

Abstract
IL-13 is a central mediator of allergic inflammation. The single nucleotide polymorphism IL13-1112C>T (rs1800925) is associated with allergic phenotypes in ethnically distinct populations, but the underlying mechanism(s) remain unknown. Using in vivo, in vitro, and in silico analysis, we show that the IL13-1112T allele enhanced IL13 promoter activity in primary human and murine CD4+ Th2 lymphocytes. Increased expression of IL13-1112T in Th2 cells was associated with the creation of a Yin-Yang 1 binding site that overlapped a STAT motif involved in negative regulation of IL13 expression and attenuated STAT6-mediated transcriptional repression. Because IL-13 secretion was increased in IL13-1112TT homozygotes, we propose that increased expression of IL13-1112T in vivo may underlie its association with susceptibility to allergic inflammation. Interestingly, IL13-1112T had opposite transcriptional effects in nonpolarized CD4+ T cells, paralleled by distinct patterns of DNA-protein interactions at the IL13 promoter. Our findings suggest the nuclear milieu dictates the functional outcome of genetic variation.
Allergic diseases have a strong genetic component. Genome-wide linkage studies have identified several loci associated with increased susceptibility to allergy and/or asthma, including one on chromosome 5q.31 that contains the Th2 cytokines IL5, IL13, and IL4 (1). IL13 is a strong candidate for allergic disease. Elegant studies in animal models have shown that IL-13 is sufficient to mediate all of the cardinal features of Th2 inflammation in the lung, such as airway hyperresponsiveness, inflammatory cell infiltration, mucus hypersecretion, and airway fibrosis (2, 3). In humans, IL-13 and its receptors are highly expressed in the respiratory tract of patients with asthma and rhinitis (4, 5). The primary sources of IL-13 are Th2 cells, but eosinophils, mast cells, and basophils also produce this cytokine (6). IL-13 stimulates human B cells to synthesize IgE (7), a key effector in Th2-mediated disease, while suppressing the expression of CD14 (8), a molecule involved in protection from atopy (9). IL13 is expressed in the placenta (10) and is actively secreted by T cells in the neonatal period (11), a time critical for susceptibility to allergic disease.
Analysis of genetic variation across the IL13 locus in European ancestry subjects identified a block of common single nucleotide polymorphisms (SNPs)6 in complete linkage disequilibrium (LD) extending from IL13-1923C>T in the third intron to IL13-2749C>T in the 3′ untranslated region (12). This block includes IL13-2044G>A, a coding SNP resulting in the expression of a gain-of-function variant (IL-13 R130Q; Ref. 13) strongly associated with allergic inflammation in several ethnically distinct populations (14). This polymorphism is also in high, albeit not complete, LD with two promoter polymorphisms, IL13-1512A>C and IL13-1112C>T (rs1800925, also referred to as −1055 and −1111) (12). The IL13-1112TT genotype was more prevalent in individuals with asthma and atopic dermatitis and has been associated with increased risk of sensitization to food and outdoor allergens in several studies (15-18). Associations between the IL13-1112T allele and allergic phenotypes, such as high IgE serum levels, bronchial hyperresponsiveness and positive skin tests, have also been demonstrated (12, 15, 18).
Although these results strongly suggest that genetic dysregulation of IL13 expression and/or function may be a critical determinant of susceptibility to allergy and asthma, the extensive LD at the IL13 locus prevents the tools of genetic epidemiology from deciphering the contribution of individual polymorphisms to increased disease risk. Stratified analysis of IL13 haplotypes in a large Caucasian population did suggest an effect of IL13-1112C>T on IgE levels independent of IL13-2044G>A (19), but functional studies are required to characterize the impact of IL13-1112C>T on the regulation of IL13 expression.
To this purpose, we used a combination of in vivo, in vitro, and in silico approaches. We show herein that IL13-1112C>T is a functional polymorphism that results in increased IL13 transcription in primary Th2 cells through Yin-Yang 1 (YY1)-dependent attenuation of STAT6-mediated promoter repression. Analyses in a large population confirmed that IL-13 secretion was increased in IL13-1112TT homozygotes, suggesting that increased expression of the IL13-1112T allele may underlie its association with increased susceptibility to allergic inflammation.
Materials and Methods
Phylogenetic shadowing
The IL13 promoter was sequenced in genomic DNA samples (Coriell Cell Repository) from 12 primate species consisting of hominoids. Old World and New World monkeys. PCR primers for amplifying the primate promoters were designed based on the human IL13 sequence (GenBank accession No. AC004041 and L42080). Phylogenetic shadowing (20) was conducted using a multiple sequence alignment of human and primate sequences by Clustal W, and conservation plots were generated with eShadow (www.eshadow.dcode.org) (21). Settings were a 25-bp sliding window, 800-bp layer, default Hidden Markov Model Islands (0.85/0.77/0.1), and a divergence threshold (DT) of 15% variation within a minimum length of 70 bp (DT 15/70).
