

Abstract
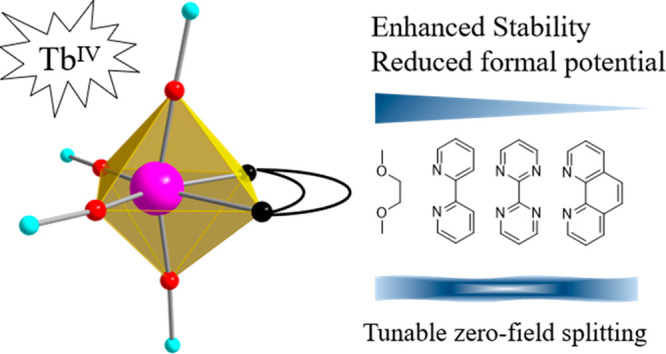
Coordination chemistry of rare-earth elements has been dominated by the +3 oxidation state. Complexes with higher-valence lanthanide ions are synthetically challenging but are of fundamental research interest and significance as advanced molecular materials. Herein, four tetravalent terbium complexes (2–5) of the common formula [Tb(OSiPh3)4L] (L = ethylene glycol dimethyl ether (DME), 2,2’-bipyridine (bpy), 2,2’-bipyrimidine (bpym), and 1,10-phenanthroline (phen)) are reported. Crystallographic analyses reveal in each of these complexes a hexacoordinate Tb(IV) ion situated in a distorted octahedral coordination environment formed by four triphenylsiloxido ligands and a bidentate chelating ligand. The use of chelating ligands enhances the stability of the resulting complexes over their THF solvate precursor. More significantly, the aromatic N-chelating ligands have been found to tune effectively the electronic structures of the complexes, as evidenced by the sizable potential shifts observed for the quasi-reversible redox Tb(IV/III) process and by the changes in their absorption spectra. The experimental findings are augmented with quantum theoretical calculations in which the ligand π-donation to the 5d orbitals of the Tb(IV) center is found to be primarily responsible for stability enhancement and the corresponding changes of physical properties observed. Magnetic measurements and results from electron paramagnetic resonance studies produced small absolute values of zero-field splittings of these complexes, ranging from 0.1071(22) to 1.1484(112) cm–1 and comparable to the values reported for analogous Tb(IV) complexes.
Keywords: Tetravalent Terbium Ion, Chelating Ligand, Formal Potential, Stability, Magnetic Property
Introduction
Complexes with their metal ions in unusual oxidation states are of interest for fundamental research and applications of practical significance. For example, high-valence iron species play a major role in oxidative reactions in nature,1 while actinide ions in their unconventionally high oxidation states enable their effective separation from a gamut of coexisting rare earth ions.2,3 In this context, the coordination chemistry of the lanthanide (Ln) ions in unconventional oxidation states is of particular relevance and of high interest to researchers interested in theoretical and computational chemistry, synthetic and structural chemistry, physical properties and chemical reactivity, and materials applications of the lanthanides. However, the chemistry of the lanthanides is dominated by the +3 oxidation state, with the few exceptions of Ce(IV), Sm(II), Eu(II), and Yb(II) being reasonably readily accessible.4−10 With the pioneering work by Lappert and co-workers11 and the following efforts by Evans, Mazzanti, Meyer, Long, and others, remarkable progress in the chemistry of Ln(II) ions has been achieved.12−27 The isolation of divalent complexes of all lanthanide elements was completed in 2013,23 some of which display intriguing physical properties with potential applications as high-performing single-molecule magnets26 and molecular spin qubits.27 In stark contrast, the chemistry of high-valence lanthanide ions has lagged far behind due primarily to the lack of synthetic methods or reagents and the notorious air/moisture sensitivity or reactivity of such high-valence species. In this context, the formation of Pr(V) species in the gas phase and in a solid noble-gas matrix is noteworthy.28,29 It is also of note that the Tb(IV) ion in the solid was identified more than 70 years ago,30,31 but complexes of tetravalent lanthanide other than Ce(IV) were unknown until very recently. It is the efforts led independently by Mazzanti32−35 and La Pierre36−38 that have broken new ground in the chemistry of tetravalent lanthanide complexes.39
A number of representative Tb(IV) complexes are collected in time order as shown in Scheme 1, the first being [Tb(OSi(OtBu)3)4] (A) reported by Mazzanti and co-workers with its pentacoordinate Tb(IV) center situated in a coordination sphere formed by four OSi(OtBu)− ligands.32 La Pierre et al. reported [Tb(NP(1,2-bis-tBu-diamidoethane)(NEt2))4] (B) stabilized by phosphinimine ligands (PN*) in the same year.36,37 Shortly after, Mazzanti and co-workers reported the first Pr(IV) complex [Pr(OSiPh3)4(MeCN)2]35 by adopting the same procedure for the preparation of its Tb(IV) congeners [Tb(OSiPh3)4(L)2] (L = CH3CN (C) or THF (1)) and [Tb(OSiPh3)4(L)] (L = O=PPh3 (D) or O=PEt3 (E)).33,34 Out of the small number of tetravalent lanthanide complexes, these of Pr(IV) and Tb(IV), featuring, respectively, 4f1 and 4f7 electron configurations, are magnetically attractive, both capable of providing a pure nuclear-spin environment and hyperfine coupling between nuclear and electronic spins. Potential applications as single-molecule magnets and molecular spin qubits can be envisioned.40−44 It is thus important to develop tetravalent Pr(IV) and Tb(IV) complexes with enhanced stability and to study their physicochemical properties with an eye on the aforementioned applications.
Scheme 1. Structures of the Tb(IV) Complex in the Literature.

The rightmost one represents the generic structure of the chelates in this work.
