influence of the P–Cu–P bite angle and intramolecular π-stacking interactions in governing the light absorption and emission characteristics. For the highly rigid binuclear complex 81 (Figure 16b, 81), it exhibited minimal planar distortion in the excited state, with the P–Cu–P bite angle remaining nearly unchanged. Additionally, strong intermolecular and intramolecular π-stacking interactions were present. These factors contributed to a reduced distribution of triplet-state lifetimes and an increase in fluorescence lifetimes.
In 2014, Herres-Pawlis and his team developed a series of bis(chelate) Cu(I) and Cu(II) guanidine–quinoline complex cations [CuII(TMGqu)2]+ and 82 (Figure 16b, 82).76 These compounds demonstrated a remarkable characteristic where their structures exhibited close similarities with a coordination polyhedron lying between tetrahedral and square-planar environments. They found that the coordination geometry was not solely determined by solid-state effects but rather arose from an inherent balance between electronic and spatial factors. Cu(II/I) complexes undergo MLCT and ligand-to-metal charge-transfer (LMCT) processes, resulting in resonance occurring at approximately the same energy. The resonance involved the coupling of Cu-Ngua stretching and chelate angle torsion with MLCT from CuI to CuII and LMCT from CuII to CuI. Therefore, the concept of ligand restrictions imposed upon Cu coordination provides valuable guidance to the generation of electron transfer models. Based on these findings, they utilized guanidine and quinoline in ligand design to achieve a constrained geometric shape that allowed only minor structural changes upon photoexcitation, resulting in very short MLCT lifetimes.77 The donor interplay between guanidine and quinoline units, along with the steric hindrance of guanidine, played a vital role in achieving the constrained coordination.
As research on dipyridyl ligands in Cu complexes continues to advance, the diverse range of heteroleptic diimine-diphosphine Cu(I) complexes has highlighted their advantageous role in modulating optical properties.78 In the past, two main optimization strategies have been employed to overcome the limitations of copper complexes in terms of their visible light absorption capacity. The first approach involves the introduction of bridging ligands or carbene ligands to enhance the absorption capability through π-conjugation.79 The second approach is focused on introducing fused aromatic rings into the diimine backbone, leading to a redshift in the absorption peak and an extension of the π-electron system.80 However, these methods can have a negative impact on the oxidation–reduction potentials and excited-state lifetimes of Cu(I) complexes. In 2021, Tschierlei et al. expanded the π-electron system of 1,10-phenanthroline by selectively introducing various types of phenyl and alkynyl substituents at the 5 and 6 positions.81 They employed a diverse range of substituents to investigate the effects of electron-withdrawing and electron-donating groups, as well as different spatial requirements, on the fluorescence lifetimes of Cu(I) complexes. Phenyl-based substituents have a significant impact on the emission lifetime of Cu(I) complexes. Specifically, both phenyl-based substituents (83) and para-fluorophenyl-based substituents (84) contribute to an extended emission lifetime of Cu(I) complexes, reaching up to 504 ns (Figure 16b, 83–84). With an increase in steric bulk, the longer lifetime decreases while the shorter one increases. This observation suggested that a significant portion of the excited state was likely localized on the distal substituents positioned at the 5,6 positions of the diimine ligands. The absence of steric bulk potentially enabled greater orbital overlap between the diimine unit and the substituent as well as less-hindered rotation around the substituent axis in the excited state.
It has been established that the spatial strain around the Cu(I) ion in Cu(I) complexes with α-substituted diimine ligands significantly influences the properties of the excited state. In general, larger substituents lead to longer emission lifetimes and higher quantum yields. Nevertheless, the stability of the coordination scaffold may be compromised if the steric strain induced by R becomes too significant. Pellegrin et al. investigated a strategy to finely adjust the steric strain around Cu(I) in order to achieve a delicate balance between a high emission quantum yield and stability in a copper(I) complex with substantial bulkiness.82 They devised and characterized the unsymmetrical ligand 2-isopropyl-9-tert-butyl-1,10-phenanthroline (L1) along with its corresponding complex 85 (Figure 16b, 85). The main experimental observations revealed that 85 adopted a rigid tetrahedral geometry in the ground state, closely resembling that of 80, while exhibiting intermediate stability between 86 (Figure 16b, 86) and 80. However, due to the nonsymmetrical nature of ligand L1, the emission lifetime of 85 (127 ns) was comparatively shorter and smaller than that of both 85 (342 ns) and 80 (3260 ns).
4.2. Fluorescence Lifetime Control of Other Metal Complexes
Three-coordinate pyridine derivatives, such as terpyridine ligands, play a crucial role in metal coordination chemistry. Their unique structure, characterized by three nitrogen atoms binding to a central metal ion, allows for the formation of stable and selective metal complexes, enabling diverse applications in a luminous metal complex.83 From a structural perspective, similar to terpyridine ligands, acridine is also an N-heterocycle with a π-conjugated planar structure. Hence, it can serve as a ligand for metal coordination complexes, offering potential applications in catalysis, sensing, and materials science. However, despite the ability of acridines to form coordination complexes with diverse metal ions, their coordination affinity is relatively weak, and their selectivity or versatility in binding to specific metal ions is limited. These drawbacks hinder their potential applications as fluorescent probes. In 2022, Qian et al. incorporated an acridine unit into a terpyridine (TPy) framework to generate a novel acridine-TPy bidentate ligand (AcTPy).84 This design was motivated by the fact that the N-heteroaromatic ligand [2,2′:6′,2″] TPy possessed a convergent N,N′,N′′-chelating donor group, enabling robust coordination interactions with a wide range of transition-metal ions. The zinc–metalated AcTPy (87) complexes exhibited yellow fluorescence with luminescent emission bands ranging from 500 to 570 nm and a fluorescence lifetime of 4.37 ns, distinguishing them from other metal ions (Figure 17, 87). Due to the protonation reaction of the N-heterocycle of acridine, the fluorescence lifetime of 87 increased under acidic conditions.