T cell isolation and culture
Human naive cord blood CD45RO−CD4+ T cells were obtained by negative selection and depletion of memory T cells using Ab against CD45RO (Miltenyi Biotec). Purity was typically ≥95%. All cytokines and mAbs were from R&D Systems, unless otherwise specified. Cells were primed for 4 days with plate-bound anti-CD3 mAb (UCHT1, 2 μg/ml) and soluble anti-CD28 mAb (1 μg/ml) under Th2 polarizing conditions (recombinant human (rh) IL-2, 5 ng/ml; rhIL-4, 4 ng/ml; neutralizing Abs to IL-12, 2 μg/ml; and IFN-γ, 2 μg/ml). Cells were rested for 3 days in rhIL-2 (5 ng/ml), rhIL-4 (4 ng/ml), and anti-IL-12 mAb (1 μg/ml). For subsequent weeks, cells were restimulated for 4 days on anti-CD3-coated plates with IL-4 (20 ng/ml) and anti-IL-12 mAb. Experiments were performed on day 16 or 21. Th2 cell polarization was assessed by intracellular cytokine staining.
Jurkat T cells (clone E6-1) and murine D10.G4.1 cells were obtained from American Type Culture Collection. The D10.G4.1 Th2 clone was cultured in RPMI 1640 (Cellgro) supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 0.05 mM 2-ME, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and murine rIL-2 (5 U/ml). Cells were restimulated with 100 μg/ml conalbumin (Sigma-Aldrich) and irradiated syngeneic AKR/J splenocytes every 3 wk. Experiments were conducted 6–12 days after restimulation.
Vector construction and transient transfections
A 2666-bp IL13 promoter construct (−1112/Luc; numbering relative to the IL13 ATG) was generated using genomic DNA obtained from an IL13-1112C homozygote as PCR template and cloned into the pGL3 Basic luciferase reporter vector (Promega). QuickChange site-directed mutagenesis (Stratagene) was performed to create a construct with a T at position −1112 (−1112T/Luc), to represent the minor allele, as well as the −1112C STATmut/Luc and −1112T STATmut/Luc constructs that contained a 3-bp mutation (TTC/gca, −1123/−1121) abolishing the STAT motif. Fidelity of PCR reactions was confirmed by sequencing.
Th2 cells (2 × 104/μl) were transiently nucleofected with the −1112/Luc reporter constructs (1 μg) and pRL-TK (Promega: 50 ng) using program T23 (Amaxa). Efficiencies ≥45% were achieved, as determined by flow cytometric analysis of cells transfected with pCMV-GFP (eGFP_C1; BD Clontech). Freshly isolated CD4+ T cells (1.25 × 104 cells/μl) were transiently nucleofected with the −1112/Luc reporter constructs (5 μg) and pRL-TK (5 ng) using program U14 (Amaxa) and achieving ≥75% transfection efficiency. Jurkat T cells (5 × 106) were transiently cotransfected with the −1112/Luc reporter construct (10 μg), STAT6 (TPU-388, 1–10 μg) or STAT1 (a gift from Dr. P. Rothman, Columbia University, New York, NY; 10 μg) expression vectors and pRL-TK (10 ng) by square wave electroporation using one pulse of 50 ms, 240 V (BTX, ECM 830, Genetronics). These conditions resulted in transfection efficiency ≥40%. In all experiments, cells were stimulated with PMA (20 ng/ml) and ionomycin (1 μM) immediately after transfection. IL-4 (10 ng/ml) was added to the STAT6/STAT1 cotransfections. Luciferase activity was assessed after 16–18 h incubation using the Dual Luciferase Assay (Promega) according to the manufacturer’s instructions. Protein concentration was determined with the bicinchoninic acid protein assay kit (Pierce). Results were normalized for transfection efficiency (as determined by Renilla luciferase activity) and protein concentrations, and expressed as relative luciferase activity (RLA; luciferase counts per microgram of protein).
Murine D10.G4.1 cells (2 × 106 cells/sample) were nucleofected with IL13-1112/Luc reporter constructs or pGL3 Basic (2 μg) and pRL-TK (50 ng) using program T01 (Amaxa). When indicated, a neutralizing anti-murine IL-4 mAb or an IgG1 isotype control (10 μg; BD Pharmingen) were added during the culture. Luciferase activity was assessed 16–18 h after nucleofection. Results were expressed as fold increase in the RLA detected in unstimulated cells transfected with IL13 reporter constructs relative to cells transfected with pGL3 Basic.