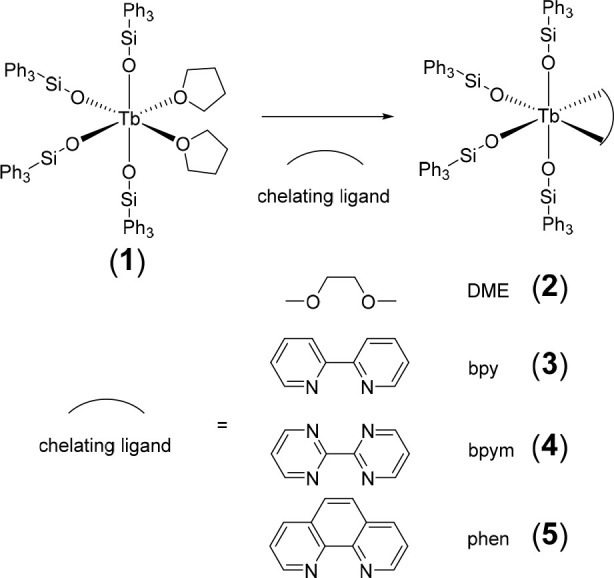
Ligand substitution was studied by Mazzanti and co-workers who found that the coordinated solvent molecules in [Tb(OSiPh3)4(L)2] (L = CH3CN or THF) can be replaced by phosphinoxide ligands,33,34 leading to the enhanced stability of the resulting complexes due to the strong π(O–Tb) interaction from phosphinoxide ligands. Inspired by these literature reports and in hopes of producing tetravalent lanthanide complexes with even further enhanced stability for property studies, we explore in this work the replacement of the coordinated tetrahydrofuran (THF) molecules in Tb(OSiPh3)4(THF)2 (1)—one of the early examples of Tb(IV) complexes—with a number of O- and N-chelating ligands. Specifically, the syntheses and crystal structures of four new tetravalent Tb(IV) complexes (2–5, Scheme 2) are reported, with each featuring four triphenylsiloxido ligands and a bidentate chelating ligand, including ethylene glycol dimethyl ether (DME), 2,2′-bipyridine (bpy), 2,2′-bipyrimidine (bpym), and 1,10-phenanthroline (phen). Property studies by cyclic voltammetry, absorption spectroscopy, and DFT calculations reveal enhanced stability of the chelates over the precursor complex with two coordinated THF molecules and collectively point to the stabilizing effect of the chelating ligands. Magnetic measurements and studies by electron paramagnetic resonance (EPR) spectroscopy indicate that the absolute values of zero-field splitting for these complexes are relatively small.
Scheme 2. Syntheses of 2–5 by Ligand Exchange of Tb(OSiPh3)4(THF)2 (1) with the Chelating Ligands Shown.
Results and Discussion
Syntheses and Structural Characterization
Complex 1 was obtained by adopting a literature procedure:34 Oxidizing the trivalent precursor Tb(OSiPh3)3(THF)3 (TbPh3)45 with [(C6H4Br)3N][SbCl6] in the presence of KOSiPh3, followed by recrystallization from THF at −30 °C, afforded the desired product. LiOSiPh3 or NaOSiPh3 can be used in place of KOSiPh3, but the desired tetravalent complex was not formed without this additional equivalent of siloxido to balance the extra positive charge of the newly generated Tb(IV).33,34 Complexes 2–5 were obtained by ligand exchange of 1 with its coordinated THF being replaced by DME, bpy, bpym, and phen, respectively (Scheme 2).
The solid-state structures of the four new complexes were established by single-crystal X-ray diffraction studies (Table S1). As shown in Figure 1, each of the four new complexes features a hexacoordinated Tb(IV) ion situated in a distorted octahedral coordination sphere formed by four Ph3SiO– ligands and a unique bidentate chelating ligand. Overall, these new complexes are structurally similar to the previously reported 1(34) and [Tb(OSiPh3)4(CH3CN)2] (C)33 with two cis-disposed nonsiloxido ligands. Except for solvent molecules of recrystallization, no ions of any kind are present in the complete crystal structure, which is consistent with the complexes being electrically neutral. Bond lengths and angles of interest are summarized in Table 1. The Tb–Osiloxido bonds of the new complexes are comparable with these of the Tb(IV) complexes previously reported,32−34 but shorter than these of the trivalent precursor TbPh3. Ranging from 65.09(4)° to 66.87(14)°, the O–Tb–O or N–Tb–N angles associated with the chelating ligands in 2–5 are significantly smaller than the OTHF–Tb–OTHF angle in 1 (86.10(12)°), due presumably to the enhanced rigidity of the bidentate chelating ligands. Correspondingly, the angles between the two siloxido ligands that are coplanar with the chelating ligands, ranging from 100.10(4)° to 108.13(10)°, are significantly larger than the corresponding angle of 96.02(11)° in 1 (Table S2). This scenario is entirely understandable as the less crowded coordination of the chelating ligand makes room for a more relaxed disposition of the coplanar siloxido ligands. The remaining two siloxido ligands are “steered away” due to steric repulsion by the equatorial siloxido ligands, resulting in a pronounced deviation of the axial coordination motif from linearity (157.14(12)° to 165.07(10)°) (Table S2). Two phenyl groups on each of the axial siloxido ligands are disposed in such a way that strong face-to-face π–π interactions are formed with the aromatic rings of the N-chelating ligand (Figures 1e, S1, S2; Tables S3–S5) The distortion of the coordination geometry from a perfect octahedron was estimated by continuous shape measures analysis46 to be 0.654, 1.041, 1.297, 1.330, and 1.185 for 1–5, respectively (Table S6). Albeit small, such deformation of the coordination polyhedra can perturb the electronic structure of a lanthanide complex, leading to significant changes in magnetic properties.47,48
Figure 1.

Stick-and-ball depiction of the crystal structures of (a) 2, (b) 3, (c) 4, and (d) 5, other atoms are omited for clarity; (e) π–π interactions between the phenyl groups of the axial siloxido ligands and the aromatic rings of the phen ligand in 5; yellow hexagons highlight the aromatic groups involved (color code: Tb, pink; O, red; Si, orange; C, gray; N, blue).
Table 1. Selected Bond Lengths (Å) and Angles (deg) of TbPh3 and 1–5.
| Tb–Osiloxido | Tb–O/N (L)a | O/N (L)–Tb–O/N (L) | |
|---|---|---|---|
| TbPh3 | 2.135(3)–2.145(3) | 2.441(3)–2.484(4) | |
| 1 | 2.043(2)–2.079(2) | 2.400(2) | 86.10(12) |
| 2 | 2.032(3)–2.084(3) | 2.439(3)–2.443(3) | 66.81(11) |
| 3 | 2.039(4)–2.094(4) | 2.462(5)–2.473(4) | 65.38(14) |
| 4 | 2.027(1)–2.078(1) | 2.497(1)–2.503(1) | 65.09(4) |
| 5 | 2.044(2)–2.085(2) | 2.461(3) | 66.87(14) |
O/N (L) indicates the coordinating atoms (O in 1 and 2; N in 3–5 of the neutral ligand L).
