

Abstract
Aims
Aortic aneurysm and dissection (AAD) are high-risk cardiovascular diseases with no effective cure. Macrophages play an important role in the development of AAD. As succinate triggers inflammatory changes in macrophages, we investigated the significance of succinate in the pathogenesis of AAD and its clinical relevance.
Methods and results
We used untargeted metabolomics and mass spectrometry to determine plasma succinate concentrations in 40 and 1665 individuals of the discovery and validation cohorts, respectively. Three different murine AAD models were used to determine the role of succinate in AAD development. We further examined the role of oxoglutarate dehydrogenase (OGDH) and its transcription factor cyclic adenosine monophosphate-responsive element-binding protein 1 (CREB) in the context of macrophage-mediated inflammation and established p38αMKO Apoe –/– mice. Succinate was the most upregulated metabolite in the discovery cohort; this was confirmed in the validation cohort. Plasma succinate concentrations were higher in patients with AAD compared with those in healthy controls, patients with acute myocardial infarction (AMI), and patients with pulmonary embolism (PE). Moreover, succinate administration aggravated angiotensin II-induced AAD and vascular inflammation in mice. In contrast, knockdown of OGDH reduced the expression of inflammatory factors in macrophages. The conditional deletion of p38α decreased CREB phosphorylation, OGDH expression, and succinate concentrations. Conditional deletion of p38α in macrophages reduced angiotensin II-induced AAD.
Conclusion
Plasma succinate concentrations allow to distinguish patients with AAD from both healthy controls and patients with AMI or PE. Succinate concentrations are regulated by the p38α–CREB–OGDH axis in macrophages.
Keywords: Aortic aneurysm and dissection, Macrophage, Oxoglutarate dehydrogenase, Succinate
Graphical Abstract
See page 4386 for the editorial comment for this article ‘Breaking the cycle: Succinate in aortic diseases’, by R. Bhandari and S.J. Cameron, https://doi.org/10.1093/eurheartj/ehab514.
Translational perspective
Plasma succinate concentration in patients with aortic diseases including aortic dissection and aortic aneurysm is high. This parameter may be used as a pre-hospital diagnostic auxiliary index to distinguish aortic diseases from healthy controls, acute myocardial infarction and pulmonary embolism. Increased plasma succinate leads to dysfunction of mitochondria and increased reactive oxygen species levels in vasculature, thereby aggravating aortic aneurysm and dissection (AAD). We observed that inhibition of the p38α–cyclic adenosine monophosphate-responsive element-binding protein 1–oxoglutarate dehydrogenase axis in macrophages reduced their release of succinate, inhibiting the progression of AAD. Therefore, inhibiting succinate-related pathways such as reducing the plasma succinate content or inhibiting succinate-induced mitochondrial dysfunction may be used as potential therapeutic targets for AAD.
Introduction
Aortic aneurysm and dissection (AAD) have a prevalence of 1.3–8% and are associated with high mortality due to acute aortic complications.1–3 Sporadic AAD are associated with smoking, hypertension, old age, and male gender. The pathogenesis of AAD is associated with inflammation, apoptosis, smooth muscle cell phenotype switch, and degradation of the extracellular matrix.4 , 5 There are no effective drugs to prevent the occurrence and development of AAD.6 Therefore, it is essential to explore potential therapeutic targets for AAD.
Succinate plays critical roles in a variety of physiological and pathophysiological conditions. It accumulates in the myocardium during myocardial ischaemia and is rapidly oxidized to generate reactive oxygen species (ROS) with the occurrence of reperfusion, which led to the death of myocardial cells and aggravated ischaemia–reperfusion injury.7 Dimethyl malonate, by inhibiting succinate dehydrogenase, improved ischaemia–reperfusion injury during myocardial infarction in mice and pigs.7 , 8 Succinate also plays an important role in maintaining glucose homeostasis. For example, succinate contributes to intestinal gluconeogenesis and accelerates adipose tissue thermogenesis.9 , 10 Succinate released from muscles during exercise contributes to the formation of extracellular matrix and muscle regeneration.11 Furthermore, succinate plays an important role in the pathophysiological process of diseases such as diabetes, chronic neuroinflammation, osteoclastogenesis, rheumatoid arthritis, systemic lupus erythematosus, and intestinal type 2 immunity.12–18 However, the contribution of succinate to AAD and its clinical relevance remain unclear.
Macrophages are known to display a diverse array of metabolic characteristics that depend upon their functional states.19 In M1 inflammatory macrophages, glycolysis is enhanced, whereas the tricarboxylic acid (TCA) cycle is interrupted at several key points, thereby leading to the accumulation of TCA intermediate products. Succinate is an important intermediate product of the TCA cycle that accumulates in M1 pro-inflammatory macrophages.20 Succinate transported from mitochondria to the cytoplasm inhibits proline hydroxylase activity, increases the levels of hypoxia-inducible factor 1-alpha, and induces the expression of interleukin (IL)-1β. In addition, succinate in mitochondria generates a large amount of ROS through oxidation by succinate dehydrogenase, promoting M1 polarization.21
In our current study, we found that plasma succinate concentrations were positively correlated with human aortic diseases and succinate plays an important role in the pathogenesis of AAD as demonstrated in three well-established murine AAD models.
Methods
Additional details about the methods are available in the Supplementary material online. The corresponding author can be contacted for access to experimental data associated with this study.
Study population
The study was approved by the Institutional Review Board of the Tongji Hospital (Tongji Medical College, Wuhan, China) and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. Informed consent was obtained from all participants. The study was a two-stage (the discovery stage and the replication stage) case–control study and a total of 1705 participants were included. Patients with aortic diseases, including aortic dissection (AD) and abdominal aortic aneurysm (AAA), acute myocardial infarction (AMI), or pulmonary embolism (PE), were recruited from Tongji Hospital, Tongji Medical College in Wuhan, China, between January 2014 and September 2020. Blood samples were collected before emergency surgery and mostly are the first day of admission. Because of limitation of PE cases, all patients with PE were included in our cohort. Healthy control subjects who had no significant systemic diseases (e.g. ischaemic heart disease, cancer, pulmonary disease, or infectious diseases) were recruited from the communities of Wuhan and Tongji Hospital. All blood samples were collected and plasma was separated by centrifugation immediately and stored at –80°C until analysis.