EMSA
To prepare nuclear extracts, cells (10 × 106) were pelleted, washed in cold 1× PBS and resuspended in cold buffer A (10 mM HEPES, 1.2 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM PMSF, 5 mM β-glycerophosphate, 1 mM benzamidine, 1 mM orthosodium vanadate, 1 mM sodium fluoride, 10 μg/ml antipain, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin; 120 μl). Cell lysis was monitored by trypan blue, and once swollen, cells were centrifuged at 14,000 rpm for 30 s at 4°C. Nuclear pellets were resuspended in an appropriate volume of ice-cold buffer C (420 mM NaCl, 20 mM HEPES, 25% glycerol, 1.5 mM MgCl2, 10 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 5 mM β-glycerophosphate, 1 mM benzamidine, 1 mM orthosodium vanadate, 1 mM sodium fluoride, 10 μg/ml antipain, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin), incubated on ice for 20 min, and centrifuged at 14,000 rpm for 25 min at 4°C. Supernatants containing nuclear proteins were recovered, and aliquots were frozen immediately and stored at −80°C.
Gel shift assays were performed using a 31-bp oligonucleotide probe (5′-GGACTTCTAGGAAAACGAGGGAAGAGCAGGA, position −1127/−1097 of the IL13 5′-flanking region) with either a C or a T at position −1112 (underlined). Double-stranded oligonucleotides were 5′-end-labeled with [γ-32P]ATP. The binding reaction included nuclear extract (5 μg), 100 mM NaCl, 10% glycerol, 50 ng/μl poly(deoxyinosinate-deoxycytidylate) (Pierce Chemical Co.), 1× binding buffer (10 mM Tris, 0.5 mM MgCl2, 1 mM EDTA, and 1 mM DTT), and 1 μl of probe (0.5–1 ng), and was incubated for 30 min on ice. Competition or supershift were performed by preincubating nuclear proteins with unlabeled oligonucleotides (50- to 100-fold molar excess) or transcription factor-specific Abs (4 μg) for 30 min before addition of the probe. The following oligonucleotides were used as competitors: STAT3N, AGAGTTTCCCAGAAGGATG (−889/−910 of the IL13 promoter); STATmut, GGACGCATAGGAAAACGAGGGAAGAGCAGGA; Oct-1, TGTCGAATGCAAATCACTAGAA; NFAT IL2, CGGAGGAAAAACTGTTTCATACAGAAGGCGTG. Abs against STAT1 (E-23 or M-22), STAT6 (S-20), YY1 (C-20), STAT2 (C-20), STAT3 (H-190), STAT4 (H-119), STAT5a (L-20), and STAT5b (N-20) were obtained from Santa Cruz Biotechnology. Reactions were loaded onto 5% nondenaturing polyacrylamide gels and run for 5–6 h at 18–19 mA with 0.5× Tris-buffered EDTA at 4°C.
Chromatin immunoprecipitation (ChIP)
Human naive peripheral blood CD4+ T cells, differentiated for 2 wk under Th2 conditions, were cultured in the presence or absence of PMA (20 ng/ml) and ionomycin (1 μM) for 3 h. Cells (5 × 107 per sample) were fixed for 10 min at 37°C with 1% formaldehyde. After incubation, glycine was added to a final concentration of 125 mM. Cells were washed twice with ice raid PBS and resuspended at 5 × 107/ml in ChIP lysis buffer (Upstate Biotechnology) supplemented with 1× EDTA-free protease inhibitor mixture (Roche) and 1 mM PMSF. Chromatin was sheared by sonication to an average length of 600 bp (six pulses for 10 s each at 30% maximum output; Microson XL200) and diluted 2-fold in ChIP dilution buffer (Upstate Biotechnology) supplemented with protease inhibitors as above. One-twentieth of the total sample was removed to be used as input DNA. Before immunoprecipitation, samples were precleared with salmon sperm DNA/protein A agarose slurry (Upstate Biotechnology) for 30 min. Soluble chromatin was immunoprecipitated overnight at 4°C with 10 μg of anti-STAT6 (sc-621) or anti-YY1 (sc-281) (both from Santa Cruz Biotechnology) or control mouse IgG1 (M5284; Sigma-Aldrich). Chromatin-Ab complexes were collected with salmon sperm DNA-protein A agarose beads and washed sequentially with low salt, high salt, LiCl, and Tris-EDTA (ChIP Assay Kit; Upstate Biotechnology). Complexes were then eluted (1% SDS and 0.1 M NaHCO3), incubated at 65°C overnight to reverse cross-links, and deproteinated with proteinase K. DNA samples were purified by phenol-chloroform extraction followed by ethanol precipitation in the presence of glycogen (20 μg) and resuspended in 20 μl of nuclease-free water. PCR amplification of a 202-bp amplicon encompassing position −1112 of IL13 was performed with primers 1112ChiPF (5′-GGGTAGGGGAGAAATCTTGACATC) and 1112ChIPR (5′-ATCAACCCCTGCCGTCTGG). Amplification of CNS-1 was performed using primers CNS1ChIPF (5′-CACAGCGTCGTTCAGAAACAC) and CNS1ChIPR (5′-CAGCCCCCGCACAGTTG) to yield a 152-bp amplicon. PCR was performed under the following conditions: 95°C for 15 min followed by 30 cycles at 95°C for 30 s, 57°C for 30 s, and 72°C for 45 s, ending with a final extension at 72°C for 5 min.
