

Abstract
Purpose:
Ocular coloboma arises from genetic or environmental perturbations that inhibit optic fissure (OF) fusion during early eye development. Despite high genetic heterogeneity, 70% to 85% of patients remain molecularly undiagnosed. In this study, we have identified new potential causative genes using cross-species comparative meta-analysis.
Methods:
Evolutionarily conserved differentially expressed genes were identified through in silico analysis, with in situ hybridization, gene knockdown, and rescue performed to confirm spatiotemporal gene expression and phenotype. Interrogation of the 100,000 Genomes Project for putative pathogenic variants was performed.
Results:
Nine conserved differentially expressed genes between zebrafish and mouse were identified. Expression of zebrafish ank3a, bmpr1ba/b, cdh4, and pdgfaa was localized to the OF, periocular mesenchyme cells, or ciliary marginal zone, regions traversed by the OF. Knockdown of ank3, bmpr1b, and pdgfaa revealed a coloboma and/or microphthalmia phenotype. Novel pathogenic variants in ANK3, BMPR1B, PDGFRA, and CDH4 were identified in 8 unrelated coloboma families. We showed BMPR1B rescued the knockdown phenotype but variant messenger RNAs failed, providing evidence of pathogenicity.
Conclusion:
We show the utility of cross-species meta-analysis to identify several novel coloboma disease-causing genes. There is a potential to increase the diagnostic yield for new and unsolved patients while adding to our understanding of the genetic basis of OF morphogenesis.
Keywords: Coloboma, Congenital eye defects, Disease genetics, Genomic sequencing, Microphthalmia
Introduction
Formation of the vertebrate eye involves coordinated assembly of cell types from different embryological origins and a series of tissue rearrangements. This complex process begins with the specification of eye field cells in the anterior neuroectoderm, followed by lateral migration and evagination toward the overlying surface ectoderm to form 2 optic vesicles.1,2 The interaction between the optic vesicles and the surface ectoderm induces the specification of lens placode, as well as initiates invagination of the optic vesicle to form a double-layered optic cup.3 The inner and outer layers of the optic cup give rise to the neural retina and the retinal pigment epithelium, respectively. An opening on the ventral surface of the optic cup leads to the formation of an optic fissure (OF), which enables the periocular mesenchyme (POM), composed of mesoderm and neural crest cells, to enter the developing eye and give rise to the hyaloid vasculature.4–6
The apposing edges of the OF begin to fuse bidirectionally from the midpoint proximally and distally, so the iris and the future optic nerve head fuse last. This process results in a continuous layered retina with no gaps. Unlike other epithelial fusion processes, which occur between the apical sides of cells, OF fusion begins with the apposition of the basal cell surface requiring breakdown of the basal lamina.4,7,8 An increasing number of reports show that the POM cells play a role in OF fusion, and although there is no clear mechanistic explanation, abrogation of POM specification or migration can lead to persistence of the OF.4
Partial or complete failure of OF fusion is known as ocular coloboma (OC) and in humans, it can lead to an inferonasal gap in the iris, ciliary body, choroid, retinal pigment epithelium, retina, and/or optic disc. This congenital eye defect has a prevalence of 7.5 in 10,000 births and accounts for 10% of childhood blindness worldwide.5 OC can be found in isolation, in association with other ocular malformations (complex), or as part of a syndrome with systemic features. It forms part of the clinical spectrum together with microphthalmia and anophthalmia (collectively known by the acronym MAC), with significant mixed ocular phenotypes and approximately 100 shared causative genes.6 Despite no notable environmental factors in pregnancy, diagnostic rates for patients remain around 10% on enriched targeted exome gene panels.9 Genome sequencing (GS) approaches bring the promise of increasing molecular diagnosis by a further 25% to 40%.10
Significant identification of genes involved in OF fusion and OC have originated from animal studies, including mice, chick, Xenopus, and zebrafish. We recently generated a temporal transcriptome of the developing OF in wild-type zebrafish to further our understanding of genes involved in tissue fusion events.11
To further identify evolutionarily conserved genes involved in OF morphogenesis and to aid the discovery of novel OC candidate genes, we undertook in silico cross-species comparative meta-analysis, mining mouse and zebrafish transcriptomes to identify conserved differentially expressed genes (DEGs).11,12 These genes were further studied using in situ hybridization to ascertain the spatiotemporal gene expression pattern in zebrafish, resulting in 8 potential candidates: ank3a, bmpr1ba, bmpr1bb, cdh4, nr2f2, pdgfaa, and pdgfab. We showed that knockdown of candidate genes can lead to failure of OF closure. Furthermore, we leveraged GS data from the 100,000 Genomes Project (launched in the United Kingdom in 2012 to improve diagnosis and clinical management of those with rare diseases or cancer)13 for variants in the aforementioned genes from patients with human phenotype ontology terms encompassing MAC. We found novel pathogenic variants in ANK3, BMPR1B, CDH4, and PDGFRA. In vivo zebrafish assays were performed, combining gene knockdown and human messenger RNA (mRNA) rescue to assess specificity of targeting. Variants reported in BMPR1B were recapitulated, and the variant mRNA failed to rescue the phenotype. We show that our approach can identify new candidate genes, which may now be included in diagnostic gene panels.
Materials and Methods
Zebrafish husbandry
Adult zebrafish (wild-type, AB-strain) were maintained at the University College London main campus zebrafish facility under standard husbandry conditions, and embryos were obtained by natural spawning. Wild-type and mutant embryos were raised at 28.5 °C and staged according to Kimmel et al.14 To minimize variations in staging, embryos were collected every 30 minutes and kept in separate clutches according to their time of fertilization. When necessary, pigmentation development was inhibited by incubating embryos in 0.2 mM phenylthiourea 24 hours post fertilization (hpf).