Cyclic Voltammetry
Redox properties of the complexes were studied by cyclic voltammetry (CV). The voltammograms are each characterized by a single pair of redox events, exhibiting a quasi-reversible redox process. As shown by the data collected in Table 2 (Figure 2), the reduction (Epc) and oxidation (Epa) potentials both decrease upon chelation of the Tb(IV) center, with the peaks shifting from 0.020 and 0.582 V of 1 to −0.278 and 0.078 V for 5, respectively. The Epa of 0.078 V for 5 is the smallest among all Tb(IV) siloxido complexes, and is only higher than that of B, a tetravalent terbium complex with a ligand of strong π character.32−34,36
Table 2. Electrochemical Data for the Tb(III/IV) Peak Couple of 1–5 vs Fc/Fc+ (Fc = Ferrocene) in dichloromethane at a Sweep Rate of 500 mV s–1.
| Complex | Epc (V) | Epa (V) | E° (V) | ΔE (V) | Ipa/Ipc |
|---|---|---|---|---|---|
| 1 | 0.020 | 0.582 | 0.301 | 0.562 | 0.894 |
| 2 | –0.017 | 0.346 | 0.165 | 0.363 | 0.996 |
| 3 | –0.164 | 0.223 | 0.030 | 0.387 | 0.823 |
| 4 | –0.208 | 0.266 | 0.029 | 0.474 | 0.792 |
| 5 | –0.278 | 0.078 | –0.100 | 0.356 | 1.093 |
Figure 2.
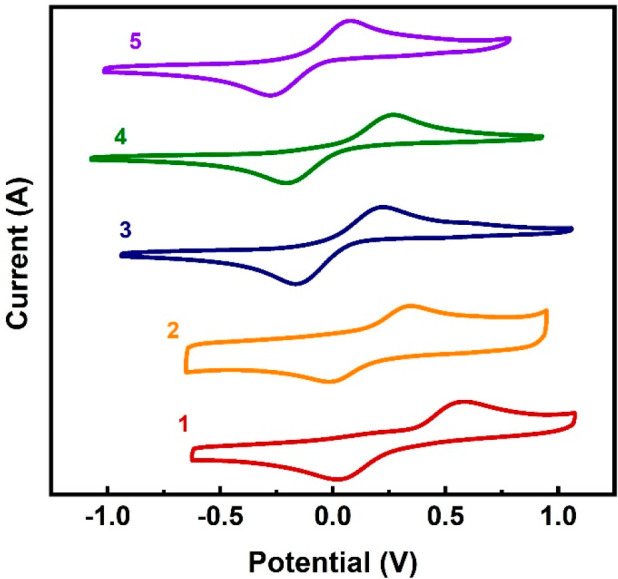
Cyclic voltammograms of 1–5 (1 mM in dichloromethane) with [NnBu4][B(C6F5)4] (0.1 mM) as supporting electrolyte at a sweep rate of 500 mV s–1 vs Fc/Fc+.
The peak separations (ΔE) for 2–5 is smaller than that of 1 by about 0.1–0.2 V, and even smaller than these of the other previously reported Tb(IV) complexes (Table 2),32−34,36 reflecting well the enhanced stability of the chelates during the redox process. The relationship between the peak current and the square root of the scan rate was found to be nearly linear, and the ΔE increased with increasing scan rate, suggesting that Tb(III/IV) redox reactions are diffusion-controlled. For 2–5, the ratio of peak currents (Ipa/Ipc) ranges from 0.8 to 1.2, indicating that the reduced species were mostly reoxidized upon reversal of the scan direction, exhibiting good chemical reversibility (Figures S5–S12). In contrast, the peak current ratio of 1 varies more sensitively upon change of scan rate (Figures S3 and S4, Table S7). This result suggests that the redox process of 1 is more complex, possibly involving dissociation and recoordination of the THF ligands, a scenario corroborated by the large separation between the reduction and oxidation peaks observed for 1.
The formal potential (E°), a good reflection of the relative thermodynamic stability of the Tb(IV) and Tb(III) redox states,49−51 ranges from −0.1 V of 5 to 0.301 V of 1 (Table 2). In other words, the use of chelating ligands is propitious to enhancing the thermodynamic stability of a complex, which is completely expected. Soundly supporting this conclusion is the shift of E° from 0.301 V for 1 to 0.165 V for 2 with the mere substitution of two coordinated THF molecules with a single DME ligand. It appears that introduction of a conjugated chelating ligand can further enhance the stability of the complex as reflected by the further reduced E°, to 0.030, 0.029, and −0.100 V for 3–5, respectively (Table 2). Complex 5 emerged as the most thermodynamically stable among complexes 1–5, probably owing to the additional conjugated phenyl ring relative to the 2,2′-bipyridine ligand.
UV–Vis Spectra
The UV–vis spectra of 1–5 were collected immediately following dissolution in toluene (Figure 3) and dichloromethane (Figure S13). The absorptions of 1 and 2 in toluene, spanning between 285 and 575 nm, show maxima at ca. 382 nm, while the absorptions of the N-chelates (3–5) covers the 320–550 nm range with maxima at ca. 371 nm. The spectra obtained with dichloromethane solutions are essentially the same as these with the toluene solutions with only a <5 nm shift of the absorption maxima (Table 3). The molar absorptions, ranging from 3300 to 4200 M–1 cm–1 for 1–5, are comparable to the values reported for analogous Tb(IV) siloxido complexes32−34 and an electrochemically generated Tb(IV) species.52,53
Figure 3.
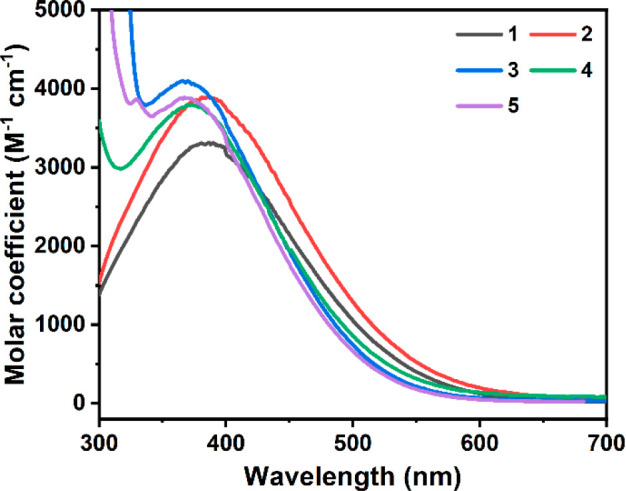
UV–vis absorption spectra of 1–5 in toluene at room temperature.