Figure 17.

Structures of other metal complexes. The special ligand of compound 87 consisted of a terpyridine linked to an acridine unit.
Transition-metal ions can be doped into the large caves of g-C3N4 by coordination bonding with the negatively charged nitrogen atoms in the g-C3N4 particles. In 2020, Qian et al. developed graphite-like carbon nitride (g-C3N4)-based nanocomposites with zinc terpyridyl (ZnBTPy) coordination polymers (CPs).85 Following the surface modification of O-g-C3N4 nanocores with P68, a slight decrease in the fluorescence lifetime was observed (Figure 17, 88). The reduction in emission lifetime can be attributed to the facilitated transfer of excited charge carriers (electrons) from the conductive band of O-g-C3N4 to the excited state of the ZnBTPy CPs.
It is demonstrated that the energy difference between the first singlet and triplet excited states of thioxanthone (TX) was less than 0.3 eV.86 This characteristic facilitated the reverse intersystem crossing process from the triplet state to the singlet state, holding great potential for the advancement of highly efficient luminescent emitters. The characterization of the first singlet excited-state lifetimes and intersystem crossing rates provide valuable insights into the excitation behavior of these metal complexes, thereby aiding in the understanding of the distribution of their fluorescence lifetimes.87 In 2019, Qian et al. have demonstrated photophysical properties of new TX-polypyridyl photoinitiators and their metal complexes in various solutions.88 The rapid and reversible intersystem crossing rate in this system may be affected by the energy-level relationship between the T1-TX and S1-TX states, particularly in high polar solvents like methanol. In such solvents, where the energy level of the T1-TX state could exceed that of the S1-TX state, a decreased energy transfer efficiency from the S1-TX to T1-TX state was anticipated, leading to reduced fluorescent emission from the Ln3+ ions and increased fluorescent emission from the TX aromatic rings. The short fluorescence lifetime for the TXs and their Zn/Fe/Ni-complexes was in the range of 0.21–1.12 ns, in agreement with previous reports.89,90 The fluorescent emission decay of the central Ln3+ ions in the Ln-TXOBPy and Ln-TXOTPy complexes was measured, and the estimated emission lifetime ranged from 0.3 to 0.8 ms. Due to the energy transfer to Ln3+ ions via intersystem crossing, the emission lifetime of Ln3+ ions was typically significantly longer compared to that of commonly employed organic dyes.
5. Summary and Outlook
Fluorescence lifetime was defined as the average time the molecule spends in the excited state. Importantly, as a crucial characteristic in fluorescence systems, its value is not affected by concentration. Different fluorophores have distinct fluorescence lifetimes, which makes fluorescence lifetime have broader potential applications in imaging. Fluorescence lifetime modulation can broaden the range of fluorescence lifetimes for fluorophores, thereby promoting the development of fluorescence lifetime imaging. This review provides a comprehensive summary and analysis of fluorescence lifetime modulation techniques for small molecules, quantum dots, and metal complexes, primarily focusing on the control of nonradiative pathways of fluorophores. However, due to the diverse characteristics and modulation mechanisms of different types of fluorophores, it is essential to comprehensively consider their structures and fluorescence emission mechanisms. For instance, the fluorescence lifetime of small molecules and metal complexes closely correlates with the conformational changes in their excited states, while the fluorescence lifetime of quantum dots is influenced by parameters associated with carrier recombination efficiency. At present, there is limited research on fluorescence lifetime modulation, leaving considerable scope for exploring the chemical principles underlying fluorescence lifetimes. The investigation of fluorescence lifetime modulation offers a more comprehensive understanding of the changes in conformation and electron distribution associated with electronic energy-level transitions. Accurate fluorescence lifetime modulation can yield a wide range of fluorescence lifetimes, contributing to the advancement of multicomponent imaging techniques. With the continuous advancement of technology and profound research, fluorescence lifetime modulation holds a highly promising future in the field of fluorescence imaging. Subsequent investigations will further explore additional strategies and approaches for fluorescence lifetime modulation, aiming to achieve more precise and efficient imaging and detection. Future studies are expected to explore additional strategies and methodologies for achieving more precise and efficient imaging and detection.
Acknowledgments
This work is partially supported by the Research Center for Industries of the Future (RCIF) at Westlake University (X.Z.).
Glossary
Vocabulary
- fluorescence lifetime
The average time the molecule spends in the excited state.
- nonradiative process
Transitions between energy levels which are not associated with the emission.
- small molecule
A low molecular weight organic compound.
- quantum dot
Semiconductor particle a few nanometers in size.
- metal complex
A coordination complex consisting of a central metal atom or ion and a surrounding array of bound molecules or ions known as ligands or complexing agents.
Biographies
Jianan Dai received her M.S. from Nanjing Normal University in 2022. Now she is a doctoral student at Westlake University. Her current research interests focus on BODIPY-based molecular rotors to provide insights into viscosity changes of organelles.
Xin Zhang is the Professor of Chemistry and Cell Biology at the Westlake University, Hangzhou. Prior to joining the faculty at Westlake, Zhang was the Paul & Mildred Berg Early Career Professor and associate professor of chemistry and of biochemistry and molecular biology at the Pennsylvania State University. Zhang’s research is focused on the chemistry of biological aggregates formed by proteins and RNAs under physiological and pathological conditions. Zhang’s independent work has received multiple honors and awards, including the Priestley Prize for undergraduate teaching in chemistry, Kavli Fellow of the National Academy of Sciences USA, CAPA Distinguished Junior Faculty Award, NSF CAREER award, NIGMS MIRA, Pew Scholar in the Biomedical Sciences, Scialog Fellowship, Sloan Research Fellowship, the Lloyd and Dottie Huck Early Career Award, and the Burroughs Wellcome Fund Career Award at the Scientific Interface.