Statistical analyses
Data were analysed using R (version 3.6.3; R Foundation, Vienna, Austria) and GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA). Continuous variables with normal distribution are expressed as the mean ± standard deviation (SD). Continuous variables that did not pass the normality test are expressed as the median with interquartile range (IQR). Categorical variables are expressed as numbers and percentages (%). Comparisons between groups were performed using the Student’s t-test or Wilcoxon test for quantitative variables and the Fisher’s exact test or the Chi-squared test for categorical variables.
Results
Untargeted metabolomics reveals elevated plasma succinate concentration in patients with aortic disease
In the discovery cohort, a total of 20 patients with aortic disease, including 5 Type A AD, 7 Type B AD and 8 AAA, and 20 age-, sex-, and comorbidity-matched (including smoking and drinking status, as well as history of hypertension and diabetes; Supplementary material online, Table S1) healthy individuals were enrolled and performed untargeted metabolomics by using Q Exactive HFX orbitrap (Thermo, CA, USA) (Supplementary material online, Figure S1A–I). Plasma concentrations of 119 metabolites were significantly different [absolute log2 (fold change) > 1, false discovery rate (FDR) < 0.05] between all patients with aortic diseases and healthy controls (Figure 1A), and a total of 28 metabolites were significant in each subgroups (Supplementary material online, Figure S1C–G). Of these, succinate was the most upregulated metabolite (FDR = 1.23 × 10–8, fold change = 2.34). This result was also verified by using targeted mass spectrometry (Supplementary material online, Figure S1J).
Figure 1.

High level of succinate could increase the diagnostic performance for aortic disease. (A) Volcano plot of plasma metabolites in all patients vs. healthy controls. (B) Distribution of succinate according to disease status in the validation cohort, before propensity score-matching. (C) Forest plots showing the adjusted per-standard deviation odds ratio (95% confidence interval) for plasma succinate levels associated with aortic diseases in subgroups using the integration of discovery and validation cohort data. Binary logistic regression was adjusted for age, sex, smoking and drinking status, history of hypertension, diabetes, or systolic blood pressure, and levels of glucose, total cholesterol, and triglycerides. (D) Decision curve analysis: aortic diseases vs. healthy controls for baseline, succinate, and the combination of baseline and succinate using the integration of discovery and validation cohort data. Baseline refers to age, sex, smoking and drinking status, and history of hypertension, diabetes, and systolic blood pressure. (E) Distribution of succinate plasma levels in patients with aortic diseases, acute myocardial infarction, and pulmonary embolism and in healthy controls. (F) Receiver operating characteristic curves: patients with aortic diseases, acute myocardial infarction, and pulmonary embolism vs. healthy controls for succinate. (G) Receiver operating characteristic curves: acute myocardial infarction and pulmonary embolism vs. aortic diseases for succinate.
In the validation set, data were available for 1665 individuals, including 700 aortic disease cases (241 type A AD, 374 type B AD, and 85 AAA), 199 AMI cases, 74 PE cases, and 692 healthy controls. The baseline characteristics and clinical parameters are shown in Supplementary material online, Table S2. Succinate concentrations were elevated in patients with aortic diseases (median: 35.15 μmol/L, IQR: 23.10–58.42) compared with those in healthy controls (median: 15.30 μmol/L, IQR: 10.67–23.10; Figure 1B, P < 2.22 × 10–16). In agreement with the results in the discovery set, succinate levels were higher in patients with type A AD (median: 38.00 μmol/L, IQR: 24.70–62.80), type B AD (median: 33.60 μmol/L, IQR: 23.15–58.05), and AAA (median: 29.8 μmol/L, IQR: 20.40–54.70). As a significant difference was noted for most of the clinical variables between the patients and healthy subjects, we used propensity score matching to minimize differences in baseline characteristics: 190 aortic disease cases were matched at a 1:1 ratio with 190 healthy individuals. After this refinement, baseline characteristics were comparable between the two groups (Supplementary material online, Table S3), whereas a significant association was still found between plasma succinate concentrations and aortic disease status (Supplementary material online, Figure S2; P < 2.22 × 10–16).
To investigate potential clinical implications of succinate, we conducted an integrated analysis of the two-stage case–control cohort data and found individuals with higher plasma succinate concentrations presented with significantly increased risk of aortic diseases. Each SD of log-transformed plasma succinate concentration was associated with a 3.64-fold [95% confidence interval (CI): 2.94–4.52], 3.71-fold (95% CI: 2.81–4.92), 3.24-fold (95% CI: 2.53–4.15), and 2.49-fold (95% CI: 1.69–3.59) increment in the odds ratio (OR) of all types of aortic disease, type A AD, type B AD, and AAA, respectively (Supplementary material online, Figure S3A). Moreover, plasma succinate concentrations remained an independent predictor even after adjustments for common risk factors (age, sex, smoking and drinking habits, history of hypertension and diabetes, and systolic blood pressure) (adjusted model 1, per SD OR: 4.35, 95% CI: 3.64–5.25, all aortic diseases), or for the concentrations of glucose, lipids (total cholesterol and triglycerides) and different indicators of disease severity and organ injury (white blood cell count, alanine transaminase, aspartate transaminase, creatinine, and estimated glomerular filtration rate) (adjusted model 2, per SD OR: 3.91, 95% CI: 3.15–4.94, all aortic diseases). In general, the associations between plasma succinate concentrations and all aortic diseases were consistent across different risk strata subgroups defined by age, sex, smoking and drinking status, or history of hypertension (Figure 1C).
Diagnostic performance of plasma succinate concentrations for aortic diseases
To determine the utility of plasma succinate concentrations as a potential biomarker for all aortic diseases, the predictive ability of succinate was evaluated by using the area under the receiver operating characteristic curve (AUROC) that gave a value of 0.83 (95% CI: 0.81–0.85, P = 0.006, Supplementary material online, Figure S3B) with better discriminative performance than baseline module (AUROC = 0.79, 95% CI: 0.76–0.81, reference), which included age, sex, smoking, drinking, history of hypertension, and diabetes, and systolic blood pressure. Furthermore, the combined AUROC of these two factors was statistically higher (AUROC = 0.87, 95% CI: 0.86–0.89, P < 0.0001).
In addition, the decision curve analysis (DCA) is a novel method used to evaluate alternative diagnostic strategies that has advantages over the AUROC method. The DCA curves obtained for baseline, succinate, and baseline plus succinate conditions are presented in Figure 1D. Compared with the baseline condition, succinate alone modestly elevated the net benefit; however, the combination showed significantly higher net benefits, indicating that plasma succinate test has a potential clinical application value.