Table 3. Experimental and TDDFT Results (nm) of the Absorption Maxima for 1–5.
| Complexes | Dichloromethane | Toluene | Excitation energy | Assignments |
|---|---|---|---|---|
| 1 | 382 | 385 | 388 | LMCT |
| 2 | 383 | 384 | 384 | LMCT |
| 3 | 370 | 370 | 378/373 | LLCT/LMCT |
| 4 | 374 | 373 | 377/375 | LLCT/LMCT |
| 5 | 370 | 370 | 376/374 | LLCT/LMCT |
To rationalize the blue shift of absorption maxima of the N-chelates (3–5) with respect to these of 1 and 2, TDDFT calculations were performed, and the results are summarized in Tables 3 and S13.54 The computed UV–vis spectra (Figure S15) are in excellent agreement with the experimental ones. The broad bands of 1 and 2 are attributed exclusively to the ligand-to-metal charge transfer (LMCT) from the ligand-dominant molecular orbitals (MOs) to the Tb 4f orbitals (Figure S16). In comparison, the absorption maxima of 3–5 can be characterized as a dominant LMCT with an appreciable ligand-to-ligand charge transfer (LLCT) contribution (Figure S17). Specifically, these LLCT peaks can be assigned to ligand-dominant π → π* electronic transitions of these N-chelated ligands, leading to the blue-shifted absorption and demonstrating the stabilization of the π-orbitals upon coordination with the aromatic N-chelating ligand (Figure S18).55
The solution stability of the Tb(IV) complexes was evaluated with their UV–vis spectra collected over a period of 2 weeks in toluene (Figure 4) and 1 week in dichloromethane (Figure S14) under an argon atmosphere. In toluene, the solutions of 1 and 2 decolored completely after 72 h, whereas 39%, 17%, and 49% of the characteristic absorption were retained after 120 h for the solutions of 3–5, respectively. The enhancement of solution stability of 3–5 can be attributed to the strong intramolecular π–π interactions, as mentioned above. It should be noted that the previously reported complexes D and E survived in a 96 h UV–vis experiment;34 the impressive stability may be attributed, at least partly, to the shielding of the Tb(IV) center by the bulky ligands.
Figure 4.

Time-dependent UV–vis spectra of 1–5 (a–e) in toluene at room temperature.
DFT Calculations
The bonding interactions of the Tb(IV) center with both the siloxido and neutral ligands were studied by using DFT calculations. The theoretical and computational details are given in the SI file. The metric values of Wiberg56 and Mayer57 bond orders obtained are collected in Table S12. The average Tb-L bond orders, ranging from 0.17 to 0.25, are much smaller than those of the Tb–Osiloxido bonds (0.45–0.60), indicating the significant donor–acceptor character of the coordinate bond. Further analyses by EDA-NOCV58,59 reveal the remarkable thermostability of the N-chelates (3–5) and the tuning of the Tb-L bond interactions by the chelating ligands (Table 4). For 1 and 2, it has been found that the donation from the occupied sp-hybrid orbital of the THF/DME O atom to the empty 5d orbitals of TbIV is dominant (23.2 kcal/mol for 1, 20.2 kcal/mol for 2), while the π-donation is much less significant (< ∼5 kcal/mol). The opposite is, not surprisingly, observed for the N-chelates: The N-to-TbIV π-donations are found to be 14.3, 13.3, and 11.6 kcal/mol for 3, 4, and 5, respectively. This difference in π donation of the neutral ligands to Tb(IV) is presumably the determining factor in the observed stability of the N-chelates with respect to their O-chelating cognates.
Table 4. Energies of the σ- and π-Donation from the Neutral Ligand L to the Tb(IV) Center Produced by EDA-NOCV Analysisa.
| Energy terms | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| ΔEint | –49.90 | –43.87 | –56.54 | –61.03 | –62.56 |
| ΔEorb(L→Tb σ donation) | –23.24 | –20.15 | –29.62 | –21.37 | –27.09 |
| ΔEorb(L→Tb π donation) | –3.68 | –4.94 | –14.33 | –13.26 | –11.55 |
All energies are given in kcal/mol.
Magnetic Studies
The electronic structures of the complexes were further investigated by magnetic measurements and EPR. Static magnetic susceptibilities were measured under an applied DC field of 1000 Oe with cooling from 300 to 2 K (Figure 5). The χT values at 300 K are 7.81, 7.75, 7.98, 7.85, and 8.02 cm3 K mol–1 for 1–5, respectively, significantly smaller than the value of 11.82 cm3 K mol–1 for the mononuclear Tb(III) complex but in good agreement with the values reported for other Tb(IV) complexes.32,36 The χT decreases slowly with the lowering of temperature to ca. 20 K, at which a sudden drop occurs, reaching a minimum of 1.72, 6.68, 3.37, 6.88, and 6.79 cm3 K mol–1 for 1–5, respectively. The drop in the low-temperature region is indicative of varied zero-field splitting. Field-dependent magnetizations of complexes 1–5 were subsequently measured at low temperatures with the field up to a maximum of 7 T (inset in Figure 5). The maximum magnetization values at 2 K and 7 T were found to be 6.77, 6.39, 6.68, 6.62, and 6.54 μB for 1–5, respectively, close to the saturation magnetization value of 7 μB calculated for the 4f7 electronic configuration. The temperature- and field-dependent magnetizations were fitted with the program PHI,60 giving the isotropic Landé g-factor (g), axial zero-field splitting (D), and rhombic zero-field splitting (EZFS) as collected in Table 5.
Figure 5.