Analysis of IL-13 secretion by IL13-1112 genotype
Peripheral blood samples were obtained from women at the time of their enrollment in the Infant Immune Study (22), at about the eighth month of pregnancy. PBMC (2 × 106 cells/ml) were incubated in the presence or absence of Con A (10 μg/ml) and PMA (10 ng/ml: Sigma-Aldrich). After 18–24 h, supernatants were harvested and stored at − 70°C. IL-13 levels were assayed using a commercially available ELISA kit (Diaclone Research). Genotyping for IL13-1112C>T and IL132044G>A was performed as previously described (12).
Statistical analysis
The relationship between IL13-1112 genotypes and IL-13 production was assessed by one-way ANOVA, whereas the differences in IL-13 levels between individual genotypes were assessed with a Bonferroni multiple comparison test. Statistical significance of differences in fold induction between transfected vectors was determined using the Wilcoxon two-sample test.
Human subjects
Cord blood was obtained from anonymous cesarean deliveries. Peripheral blood was drawn from healthy nonallergic subjects after written informed consent was obtained. All protocols were approved by the Institutional Review Board of the University of Arizona.
Results
IL13-1112T is the ancestral allele and resides within a putative primate-specific cis-regulatory element
To investigate the functional relevance of IL13-1112C>T to IL13 regulation, we initially relied on comparative genomic analysis. Genomic segments strongly conserved during evolution frequently exhibit regulatory properties (20), implying that SNPs located in such regions are more likely to be functional. A human/mouse sequence alignment revealed poor conservation of the region containing IL13-1112C>T (Fig. 1). However, comparisons among distant species, although effective for the identification of highly constrained elements, invariably miss more recent changes in DNA sequence, such as those accounting for biological traits unique to primates (23). Therefore, we turned to phylogenetic shadowing, an approach that was recently developed to analyze sequence conservation profiles among closely related species (20) and accurately predicted primate-specific exons and regulatory elements arisen after divergence from the mouse lineage (21).