Aortic diseases, AMI, and PE are potentially fatal conditions that share similar symptoms but require different treatment strategies. Therefore, it is vital for clinicians to distinguish these three diseases. Thus, we enrolled 199 patients with AMI and 76 patients with PE to find whether any biochemical parameters could reliably distinguish aortic diseases from AMI and PE (Supplementary material online, Table S4). Plasma succinate concentrations were increased significantly in patients with aortic diseases compared with those in patients with AMI (median: 12.0 μmol/L, IQR: 8.23–16.85, P < 2.22 × 10–16) and PE (median: 15.45 μmol/L, IQR: 11.22–26.8, P < 2.22 × 10–16) (Figure 1E). The AUROC values for patients with AMI and PE vs. healthy controls were 0.63 (0.59–0.67) and 0.53 (0.46–0.60), respectively (Figure 1F). The AUROC values in patients with aortic disease vs. patients with AMI and PE were 0.91 (0.88–0.93) and 0.80 (0.73–0.86), respectively (Figure 1G). Therefore, plasma succinate concentrations had a positive diagnostic performance and allowed differentiation of patients with aortic disease from healthy controls and patients with AMI and PE.
Beta-blocker therapy is routine in AAD and could suppress mitochondrial metabolism, potentially leading to the accumulation of succinate. We then assessed cross-sectional relationships between blood succinate levels and presence of AD, stratified by those who had vs. had not received beta-blocker therapy (Supplementary material online, Table S5). While blood succinate levels were noted to be elevated in subjects having AD, there was no difference among those who had vs. had not received beta-blocker therapy (all patients, P = 0.27, Type A AD, P = 0.49, Type B AD, P = 0.37, Supplementary material online, Figure S4).
Succinate aggravates AAD formation in mice
To determine the role of succinate in AAD, we administered succinate or vehicle control to mice and constructed BAPN/angiotensin II (Ang II)-induced AAD model (Supplementary material online, Figure S5A). Succinate administration increased the mortality rate (41.7% vs. 71.4%, Figure 2A and B), AAD incidence (67% vs. 100%, Figure 2C), and also appeared to increase the diameter of ascending aorta, aortic arch and descending aorta at 31 days, but with only three animals remaining in the succinate group, statistical significance is not reported. Compared with the vehicle control group, succinate administration increased aortic pathology as showed by haematoxylin and eosin (H&E) staining and Elastic Van Gieson (EVG) staining (Supplementary material online, Figure S5B). Succinate administration increased the level of ROS in the vasculature (Supplementary material online, Figure S5C and D).
Figure 2.

Supplementation with succinate aggravates aortic aneurysm and dissection in vivo. (A–D) Four-week-old male mice were administered with 0.25% BAPN (wt/vol) for 28 days with or without 1.5% sodium succinate (wt/vol) and then infused with saline or angiotensin II (1000 ng/kg/min) for 3 days: vehicle + BAPN/angiotensin II, n = 12; and 1.5% succinate + BAPN/angiotensin II, n = 14. (A) Representative morphology of the aortas for each group. Scale bar = 2 mm. (B) Survival curve and number at risk, log-rank test. (C) Incidence of aortic aneurysm and dissection for each group. (D) Measurement of the maximal aortic diameter ex vivo. n = 7 for mice administered BAPN/angiotensin II and n = 3 for 1.5% succinate + BAPN/angiotensin II (mice died of aortic rupture were not included). (E–G) Nine-week-old Apoe–/– male mice were infused with saline or angiotensin II (1000 ng/kg/min) for 28 days with or without 1.5% sodium succinate (wt/vol): vehicle + angiotensin II, n = 10; and 1.5% succinate + angiotensin II, n = 10. (E) Representative morphology of the aortas from different groups of mice. Scale bar = 2 mm. (F) Incidence of aortic aneurysm and dissection in different groups of mice. (G) Measurement of the maximal aortic diameter ex vivo. n = 6 for mice administered angiotensin II and n = 9 for 1.5% succinate + angiotensin II (mice died of aortic rupture were not included). H–J, Three-week-old male mice were administered with 0.25% BAPN (wt/vol) for 28 days with or without 1.5% sodium succinate (wt/vol): vehicle + BAPN, n = 22; and 1.5% succinate + BAPN, n = 22. (H) Representative morphology of the aortas from the different groups of mice. Scale bar = 2 mm. (I) Incidence of aortic aneurysm and dissection from the different groups of mice. (J) Measurements of the maximal aortic diameter ex vivo. n = 19 for mice administered BAPN and n = 18 for 1.5% succinate + BAPN (mice died of aortic rupture were not included). Apoe–/–, apolipoprotein E-deficient; Asc, ascending aorta; Des, descending aorta.
In a second AAD model, we administered succinate or vehicle control to Apoe − / − mice and then infused Ang II for 28 days (Supplementary material online, Figure S6A and B). Succinate administration elevated AAD incidence from 60% in Ang II-infused control animals to 100% in Ang II-infused succinate-supplemented mice of AAD (Figure 2E and F). Succinate-administered mice exhibited a significant increase in the maximal aortic diameter compared with control mice (ascending aorta: 1.33 ± 0.18 vs. 1.67 ± 0.29 mm, P = 0.0301; aortic arch: 1.34 ± 0.11 vs. 1.65 ± 0.25 mm, P = 0.0138; abdominal: 1.66 ± 0.84 vs. 1.06 ± 0.38 mm, P = 0.0490; Figure 2G). Similar changes were observed in mice infused with Ang II for 14 days (Supplementary material online, Figure S7). H&E staining revealed that aortic medial thickness increased significantly, whereas EVG staining showed that elastic tissue fibers had increased fragmentation in succinate-treated animals (Supplementary material online, Figure S6C). Moreover, immunohistochemistry revealed that the levels of pro-inflammatory factors such as IL-1β, matrix metalloproteinase-9 (MMP9), IL-6, and tumour necrosis factor (TNF)-α were increased in the aortic tissues of succinate-administered mice (Supplementary material online, Figure S6D and E). Similarly, we also found increased ROS in succinate-administered Apoe − / − mice after Ang II-infused for 3 days (Supplementary material online, Figure S6F and G). Notably, we further confirmed that succinate supplementation aggravated BAPN-induced AAD (Figure 2H). We found that the incidence of AAD increased in mice receiving water containing 1.5% succinate (Figure 2I), and aortic diameters were increased (Figure 2J). Elastin integrity was decreased in succinate-administered animals (Supplementary material online, Figure S8B and C). VSMC-specific smooth muscle α-actin, calponin, and SM22α (smooth muscle 22 α) were also decreased in succinate-administered mice (Supplementary material online, Figure S8D). In line with the above results, succinate administration aggravated the manifestations of elastase-induced AAA in vivo (Supplementary material online, Figure S9).