Author Contributions
Jianan Dai writing-original draft, writing-review and editing; Xin Zhang funding acquisition, supervision, writing-review and editing.
The authors declare no competing financial interest.
References
- Chen L.-C.; Lloyd W. R.; Chang C.-W.; Sud D.; Mycek M.-A. Fluorescence Lifetime Imaging Microscopy for Quantitative Biological Imaging. Methods Cell Biol. 2013, 114, 457–488. 10.1016/B978-0-12-407761-4.00020-8. [DOI] [PubMed] [Google Scholar]
- Frei M. S.; Tarnawski M.; Roberti M. J.; Koch B.; Hiblot J.; Johnsson K. Engineered HaloTag Variants for Fluorescence Lifetime Multiplexing. Nat. Methods 2022, 19 (1), 65–70. 10.1038/s41592-021-01341-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei M. S.; Koch B.; Hiblot J.; Johnsson K. Live-Cell Fluorescence Lifetime Multiplexing Using Synthetic Fluorescent Probes. ACS Chem. Biol. 2022, 17 (6), 1321–1327. 10.1021/acschembio.2c00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKABAYASHI T.; OHTA N. Sensing of Intracellular Environments by Fluorescence Lifetime Imaging of Exogenous Fluorophores. Anal. Sci. 2015, 31, 275–285. 10.2116/analsci.31.275. [DOI] [PubMed] [Google Scholar]
- Rotkiewicz K.; Grellmann K. H.; Grabowski Z. R. Reinterpretation of the Anomalous Fluorescence of p-N,N-dimethylamino-benzonitrile. Chem. Phys. Lett. 1973, 19, 315–318. 10.1016/0009-2614(73)80367-7. [DOI] [Google Scholar]
- Lavis L. D.; Raines R. T. Bright Building Blocks for Chemical Biology. ACS Chem. Biol. 2014, 9 (4), 855–866. 10.1021/cb500078u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beija M.; Afonso C. A.; Martinho J. M. Synthesis and Applications of Rhodamine Derivatives as Fluorescent Probes. Chem. Soc. Rev. 2009, 38 (8), 2410–2433. 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]
- Zhang X.-F.; Zhang Y.; Liu L. Fluorescence Lifetimes and Quantum Tields of Ten Rhodamine Derivatives: Structural Effect on Emission Mechanism in Different Solvents. J. Lumin. 2014, 145, 448–453. 10.1016/j.jlumin.2013.07.066. [DOI] [Google Scholar]
- Grimm J. B.; English B. P.; Chen J.; Slaughter J. P.; Zhang Z.; Revyakin A.; Patel R.; Macklin J. J.; Normanno D.; Singer R. H.; Lionnet T.; Lavis L. D. A General Method to Improve Fluorophores for Live-cell and Single-Molecule Microscopy. Nat. Methods 2015, 12 (3), 244–250. 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. F.; Zhang J.; Lu X. The Fluorescence Properties of Three Rhodamine Dye Analogues: Acridine Red, Pyronin Y and Pyronin B. J. Fluoresc. 2015, 25 (4), 1151–1158. 10.1007/s10895-015-1610-5. [DOI] [PubMed] [Google Scholar]
- Lv X.; Gao C.; Han T.; Shi H.; Guo W. Improving the Quantum Yields of Fluorophores by Inhibiting Twisted Intramolecular Charge Transfer Using Electron-Withdrawing Group-Functionalized Piperidine Auxochromes. Chem. Commun. 2020, 56 (5), 715–718. 10.1039/C9CC09138F. [DOI] [PubMed] [Google Scholar]
- Mitronova G. Y.; Belov V. N.; Bossi M. L.; Wurm C. A.; Meyer L.; Medda R.; Moneron G.; Bretschneider S.; Eggeling C.; Jakobs S.; Hell S. W. New Fluorinated Rhodamines for Optical Microscopy and Nanoscopy. Chem.-Eur. J. 2010, 16 (15), 4477–4488. 10.1002/chem.200903272. [DOI] [PubMed] [Google Scholar]
- Savarese M.; Aliberti A.; De Santo I.; Battista E.; Causa F.; Netti P. A.; Rega N. Fluorescence Lifetimes and Quantum Yields of Rhodamine Derivatives: New Insights from Theory and Experiment. J. Phys. Chem. A 2012, 116 (28), 7491–7497. 10.1021/jp3021485. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Zhang G.; Wang J.; Sun S.; Zhang Z. The mechanisms of Large Stokes Shift and Fluorescence Quantum Yields in Anilino Substituted Rhodamine Analogue: TICT and PICT. Comput. Theor. Chem. 2016, 1095, 44–53. 10.1016/j.comptc.2016.09.017. [DOI] [Google Scholar]
- Berezin M. Y.; Achilefu S. Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 2010, 110, 2641–2684. 10.1021/cr900343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Jackson W. R.; Choi C. Y.; Bergmark W. R. Solvent Effects on Emission Yield and Lifetime for Coumarin Laser Dyes. Requirements for a Rotatory Decay Mechanism. J. Phys. Chem. 1985, 89, 294–300. 10.1021/j100248a024. [DOI] [Google Scholar]