χT versus T plot of 1–5 (a-e) under 1000 Oe dc field. Inset: The field-dependent magnetization plots were at 2, 3, and 5 K. Solid lines are best fits with PHI.60
Table 5. Crystal Field Parameters of 1–5.
| Magnetization |
EPR |
|||||
|---|---|---|---|---|---|---|
| Complex | g | D | |EZFS| | g | D | |EZFS| |
| 1 | 2.0067 | –0.1103 | 0.0066 | 1.9958 | 0.2130 | 0.0040 |
| 2 | 2.0097 | 1.1484 | 0.0108 | 1.9958 | 0.2117 | 0.0043 |
| 3 | 2.0020 | –0.1071 | 0.0084 | 1.9958 | 0.2100 | 0.0040 |
| 4 | 1.9953 | 0.7453 | 0.0076 | 1.9958 | 0.2050 | 0.0019 |
| 5 | 2.0060 | 0.4376 | 0.0020 | 1.9958 | 0.2127 | 0.0040 |
Electron Paramagnetic Resonance Studies
Another important technique to evaluate the D, EZFS, and g values for magnetically active complexes is electron paramagnetic resonance (EPR) spectroscopy. The X-band (9.36 GHz) continuous-wave EPR spectra for polycrystalline samples for 1–5, shown in Figure 6, were collected at 100 K. The corresponding g and D values (Table 5) were obtained via simulation.61 While the g values (1.9985) are identical for all complexes, the D values, all around 0.2 cm–1, are discernibly different. These D, EZFS, and g values, ranging from 0.1071(22) to 1.1484(112) cm–1, were obtained by both fitting of the magnetization data and by EPR simulations; they are relatively small, which is consistent with the results obtained using other Tb(IV) complexes.32,36,62
Figure 6.
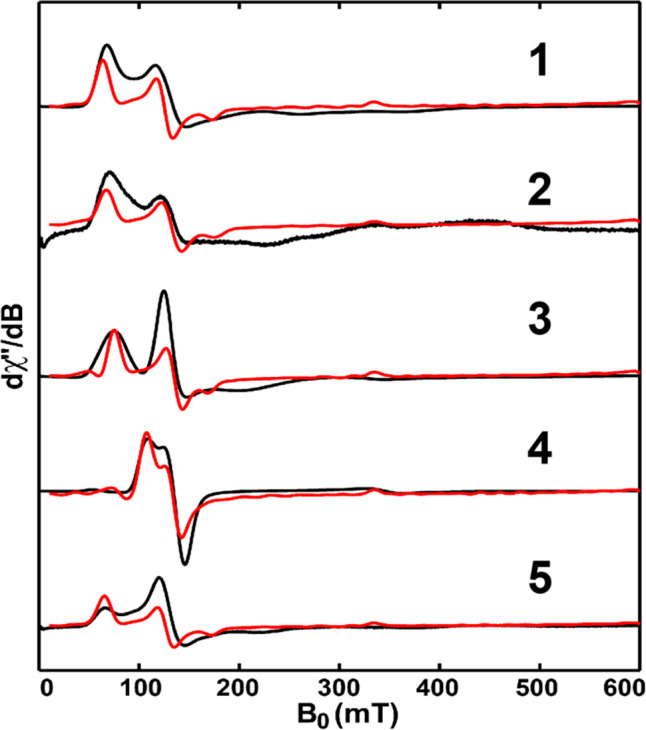
Experimental (black traces, measured at 9.36 GHz and 100 K) and simulated (red traces) X-band EPR spectra of solid 1–5.
Conclusion
Herein, the syntheses and crystallographic structure determination of four Tb(IV) siloxido complexes with O- and N-based chelating ligands were reported together with the experimental and computational studies of their physical properties. The chelating ligands enhance the stability of the resulting complexes, as expected. More significantly, aromatic N-based chelating ligands have been found to tune effectively the electronic structures of the complexes, as evidenced by the sizable potential shifts observed for the quasi-reversible redox Tb(IV/III) process. Corresponding differences in the absorption spectra between the complexes in the comparison group provide further support for the tuning effect by the chelating ligands. The experimental findings are augmented with DFT calculations in which the ligand π-donation to the 5d orbitals of Tb(IV) center is primarily responsible for the stability enhancement and corresponding physical properties changes observed. The results by both fitting of the magnetization data and EPR simulations produced relatively small absolute values of zero-field splitting anticipated for a Tb(IV) ion situated in a distorted octahedral coordination geometry.
Acknowledgments
We thank Professor Zewei Quan for the use of the UV–vis-NIR spectrometer and Mr. Lei Li for assistance with the experiments and discussion of the results.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/prechem.3c00065.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Natural Science Foundation of China (92261203, 22101116, 22033005 and 21971106), Key Laboratory of Rare Earth Chemistry of Guangdong Higher Education Institutes (2022KSYS006), the Stable Support Plan Program of Shenzhen Natural Science Fund (20200925161141006), Shenzhen Fundamental Research Program (JCYJ20220530115001002), and Postdoctoral Scientific Research Fund for staying at (coming to) Shenzhen (K21217520). The calculations were performed with SUSTech supercomputer and were supported by Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002).
The authors declare no competing financial interest.
Supplementary Material
References
- Martinez J. L.; Lutz S. A.; Yang H.; Xie J.; Telser J.; Hoffman B. M.; Carta V.; Pink M.; Losovyj Y.; Smith J. M. Structural and spectroscopic characterization of an Fe(VI) bis(imido) complex. Science 2020, 370 (6514), 356–359. 10.1126/science.abd3054. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Lu J.-B.; Dong X.; Yan Q.; Feng X.; Hu H.-S.; Wang S.; Chen J.; Li J.; Xu C. Ultra-Efficient Americium/Lanthanide Separation through Oxidation State Control. J. Am. Chem. Soc. 2022, 144 (14), 6383–6389. 10.1021/jacs.2c00594. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Li A.; Li K.; Wang Z.; Xu X.; Wang Y.; Sheridan M. V.; Hu H.-S.; Xu C.; Alekseev E. V.; Zhang Z.; Yan P.; Cao K.; Chai Z.; Albrecht-Schönzart T. E.; Wang S. Ultrafiltration separation of Am(VI)-polyoxometalate from lanthanides. Nature 2023, 616 (7957), 482–487. 10.1038/s41586-023-05840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungur L.Introduction to the electronic structure, luminescence, and magnetism of lanthanides. In Lanthanide-Based Multifunctional Materials; Martín-Ramos P.; Ramos Silva M., Eds.; Elsevier: 2018; pp 1–58. 10.1016/B978-0-12-813840-3.00001-6. [DOI] [Google Scholar]