Inhibition of oxoglutarate dehydrogenase reduces macrophage-mediated inflammation
Tannahill et al.20 revealed that succinate concentration increased in macrophages incubated with lipopolysaccharide (LPS), which promoted IL-1β expression and enhanced pro-inflammatory macrophage responses. Macrophages can release succinate into the extracellular environment.15 In the TCA cycle, succinate is synthesized by oxidative decarboxylation of alpha-ketoglutarate mediated by oxoglutarate dehydrogenase (OGDH) (Figure 3A). Interestingly, it has been proven that OGDH activity in macrophages increased significantly after LPS stimulation, which promoted succinate accumulation.22 Here, we found that OGDH was increased significantly in macrophages within AAD tissue sections, suggesting that OGDH might play an important role in AAD (Figure 3B and Supplementary material online, Figure S10). Importantly, knockdown of the Ogdh gene via siRNA attenuated inflammation in bone marrow-derived macrophages (BMDMs) stimulated with LPS for 24 h, reducing the expression of Il1b and Nos2 at the mRNA level (Figure 3C and D), as well as of OGDH and pro-IL-1β at the protein level (Figure 3E). The concentration of succinate decreased significantly after Ogdh knockdown (Figure 3F). Moreover, the addition of succinyl phosphonate, a specific inhibitor of OGDH, downregulated the expression of Il1b and Nos2 at the mRNA level and the content of pro-IL-1β at the protein level (Figure 3G–I).
Figure 3.

Inhibition of oxoglutarate dehydrogenase (OGDH) reduces the expression of inflammatory factors in macrophages. (A) Schematic diagram of the tricarboxylic acid cycle. B, Representative immunofluorescence staining for F4/80 and OGDH in the suprarenal abdominal aortas from NaCl- and angiotensin II-infused mice. Scale bar = 50 μm. Arrows indicate macrophages expressing OGDH. (C and D) Differentially expressed genes were validated by qRT-PCR in bone marrow-derived macrophages stimulated with lipopolysaccharide after transfection with negative control (NC) or siOGDH. Data are pooled from three independent experiments. n = 9 per group. (E) Representative western blot of pro-IL-1β in lipopolysaccharide-stimulated bone marrow-derived macrophages after transfection with NC or siOGDH. (F) Relative succinate abundance in lipopolysaccharide-stimulated bone marrow-derived macrophages after transfection with NC or siOGDH. Data are pooled from three independent experiments. n = 9 per group. (G and H) Differentially expressed genes were validated by qRT-PCR in bone marrow-derived macrophages stimulated with lipopolysaccharide with or without the addition of succinyl phosphonate. Data are pooled from three independent experiments. n = 9 per group (G and H). (I) Representative western blot of pro-IL-1β in bone marrow-derived macrophages stimulated with lipopolysaccharide with or without the addition of succinyl phosphonate.
CREB controls Ogdh transcription
We then focused on the transcriptional regulation of Ogdh expression in macrophages. According to the chromatin immunoprecipitation (ChIP)-seq database (http://rna.sysu.edu.cn/chipbase3/)23, transcription factor cyclic adenosine monophosphate-responsive element-binding protein 1 (CREB) could bind to the promoter region of Ogdh (Figure 4A). In the dual-luciferase reporter assay performed to validate these in silico data, overexpression of CREB resulted in the activation of the Ogdh promoter compared with the results obtained with vector-control (Figure 4B). CREB bound to the promoter region of Ogdh in BMDMs according to the ChIP assay (Figure 4C and D). To prove that CREB regulates OGDH expression in macrophages, we knocked down the Creb1 gene using siRNA, which decreased OGDH and pro-IL-1β expression levels in LPS-treated BMDMs (Figure 4E). Similar results were obtained in BMDMs incubated with the selective CREB inhibitor 666-15 (Supplementary material online, Figure S11A).
Figure 4.

Cyclic AMP-responsive element-binding protein 1 (CREB) regulates oxoglutarate dehydrogenase OGDH expression via direct promoter binding. (A) CREB-binding motifs. (B) Luciferase reporter analysis in 293T cells. Results are presented as the mean ± standard deviation. Data are pooled from three independent experiments. n = 11 per group. (C) Chromatin immunoprecipitation-seq assay of the occupancy of OGDH gene promoters by CREB (data from GSM2663863 dataset). (D) Chromatin immunoprecipitation assay: binding of CREB to the OGDH gene promoter in bone marrow-derived macrophages. Three independent experiments were performed. (E) Representative western blot of OGDH in bone marrow-derived macrophages stimulated with lipopolysaccharide for 24 h with or without addition of siCREB. Three independent experiments were performed. (F) Representative immunofluorescence staining for F4/80 and phosphorylated CREB in the suprarenal abdominal aortas from control and aortic aneurysm and dissection mice. Scale bar = 50 μm.
We also found that the levels of phosphorylated CREB (p-CREB) in macrophages were increased significantly in AAD (Figure 4F and Supplementary material online, Figure S11B–E). Furthermore, expression levels of CREB and OGDH showed remarkable positive correlation in three independent public aortic disease databases (Supplementary material online, Figure S11F–H).