FIGURE 1.
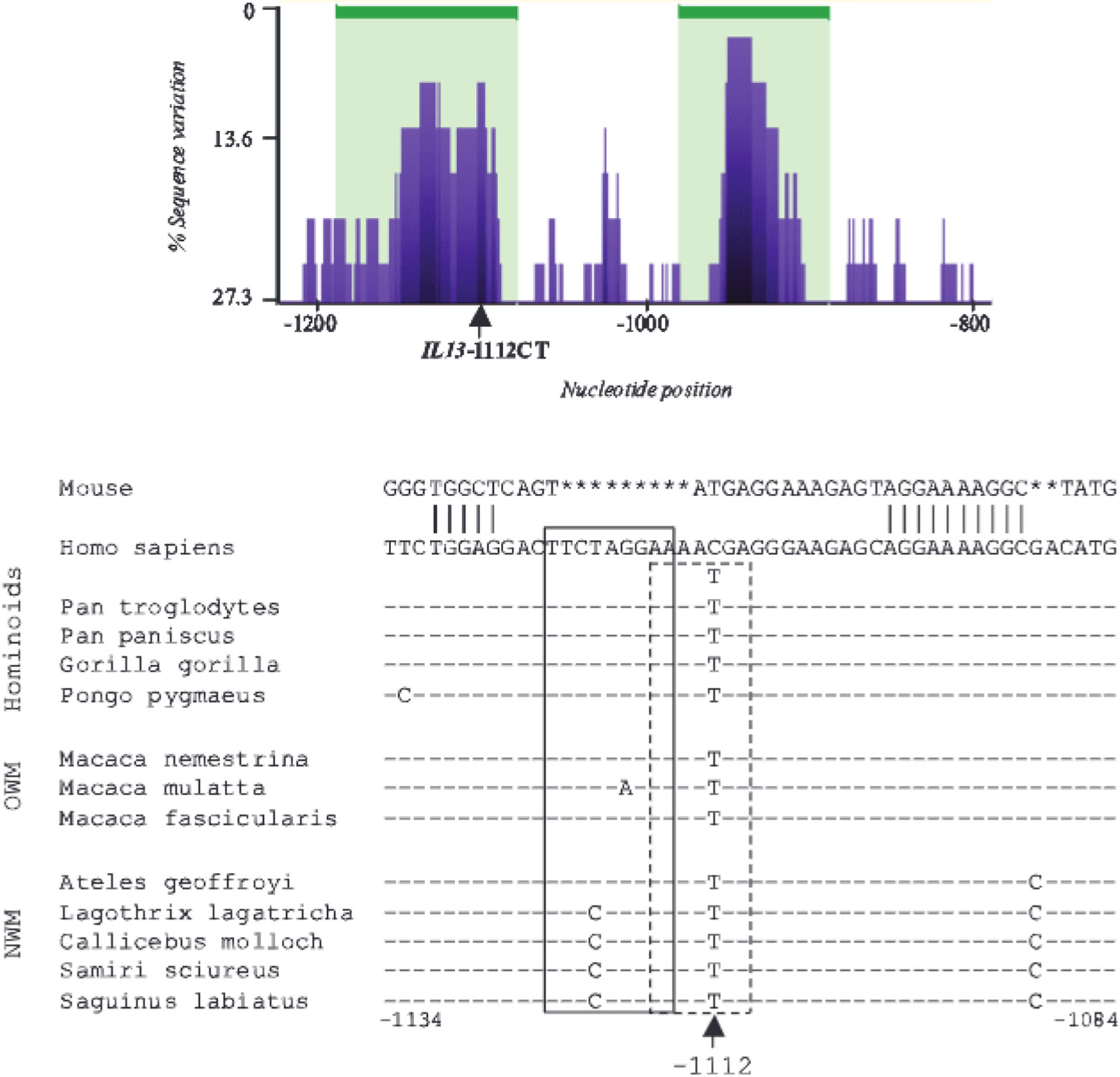
IL13-1112T is the ancestral allele and resides within a putative primate-specific cis-regulatory element. Top, eShadow conservation plot (www.eshadow.dcode.org) for the region surrounding IL13-1112C>T (arrow) in humans and 12 primate species representative of distinct clades. Peaks and valleys correspond to regions of low and high variation, respectively, with 0% variation = 100% sequence identity. Areas shaded in green represent regions below the DT (15/70). Bottom, Sequence alignment for the region surrounding IL13-1112C>T (arrow) in mice, humans, and primates. Numbering is relative to the human IL13 ATG. The pairwise alignment of human (GcnBank accession No. NC_000005:132016718-132031715) and mouse (GenBank accession No. NC_000077:53392544-5337166) promoter sequences for the IL13 –1112 region was performed with AlignX, a feature of the Vector NTI suite, which is based on the Clustal W algorithm. The dashes and asterisks mark conserved positions and gaps in the sequence, respectively. The STAT6 and YY1 binding motifs revealed by the DNA-protein interaction analysis discussed later in this paper are boxed by continuous or dashed lines, respectively. OWM, Old World monkeys; NWM, New World monkeys.
A 2-kb region of the IL13 promoter was sequenced in 12 primate species representative of the main clades of hominoids. Old World and New World monkeys. Analysis of multiple sequence alignments using eShadow (21) showed that IL13-1112C>T falls within a peak of high intraprimate conservation that spans ~80 bp and predicts the existence of a primate-specific cis-regulatory element (Fig. 1, top). This element maps to the vicinity of a region that exhibits constitutive DNA hypomethylation and hypersensitivity to DNase I digestion in human naive, Th1 and Th2 CD4+ T cells (R. B. Webster, Y. Rodriguez, W. Klimecki, and D. Vercelli, in press), suggesting this region may be endowed with regulatory properties. These findings provide indirect but suggestive evidence for a potential role of IL13-1112C>T in the regulation of IL13 expression. All 12 nonhuman primate species had a T at position −1112 (Fig. 1, bottom). Therefore, the disease-associated IL13-1112T allele is the ancestral allele.