- Wang Y.; Huang W. Chemistry of non-traditional oxidation states of rare earth metals. Sci. China Chem. 2020, 50 (11), 1504–1559. 10.1360/SSC-2020-0154. [DOI] [Google Scholar]
- Su J.; Hu S.; Huang W.; Zhou M.; Li J. On the oxidation states of metal elements in MO3– (M = V, Nb, Ta, Db, Pr, Gd, Pa) anions. Sci. China Chem. 2016, 59 (4), 442–451. 10.1007/s11426-015-5481-z. [DOI] [Google Scholar]
- Schulz A.; Liebman J. F. Paradoxes and paradigms: high oxidation states and neighboring rows in the periodic table - Lanthanides, Actinides, Exotica and Explosives. Struct. Chem. 2008, 19 (4), 633–635. 10.1007/s11224-008-9336-5. [DOI] [Google Scholar]
- Vent-Schmidt T.; Riedel S. Investigation of Praseodymium Fluorides: A Combined Matrix-Isolation and Quantum-Chemical Study. Inorg. Chem. 2015, 54 (23), 11114–11120. 10.1021/acs.inorgchem.5b01175. [DOI] [PubMed] [Google Scholar]
- Sroor F. M. A.; Edelmann F. T.. Lanthanides: Tetravalent Inorganic. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Wiley: Hoboken, NJ, 2012. 10.1002/9781119951438.eibc2034. [DOI] [Google Scholar]
- Lucena A. F.; Lourenço C.; Michelini M. C.; Rutkowski P. X.; Carretas J. M.; Zorz N.; Berthon L.; Dias A.; Conceição Oliveira M.; Gibson J. K.; Marçalo J. Synthesis and hydrolysis of gas-phase lanthanide and actinide oxide nitrate complexes: a correspondence to trivalent metal ion redox potentials and ionization energies. Phys. Chem. Chem. Phys. 2015, 17 (15), 9942–9950. 10.1039/C5CP00515A. [DOI] [PubMed] [Google Scholar]
- Hitchcock P. B.; Lappert M. F.; Maron L.; Protchenko A. V. Lanthanum Does Form Stable Molecular Compounds in the + 2 Oxidation State. Angew. Chem. Inter. Ed. 2008, 47 (8), 1488–1491. 10.1002/anie.200704887. [DOI] [PubMed] [Google Scholar]
- Kotyk C. M.; Fieser M. E.; Palumbo C. T.; Ziller J. W.; Darago L. E.; Long J. R.; Furche F.; Evans W. J. Isolation of + 2 rare earth metal ions with three anionic carbocyclic rings: bimetallic bis(cyclopentadienyl) reduced arene complexes of La2+ and Ce2+ are four electron reductants. Chem. Sci. 2015, 6 (12), 7267–7273. 10.1039/C5SC02486B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieser M. E.; Palumbo C. T.; La Pierre H. S.; Halter D. P.; Voora V. K.; Ziller J. W.; Furche F.; Meyer K.; Evans W. J. Comparisons of lanthanide/actinide + 2 ions in a tris(aryloxide)arene coordination environment. Chem. Sci. 2017, 8 (11), 7424–7433. 10.1039/C7SC02337E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. P.; Maron L.; Scopelliti R.; Mazzanti M. Reduction of a Cerium(III) Siloxide Complex To Afford a Quadruple-Decker Arene-Bridged Cerium(II) Sandwich. Angew. Chem. Inter. Ed. 2017, 56 (49), 15663–15666. 10.1002/anie.201709769. [DOI] [PubMed] [Google Scholar]
- Jenkins T. F.; Woen D. H.; Mohanam L. N.; Ziller J. W.; Furche F.; Evans W. J. Tetramethylcyclopentadienyl Ligands Allow Isolation of Ln(II) Ions across the Lanthanide Series in [K(2.2.2-cryptand)][(C5Me4H)3Ln] Complexes. Organometallics 2018, 37 (21), 3863–3873. 10.1021/acs.organomet.8b00557. [DOI] [Google Scholar]
- Palumbo C. T.; Darago L. E.; Windorff C. J.; Ziller J. W.; Evans W. J. Trimethylsilyl versus Bis(trimethylsilyl) Substitution in Tris(cyclopentadienyl) Complexes of La, Ce, and Pr: Comparison of Structure, Magnetic Properties, and Reactivity. Organometallics 2018, 37 (6), 900–905. 10.1021/acs.organomet.7b00881. [DOI] [Google Scholar]
- Palumbo C. T.; Halter D. P.; Voora V. K.; Chen G. P.; Chan A. K.; Fieser M. E.; Ziller J. W.; Hieringer W.; Furche F.; Meyer K.; Evans W. J. Metal versus Ligand Reduction in Ln3+ Complexes of a Mesitylene-Anchored Tris(Aryloxide) Ligand. Inorg. Chem. 2018, 57 (5), 2823–2833. 10.1021/acs.inorgchem.7b03236. [DOI] [PubMed] [Google Scholar]
- Ryan A. J.; Darago L. E.; Balasubramani S. G.; Chen G. P.; Ziller J. W.; Furche F.; Long J. R.; Evans W. J. Synthesis, Structure, and Magnetism of Tris(amide) [Ln{N(SiMe3)2}3]1- Complexes of the Non-traditional + 2 Lanthanide Ions. Chem. -Eur. J. 2018, 24 (30), 7702–7709. 10.1002/chem.201800610. [DOI] [PubMed] [Google Scholar]
- Angadol M. A.; Woen D. H.; Windorff C. J.; Ziller J. W.; Evans W. J. tert-Butyl(cyclopentadienyl) Ligands Will Stabilize Nontraditional + 2 Rare-Earth Metal Ions. Organometallics 2019, 38 (5), 1151–1158. 10.1021/acs.organomet.8b00941. [DOI] [Google Scholar]
- McClain K. R.; Gould C. A.; Marchiori D. A.; Kwon H.; Nguyen T. T.; Rosenkoetter K. E.; Kuzmina D.; Tuna F.; Britt R. D.; Long J. R.; Harvey B. G. Divalent Lanthanide Metallocene Complexes with a Linear Coordination Geometry and Pronounced 6s-5d Orbital Mixing. J. Am. Chem. Soc. 2022, 144 (48), 22193–22201. 10.1021/jacs.2c09880. [DOI] [PubMed] [Google Scholar]
- Anderson-Sanchez L. M.; Yu J. M.; Ziller J. W.; Furche F.; Evans W. J. Room-Temperature Stable Ln(II) Complexes Supported by 2,6-Diadamantyl Aryloxide Ligands. Inorg. Chem. 2023, 62 (2), 706–714. 10.1021/acs.inorgchem.2c02167. [DOI] [PubMed] [Google Scholar]