Phosphorylation of CREB by p38α increases succinate production and promotes inflammatory reactions of macrophages
CREB is a widely expressed transcription factor whose activity is regulated by a variety of kinases. Previous research reported that, under LPS stimulation, CREB has been shown to be phosphorylated by p38α24 and we reproduced those observations (Supplementary material online, Figure S12). We further investigated the role of p38α in the regulation of succinate production. First, p38α myeloid-deleted mice (p38αMKO mice) were obtained by crossing p38α-flox mice (p38αfl/fl mice) with LysM-Cre mice (Figure 5A). In LPS-stimulated BMDMs from p38αMKO mice, the levels of phosphorylated CREB were lower than in LPS-treated BMDMs from p38αfl/fl animals. These results support the notion that p38α phosphorylates CREB after LPS stimulation (Figure 5A). Immunoprecipitation experiments showed that p38α binds to CREB (Figure 5B). p38α knockout downregulated OGDH at both the mRNA and protein levels (Figure 5C and D) and decreased the content of succinate in BMDMs (Figure 5E). We also found that the expression of pro-IL-1β in p38αMKO BMDMs was lower than in p38αfl/fl BMDMs (Figure 5F). p38α deletion impacted expression of other inflammatory factors in BMDMs stimulated by LPS: we detected decreases in the expression levels of genes encoding macrophage pro-inflammatory factors, such as Nos2, Hif1a, Il1b, and Ccl2 (Figure 5G). The proportion of M1 pro-inflammatory (CD86 positive) macrophages (Figure 5H) and the level of intracellular ROS (Figure 5I) were lower in p38αMKO BMDMs than in p38αfl/fl BMDMs. In addition, we constructed a different p38α-deficient macrophage cell line by using CRISPR-Cas9 technology (Supplementary material online, Figure S13A and B). Overall, p38α deletion in RAW264.7 cells reduced their inflammatory potential (Supplementary material online, Figure S13C–I).
Figure 5.

p38α affects macrophage inflammatory phenotype by regulating the expression of OGDH and succinate via CREB phosphorylation. (A) Representative western blot of phosphorylated p38α (p-p38α), total p38α, p-CREB, and total CREB in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide for 0, 7.5, 15, 30, 60, or 120 min. (B) The co-immunoprecipitation assay of CREB and p38α in bone marrow-derived macrophages. (C) Oxoglutarate dehydrogenase (Ogdh) mRNA level was measured by qRT-PCR in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. Data are pooled from three independent experiments. n = 9 per group. (D) Representative western blot of OGDH in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. (E) Relative succinate abundance in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. Data are pooled from three independent experiments. n = 9 per group. (F) Representative western blot of pro-IL-1β in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. (G) Expression levels of Nos2, Il1b, and Ccl2 determined by qRT-PCR in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. (H) Percentage of CD86+ cells determined by flow cytometry in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. Data are pooled from three independent experiments. n = 9 per group. (I) Dihydroethidium staining of reactive oxygen species in p38αfl/fl and p38αMKO bone marrow-derived macrophages stimulated with lipopolysaccharide. Scale bar = 50 μm.
Figure 6.

Macrophage-specific p38α deficiency attenuates angiotensin II-induced aortic aneurysm and dissection. (A) Representative immunofluorescence staining for CD68 (marker of macrophages) and p38α in the aortas from healthy individuals and patients with aortic aneurysm and dissection. (B) Measurement of the maximal suprarenal aortic diameters ex vivo from elastase-treated male mice injected with either SB203580 or vehicle. n = 7 for mice administered vehicle and n = 6 for mice administered SB203580. (C–I) 14-weeks-old p38αfl/fl Apoe−/− and p38αMKO Apoe−/− male mice were infused with either saline or angiotensin II (1000 ng/kg/min) for 28 d. p38αfl/fl Apoe−/− + saline, n = 4; p38αMKO Apoe−/− + saline, n = 4; p38αfl/fl Apoe−/− + angiotensin II, n = 26; and p38αMKO Apoe−/− + angiotensin II, n = 27. (C) Representative morphology of the abdominal aortas from the different groups of mice. Scale bar = 2 mm. (D) Incidence of aortic aneurysm and dissection in different groups of mice. (E) Measurement of the maximal suprarenal aortic diameters ex vivo in different groups of mice. p38αfl/fl Apoe−/− + angiotensin II, n = 16; p38αMKO Apoe−/− + angiotensin II, n = 19 (mice died of aortic rupture were not included). Representative images of haematoxylin and eosin (F; scale bar = 100 μm) and Elastic Van Gieson (G; scale bar = 200 and 50 μm, n = 7 per group) staining in the suprarenal abdominal aortas of mice, and the grade of elastin degradation. (H) Representative immunofluorescence staining images for F4/80 and CD206 in the suprarenal abdominal aortas from the different groups of mice. Scale bar = 100 μm. (I) Representative immunohistochemical staining images for matrix metalloproteinase-9, IL-1β, IL-6, monocyte chemoattractant protein-1, and tumour necrosis factor-alpha in the suprarenal abdominal aortas from the different groups of mice Scale bar = 50 μm.
p38α deletion in macrophages attenuates the formation of AAD
We found that p38α expression was significantly increased in human AAD tissues (Supplementary material online, Figure S14A–D). Furthermore, we confirmed that p38α was specifically enriched in macrophages (Figure 6A and Supplementary material online, Figure S14E–H). Therefore, we explored the role of p38α in the progression of AAD. We treated wild-type mice with the specific p38α inhibitor SB203580 (Supplementary material online, Figure S15A). This compound inhibited the progression of AAA induced by elastase (Figure 6B and Supplementary material online, Figure S15B–D). Next, we crossed p38αMKO and p38αfl/fl mice with Apoe– / − mice to obtain p38αMKO Apoe– / − and p38αfl/fl Apoe– / − mice. The incidence of AAD decreased in p38αMKO ApoE− / − mice infused with Ang II (Figure 6C and D), and their aortic diameters were smaller than in their wild-type littermates (Figure 6E). H&E staining revealed that the wall thickness and the loss of smooth muscle cells were less pronounced in p38αMKO Apoe– / − mice than in their wild-type littermates (Figure 6F). EVG staining revealed that the knockout of p38α in macrophages prevented the degradation of elastin (Figure 6G).
Macrophage infiltration in AAD, especially that of M1 pro-inflammatory macrophages (F4/80+/CD206–), was lower in p38αMKO Apoe– / − mice (Figure 6H), indicating weaker aortic inflammation. A similar phenomenon was also observed in mice infused with Ang II for either 3 or 7 days (Supplementary material online, Figure S16). Moreover, immunohistochemistry data showed reduced aortic expression levels of inflammatory factors such as MMP9, IL-6, monocyte chemoattractant protein-1 MCP1, TNF-α, and IL-1β in p38αMKO Apoe– / − mice compared with those in p38αfl/fl Apoe– / − mice (Figure 6I). Our findings were also supported by the observations in other AAA models: deletion of p38α in macrophages attenuated AAA induced by elastase (Supplementary material online, Figure S17).