- Wang C.; Jiang W.; Tan D.; Huang L.; Li J.; Qiao Q.; Yadav P.; Liu X.; Xu Z. Monitoring Amyloid Aggregation via a Twisted Intramolecular Charge Transfer (TICT)-Based Fluorescent Sensor Array. Chem. Sci. 2023, 14 (18), 4786–4795. 10.1039/D2SC06710B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Qiao Q.; Tian W.; Liu W.; Chen J.; Lang M. J.; Xu Z. Aziridinyl Fluorophores Demonstrate Bright Fluorescence and Superior Photostability by Effectively Inhibiting Twisted Intramolecular Charge Transfer. J. Am. Chem. Soc. 2016, 138 (22), 6960–6963. 10.1021/jacs.6b03924. [DOI] [PubMed] [Google Scholar]
- Sasaki S.; Drummen G. P. C.; Konishi G.-i. Recent Advances In Twisted Intramolecular Charge Transfer (TICT) Fluorescence and Related Phenomena in Materials Chemistry. J. Mater. Chem. C 2016, 4 (14), 2731–2743. 10.1039/C5TC03933A. [DOI] [Google Scholar]
- Ye Z.; Yang W.; Wang C.; Zheng Y.; Chi W.; Liu X.; Huang Z.; Li X.; Xiao Y. Quaternary Piperazine-Substituted Rhodamines with Enhanced Brightness for Super-Resolution Imaging. J. Am. Chem. Soc. 2019, 141 (37), 14491–14495. 10.1021/jacs.9b04893. [DOI] [PubMed] [Google Scholar]
- Wang C.; Chi W.; Qiao Q.; Tan D.; Xu Z.; Liu X. Twisted Intramolecular Charge Transfer (TICT) and Twists Beyond TICT: From Mechanisms to Rational Designs of Bright and Sensitive Fluorophores. Chem. Soc. Rev. 2021, 50 (22), 12656–12678. 10.1039/D1CS00239B. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xiao Y.; Qi J.; Qu J.; Kim B.; Yue X.; Belfield K. D. Long-Wavelength, Photostable, Two-Photon Excitable BODIPY Fluorophores Readily Modifiable for Molecular Probes. J. Org. Chem. 2013, 78 (18), 9153–9160. 10.1021/jo401379g. [DOI] [PubMed] [Google Scholar]
- Osmialowski B.; Zakrzewska A.; Jedrzejewska B.; Grabarz A.; Zalesny R.; Bartkowiak W.; Kolehmainen E. Influence of Substituent and Benzoannulation on Photophysical Properties of 1-Benzoylmethyleneisoquinoline Difluoroborates. J. Org. Chem. 2015, 80 (4), 2072–2080. 10.1021/jo502244j. [DOI] [PubMed] [Google Scholar]
- Zhang X.-F.; Su N.; Lu X.; Jia W. Benzoate-modified rhodamine dyes: Large Change in Fluorescence Properties due to Photoinduced Electron Transfer. J. Lumin. 2016, 179, 511–517. 10.1016/j.jlumin.2016.07.031. [DOI] [Google Scholar]
- Zhang X.-F.; Zhang Y.-K. Long-Lived and Largely Red-Shifted Photoluminescence of Solid-State Rhodamine Dyes: Molecular Exciton Coupling and Structural Effect. J. Lumin. 2015, 166, 215–221. 10.1016/j.jlumin.2015.05.048. [DOI] [Google Scholar]
- Cai X.; Liu B. Aggregation-Induced Emission: Recent Advances in Materials and Biomedical Applications. Angew. Chem., Int. Ed. Engl. 2020, 59 (25), 9868–9886. 10.1002/anie.202000845. [DOI] [PubMed] [Google Scholar]
- Mei J.; Leung N. L.; Kwok R. T.; Lam J. W.; Tang B. Z. Aggregation-Induced Emission: Together We Shine, United We Soar!. Chem. Rev. 2015, 115 (21), 11718–11940. 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- Liu S.; Wang B.; Yu Y.; Liu Y.; Zhuang Z.; Zhao Z.; Feng G.; Qin A.; Tang B. Z. Cationization-Enhanced Type I and Type II ROS Generation for Photodynamic Treatment of Drug-Resistant Bacteria. ACS Nano 2022, 16 (6), 9130–9141. 10.1021/acsnano.2c01206. [DOI] [PubMed] [Google Scholar]
- Sun H.; Chen S.; Zhong A.; Sun R.; Jin J.; Yang J.; Liu D.; Niu J.; Lu S. Tuning Photophysical Properties via Positional Isomerization of the Pyridine Ring in Donor-Acceptor-Structured Aggregation-Induced Emission Luminogens Based on Phenylmethylene Pyridineacetonitrile Derivatives. Molecules 2023, 28 (7), 3282. 10.3390/molecules28073282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Su H.; Kwok R. T. K.; Hu X.; Zou H.; Luo Q.; Lee M. M. S.; Xu W.; Lam J. W. Y.; Tang B. Z. Rational Design of a Water-Soluble NIR AIEgen, and Its Application in Ultrafast Wash-Free Cellular Imaging and Photodynamic Cancer Cell Ablation. Chem. Sci. 2018, 9 (15), 3685–3693. 10.1039/C7SC04963C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung J. C. K.; Forman A.; Jockusch R. A. The Effect of Methylation on the Intrinsic Photophysical Properties of Simple Rhodamines. Phys. Chem. Chem. Phys. 2019, 21 (20), 10261–10271. 10.1039/C9CP00730J. [DOI] [PubMed] [Google Scholar]