Morpholino embryo microinjection and mRNA rescue
Translational blocking morpholinos (MOs) were used to knockdown endogenous zebrafish genes (Gene Tools, LLC). One to 2-cell stage embryos were coinjected with 10 nL of a mix of GFP mRNA (50 pg) and MOs in the amounts indicated: ank3a (0.33 pmol, 5′-TTTTTTCTTTGGCTTCT TCACTCAT-3′), ank3b (0.25 pmol, 5′-TTTTTTCTTTGG CTTCTTCACTCAT-3′), bmpr1ba (2 pmol, 5′-CATCGTG CGTCTGCAGAACTCCAGT-3′), bmpr1bb (1 pmol, 5′-TC CGTACCCGGAGCTCCATCCCTGA-3′), cdh4 (0.1 pmol, 5′-CATGTTTAACGTTACGGGCTTGTCT-3′), pdgfaa (0.2 pmol, 5′-GTGGTCCAAAAGTGTCCCAGCGCAG-3′), pdgfab (0.2 pmol, 5′-CAATGTCAATTTCCCACCCAGCA AG-3′), and control (0.25 pmol, 5′-TGTTGAAATCAG CGTGTTCAAG-3′). Coinjection of p53 ATG-MO was used to control for p53-dependent neural toxicity. Each MO was injected into approximately 100 embryos, and the surviving ones were collected. For rescue, plasmid containing wild-type or variant BMPR1B was linearized by AscI (New England BioLabs) and heat inactivated for 20 minutes at 80 °C. Linearized complementary DNA was purified using QIAquick PCR purification kit (QIAGEN). Capped mRNAs were transcribed in vitro using MEGAscript T7 Transcription Kit (AM1334, ThermoFisher Scientific) for each template and purified using MEGAclear Transcription Clean-Up Kit (AM1908, ThermoFisher Scientific). In vitro synthesized capped mRNAs were individually coinjected with the MO for rescue experiments (75 pg).
In situ hybridization and immunohistochemistry
Whole mount in situ hybridization was performed using digoxigenin (DIG)-labelled RNA probes according to standard protocols.15 Probes were synthesized using T7 or T3 RNA polymerases (Promega) according to manufacturers’ instructions and supplied with DIG labelled UTP (Roche). Probes were detected with anti-DIG-AP (1:5000, 110932 74910 Roche) or anti-DIG-POD (1:1000, 11207733910 Roche) antibodies and developed using NBT/BCIP mix (Roche) for regular microscopy or CY-3 tyramide substrate for confocal analysis. Laminin subunit alpha 1 antibody (L9393, Sigma-Aldrich) was used at 1 per 100 for whole mount immunohistochemistry using 4′,6-diamidino-2-phenylindole counterstain at 2 μM when incubating with antimouse Alexa 568 secondary antibody (A-11011, ThermoFisher Scientific) as previously described.4 Confocal imaging was performed on a Leica TCS SP8 confocal microscope. Image analysis measurement was performed using Fiji.16
Eye measurement and statistics
Horizontal eye diameter was determined by imaging lateral views of anaesthetised larvae at 3 days after fertilization. Images were analyzed using Fiji, and for each group, the mean ± SD was reported. Data were compared using unpaired t tests or Mann-Whitney tests; P < .05 was considered to be statistically significant. For animal studies, no randomization was used and no blinding was done. The investigators were not blinded to the group allocation during experiments and/or when assessing the outcome. The variance was similar between the groups that were statistically compared.
Patient involvement and GS
All studies conducted were approved by Moorfields Eye Hospital and the National Research Ethics Committee and were conducted in adherence to the tenets of the Declaration of Helsinki. Patients and relatives gave written informed consent for participation in this study through either the Genetic Study of Inherited Eye Disease (REC reference 12/LO/0141) or Genomics England 100,000 Genomes project (REC reference 14/EE/1112). GS was performed through the Genomics England 100,000 Genomes Project (GE100KGP) as previously described.17
Results
Comparison of conserved gene expression in vertebrate data sets
Our previous zebrafish RNA sequencing analysis established temporal and spatial expression of genes during OF closure.11 We identified 178 DEGs at prefusion time point (32 hpf), 78 at perifusion time point (48 hpf), and 62 at postfusion time point (56 hpf) through comparison of tissue isolated from the OF region and dorsal retina. For cross-species comparison, we used a microarray data set quantifying gene expression in mouse retinal tissue margins spanning the OF between E10.5 and E12.5,12 identifying 168 significantly DEGs.
Conservation across species within the DEG groups was confirmed and commonalities identified. Representation of common genes across species and temporal changes are shown in Figure 1 and detailed in Supplemental Table 1. We identified 9 genes that showed concordant differential expression across mouse and zebrafish OF and dorsal retina: ank3a, bmpr1b, cdh4, gadd45a, nedd9, nr2f2, pdgfa, smoc1, and zbtb4. Initially, we searched for biological significance, inspecting variant, knockout, and expression databases (Mouse Genome Informatics and The Zebrafish Information Network). We further queried clinical databases to identify genes previously associated with human variants and the MAC disease spectrum (ClinVar and Genetic Home Reference). Smoc1 has been previously identified as a causative gene for human anophthalmia and microphthalmia,18–20 and zebrafish MO knockdown experiments generated a small eye phenotype but lacked a coloboma.19 Knockdown of zbtb4 in zebrafish embryos showed high embryonic mortality (90%) with surviving morphants exhibiting early developmental defects, including microcephaly, which would prevent potential identification of eye defects.21 Therefore, both genes were excluded from further analysis.
Figure 1. Venn diagram of the significantly regulated genes identified in mouse and zebrafish optic fissure developmental time points.

The figure shows comparisons between differentially expressed gene lists of mouse and zebrafish optic fissure vs retinal tissue, with the numeric value indicating the number of genes at each intersection. Identified commonalities of genes are represented in each box, with colors representing each data set intersection. hpf, hours post fertilization.
Phenotypic analysis of candidate gene knockdown reveals OC
To characterize loss of function (LoF), we individually knocked down the zebrafish candidate genes using translational blocking MO antisense oligonucleotides, including ank3a, ank3b, bmpr1ba, bmpr1bb, pdgfaa, and cdh4. At 56 hpf, OF fusion is complete after remodeling of the basal lamina of the apposing edges of the ventral retina with disassembly of the laminin rich extracellular matrix. Coloboma phenotypes can have a variable severity, and hence, laminin immunostaining was performed at 76 hpf to exclude the possible effect of nonspecific delay in experimental conditions. Knockdown of ank3a (Figure 2B; 79% penetrance, n = 12 embryos analyzed from 2 independent experiments) and ank3b (Figure 2C; 75% penetrance, n = 19 embryos) or both genes simultaneously (75% penetrance, n = 20) resulted in microphthalmia and penetrant coloboma phenotype. Laminin localization showed the position of the apposing edges of the OF. The mean eye diameter of 76 hpf ank3a, ank3b, and ank3a/ank3b morphants were 273 ± 27 μm, 252 ± 31 μm, and 283 ± 24 μm, respectively, compared with 308 ± 16 μm (n = 28) in wild-type larvae at the same timepoint (P < .0001 for all 3 morphant groups).