To definitely prove that the relief of AAA induced by p38 knockout was due to the decrease of succinate levels, we examined the effect of succinate supplementation in p38 knockout mice. In p38αMKO BMDMs, concentrations of pro-IL-1β and IL-1β in the supernatant increased significantly after pre-treatment with 5 mM succinate (Supplementary material online, Figure S18A and B). In our further in vivo experiments, p38αMKO Apoe– / − mice received water containing 1.5% succinate (Supplementary material online, Figure S18C), which significantly increased AAD incidence rate and maximal aortic diameter (Supplementary material online, Figure S18D–F) compared with those in mice that received normal water. In addition, wall thickness and levels of pro-inflammatory factors increased and elastin integrity decreased in succinate-supplemented mice (Supplementary material online, Figure S18G and H). Notably, we further confirmed that the administration of succinate aggravated elastase-induced AAA (Supplementary material online, Figure S19).
Discussion
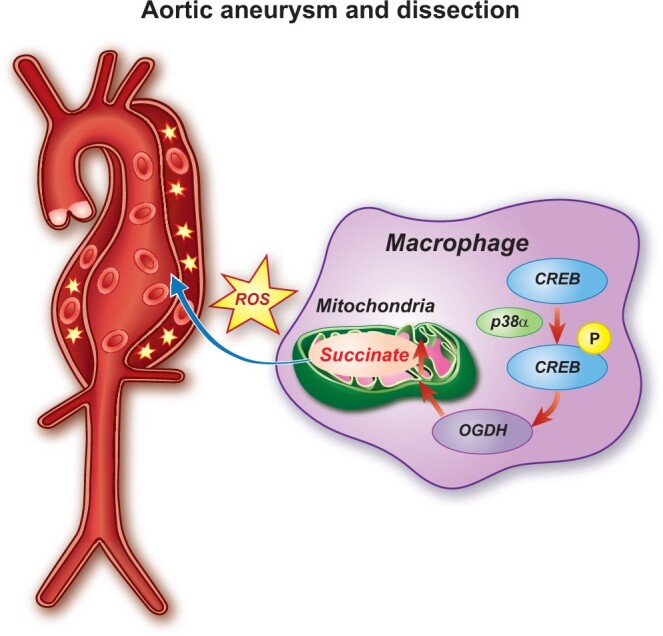
AAD is becoming more prevalent among the general population, and mortality rates associated with its rupture are high. Here, we found that plasma succinate concentrations were increased in patients with aortic diseases (AD and AAA) using untargeted metabolomics and targeted mass spectrometry. Furthermore, plasma succinate concentrations can be used as a biomarker for pre-hospital AAD diagnosis and allow reliable differentiation from AMI and PE when patients present with chest pain. We also found that succinate administration increased the mitochondrial ROS level in the vasculature, further aggravated Ang II-, BAPN- and BAPN/Ang II-induced AAD. We discovered that the p38α–CREB–OGDH axis regulates the production of succinate in macrophages. Deletion of p38α could inhibit the progression of AAD (Graphical abstract). Therefore, plasma succinate can be used both as a biomarker for AAD diagnosis and a potential target for AAD therapeutics.
In the quest for biomarkers of cardiovascular disease, although not perfect, high-throughput -omics technologies have allowed for the screening of a large cohort of samples to identify potentially applicable circulating markers. In conjunction with functional assessment, unbiased, untargeted metabolomics has gained traction as a discovery platform to identify plasma metabolites that could serve as novel markers of cardiovascular diseases.25 , 26 In the field of AAD, an increasingly prevalent disease with high mortality, metabolic profiling studies are lacking, which hinders our understanding of the biology of signature metabolites and their contribution to the pathogenesis of AAD.
Toward that end, our study is the first to apply untargeted metabolomics to delineate the metabolic landscape of patients with AAD. Very excitingly, we identified succinate as the most significantly upregulated metabolite in the plasma of patients with AAD when compared with healthy controls. Importantly, as shown in Figure 1E– G, succinate plasma level may not only serve as an independent circulating marker for AAD, but can also be utilized as an auxiliary indicator for pre-hospital diagnosis to differentiate patients with AAD from patients with AMI and PE who present similar symptoms. We recognize that computed tomography and magnetic resonance imaging remains the preferred imaging modalities for AAD diagnosis and monitoring; however, they are less applicable for mass screening and large-scale trials. While current succinate measurement by mass spectrometry as performed in our study may limit its practical utility in clinic, a point-of-care testing can potentially be developed for plasma succinate detection in the future for emergency usage. Of note, beta-blocker therapy did not increase the concentration of succinate in the current study. Given the size of this data set, there still remains a possibility that acute administration of beta-blockers or other therapeutics makes a partial contribution to the increase in circulating succinate concentration at the time of surgical repair. This will deserve future confirmation studies using larger patient cohorts of AAD.
Our study also highlights a previously unknown causal role of high plasma succinate in driving AAD and may be exploited as a target for therapy development against AAD. Accumulating studies have implicated a pathological role for succinate in the development of a variety of diseases such as AMI, autoimmune diseases, diabetes, and chronic neuroinflammation. In light of our findings, we postulated that high plasma succinate concentration in patients with AAD plays a promotional role in the development of AAD. As unequivocally demonstrated in three AAD murine models (Ang II, BAPN, and BAPN/Ang II), succinate administration to mice markedly aggravated the AAD phenotype. Thus, we believe that plasma succinate levels can not only be used as a biomarker to discriminate AAD from healthy control and patients with AMI and PE but also be utilized as a potential target for AAD treatment.
The cellular source of succinate has become increasingly clear. Previous studies have reported that succinate accumulated in inflammatory macrophages and can be released to the extracellular milieu, such as in rheumatoid arthritis, chronic neuroinflammation, and systemic lupus erythematosus, further accelerates the progression of diseases.13 , 15 , 17 According to the above studies, inflammatory cells, especially macrophages, accumulate in AAD tissues and may release succinate into the blood during AAD, causing an increase in plasma succinate level, which in turn, aggravates the progression of AAD. With that in mind, we focused our attention on macrophages to determine the requisite importance of succinate and the mechanistic basis of succinate regulation, in the promotion of AAD. To address this, we hypothesized that disruption of key pathways of succinate production in macrophages could inhibit AAD development. The activity of OGDH increases in inflammatory macrophages.22 In line with our hypothesis, we found that knockdown of the OGDH gene can significantly inhibit the accumulation of succinate and secretion of inflammatory factors in inflammatory macrophages (Figure 3C–E). The expression of OGDH in patients with AAD also increased significantly. These results suggest that OGDH is a potential therapy target for AAD.