- Qiu S.; Zhang Z.; Wu Y.; Tong F.; Chen K.; Liu G.; Zhang L.; Wang Z.; Qu D.-H.; Tian H. Vibratile Dihydrophenazines with Controllable Luminescence Enabled by Precise Regulation of π-Conjugated Wings. CCS Chem. 2022, 4 (7), 2344–2353. 10.31635/ccschem.021.202101193. [DOI] [Google Scholar]
- Kotani R.; Liu L.; Kumar P.; Kuramochi H.; Tahara T.; Liu P.; Osuka A.; Karadakov P. B.; Saito S. Controlling the S(1) Energy Profile by Tuning Excited-State Aromaticity. J. Am. Chem. Soc. 2020, 142 (35), 14985–14992. 10.1021/jacs.0c05611. [DOI] [PubMed] [Google Scholar]
- Luo J.; Xie Z.; Lam J. W.; Cheng L.; Chen H.; Qiu C.; Kwok H. S.; Zhan X.; Liu Y.; Zhu D.; Tang B. Z. Aggregation-Induced Emission of 1-Methyl-1,2,3,4,5-Pentaphenylsilole. Chem. Commun. 2001, (18), 1740–1741. 10.1039/b105159h. [DOI] [PubMed] [Google Scholar]
- Sadowski B.; Su S.-H.; Lin T.-C.; Lohrey T. D.; Deperasińska I.; Chou P.-T.; Gryko D. T. The Influence of Tetraphenylethylene Moieties on the Emissive Properties of Dipyrrolonaphthyridinediones. J. Mater. Chem. C 2018, 6 (45), 12306–12313. 10.1039/C8TC03880E. [DOI] [Google Scholar]
- Yang Z.; Li X.; Yang K.; Yu N.; Gao R.; Ren Y. Synthesis and Unexpected Optical Properties of Ionic Phosphorus Heterocycles with P-Regulated Noncovalent Interactions. J. Org. Chem. 2023, 88 (5), 2792–2800. 10.1021/acs.joc.2c02424. [DOI] [PubMed] [Google Scholar]
- Szlapa-Kula A.; Małecka M.; Machura B. Insight Into Structure-Property Relationships of Aryl-Substituted 2,2′:6′,2″-Terpyridines. Dyes Pigm. 2020, 180, 108480. 10.1016/j.dyepig.2020.108480. [DOI] [Google Scholar]
- Belaidi H.; Rauch F.; Zhang Z.; Latouche C.; Boucekkine A.; Marder T. B.; Halet J. F. Insights into the Optical Properties of Triarylboranes with Strongly Electron-Accepting Bis(fluoromesityl)boryl Groups: When Theory Meets Experiment. ChemPhotoChem. 2020, 4 (3), 173–180. 10.1002/cptc.201900256. [DOI] [Google Scholar]
- Majumdar P.; Zhao J. 2-(2-Hydroxyphenyl)-Benzothiazole (HBT)-Rhodamine Dyad: Acid-Switchable Absorption and Fluorescence of Excited-State Intramolecular Proton Transfer (ESIPT). J. Phys. Chem. B 2015, 119 (6), 2384–2394. 10.1021/jp5068507. [DOI] [PubMed] [Google Scholar]
- Keshri S. K.; Mandal K.; Kumar Y.; Yadav D.; Mukhopadhyay P. Naphthalenediimides with High Fluorescence Quantum Yield: Bright-Red, Stable, and Responsive Fluorescent Dyes. Chem.-Eur. J. 2021, 27 (23), 6954–6962. 10.1002/chem.202100020. [DOI] [PubMed] [Google Scholar]
- dos Santos P. L.; Etherington M. K.; Monkman A. P. Chemical and Conformational Control of The Energy Gaps Involved in the Thermally Activated Delayed Fluorescence Mechanism. J. Mater. Chem. C 2018, 6 (18), 4842–4853. 10.1039/C8TC00991K. [DOI] [Google Scholar]
- Fang F.; Zhu L.; Li M.; Song Y.; Sun M.; Zhao D.; Zhang J. Thermally Activated Delayed Fluorescence Material: An Emerging Class of Metal-Free Luminophores for Biomedical Applications. Adv. Sci. 2021, 8 (24), e2102970 10.1002/advs.202102970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Sun Y.; Wang G.; Li J.; Li X.; Zhang K. TADF-Type Organic Afterglow. Angew. Chem., Int. Ed. Engl. 2021, 60 (31), 17138–17147. 10.1002/anie.202105628. [DOI] [PubMed] [Google Scholar]
- Dhali R.; Phan Huu D. K. A.; Bertocchi F.; Sissa C.; Terenziani F.; Painelli A. Understanding TADF: a Joint Experimental and Theoretical Study of DMAC-TRZ. Phys. Chem. Chem. Phys. 2021, 23 (1), 378–387. 10.1039/D0CP05982J. [DOI] [PubMed] [Google Scholar]
- Kim D. S.; Yoon S. J.; Lee K. H.; Lee J. Y.; Hong W. P. Starburst Type Benzofuroindolocarbazole Donor for High Efficiency and Long Lifetime in Thermally Activated Delayed Fluorescence Emitters. Adv. Opt. Mater. 2021, 9 (1), 2001432. 10.1002/adom.202001432. [DOI] [Google Scholar]
- Cui L. S.; Nomura H.; Geng Y.; Kim J. U.; Nakanotani H.; Adachi C. Controlling Singlet-Triplet Energy Splitting for Deep-Blue Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed. Engl. 2017, 56 (6), 1571–1575. 10.1002/anie.201609459. [DOI] [PubMed] [Google Scholar]