- Moore W. N. G.; McSorley T. J.; Vincent A.; Ziller J. W.; Jauregui L. A.; Evans W. J. van der Waals Heterostructures Based on Nanolayered Paramagnetic Tm(II) Compounds and Boron Nitride for Investigating Spin Frustration. ACS Appl. Nano Mater. 2023, 6 (8), 6461–6466. 10.1021/acsanm.3c00662. [DOI] [Google Scholar]
- MacDonald M. R.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Completing the Series of + 2 Ions for the Lanthanide Elements: Synthesis of Molecular Complexes of Pr2+, Gd2+, Tb2+, and Lu2+. J. Am. Chem. Soc. 2013, 135 (26), 9857–9868. 10.1021/ja403753j. [DOI] [PubMed] [Google Scholar]
- Woen D. H.; Chen G. P.; Ziller J. W.; Boyle T. J.; Furche F.; Evans W. J. Solution Synthesis, Structure, and CO2 Reduction Reactivity of a Scandium(II) Complex, {Sc[N(SiMe3)2]3}−. Angew. Chem. Inter. Ed. 2017, 56 (8), 2050–2053. 10.1002/anie.201611758. [DOI] [PubMed] [Google Scholar]
- Meihaus K. R.; Fieser M. E.; Corbey J. F.; Evans W. J.; Long J. R. Record High Single-Ion Magnetic Moments Through 4fn5d1 Electron Configurations in the Divalent Lanthanide Complexes [(C5H4SiMe3)3Ln]−. J. Am. Chem. Soc. 2015, 137 (31), 9855–9860. 10.1021/jacs.5b03710. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Yu J. M.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141 (33), 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- Kundu K.; White J. R. K.; Moehring S. A.; Yu J. M.; Ziller J. W.; Furche F.; Evans W. J.; Hill S. A 9.2-GHz clock transition in a Lu(II) molecular spin qubit arising from a 3,467-MHz hyperfine interaction. Nat. Chem. 2022, 14 (4), 392–397. 10.1038/s41557-022-00894-4. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Hu S.-X.; Qu H.; Su J.; Wang G.; Lu J.-B.; Chen M.; Zhou M.; Li J. Pentavalent Lanthanide Compounds: Formation and Characterization of Praseodymium(V) Oxides. Angew. Chem. Inter. Ed. 2016, 55 (24), 6896–6900. 10.1002/anie.201602196. [DOI] [PubMed] [Google Scholar]
- Hu S.-X.; Jian J.; Su J.; Wu X.; Li J.; Zhou M. Pentavalent lanthanide nitride-oxides: NPrO and NPrO– complexes with N≡Pr triple bonds. Chem. Sci. 2017, 8 (5), 4035–4043. 10.1039/C7SC00710H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen D.; Koehler W.; Katz J. Higher Oxides of the Lanthanide Elements. Terbium Dioxide. J. Am. Chem. Soc. 1951, 73, 1475–1479. 10.1021/ja01148a020. [DOI] [Google Scholar]
- MacChesney J.; Williams H.; Sherwood R.; Potter J. Preparation and low temperature magnetic properties of the terbium oxides. J. Chem. Phys. 1966, 44 (2), 596–601. 10.1063/1.1726730. [DOI] [Google Scholar]
- Palumbo C. T.; Zivkovic I.; Scopelliti R.; Mazzanti M. Molecular complex of Tb in the + 4 oxidation state. J. Am. Chem. Soc. 2019, 141 (25), 9827–9831. 10.1021/jacs.9b05337. [DOI] [PubMed] [Google Scholar]
- Willauer A. R.; Palumbo C. T.; Scopelliti R.; Zivkovic I.; Douair I.; Maron L.; Mazzanti M. Stabilization of the oxidation state + IV in siloxide-supported terbium compounds. Angew. Chem. Inter. Ed. 2020, 59 (9), 3549–3553. 10.1002/anie.201914733. [DOI] [PubMed] [Google Scholar]
- Willauer A. R.; Douair I.; Chauvin A.-S.; Fadaei-Tirani F.; Bünzli J.-C. G.; Maron L.; Mazzanti M. Structure, reactivity and luminescence studies of triphenylsiloxide complexes of tetravalent lanthanides. Chem. Sci. 2022, 13 (3), 681–691. 10.1039/D1SC05517H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willauer A. R.; Palumbo C. T.; Fadaei-Tirani F.; Zivkovic I.; Douair I.; Maron L.; Mazzanti M. Accessing the + IV Oxidation State in Molecular Complexes of Praseodymium. J. Am. Chem. Soc. 2020, 142 (12), 5538–5542. 10.1021/jacs.0c01204. [DOI] [PubMed] [Google Scholar]
- Rice N. T.; Popov I. A.; Russo D. R.; Bacsa J.; Batista E. R.; Yang P.; Telser J.; La Pierre H. S. Design, isolation, and spectroscopic analysis of a tetravalent terbium complex. J. Am. Chem. Soc. 2019, 141 (33), 13222–13233. 10.1021/jacs.9b06622. [DOI] [PubMed] [Google Scholar]
- Rice N. T.; Popov I. A.; Russo D. R.; Gompa T. P.; Ramanathan A.; Bacsa J.; Batista E. R.; Yang P.; La Pierre H. S. Comparison of tetravalent cerium and terbium ions in a conserved, homoleptic imidophosphorane ligand field. Chem. Sci. 2020, 11 (24), 6149–6159. 10.1039/D0SC01414A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompa T. P.; Ramanathan A.; Rice N. T.; La Pierre H. S. The chemical and physical properties of tetravalent lanthanides: Pr, Nd, Tb, and Dy. Dalton Trans. 2020, 49 (45), 15945–15987. 10.1039/D0DT01400A. [DOI] [PubMed] [Google Scholar]
- Li N.; Zhang W.-X. Molecular Complexes of Emerging Tetravalent Rare-Earth Metals. Chin. J. Chem. 2020, 38 (11), 1449–1450. 10.1002/cjoc.202000258. [DOI] [Google Scholar]
- Borah A.; Murugavel R. Magnetic relaxation in single-ion magnets formed by less-studied lanthanide ions Ce(III), Nd(III), Gd(III), Ho(III), Tm(II/III) and Yb(III). Coord. Chem. Rev. 2022, 453, 214288. 10.1016/j.ccr.2021.214288. [DOI] [Google Scholar]
- Ding Y.-S.; Deng Y.-F.; Zheng Y.-Z. The Rise of Single-Ion Magnets as Spin Qubits. Magnetochemistry 2016, 2 (4), 40. 10.3390/magnetochemistry2040040. [DOI] [Google Scholar]