In addition to inflammatory cells, succinate accumulates in ischaemic tissues such as heart and brain.7 Aortic hypoxic injury may also occur in diseased states where hypoxia-inducible factor-1α was previously found to be involved AAD development.27 This suggests that a relatively hypoxic region is present in the AAD aortic wall which is likely to be associated with succinate accumulation.28 Therefore, the release of aortic sources of succinate from AAD would be expected to occur.
In addition to the host, the gut microbiota is an important source of succinate.29 Increased abundance of succinate-producing microbes is positively correlated with elevated plasma succinate concentration and an increased risk of obesity.12 However, in our study, it cannot be ruled out whether elevated succinate plasma levels in patients with AAD are related to the altered composition of their gut microbiota or destruction of the integrity of intestinal barrier. Hence, further experiments are needed to determine the major source of succinate in patients with AAD.
In searching for the potential mechanisms by which succinate promotes AAD, we turned to oxidative stress as it has been implicated in the development of AAD. It is known that succinate can be taken up directly by cells to exert its biological functions, such as promoting intestinal gluconeogenesis and adipose thermogenesis.9 , 10 Indeed, in our study, administration of succinate increased ROS level in AAD tissues. These results support the notion that increased plasma succinate levels in patients with AAD lead to the increased uptake of extracellular succinate by macrophages or smooth muscle cells. The succinate is then oxidized by succinate dehydrogenase to produce a large amount of ROS that aggravates AAD. Thus, it is conceivable that dimethyl malonate, a competitive inhibitor of succinate dehydrogenase, can be used to reduce the generation of ROS and alleviate AAD.
SUCNR1, the receptor of succinate, is widely expressed in a variety of cells of the immune system, nervous system, heart, and muscle. Whether the expression or function of SUCNR1 is altered during the pathophysiology of AAD is also unknown. The EC50 of succinate required for the activation of SUCNR1 is ∼17–56 μM.30 Our study determined plasma succinate concentrations to be a median of 15.3 μM in healthy individuals. However, its level can be as up to 35.2 μM in patients with AAD. These high plasma succinate concentrations can be expected to cause SUCNR1 activation and signal transduction in macrophages and smooth muscle cells. In our future study, we will focus on verifying whether the succinate–SUCNR1 axis is involved in the progression of AAD. Using succinate-neutralizing antibodies to maintain normal plasma succinate concentration may be another possible way to inhibit AAD progression.31
In summary, our findings reveal that succinate plays an important role in the pathogenesis of AAD and can potentially be used as a new biomarker for diagnosis. In mice, inhibiting the p38α–CREB–OGDH axis in macrophages is sufficient to mitigate AAD. These data provide a rationale for targeting succinate-related pathways as a new target for AAD treatment.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgements
We thank Hao Ying for providing the p38αfl/fl mouse model.
Funding
This work was supported by the National Natural Science Foundation of China projects (No. 91639108, No. 81770272, No. 81970425, No. 91439203, No. 91839302, and No. 81901235), the National Key Research and Development Program of China (2020YFA0803700), and China Postdoctoral Science Foundation Grant (No. 2020T130004ZX).
Conflict of interest: none declared.
Data availability
All original data used for this study are available from the corresponding authors upon reasonable request.
Contributor Information
Hongtu Cui, The Institute of Cardiovascular Sciences and Institute of Systems Biomedicine, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Sciences of Ministry of Education, Health Sciences Center, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100871, China.
Yanghui Chen, Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang Avenue NO.1095, Qiaokou District, Wuhan 430000, China.
Ke Li, Beijing Tiantan Hospital, China National Clinical Research Center of Neurological Diseases, Advanced Innovation Center for Human Brain Protection, The Capital Medical University, Nan Si Huan Xi Lu NO.119, Fengtai District, Beijing 100050, China.
Rui Zhan, The Institute of Cardiovascular Sciences and Institute of Systems Biomedicine, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Sciences of Ministry of Education, Health Sciences Center, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100871, China.
Mingming Zhao, The Institute of Cardiovascular Sciences and Institute of Systems Biomedicine, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Sciences of Ministry of Education, Health Sciences Center, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100871, China.
Yangkai Xu, The Institute of Cardiovascular Sciences and Institute of Systems Biomedicine, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Sciences of Ministry of Education, Health Sciences Center, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100871, China.
Zhiyong Lin, Cardiology Division, Emory University School of Medicine, 100 Woodruff Circle, Atlanta, GA 30322, USA.
Yi Fu, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Peking University, Key Laboratory of Molecular Cardiovascular Science, Ministry of Education, Xueyuan Road NO.38, Haidian District, Beijing 100191, China.
Qihua He, Center of Medical and Health Analysis, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100191, China.
Paul C Tang, Department of Cardiac Surgery, Frankel Cardiovascular Center, The University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA.
Ienglam Lei, Department of Cardiac Surgery, Frankel Cardiovascular Center, The University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA.
Jifeng Zhang, Department of Internal Medicine, The University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA.
Chenze Li, Department of Cardiology, Zhongnan Hospital of Wuhan University, Donghu Road NO.169, Wuchang District, Wuhan, China.
Yang Sun, Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang Avenue NO.1095, Qiaokou District, Wuhan 430000, China.
Xinhua Zhang, Department of Biochemistry and Molecular Biology, The Key Laboratory of Neural and Vascular Biology, Ministry of Education, Hebei Medical University, Zhongshan East Road NO.361, Shijiazhuang, Shijiazhuang 050017, China.
Tiffany Horng, ShanghaiTech University, Yueyang Road NO.319, Xuhui District, Shanghai 201210, China.
Hong S Lu, Department of Physiology, Saha Cardiovascular Research Center, University of Kentucky, South Limestone, Lexington, KY 40536-0298, USA.
Y Eugene Chen, Department of Cardiac Surgery, Frankel Cardiovascular Center, The University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA; Department of Internal Medicine, The University of Michigan, 500 S. State Street, Ann Arbor, MI 48109, USA.
Alan Daugherty, Department of Physiology, Saha Cardiovascular Research Center, University of Kentucky, South Limestone, Lexington, KY 40536-0298, USA.
Daowen Wang, Division of Cardiology, Department of Internal Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Jiefang Avenue NO.1095, Qiaokou District, Wuhan 430000, China.