- Geng Y.; D’Aleo A.; Inada K.; Cui L. S.; Kim J. U.; Nakanotani H.; Adachi C. Donor-σ-Acceptor Motifs: Thermally Activated Delayed Fluorescence Emitters with Dual Upconversion. Angew. Chem., Int. Ed. Engl. 2017, 56 (52), 16536–16540. 10.1002/anie.201708876. [DOI] [PubMed] [Google Scholar]
- Kawasumi K.; Wu T.; Zhu T.; Chae H. S.; Van Voorhis T.; Baldo M. A.; Swager T. M. Thermally Activated Delayed Fluorescence Materials Based on Homoconjugation Effect of Donor-Acceptor Triptycenes. J. Am. Chem. Soc. 2015, 137 (37), 11908–11911. 10.1021/jacs.5b07932. [DOI] [PubMed] [Google Scholar]
- Tsujimoto H.; Ha D. G.; Markopoulos G.; Chae H. S.; Baldo M. A.; Swager T. M. Thermally Activated Delayed Fluorescence and Aggregation Induced Emission with Through-Space Charge Transfer. J. Am. Chem. Soc. 2017, 139 (13), 4894–4900. 10.1021/jacs.7b00873. [DOI] [PubMed] [Google Scholar]
- Shao S.; Hu J.; Wang X.; Wang L.; Jing X.; Wang F. Blue Thermally Activated Delayed Fluorescence Polymers with Nonconjugated Backbone and Through-Space Charge Transfer Effect. J. Am. Chem. Soc. 2017, 139 (49), 17739–17742. 10.1021/jacs.7b10257. [DOI] [PubMed] [Google Scholar]
- Auffray M.; Kim D. H.; Kim J. U.; Bencheikh F.; Kreher D.; Zhang Q.; D’Aleo A.; Ribierre J. C.; Mathevet F.; Adachi C. Dithia[3.3]paracyclophane Core: A Versatile Platform for Triplet State Fine-Tuning and Through-Space TADF Emission. Chem.-Asian J. 2019, 14 (11), 1921–1925. 10.1002/asia.201900401. [DOI] [PubMed] [Google Scholar]
- Hou X.; Li Y.; Qin H.; Peng X. Effects of Interface-Potential Smoothness and Wavefunction Delocalization on Auger Recombination in Colloidal CdSe-based Core/Shell Quantum Dots. J. Chem. Phys. 2019, 151 (23), 234703. 10.1063/1.5125940. [DOI] [PubMed] [Google Scholar]
- Pu C.; Dai X.; Shu Y.; Zhu M.; Deng Y.; Jin Y.; Peng X. Electrochemically-Stable Ligands Bridge The Photoluminescence-Electroluminescence Gap of Quantum Dots. Nat. Commun. 2020, 11 (1), 937. 10.1038/s41467-020-14756-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Shu Y.; Qin H.; Su B.; Peng X. Quantum Dots with Highly Efficient, Stable, and Multicolor Electrochemiluminescence. ACS Cent. Sci. 2020, 6 (7), 1129–1137. 10.1021/acscentsci.0c00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H.; Wang Y.; Liu S.; Zhu M.; Pu C.; Lin S.; Qin H.; Peng X. Delocalized Surface Electronic States on Polar Facets of Semiconductor Nanocrystals. ACS Nano 2020, 14 (12), 16614–16623. 10.1021/acsnano.0c07176. [DOI] [PubMed] [Google Scholar]
- Lv L.; Liu S.; Li J.; Lei H.; Qin H.; Peng X. Synthesis of Weakly Confined, Cube-Shaped, and Monodisperse Cadmium Chalcogenide Nanocrystals with Unexpected Photophysical Properties. J. Am. Chem. Soc. 2022, 144 (37), 16872–16882. 10.1021/jacs.2c05151. [DOI] [PubMed] [Google Scholar]
- Gao X.; Yang L.; Petros J. A.; Marshall F. F.; Simons J. W.; Nie S. In Vivo Molecular and Cellular Imaging with Quantum Dots. Curr. Opin. Biotechnol. 2005, 16 (1), 63–72. 10.1016/j.copbio.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Tang R.; Lee H.; Achilefu S. Induction of pH Sensitivity on the Fluorescence Lifetime of Quantum Dots by NIR Fluorescent Dyes. J. Am. Chem. Soc. 2012, 134 (10), 4545–4548. 10.1021/ja300276s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G. X.; Gu M. M.; Zhang J. R.; Zhu J. J. Preparation and Bioapplication of High-Quality, Water-Soluble, Biocompatible, and Near-Infrared-Emitting CdSeTe Alloyed Quantum Dots. Nanotechnology 2009, 20 (41), 415103. 10.1088/0957-4484/20/41/415103. [DOI] [PubMed] [Google Scholar]
- Elibol E. Synthesis of Near Unity Photoluminescence CdSeTe Alloyed Quantum Dots. J. Alloys Compd. 2020, 817, 152726. 10.1016/j.jallcom.2019.152726. [DOI] [Google Scholar]
- Cichy B.; Rich R.; Olejniczak A.; Gryczynski Z.; Strek W. Two Blinking Mechanisms in Highly Confined AgInS2 and AgInS2/ZnS Quantum Dots Evaluated by Single Particle Spectroscopy. Nanoscale 2016, 8 (7), 4151–4159. 10.1039/C5NR07992F. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Mohs A. M.; Nie S. Tuning the Optical and Electronic Properties of Colloidal Nanocrystals by Lattice Strain. Nat. Nanotechnol. 2009, 4 (1), 56–63. 10.1038/nnano.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ca N. X.; Hien N. T.; Luyen N. T.; Lien V. T. K.; Thanh L. D.; Do P. V.; Bau N. Q.; Pham T. T. Photoluminescence Properties of CdTe/CdTeSe/CdSe Core/Alloyed/Shell Type-II Quantum Dots. J. Alloys Compd. 2019, 787, 823–830. 10.1016/j.jallcom.2019.02.139. [DOI] [Google Scholar]