- Liu Z.; Wang Y.-X.; Fang Y.-H.; Qin S.-X.; Wang Z.-M.; Jiang S.-D.; Gao S. Electric field manipulation enhanced by strong spin-orbit coupling: promoting rare-earth ions as qubits. Nat. Sci. Rev. 2020, 7 (10), 1557–1563. 10.1093/nsr/nwaa148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z.; Dong B.-W.; Liu Z.; Liu J.-J.; Su J.; Yu C.; Xiong J.; Shi D.-E.; Wang Y.; Wang B.-W.; Ardavan A.; Shi Z.; Jiang S.-D.; Gao S. Endohedral Metallofullerene as Molecular High Spin Qubit: Diverse Rabi Cycles in Gd2@C79N. J. Am. Chem. Soc. 2018, 140 (3), 1123–1130. 10.1021/jacs.7b12170. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Huang H.; Wang Y.-X.; Dong B.-W.; Sun B.-Y.; Jiang S.-D.; Gao S. Amination of the Gd@C82 endohedral fullerene: tunable substitution effect on quantum coherence behaviors. Chem. Sci. 2020, 11 (39), 10737–10743. 10.1039/D0SC02182B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeary M. J.; Coan P. S.; Folting K.; Streib W. E.; Caulton K. G. Yttrium and lanthanum silyloxide complexes. Inorg. Chem. 1991, 30 (8), 1723–1735. 10.1021/ic00008a011. [DOI] [Google Scholar]
- Alvarez S.; Avnir D.; Llunell M.; Pinsky M. Continuous symmetry maps and shape classification. The case of six-coordinated metal compounds. New J. Chem. 2002, 26 (8), 996–1009. 10.1039/b200641n. [DOI] [Google Scholar]
- Yu K.-X.; Ding Y.-S.; Han T.; Leng J.-D.; Zheng Y.-Z. Magnetic relaxations in four-coordinate Dy(iii) complexes: effects of anionic surroundings and short Dy-O bonds. Inorg. Chem. Fron. 2016, 3 (8), 1028–1034. 10.1039/C6QI00117C. [DOI] [Google Scholar]
- Petersen J. B.; Ding Y.-S.; Gupta S.; Borah A.; McInnes E. J. L.; Zheng Y.-Z.; Murugavel R.; Winpenny R. E. P. Electron Paramagnetic Resonance Spectra of Pentagonal Bipyramidal Gadolinium Complexes. Inorg. Chem. 2023, 62 (21), 8435–8441. 10.1021/acs.inorgchem.3c01227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirneise L.; Langmann J.; Zitzer G.; Ude L.; Maichle-Mössmer C.; Scherer W.; Speiser B.; Anwander R. Tuning Organocerium Electrochemical Potentials by Extending Tris(cyclopentadienyl) Scaffolds with Terminal Halogenido, Siloxy, and Alkoxy Ligands. Organometallics 2021, 40 (11), 1786–1800. 10.1021/acs.organomet.1c00276. [DOI] [Google Scholar]
- Friedrich J.; Qiao Y.; Maichle-Mössmer C.; Schelter E. J.; Anwander R. Redox-enhanced hemilability of a tris(tert-butoxy)siloxy ligand at cerium. Dalton Trans. 2018, 47 (30), 10113–10123. 10.1039/C8DT01878B. [DOI] [PubMed] [Google Scholar]
- Bogart J. A.; Lippincott C. A.; Carroll P. J.; Booth C. H.; Schelter E. J. Controlled Redox Chemistry at Cerium within a Tripodal Nitroxide Ligand Framework. Chem.—Eur. J. 2015, 21 (49), 17850–17859. 10.1002/chem.201502952. [DOI] [PubMed] [Google Scholar]
- Hobart D. E.; Samhoun K.; Young J. P.; Norvell V. E.; Mamantov G.; Peterson J. R. Stabilization of praseodymium(IV) and terbium(IV) in aqueous carbonate solution. Inorg. Nucl. Chem. Lett. 1980, 16 (5), 321–328. 10.1016/0020-1650(80)80069-9. [DOI] [Google Scholar]
- Hoefdraad H. E. Charge-transfer spectra of tetravalent lanthanide ions in oxides. J. Inorg. Nucl. Chem. 1975, 37 (9), 1917–1921. 10.1016/0022-1902(75)80915-8. [DOI] [Google Scholar]
- Gritsenko O. V.; Schipper P. R. T.; Baerends E. J. Approximation of the exchange-correlation Kohn-Sham potential with a statistical average of different orbital model potentials. Chem. Phys. Lett. 1999, 302 (3), 199–207. 10.1016/S0009-2614(99)00128-1. [DOI] [Google Scholar]
- Kinnunen T.-J. J.; Haukka M.; Nousiainen M.; Patrikka A.; Pakkanen T. A. Electron withdrawing and electron donating effects of 4,4′-bipyridine substituents on ruthenium mono(bipyridine) complexes. J. Chem. Soc., Dalton Trans. 2001, (18), 2649–2654. 10.1039/b103067c. [DOI] [Google Scholar]
- Wiberg K. B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24 (3), 1083–1096. 10.1016/0040-4020(68)88057-3. [DOI] [Google Scholar]
- Mayer I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97 (3), 270–274. 10.1016/0009-2614(83)80005-0. [DOI] [Google Scholar]
- Michalak A.; Mitoraj M.; Ziegler T. Bond Orbitals from Chemical Valence Theory. J. Phys. Chem. A 2008, 112 (9), 1933–1939. 10.1021/jp075460u. [DOI] [PubMed] [Google Scholar]
- Mitoraj M. P.; Michalak A.; Ziegler T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5 (4), 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]
- Chilton N. F.; Anderson R. P.; Turner L. D.; Soncini A.; Murray K. S. PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34 (13), 1164–1175. 10.1002/jcc.23234. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178 (1), 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Gompa T. P.; Greer S. M.; Rice N. T.; Jiang N.; Telser J.; Ozarowski A.; Stein B. W.; La Pierre H. S. High-Frequency and -Field Electron Paramagnetic Resonance Spectroscopic Analysis of Metal-Ligand Covalency in a 4f7 Valence Series (Eu2+, Gd3+, and Tb4+). Inorg. Chem. 2021, 60 (12), 9064–9073. 10.1021/acs.inorgchem.1c01062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.