Lemin Zheng, The Institute of Cardiovascular Sciences and Institute of Systems Biomedicine, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Sciences of Ministry of Education, Health Sciences Center, Peking University, Xueyuan Road NO.38, Haidian District, Beijing 100871, China; Beijing Tiantan Hospital, China National Clinical Research Center of Neurological Diseases, Advanced Innovation Center for Human Brain Protection, The Capital Medical University, Nan Si Huan Xi Lu NO.119, Fengtai District, Beijing 100050, China.
References
- 1. Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011;59:1–126. [PubMed] [Google Scholar]
- 2. Wang SW, Huang YB, Huang JW, Chiu CC, Lai WT, Chen CY. Epidemiology, clinical features, and prescribing patterns of aortic aneurysm in Asian population from 2005 to 2011. Medicine (Baltimore) 2015;94:e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwöger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RSv, Vrints CJM. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2873–2926. [DOI] [PubMed] [Google Scholar]
- 4. Guo DC, Papke CL, He R, Milewicz DM. Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci 2006;1085:339–352. [DOI] [PubMed] [Google Scholar]
- 5. Lu H, Daugherty A. Aortic aneurysms. Arterioscler Thromb Vasc Biol 2017;37:e59–e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bossone E, LaBounty TM, Eagle KA. Acute aortic syndromes: diagnosis and management, an update. Eur Heart J 2018;39:739–749d. [DOI] [PubMed] [Google Scholar]
- 7. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu J, Pan H, Xie X, Zhang J, Wang Y, Yang G. Inhibiting succinate dehydrogenase by dimethyl malonate alleviates brain damage in a rat model of cardiac arrest. Neuroscience 2018;393:24–32. [DOI] [PubMed] [Google Scholar]
- 9. De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Bäckhed F, Mithieux G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab 2016;24:151–157. [DOI] [PubMed] [Google Scholar]
- 10. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, Banks AS, Haigis MC, Kajimura S, Murphy MP, Gygi SP, Clish CB, Chouchani ET. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018;560:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, Paschini M, Paulo JA, Garrity R, Laznik-Bogoslavski D, Ferreira JCB, Carl CS, Sjøberg KA, Wojtaszewski JFP, Jeppesen JF, Kiens B, Gygi SP, Richter EA, Mathis D, Chouchani ET. pH-gated succinate secretion regulates muscle remodeling in response to exercise. Cell 2020;183:62–75. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, Urpi-Sarda M, Sabater M, Pérez-Brocal V, Andrés-Lacueva C, Moya A, Tinahones FJ, Fernández-Real JM, Vendrell J, Fernández-Veledo S. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J 2018;12:1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, Booty LM, Bicci I, Balzarotti B, Volpe G, Mallucci G, Manferrari G, Donega M, Iraci N, Braga A, Hallenbeck JM, Murphy MP, Edenhofer F, Frezza C, Pluchino S. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell 2018;22:355–368. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Y, Xie C, Li X, Yang J, Yu T, Zhang R, Zhang T, Saxena D, Snyder M, Wu Y, Li X. Succinate and its G-protein-coupled receptor stimulates osteoclastogenesis. Nat Commun 2017;8:15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, Valeaux S, Gommermann N, Rubic-Schneider T, Junt T, Carballido JM. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 2016;213:1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, Erle DJ, Anderson MS, Locksley RM, Raftery D, von Moltke J. Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity 2018;49:33–41. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, Banchereau R, Xu Z, Chandra M, Chung C-H, Walters L, Baisch J, Wright T, Punaro M, Nassi L, Stewart K, Fuller J, Ucar D, Ueno H, Zhou J, Banchereau J, Pascual V. A CD4 T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med 2019;25:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ceperuelo-Mallafré V, Llauradó G, Keiran N, Benaiges E, Astiarraga B, Martínez L, Pellitero S, González-Clemente JM, Rodríguez A, Fernández-Real JM, Lecube A, Megía A, Vilarrasa N, Vendrell J, Fernández-Veledo S. Preoperative circulating succinate levels as a biomarker for diabetes remission after bariatric surgery. Diabetes Care 2019;42:1956–1965. [DOI] [PubMed] [Google Scholar]
- 19. Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol 2019;21:85–93. [DOI] [PubMed] [Google Scholar]
- 20. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O'Neill LA. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013;496:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O'Neill LA. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016;167:457–470. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, Muret C, Debackere K, Mazzone M, Huang HD, Fendt SM, Ivanisevic J, Ho PC. Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 2017;18:985–994. [DOI] [PubMed] [Google Scholar]
- 23. Zhou KR, Liu S, Sun WJ, Zheng LL, Zhou H, Yang JH, Qu LH. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res 2017;45:D43–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones DS, Jenney AP, Joughin BA, Sorger PK, Lauffenburger DA. Inflammatory but not mitogenic contexts prime synovial fibroblasts for compensatory signaling responses to p38 inhibition. Sci Signal 2018;11:eaal1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, DiDonato JA, Brown JM, Gogonea V, Lusis AJ, Garcia-Garcia JC, Hazen SL. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat Med 2018;24:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lian G, Li X, Zhang L, Zhang Y, Sun L, Zhang X, Liu H, Pang Y, Kong W, Zhang T, Wang X, Jiang C. Macrophage metabolic reprogramming aggravates aortic dissection through the HIF1α-ADAM17 pathway. EBioMedicine 2019;49:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li DY, Busch A, Jin H, Chernogubova E, Pelisek J, Karlsson J, Sennblad B, Liu S, Lao S, Hofmann P, Bäcklund A, Eken SM, Roy J, Eriksson P, Dacken B, Ramanujam D, Dueck A, Engelhardt S, Boon RA, Eckstein H-H, Spin JM, Tsao PS, Maegdefessel L. H19 induces abdominal aortic aneurysm development and progression. Circulation 2018;138:1551–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Canfora EE, Meex RCR, Venema K, Blaak EE. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol 2019;15:261–273. [DOI] [PubMed] [Google Scholar]
- 30. Krzak G, Willis CM, Smith JA, Pluchino S, Peruzzotti-Jametti L. Succinate receptor 1: an emerging regulator of myeloid cell function in inflammation. Trends Immunol 2021;42:45–58. [DOI] [PubMed] [Google Scholar]
- 31. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, Yeh CC, Peng YJ, Kuo YY, Wen HT, Lin HC, Hsiao CW, Wu KK, Kung HJ, Hsu YJ, Kuo CC. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell 2020;77:213–227. e5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data used for this study are available from the corresponding authors upon reasonable request.