Figure 2. Knockdown analysis of ventral retina and optic fissure specific genes in zebrafish.
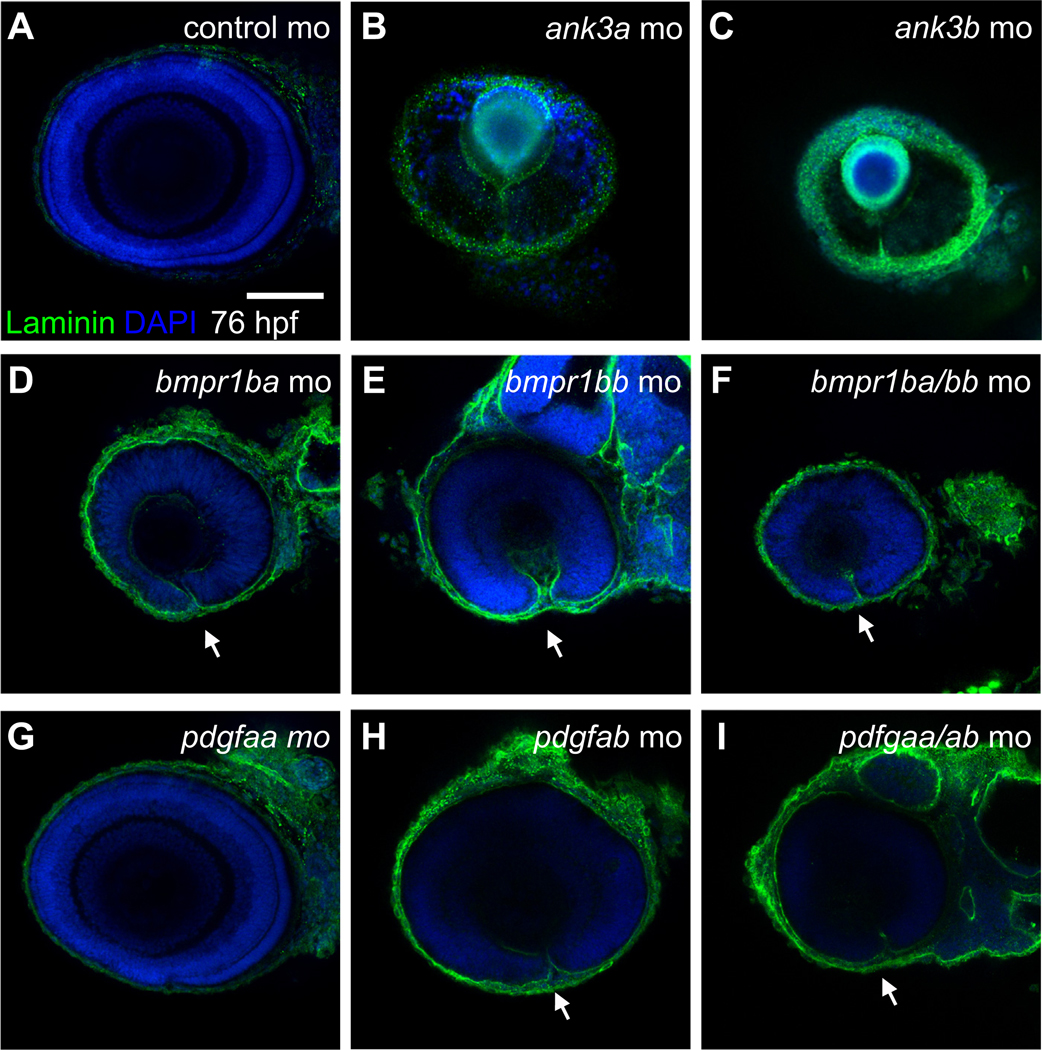
Confocal imaging of lateral views (dorsal up, anterior left) of immunofluorescence staining of laminin protein in zebrafish embryos at 76 hours post fertilization in (A) control MO or MO knockdowns of (B) ank3a, (C) ank3b, (D) bmpr1ba, (E) bmpr1bb, (F) bmpr1ba/bmpr1bb, (G) pdgfaa, (H) pdgfab, and (I) pdgfaa/pdgfab mutant embryos. Scale bar = 100 μm. MO, morpholino.
Knockdown of bmpr1ba (Figure 2D; 85% with coloboma penetrance, n = 13), bmpr1bb (Figure 2E; 80% penetrance, n = 23), or double bmpr1ba/bb (Figure 2F; 1 pmol of each MO per embryo, 91% penetrance, n = 21) showed a penetrant coloboma covering the whole ventral retina assessed by the expression of laminin remaining along the whole proximal-distal axis of the OF. Knockdown of the bmpr1ba, bmpr1bb, and double knockdown of both genes also led to microphthalmia, with a significant reduction in eye size measured at 76 hpf compared with that of wild type. The mean eye diameter of bmpr1ba, bmpr1bb, and bmpr1ba/bmp1bb morphants were 230 ± 13 μm, 273 ± 28 μm, and 249 ± 18 μm, respectively (P < .0001 for all 3 morphant groups).
Knockdown of pdgfaa did not show a significant coloboma phenotype (Figure 2G; n = 12). However, pdgfab knockdown lead to mild coloboma in 50% of the embryos, with laminin expression extending through half the distalproximal axis of the ventral retina at 76 hpf (Figure 2H; n = 21). All double pdgfaa/ab morphants displayed a more severe coloboma phenotype (Figure 2I, 0.1 pmol each MO per embryo, 100%, n = 22). The mean eye diameter of pdgfaa, pdgfab, and pdgfaa/pdgfab morphants were 302 ± 12 μm, 284 ± 19 μm, and 267 ± 24 μm, respectively (P = 0.04, P < .0001, and P < .0001 respectively) indicating microphthalmia.
cdh4 knockdown with 0.2 pmol MO per embryo led to pleiotropic developmental defects with embryonic maldevelopment and necrosis as previously described.22 To circumvent this, embryos were injected with 0.1 pmol MO, which revealed a colobomatous defect at 76 hpf (65% penetrance, n = 22, Figure S3). cdh4 morphants displayed microphthalmia with a mean eye diameter of 227 ± 19 μm (P < .0001 compared with controls).