Transcription of the IL13-1112T allele is enhanced in primary CD4+ Th2 cells, but not in nonpolarized CD4+ T cells
Because IL13-1112C>T disrupts a CpG site (Fig. 1), we explored the possibility that loss of the C at −1112 may alter DNA methylation patterns throughout the region, rendering the minor promoter variant more accessible to the transcriptional machinery. Bisulfite sequencing analysis revealed virtually identical CpG methylation profiles in CD4+ T cells of distinct IL13-1112 genotypes (data not shown), ruling out this possibility.
To examine more directly whether IL13-1112C>T affects IL13 transcription, we generated luciferase reporter constructs driven by a 2.7-kb IL13 promoter fragment carrying either the major (C) or minor (T) allele at position −1112. Initially, the −1112/Luc reporter constructs were transfected into the Jurkat T cell line, a well-established model to study transcriptional regulation of cytokine genes. Fig. 2 (left) shows that −1112C/Luc activity was upregulated 22-fold in response to T cell activation. In contrast, the −1112T/Luc construct was induced only 15-fold (n = 26; p = 0.006). Thus, IL13-1112T was significantly less active than the major allele in Jurkat T cells.
FIGURE 2.
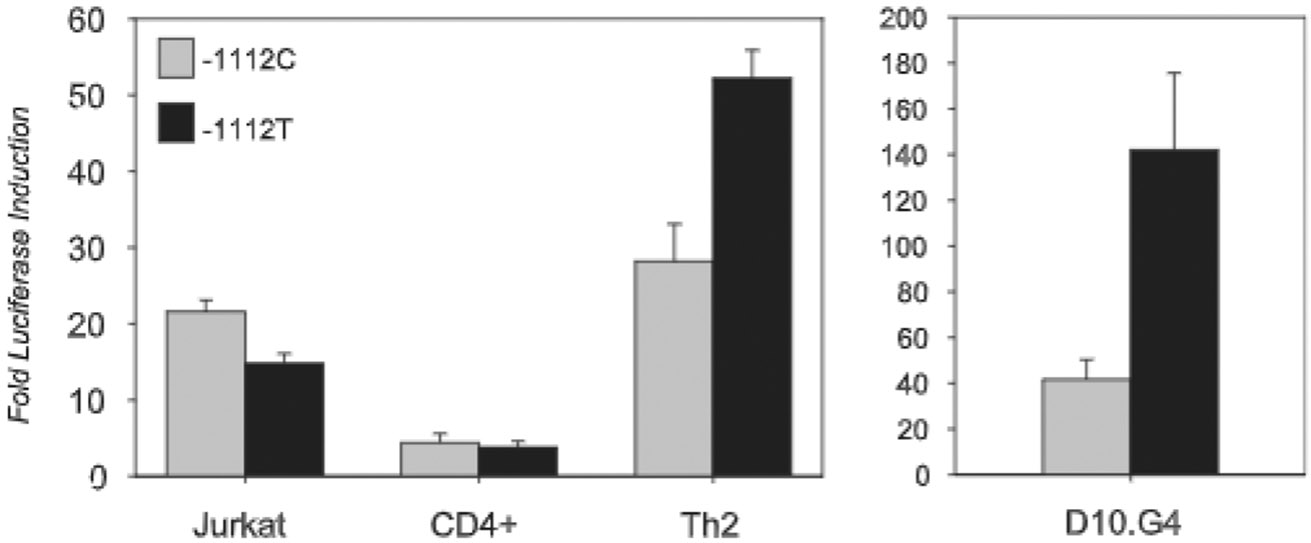
IL13-1112T enhances IL-13 promoter activity in polarized CD4+ Th2 cells but not in nonpolarized CD4+ T cells. Left, Jurkat T cells (n = 26), freshly isolated CD4+ T cells (n = 9), or in vitro polarized human Th2 cells (n = 5) were transiently transfected with 2.7-kb IL13 promoter reporter constructs carrying the major (C) or minor (T) −1112 allele. Cells were left in medium or stimulated with PMA (20 ng/ml) and ionomycin (1 μM) and harvested after 16–18 h. Results are expressed as fold-increase in RLA (mean ± SE) after stimulation. Right, Murine D10.G4.1 Th2 cells were nucleofected with pGL3 Basic or −1112 allelic variants of an IL13 reporter construct (n = 8). Cells were left unstimulatcd for 16 h after transfection. Results are expressed as fold increase in the activity of the IL13 reporter constructs relative to pGL3 Basic. Statistical significance of all results was determined using the Wilcoxon two-sample test.