- Han Z.; Ren L.; Chen L.; Luo M.; Pan H.; Li C.; Chen J. Synthesis and Optical Properties of Water-soluble CdTe1-Se Quantum Dots with Ultra-Long Fluorescence Lifetime. J. Alloys Compd. 2017, 699, 216–221. 10.1016/j.jallcom.2016.12.365. [DOI] [Google Scholar]
- Huy B. T.; Seo M.-H.; Phong P. T.; Lim J.-M.; Lee Y.-I. Facile Synthesis of Highly Luminescent Mg(II), Cu(I)-Codoped CdS/ZnSe Core/Shell Nanoparticles. Chem. Eng. J. 2014, 236, 75–81. 10.1016/j.cej.2013.09.079. [DOI] [Google Scholar]
- Zhang Z.; Luan S.; Huang K.; Zhang Y.; Shi Z.; Xie R.; Yang W. Single-Phase Dual Emissive Cu:Cds-Znse Core-Shell Nanocrystals with “Zero Self-Absorption” and Their Application in White Light Emitting Diodes. J. Mater. Chem. C 2015, 3 (15), 3614–3622. 10.1039/C5TC00374A. [DOI] [Google Scholar]
- Lien V. T. K.; Tan P. M.; Hien N. T.; Hoa V. X.; Chi T. T. K.; Truong N. X.; Oanh V. T. K.; Thuy N. T. M.; Ca N. X. Tunable Photoluminescent Cu-Doped CdS/ZnSe Type-II Core/Shell Quantum Dots. J. Lumin. 2019, 215, 116627. 10.1016/j.jlumin.2019.116627. [DOI] [Google Scholar]
- Van H. T.; Vinh N. D.; Tan P. M.; Thuy U. T. D.; Ca N. X.; Hien N. T. Synthesis and Optical Properties of Tunable Dual Emission Copper Doped CdTe1-xSex Alloy Nanocrystals. Opt. Mater. 2019, 97, 109392. 10.1016/j.optmat.2019.109392. [DOI] [Google Scholar]
- Pu C.; Ma J.; Qin H.; Yan M.; Fu T.; Niu Y.; Yang X.; Huang Y.; Zhao F.; Peng X. Doped Semiconductor-Nanocrystal Emitters with Optimal Photoluminescence Decay Dynamics in Microsecond to Millisecond Range: Synthesis and Applications. ACS Cent. Sci. 2016, 2 (1), 32–39. 10.1021/acscentsci.5b00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Liu S.; Qin H.; Zhou J.; Peng X. Oxygen Stabilizes Photoluminescence of CdSe/CdS Core/Shell Quantum Dots via Deionization. J. Mater. Chem. C 2020, 142 (9), 4254–4264. 10.1021/jacs.9b11978. [DOI] [PubMed] [Google Scholar]
- Qin W.; Shah R. A.; Guyot-Sionnest P. CdSeS_ZnS Alloyed Nanocrystal Lifetime and Blinking Studies under Electrochemical Control. ACS Nano 2012, 6 (1), 912–918. 10.1021/nn2044388. [DOI] [PubMed] [Google Scholar]
- Beaudelot J.; Oger S.; Perusko S.; Phan T. A.; Teunens T.; Moucheron C.; Evano G. Photoactive Copper Complexes: Properties and Applications. Chem. Rev. 2022, 122 (22), 16365–16609. 10.1021/acs.chemrev.2c00033. [DOI] [PubMed] [Google Scholar]
- Gothard N. A.; Mara M. W.; Huang J.; Szarko J. M.; Rolczynski B.; Lockard J. V.; Chen L. X. Strong Steric Hindrance Effect on Excited State Structural Dynamics of Cu(I) Diimine Complexes. J. Phys. Chem. A 2012, 116 (9), 1984–1992. 10.1021/jp211646p. [DOI] [PubMed] [Google Scholar]
- Razgoniaev A. O.; McCusker C. E.; Castellano F. N.; Ostrowski A. D. Restricted Photoinduced Conformational Change in the Cu(I) Complex for Sensing Mechanical Properties. ACS Macro Lett. 2017, 6 (9), 920–924. 10.1021/acsmacrolett.7b00465. [DOI] [PubMed] [Google Scholar]
- Leoni E.; Mohanraj J.; Holler M.; Mohankumar M.; Nierengarten I.; Monti F.; Sournia-Saquet A.; Delavaux-Nicot B.; Nierengarten J.-F.; Armaroli N. Heteroleptic Copper(I) Complexes Prepared from Phenanthroline and Bis-Phosphine Ligands: Rationalization of the Photophysical and Electrochemical Properties. Inorg. Chem. 2018, 57 (24), 15537–15549. 10.1021/acs.inorgchem.8b02879. [DOI] [PubMed] [Google Scholar]
- Hoffmann A.; Binder S.; Jesser A.; Haase R.; Florke U.; Gnida M.; Salomone Stagni M.; Meyer-Klaucke W.; Lebsanft B.; Grunig L. E.; Schneider S.; Hashemi M.; Goos A.; Wetzel A.; Rubhausen M.; Herres-Pawlis S. Catching an Entatic State-A Pair of Copper Complexes. Angew. Chem., Int. Ed. Engl. 2014, 53 (1), 299–304. 10.1002/anie.201306061. [DOI] [PubMed] [Google Scholar]