In silico identification of candidate gene variants in patients with ocular maldevelopment
This study was undertaken as part of the Genomics England Clinical Interpretation Partnership to analyze genomic data from the 100,000 Genomes Project.13 We screened GS data to identify putative novel genes that may cause MAC phenotypes. Using UK 100,000 Genomes Project Data Release 8.0 (November 28, 2019) we identified 325 unsolved patients suitable for screening and variant prioritization. Table 1 presents a summary of our findings. Six families were identified with a diagnosis of OC and/or microphthalmia and likely pathogenic variants in ANK3, BMPR1B, CDH4, and PDGFRA. Variants were predicted to be disease causing/damaging with low or no allele frequency in Genome Aggregation Database or the Human Genetic Variation Database, classified on the basis of the American College of Medical Genetics and Genomics/Association of Molecular Pathology (ACMG/AMP) criteria.23 The probability of a gene being LoF intolerant (pLI) scores were reported as ANK3 (pLI 1), BMPR1B (pLI 1), CDH4 (pLI 0.73), and PDGFRA (pLI 1). No putative pathogenic variants were detected in GADD45A, NEDD9, NR2F2, and PDGFA genes. Deleterious variants, including structurals and copy number variations, were not detected in genes previously known to cause MAC in these subjects (PanelApp – Genomics England, Anophthalmia or microphthalmia [Version 1.42], Ocular coloboma [Version 1.44] gene panels).
Table 1.
Summary of in silico analysis of single nucleotide variants (SNVs) identified
| Fs | HPO Terms | Gene | cDNA / GRCh38 | Protein | Exon | SIFT | PolyPhen-2 | MutationTaster | CADD | gnomAD |
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| F1 | Visual impairment, retinal coloboma, iris coloboma, chorioretinal coloboma | ANK3 |
NM_020987.5 c.11650 A>T 10:60069231:T:A |
p.(Thr3884Ser) | 37 | Damaging | Probably damaging | Disease causing | 15.12 | <0.002% |
| F2 | Visual impairment, coloboma, global developmental delay, micropenis | ANK3 |
NM_020987.5 c.3658 A>G 10:60086767:T:C |
p.(Ile1220Val) | 30 | Damaging | Damaging | Disease causing | 25.8 | <0.002% |
| F3 | Visual impairment, optic nerve coloboma | BMPR1B |
NM_001203.2 c.272 G>T 4:95115710:G:T |
p.(Arg91Ile) | 6 | Damaging | Probably damaging | Disease causing | 27.9 | <0.007% |
| F4 | Visual impairment, microphthalmia | BMPR1B |
NM_001203.2 c.1127 G>A 4:95148798:G:A |
p.(Arg376His) | 11 | Deleterious | Probably damaging | Disease causing | 32.0 | NF |
| F5 | Bilateral iris coloboma and chorioretinal coloboma | BMPR1B |
NM_001203.2 c.671 G>A 4:95129947:G:A |
p.(Arg224His) | 9 | Deleterious | Benign | Disease causing | 25.8 | <0.096% |
| F6 | Iris coloboma, chorioretinal coloboma | BMPR1B |
NM_001203.2 c.671 G>T 4:95129947:G:T |
p.(Arg224Leu) | 9 | Deleterious | Probably damaging | Disease causing | 31 | NF |
| F7 | Visual impairment, bilateral microphthalmos, bilateral retinal coloboma | PDGFRA |
NM_006206.6 c.1295 C>T 4:54272451:C:T |
p.(Thr432Met) | 9 | Damaging | Probably damaging | Disease causing | 13.27 | <0.002% |
| F8 | Iris coloboma | CDH4 |
NM_001794.5 c.1291 C>T 20:61910524:C:T |
p.(Arg431Cys) | 9 | Damaging | Benign | Disease causing | 24.5 | NF |
Screening of probands enrolled in the 100,000 Genomes Project presenting with HPO terms, including microphthalmia, anophthalmia, or coloboma, for putative damaging variants resulted in identifying 8 families. Position of the SNV in the cDNA and subsequent amino acid change along with the genomic location for the human reference genome GRCh38 are presented in the table. All variants identified were heterozygous. Outcomes of predictive algorithms SIFT, PolyPhen-2, and MutationTaster are reported, with respective scaled CADD scores. Allele frequency was identified in gnomAD (accessed January 2020). Two more Fs (F5 and F6) were detected through international collaboration.
CADD, Combined Annotation Dependent Depletion; cDNA, complementary DNA; F, family;gnomAD, Genome Aggregation Database; HPO, Human Phenotype Ontology; PolyPhen, Polymorphism Phenotyping; NF, not found; SIFT, Sorting Intolerant from Tolerant; SNV, single nucleotide variant.