Although these data suggested that IL13-1112CT is a functional SNP, the lower activity of the T allele could not be readily reconciled with its reported association with allergic phenotypes and increased asthma susceptibility (1). To reassess these findings in primary T cells, we studied transcription of the IL13-1112 alleles after nucleofection into CD4+ T cells freshly isolated from normal peripheral blood. Activation of CD4+ T cells up-regulated transcription of both allelic variants to a comparable extent (4.4- vs 3.8-fold; n = 9, p > 0.05: Fig. 2, left), a result that was again inconsistent with the association between IL13-1112T and susceptibility to Th2-dependent inflammation. However, the true transcriptional impact of the −1112 polymorphism might only become apparent within a cytokine/nuclear environment leading to high level IL13 expression. Jurkat and CD4+ T cells up-regulate IL13 mRNA levels in response to activation (≥80-fold for Jurkat cells and ≥140-fold for primary T cells, as assessed by real time RT-PCR), but only ~3% of these cells expressed detectable levels of intracellular IL-13 protein (data not shown). Thus, the majority of luciferase activity in nonpolarized CD4+ T cells was likely generated from a nuclear environment inadequate to promote optimal IL13 expression.
Because IL13 is typically expressed by polarized CD4+ Th2 cells and these cells play a critical effector role in human and experimental allergic inflammation, the transcriptional effect of IL13-1112C>T was examined in two independent, primary Th2 cell models. In one set of experiments, human neonatal naive CD45RO−CD4+ T cells were differentiated in vitro under Th2-polarizing conditions. After 2–3 wk of culture, cells acquired a highly polarized Th2 phenotype, as demonstrated by elevated levels of intracellular IL-13 and virtually absent IFN-γ (data not shown). Nucleofection of primary human Th2 cells with IL13-1112C and T reporter constructs led to significantly higher transcriptional induction of the −1112T allele in response to T cell activation (Fig. 2, left). Likewise, the −1112T variant was significantly more active in the murine D10.G4.1 Th2 clone, a well-established model of Th2 cell polarization (24) (Fig. 2, right). Our results demonstrate that the nuclear environment can dictate the transcriptional outcome of genetic variation. In the context of a Th2 milieu that drives high IL13 expression, but not within nonpolarized CD4+ T cells, the −1112T allele conferred higher activity to the IL13 promoter, consistent with the reported association between this allele and increased susceptibility to allergic inflammation (1).
IL13-1112 allele- and cell type-specific patterns of transcription factor binding parallel profiles of transcriptional activity
To identify the mechanisms underlying higher transcription of IL13-1112T in Th2 cells, we initially used EMSA analysis to compare and contrast patterns of DNA-protein interactions occurring at the IL13 −1112 promoter variants in distinct T cell nuclear environments. We reasoned that such comparisons could provide an indirect but powerful tool to tease out the interactions involved in increased transcription of the −1112T allele.
Using oligonucleotides corresponding to the C or T allelic variants of the −1127/−1097 IL13 promoter region (Fig. 3A, top), we demonstrated that nuclear extracts from activated primary Th2 cells (in which the −1112T allele was transcriptionally more active) contained two specific, slowly migrating complexes that bound both the C and the T allele (Fig. 3A, lanes 2 and 8) and were cross-competed by cold C and T 1127/−1097 IL13 oligonucleotides (Fig. 3A, lanes 3, 4, 9, and 10). The upper complex contained STAT6, because formation of the complex was abrogated by a STAT3N (lanes 6 and 12), but not an NFAT competitor (lanes 5 and 11). Furthermore, the band was supershifted by an anti-STAT6 (lanes 14 and 18), but not an anti-STAT1 (Fig. 3A, lanes 15 and 19) or an anti-C/EBPγ (Fig. 3A, lane 16) Ab. Conversely, the lower complex was formed by STAT1, because it was fully competed by a STAT3N oligonucleotide (Fig. 3A, lanes 6 and 12) and selectively supershifted by an Ab against this STAT family member (Fig. 3A, lanes 15 and 19). No other STAT proteins were found to participate in complex formation, as assessed by supershifting with Abs specific for individual family members (data not shown). The −1112T but not the C probe bound an additional fast-migrating complex that was competed by a cold −1112T, but not −1112C, oligonucleotide (Fig. 3A, lanes 9 and 10). The complex contained YY1 because it was supershifted by an anti-YY1 Ab (Fig. 3A. lane 20), but not by Abs to STAT6 or STAT1 (Fig. 3A, lanes 18 and 19). Comparison of nuclear extracts from resting and activated Th2 cells revealed that the YY1 complex bound constitutively to the −1112T allele (Fig. 3A, lane 23), whereas formation of the STAT6- and STAT1-containing complexes was dependent on T cell activation (Fig. 3A, lanes 22 and 24). Increased activity of IL13-1112T in Th2 cells was therefore paralleled by allele-specific binding of the transcription factor YY1.