- Dicke B.; Hoffmann A.; Stanek J.; Rampp M. S.; Grimm-Lebsanft B.; Biebl F.; Rukser D.; Maerz B.; Gories D.; Naumova M.; Biednov M.; Neuber G.; Wetzel A.; Hofmann S. M.; Roedig P.; Meents A.; Bielecki J.; Andreasson J.; Beyerlein K. R.; Chapman H. N.; Bressler C.; Zinth W.; Rubhausen M.; Herres-Pawlis S. Transferring the Entatic-State Principle to Copper Photochemistry. Nat. Chem. 2018, 10 (3), 355–362. 10.1038/nchem.2916. [DOI] [PubMed] [Google Scholar]
- Griffin P. J.; Charette B. J.; Burke J. H.; Vura-Weis J.; Schaller R. D.; Gosztola D. J.; Olshansky L. Toward Improved Charge Separation through Conformational Control in Copper Coordination Complexes. J. Am. Chem. Soc. 2022, 144 (27), 12116–12126. 10.1021/jacs.2c02580. [DOI] [PubMed] [Google Scholar]
- Gernert M.; Balles-Wolf L.; Kerner F.; Muller U.; Schmiedel A.; Holzapfel M.; Marian C. M.; Pflaum J.; Lambert C.; Steffen A. Cyclic (Amino)(aryl)carbenes Enter the Field of Chromophore Ligands: Expanded pi System Leads to Unusually Deep Red Emitting Cu(I) Compounds. J. Am. Chem. Soc. 2020, 142 (19), 8897–8909. 10.1021/jacs.0c02234. [DOI] [PubMed] [Google Scholar]
- Giereth R.; Reim I.; Frey W.; Junge H.; Tschierlei S.; Karnahl M. Remarkably Long-Lived Excited States of Copper Photosensitizers Containing an Extended π-system Based on an Anthracene Moiety. Sustainable Energy Fuels 2019, 3 (3), 692–700. 10.1039/C8SE00521D. [DOI] [Google Scholar]
- Doettinger F.; Yang Y.; Schmid M. A.; Frey W.; Karnahl M.; Tschierlei S. Cross-Coupled Phenyl- and Alkynyl-Based Phenanthrolines and their Effect on the Photophysical and Electrochemical Properties of Heteroleptic Cu(I) Photosensitizers. Inorg. Chem. 2021, 60 (7), 5391–5401. 10.1021/acs.inorgchem.1c00416. [DOI] [PubMed] [Google Scholar]
- Gimeno L.; Phelan B. T.; Sprague-Klein E. A.; Roisnel T.; Blart E.; Gourlaouen C.; Chen L. X.; Pellegrin Y. Bulky and Stable Copper(I)-Phenanthroline Complex: Impact of Steric Strain and Symmetry on the Excited-State Properties. Inorg. Chem. 2022, 61 (19), 7296–7307. 10.1021/acs.inorgchem.1c03901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit A. K.; Behera S. K.; Kar P. K. A terpyridine luminophore: Synthesis, Photophysics and Selective metal ion-Mediated hydrogelation. J. Mol. Struct. 2020, 1205, 127568. 10.1016/j.molstruc.2019.127568. [DOI] [Google Scholar]
- Liu J.-H.; Ma Y.-Y.; Tu T.; Qian D.-J. Fluorescent Properties and Probe Features of Newly Synthesized Acridine-Terpyridyl Bidentate Ligand and Its Metalated Complexes. J. Lumin. 2022, 244, 118765. 10.1016/j.jlumin.2022.118765. [DOI] [Google Scholar]
- Yu H.-Y.; Li H.-J.; Ma Y.-Y.; Feng Y.-X.; Qian D.-J. Interfacial Self-Assembly of Carbon Nitride-Based Nanocomposites with Zinc Terpyridyl Coordination Polymers for Photocurrent Generation and the Photocatalytic Degradation of Organic Dyes. Colloids Surf., A 2020, 596, 124702. 10.1016/j.colsurfa.2020.124702. [DOI] [Google Scholar]
- Wang H.; Xie L.; Peng Q.; Meng L.; Wang Y.; Yi Y.; Wang P. Novel Thermally Activated Delayed Fluorescence Materials-Thioxanthone Derivatives and Their Applications for Highly Efficient OLEDs. Adv. Mater. 2014, 26 (30), 5198–5204. 10.1002/adma.201401393. [DOI] [PubMed] [Google Scholar]
- Byeon C. C.; McKerns M. M.; Sun W.; Nordlund T. M.; Lawson C. M.; Gray G. M. Excited State Lifetime and Intersystem Crossing Rate of Asymmetric Pentaazadentate Porphyrin-Like Metal Complexes. Appl. Phys. Lett. 2004, 84 (25), 5174–5176. 10.1063/1.1763983. [DOI] [Google Scholar]
- Ma D.-M.; Ding A.; Guo H.; Chen M.; Qian D.-J. Luminescent Properties of Newly Synthesized Thioxanthone-Polypyridyl Derivatives and Their Metal-Organic Complexes. J. Lumin. 2019, 212, 5–13. 10.1016/j.jlumin.2019.04.010. [DOI] [Google Scholar]
- Malval J.-P.; Jin M.; Morlet-Savary F.; Chaumeil H.; Defoin A.; Soppera O.; Scheul T.; Bouriau M.; Baldeck P. L. Enhancement of the Two-Photon Initiating Efficiency of a Thioxanthone Derivative through a Chevron-Shaped Architecture. Chem. Mater. 2011, 23 (15), 3411–3420. 10.1021/cm200595y. [DOI] [Google Scholar]
- Nazir R.; Balčiünas E.; Buczyńska D.; Bourquard F.; Kowalska D.; Gray D.; Maćkowski S.; Farsari M.; Gryko D. T. Donor-Acceptor Type Thioxanthones: Synthesis, Optical Properties, and Two-Photon Induced Polymerization. Macromolecules 2015, 48 (8), 2466–2472. 10.1021/acs.macromol.5b00336. [DOI] [Google Scholar]