We had access to families recruited into the 100,000 Genome Project through Moorfields Eye Hospital and Great Ormond Street Hospital for Children. Supplemental Table 2 summarizes the clinical details of 8 unrelated families. Family 1 (nonconsanguineous, White) had a proband with no previous family history, and segregation of parents was negative, indicative of a de novo sporadic heterozygous missense variant in ANK3 NM_020987.5:c.11650A>T, p.(Thr3884Ser). Consistent with the zebrafish morphants, the patient (P1) displayed bilateral chorioretinal coloboma involving the optic discs (Figure 3A and B). The variant was classified as pathogenic on the basis of ACMG/AMP criteria with evidence levels pathogenic strong (PS)-2, pathogenic moderate (PM)-2, pathogenic supporting (PP)-3, and PP4. Family 2 (nonconsanguineous, Black) proband was a male, presenting with bilateral optic nerve hypoplasia and left optic disc coloboma, and had a heterozygous variant in ANK3 NM_020987.5:c.3658A>G, p.(Ile1220Val) identified and classified as pathogenic on the basis of ACMG/AMP criteria (PS2, PM2, PP2/3/4); the unaffected mother did not carry the variant. Family 3 (nonconsanguineous, Indian) showed an autosomal dominant inheritance with 2 affected siblings and the mother with the confirmed heterozygous missense BMPR1B variant NM_001203.2:c.272G>T, p.(Arg91Ile), and each recorded to have bilateral optic disc coloboma (Figure 3C and D). The variant was classified as pathogenic on the basis of ACMG/AMP criteria with evidence levels PS2, PM2, PP3, and PP4. Family 4 (non-consanguineous, White) consisted of a single proband and no family history, and segregation of parents was negative; a de novo sporadic heterozygous missense variant was found in BMPR1B NM_001203.2:c.1127G>A, p.(Arg376Glu). This patient had a unilateral right microphthalmia, right dense cataract with no view of the fundus, but at age 5 weeks he underwent an examination under anesthesia, which reported persistent hyperplastic primary vitreous. The variant was classified as pathogenic on the basis of ACMG/AMP criteria (PS2, PM1, PM2, PP3, and PP4). Collaboration with the National Eye Institute aided the identification of a 4-year-old White male with bilateral iris and chorioretinal coloboma harboring a variant in BMPR1B exon 7; c.671G>A, p.(Arg224His) (family 5, Figure 3E and F). The variant was classified as pathogenic on the basis of ACMG/AMP criteria (PS2, PM1, PM2, PP4). In addition, in an unrelated 10-month-old White male (family 6) with right iris and bilateral chorioretinal coloboma with a c.671G>T, p.(Arg224Leu) variant in exon 7 of BMPR1B, this variant was classified as likely pathogenic on the basis of ACMG/AMP criteria (PS2, PM2, PP2, PP3, and PP4). Two more variants were identified in families within Genomics England 100,000 Genomes Project external to the Moorfields Eye Hospital clinic; family 7 proband presented with bilateral chorioretinal coloboma and microphthalmia as well as global developmental delay with autistic behavior and harbored a heterozygous variant in PDGFRA NM_006206.6:c.1295C>T, p.(Thr432Met) classified as likely pathogenic on the basis of ACMG/AMP criteria (PS2, PM2, PP2, PP3, and PP4), and the unaffected mother was homozygous for the wild-type reference sequence. Family 8 proband presented with an iris coloboma, intellectual disability, and microcephaly, in which we identified a heterozygous variant CDH4 NM_001794.5:c.1291C>T, p.(Arg431Cys), which was classified as likely pathogenic (PS2, PM2, PP2, PP3 and PP4), and the unaffected mother was homozygous for the reference sequence. Supplemental Table 3 outlines the classification criteria.
Figure 3. Pedigrees and clinical imaging of 3 unrelated coloboma families.

A. F1 is a White British nonconsanguineous pedigree with 1 affected male patient (proband 1–3) harboring a de novo sporadic heterozygous missense variant in ANK3 NM_020987.5:c.11650A>T, p.(Thr3884Ser). B. Corresponding widefield color fundus photographs of the right and left eye of the proband aged 17 years showing bilateral inferior chorioretinal coloboma with extensive optic disc involvement (right eye is more severely affected than the left eye as indicated also by the residual best corrected visual acuity). C. F3 is an Indian nonconsanguineous pedigree with autosomal dominant inheritance; the mother and the 2 daughters were affected with a heterozygous missense variant in BMPR1B NM_001203.2:c.272G>T, p.(Arg91Ile). D. Widefield color fundus photograph of the right eye of the proband (3–4) aged 13 showed a large optic disc coloboma extending inferiorly with a region of associated retinal pigment epithelium atrophy below this. The left eye image was not available. Widefield color fundus photographs of the right and left eye of the proband (3–3) aged 18 showing bilateral optic disc coloboma sparing the macula, with irregular asymmetrical peripapillary atrophy in the right eye. E. F5 is a White Northern European nonconsanguineous pedigree with 1 affected male patient (proband 5–3) with a heterozygous missense variant in BMPR1B NM_001203.2:c.671 G>A, p.(Arg224His). Parental samples for segregation were not available. F. Anterior segment color photographs of the right and left eye of the proband aged 4 showing bilateral inferior iris coloboma and corresponding widefield color fundus photographs showing bilateral inferior chorioretinal coloboma with extensive optic disc and macular involvement. F1, family 1; F3, family 3; F5, family 5.
Human BMPR1B rescues morphant phenotype in zebrafish
To validate the consequences of the bmpr1b MO effect, we employed a rescue approach with coinjection of human BMPR1B mRNA. The severe phenotype of bmpr1ba MO was rescued through concomitant microinjection of human wild-type BMPR1B mRNA, wherein the exogenous expression of wild-type BMPR1B was able to restore eye diameter from mean 215 ± 4 μm to 250 ± 2 μm (Figure 4A). Uninjected wild-type controls had an eye diameter of 301 ± 11 μm. No rescue of the phenotype was observed in embryos coinjected with mRNA carrying the patient-specific variants identified (c.272G>T, p.[Arg91Ile], 198 ± 3 μm; c.671G>A, p.[Arg224His], 214 ± 5 μm; c.671G>T, p.[Arg224Leu], 220 ± 5 μm; and c.1127G>A, p.[Arg376Glu], 232 ± 4 μm) (Figure 4B), confirming the LoF effect of the variants on BMPR1B function. The morpholino phenotype was less severe for bmpr1bb, and although remaining significant, rescue with human BMPR1B had a less pronounced effect (Figure 4C).
Figure 4. MO knockdown and rescue experiments of bmpr1ba/b in zebrafish.

Injection of the MOs alone showed reduction in eye size and the presence of a coloboma. A. Coinjection with in vitro synthesized human BMPR1B messenger RNA (mRNA) rescues the phenotypic effects of the MO alone. Human variants identified within the Genomics England cohort were expressed and mRNAs coinjected with each MO. Scale bar 500 μm. Eye diameter was reported for (B) bmpr1ba and (C) bmpr1bb compared with wild-type uninjected; MO; MO + human wild-type BMPR1B; and variants Arg91Ile, Arg224His, Arg224Leu, and Arg376Glu. (n for bmpr1ba experiments = 23, 25, 55, 13, 21, 28, 42 and n for bmpr1bb experiments = 23, 56, 55, 32, 34, 33, 24). Unpaired t tests were used to compare data. **P < .01, ***P < .001, ****P < .0001, ns P > .05. MO, morpholino; ns, nonsignificant.
Discussion
In this investigation, we have shown that the study of genes with evolutionarily conserved expression identified by comparing model system expression data sets is a powerful approach to discover new variants involved in human diseases. It has led to the discovery of 4 novel OC genes ANK3, BMPR1B, CDH4, and PDGFRA, with pathogenic variants of diagnostic value not previously reported in this patient cohort. There is significant genetic and phenotypic heterogeneity in patients with MAC. It is rarely an isolated pure disorder but more commonly found as a mixed presentation with between 33% and 95% of cases displaying additional nonocular systemic malformations.6 Genes coordinating optic vesicle and fissure morphogenesis are time- and dose-critical. Genetic perturbations can have a global effect on ocular development with most variants associated with nonsyndromic MAC arising as a de novo sporadic .24 With these dominant heterozygous variants, variable expressivity and penetrance can be seen, and this may be linked to genetic modifiers and/or stochastic environmental factors, which influence the levels of gene expression. Unilateral ocular phenotypes were seen in patients 3–2, 4–1, and 8–1. Such phenotypic variation is not an uncommon finding in individuals with molecularly-confirmed isolated and syndromic MAC. Several hypotheses have been put forward to explain this laterality despite the presence of germline variants, including an intrinsic sensitivity of the developing eye to stressors, suggesting that a potential hypomorphic allele may be more penetrant in ocular tissues than systemically. A study identified 3 human patients with unilateral microphthalmia caused by germline TMX3 variants and postulated that the asymmetry was due to a delay in eye formation and that reduced gene dosage could be compensated for by different genes.25,26
The BMP signaling pathway has been previously related to eye malformations.6,24 BMP signaling is known to work at 2 crucial stages of eye formation, at eye field specification during gastrulation1 and the process of optic cup morphogenesis.2 For example, BMP4 LoF results in a MAC phenotype, whereas BMP7 LoF specifically results in coloboma.27,28 A recent study has reported a mouse model harboring an N-ethyl-N-nitrosourea (ENU) derived Bmpr1b variant showing optic nerve coloboma, with gliosis of the ventral retina.29 Our experiments in zebrafish show that inhibition of bmpr1ba/bb leads to embryos with coloboma and microphthalmia. This is in line with previous reports that have shown that impairment of the BMP pathway can lead to a persistent OF and morphological defects of the optic cup because it may be required for the migration of optic stalk cells into the anterior neural retina.30,31 Importantly, our study reveals that human BMPR1B performs a similar function to zebrafish protein because the human wild-type BMPR1B can compensate for the knockdown in zebrafish morphants. However, variant proteins (Arg91Ile, Arg224His, Arg224Leu, and Arg376Glu) cannot rescue the morphant phenotype, suggesting these variants interfere with BMPR1B function.
The ankyrins are a family of proteins whose key role is linking membrane-spanning proteins to the underlying actin cytoskeleton. Ankyrins have been shown to modulate cellular organization; AnkG (encoded by the Ank3 gene) is specifically required for epithelial cell polarity, proliferation, and cell survival.32 A key protein partner of AnkG is SMAD2, one of the main transcription factors in the canonical TGF-β signaling pathway, which is involved in direct signaling from TGF-β receptors in BMP pathways.31 Modulation of BMP signaling by variants in YAP1 via interaction with SMAD7, a major contributor to the BMP pathway, has been shown to result in OF closure defects, including coloboma.33 It is therefore plausible that variants in ANK3 result in altered BMP signaling through a similar manner, altering SMAD7 regulation through the interaction with SMAD2 and leading to the OC observed in this study. Further investigation of SMAD2/7 expression patterns in ank3a/b morphants would be worth evaluation.
PDGF signaling has been related to neural crest migration and vasculature formation in early development.34,35 The POM cells are neural crest derivatives that give rise to the retinal vasculature that enters through the OF and later contribute to anterior segment development.36,37 Depletion of POM cells from zebrafish optic vesicles have resulted in failure of OF closure.4 PDGF activates a cellular response through cell surface receptors PDGFR(A/B), binding to cofactors and activating signal transduction. PDGFRs are involved in several signaling pathways: RAS/MAPK, STAT3, and VEGF. Increased apoptosis on the pathway of migrating neural crest cells was observed in Pdgfra knockout mice, and reduced capacity for migration and neuronal differentiation was reported in the Pdgfrb knockout mice.38 The inhibition of PDGFA leads to OC by potentially disrupting the migration of cranial neural crest cells into the eye.
Cadherin adhesion molecules are required for eye development, and variants in CDH23 and PCDH15 can lead to inherited eye disorders such as Usher syndrome.39 Developmental eye defects such as microphthalmia, cataracts, and microspherophakia and lack of retinal lamination have been previously shown in MO knockdown of cdh4 in zebrafish.22 Reduced dose knockdown of cdh4 in this study resulted in overall normal embryo development with no evident necrosis but a resultant coloboma phenotype. Laminin expression was downregulated suggesting a regulatory role in this pathway. Laminin variants (subunits beta-1 and gamma-1) have been found to display colobomatous defects in zebrafish,40 but no corresponding human variants have been previously described with this phenotype. Of note, the proband identified in family 8 presented with intellectual disability and postnatal microcephaly; similarly, the cdh4 morphant phenotype at standard dose presented with pleiotropic developmental defects. Knockdown of cdh4 in zebrafish results in evidence of brain disorganization.22
Overall, by studying genes that show an evolutionarily conserved expression in the eye, we have generated a pipeline that has allowed the identification of new candidate disease genes. Most patients with MAC harbor de novo sporadic heterozygous variants that lead to dominant inheritance, which has implications for future generations. Improving diagnostic capabilities will allow for informed genetic counselling and family planning, such as preimplantation diagnosis or noninvasive prenatal diagnosis. Using transcriptome data sets may allow us to compare genes involved in other tissue fusion events such as the neural tube, providing further universal gene candidates. Future investigation of functional pathways may contribute to the development of preventative therapeutic interventions, similar to folic acid supplementation, which may limit the severity of tissue closure defects.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding from the Wellcome Trust 205174/Z/16/Z (M.M.), Moorfields Eye Charity (M.M.), and BIPOSA Research Award (M.M.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support. We would also like to thank Professor Trevor C. Dale from Cardiff University for kindly donating the cdh4 MO.
Footnotes
Ethics Declaration
All studies conducted were approved by Moorfields Eye Hospital and the National Research Ethics Committee and were conducted in adherence to the tenets of the Declaration of Helsinki. Informed consent and consent to publish phenotypes were obtained from all subjects or patient’s legal guardians through either the Genetic Study of Inherited Eye Disease (REC reference 12/LO/0141) or Genomics England 100,000 Genomes project (REC reference 14/EE/1112). Zebrafish were maintained according to institutional regulations for the care and use of laboratory animals under the UK Animals Scientific Procedures Act and the UCL Animal Welfare and Ethical Review Body (License no. PPL PC916FDE7). All approved standard protocols followed the guidelines of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research Ethics.
Conflict of Interest
The authors declare no conflict of interest.
Members of the Genomics England Research Consortium
J. C. Ambrose, E. L. Baple, M. Bleda, F. Boardman-Pretty, J. M. Boissiere, C. R. Boustred, M. J. Caulfield, G. C. Chan, C. E. H. Craig, L. C. Daugherty, Burca A. de, A. Devereau, G. Elgar, R. E. Foulger, T. Fowler, P. Furió-Tarí, J. M. Hackett, D. Halai, J. E. Holman, T. J. P. Hubbard, R. Jackson, D. Kasperaviciute, M. Kayikci, L. Lahnstein, K. Lawson, S. E. A. Leigh, I. U. S. Leong, F. J. Lopez, F. Maleady-Crowe, J. Mason, E. M. McDonagh, L. Moutsianas, M. Mueller, N. Murugaesu, A. C. Need, C. A. Odhams, C. Patch, D. Perez-Gil, D. Polychronopoulos, J. Pullinger, T. Rahim, A. Rendon, P. Riesgo-Ferreiro, T. Rogers, M. Ryten, K. Savage, K. Sawant, R. H. Scott, A. Siddiq, A. Sieghart, D. Smedley, K. R. Smith, A. Sosinsky, W. Spooner, H. E. Stevens, A. Stuckey, R. Sultana, E. R. A. Thomas, S. R. Thompson, C. Tregidgo, A. Tucci, E. Walsh, S. A. Watters, M. J. Welland, E. Williams, K. Witkowska, S. M. Wood, M. Zarowiecki
Website Resources
Mouse Genome Informatics. The Jackson Laboratory. Accessed November 11, 2019. http://www.informatics.jax.org/
ZFIN. University of Oregon. Accessed November 11, 2019. http://zfin.org/
ClinVar. National Center for Biotechnology Information. Accessed November 10, 2020. https://www.ncbi.nlm.nih.gov/clinvar/
Genetic Home Reference. National Library of Medicine. Accessed November 11, 2019. https://ghr.nlm.nih.gov/
SIFT. Bioinformatics Institute. Accessed November 10, 2020. https://sift.bii.a-star.edu.sg/
PolyPhen2. Brigham and Women’s Hospital, Harvard Medical School. Accessed November 10, 2020. http://genetics.bwh.harvard.edu/pph2/
MutationTaster. Berlin Institute of Health. Accessed November 10, 2020. http://mutationtaster.org/
Provean. J. Craig. Venter Institute. Accessed November 10, 2020. http://provean.jcvi.org/
gnomAD. Broad Institute. Accessed November 10, 2020. https://gnomad.broadinstitute.org
Human Genetic Variation Database. Kyoto University. Accessed November 10, 2020. https://www.hgvd.genome.med.kyoto-u.ac.jp/
Gene Expression Omnibus (GEO). National Center for Biotechnology Information. Accessed November 11, 2019. https://www.ncbi.nlm.nih.gov/geo/
Combined Annotation Dependent Depletion (CADD). University of Washington, Hudson-Alpha Institute for Biotechnology and Berlin Institute of Health. Accessed November 10, 2020. https://cadd.gs.washington.edu
Genomics England PanelApp. Accessed March 26, 2020. https://panelapp.genomicsengland.co.uk
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2021.12.014) contains supplementary material, which is available to authorized users.
Data Availability
The expression data used in this manuscript are at Gene Expression Omnibus under the accession numbers GSE159822 and GSE13103 and are currently open access.
References
- 1.Giger FA, Houart C. The birth of the eye vesicle: when fate decision equals morphogenesis. Front Neurosci. 2018;12:87. 10.3389/fnins.2018.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez-Morales JR, Cavodeassi F, Bovolenta P. Coordinated morphogenetic mechanisms shape the vertebrate eye. Front Neurosci. 2017;11:721. 10.3389/fnins.2017.00721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno-Marmol T, Cavodeassi F, Bovolenta P. Setting eyes on the retinal pigment epithelium. Front Cell Dev Biol. 2018;6:145. 10.3389/fcell.2018.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gestri G, Bazin-Lopez N, Scholes C, Wilson SW. Cell behaviors during closure of the choroid fissure in the developing eye. Front Cell Neurosci. 2018;12:42. 10.3389/fncel.2018.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel A, Sowden JC. Genes and pathways in optic fissure closure. Semin Cell Dev Biol. 2019;91:55–65. 10.1016/j.semcdb.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Harding P, Moosajee M. The molecular basis of human anophthalmia and microphthalmia. J Dev Biol. 2019;7(3):16. 10.3390/jdb7030016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geeraets R. An electron microscopic study of the closure of the optic fissure in the golden hamster. Am J Anat. 1976;145(4):411–431. 10.1002/aja.1001450402. [DOI] [PubMed] [Google Scholar]
- 8.Hero I. The optic fissure in the normal and microphthalmic mouse. Exp Eye Res. 1989;49(2):229–239. 10.1016/0014-4835(89)90093-6. [DOI] [PubMed] [Google Scholar]
- 9.Patel A, Hayward JD, Tailor V, et al. The Oculome panel test: next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology. 2019;126(6):888–907. 10.1016/j.ophtha.2018.12.050. [DOI] [PubMed] [Google Scholar]
- 10.Ma A, Yousoof S, Grigg JR, et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet Med. 2020;22(10):1623–1632. 10.1038/s41436-020-0854-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richardson R, Owen N, Toms M, Young RM, Tracey-White D, Moosajee M. Transcriptome profiling of zebrafish optic fissure fusion. Sci Rep. 2019;9(1):1541. 10.1038/s41598-018-38379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown JD, Dutta S, Bharti K, et al. Expression profiling during ocular development identifies 2 Nlz genes with a critical role in optic fissure closure. Proc Natl Acad Sci U S A. 2009;106(5):1462–1467. 10.1073/pnas.0812017106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caulfield M, Davies J, Dennys M, Elbahy L, Fowler T, Hill S, et al. (2017): The National Genomics Research and Healthcare Knowledgebase. figshare. Dataset 10.6084/m9.figshare.4530893.v5. [DOI] [Google Scholar]
- 14.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 15.Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3(1):59–69. 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 16.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor RL, Arno G, Poulter JA, et al. Association of steroid 5α-reductase type 3 congenital disorder of glycosylation with early-onset retinal dystrophy. JAMA Ophthalmol. 2017;135(4):339–347. 10.1001/jamaophthalmol.2017.0046. [DOI] [PubMed] [Google Scholar]
- 18.Okada I, Hamanoue H, Terada K, et al. SMOC1 is essential for ocular and limb development in humans and mice. Am J Hum Genet. 2011;88(1):30–41. 10.1016/j.ajhg.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abouzeid H, Boisset G, Favez T, et al. Mutations in the SPARC-related modular calcium-binding protein 1 gene, SMOC1, cause Waardenburg anophthalmia syndrome. Am J Hum Genet. 2011;88(1):92–98. 10.1016/j.ajhg.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldahmesh MA, Khan AO, Hijazi H, Alkuraya FS. Mutations in ALDH1A3 cause microphthalmia. Clin Genet. 2013;84(2):128–131. 10.1111/cge.12184. [DOI] [PubMed] [Google Scholar]
- 21.Ruzov A, Savitskaya E, Hackett JA, et al. The non-methylated DNA-binding function of Kaiso is not required in early Xenopus laevis development. Development. 2009;136(5):729–738. 10.1242/dev.025569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babb SG, Kotradi SM, Shah B, et al. Zebrafish R-cadherin (Cdh4) controls visual system development and differentiation. Dev Dyn. 2005;233(3):930–945. 10.1002/dvdy.20431. [DOI] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williamson KA, FitzPatrick DR. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet. 2014;57(8):369–380. 10.1016/j.ejmg.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Chao R, Nevin L, Agarwal P, et al. A male with unilateral microphthalmia reveals a role for TMX3 in eye development. PLoS One. 2010;5(5), e10565. 10.1371/journal.pone.0010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ragge NK, Brown AG, Poloschek CM, et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 2005;76(6):1008–1022. Published correction appears in Am J Hum Genet. 2005;77(2):334. 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyatt AW, Osborne RJ, Stewart H, Ragge NK. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum Mutat. 2010;31(7):781–787. 10.1002/humu.21280. [DOI] [PubMed] [Google Scholar]
- 28.Takenouchi T, Nishina S, Kosaki R, et al. Concurrent deletion of BMP4 and OTX2 genes, two master genes in ophthalmogenesis. Eur J Med Genet. 2013;56(1):50–53. 10.1016/j.ejmg.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Yan X, Atorf J, Ramos D, et al. Mutation in Bmpr1b leads to optic disc coloboma and ventral retinal gliosis in mice. Invest Ophthalmol Vis Sci. 2020;61(2):44. 10.1167/iovs.61.2.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heermann S, Schütz L, Lemke S, Krieglstein K, Wittbrodt J. Eye morphogenesis driven by epithelial flow into the optic cup facilitated by modulation of bone morphogenetic protein. Elife. 2015;4:e05216. 10.7554/eLife.05216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knickmeyer MD, Mateo JL, Eckert P, et al. TGFβ-facilitated optic fissure fusion and the role of bone morphogenetic protein antagonism. Open Biol. 2018;8(3):170134. 10.1098/rsob.170134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang R, Walder-Christensen KK, Lalani S, et al. Neurodevelopmental mutation of giant ankyrin-G disrupts a core mechanism for axon initial segment assembly. Proc Natl Acad Sci U S A. 2019;116(39):19717–19726. 10.1073/pnas.1909989116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson KA, Rainger J, Floyd JA, et al. Heterozygous loss-of-function mutations in YAP1 cause both isolated and syndromic optic fissure closure defects. Am J Hum Genet. 2014;94(2):295–302. 10.1016/j.ajhg.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bahm I, Barriga EH, Frolov A, Theveneau E, Frankel P, Mayor R. PDGF controls contact inhibition of locomotion by regulating N-cadherin during neural crest migration. Development. 2017;144(13):2456–2468. 10.1242/dev.147926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Espina JA, Marchant CL, De Ferrari GV, Reyes AE. Pdgf1aa regulates zebrafish neural crest cells migration through Hif-1 in an oxygen-independent manner. Mech Dev. 2018;154:203–207. 10.1016/j.mod.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Gage PJ, Rhoades W, Prucka SK, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46(11):4200–4208. 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- 37.Soules KA, Link BA. Morphogenesis of the anterior segment in the zebrafish eye. BMC Dev Biol. 2005;5:12. 10.1186/1471-213X-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishii Y, Matsumoto Y, Watanabe R, et al. Characterization of neuroprogenitor cells expressing the PDGF beta-receptor within the subventricular zone of postnatal mice. Mol Cell Neurosci. 2008;37(3): 507–518. 10.1016/j.mcn.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 39.El-Amraoui A, Petit C. Cadherins as targets for genetic diseases. Cold Spring Harb Perspect Biol. 2010;2(1):a003095. 10.1101/cshperspect.a003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J, Gross JM. Laminin beta1 and gamma1 containing laminins are essential for basement membrane integrity in the zebrafish eye. Invest Ophthalmol Vis Sci. 2007;48(6):2483–2490. 10.1167/iovs.06-1211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The expression data used in this manuscript are at Gene Expression Omnibus under the accession numbers GSE159822 and GSE13103 and are currently open access.