FIGURE 3.
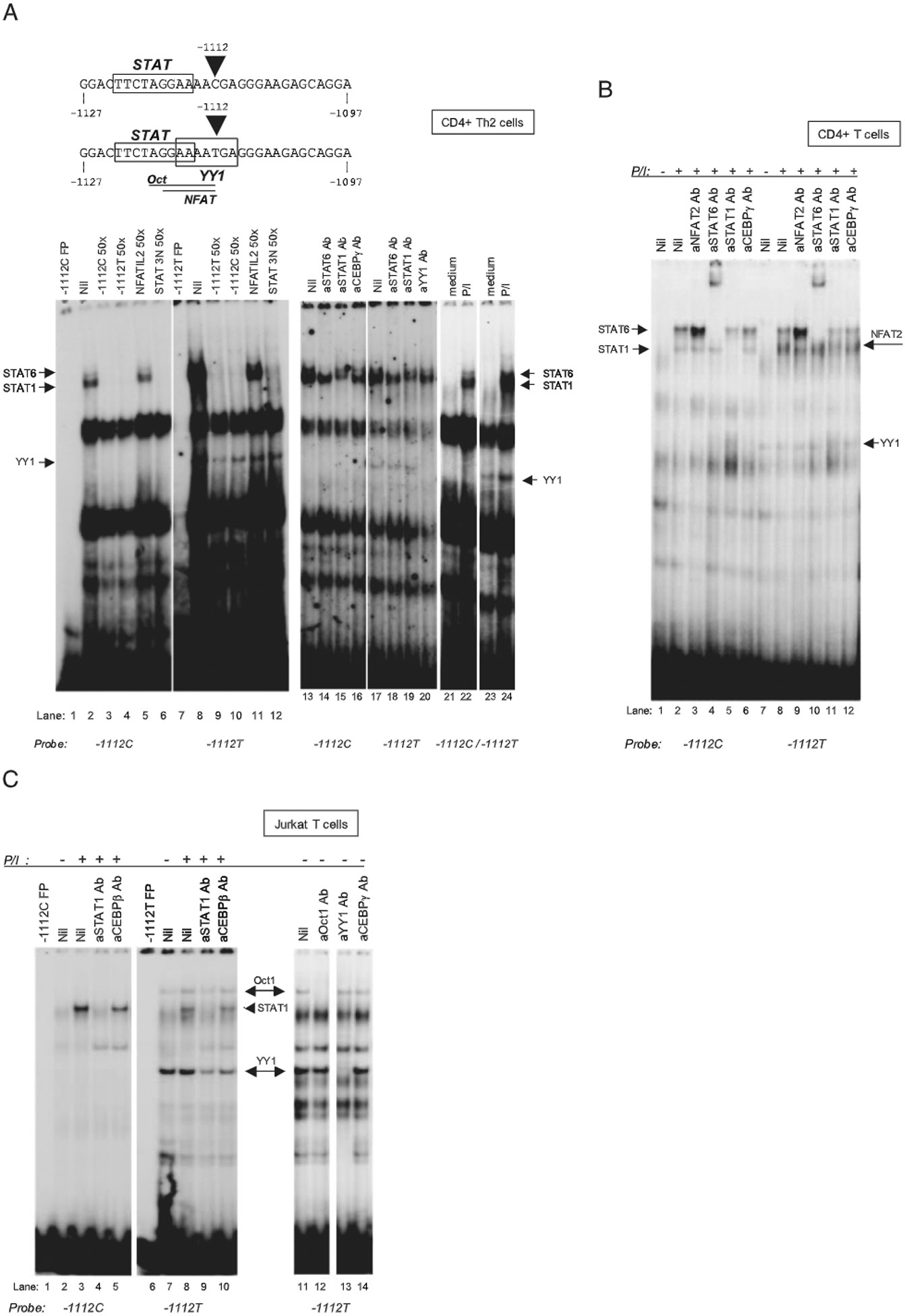
IL13-1112 allele-specific patterns of transcription factor binding in distinct T cell nuclear environments. EMSA analysis with IL13-1112C and T oligonucleotide probes (top) and nuclear extracts from primary polarized human Th2 cells (A), freshly isolated CD4+ T cells (B), and Jurkat T cells (C) cultured for 3 h in medium or PMA (P; 20 ng/ml) and ionomycin (I; 1 μM). The nuclear extracts used in lanes 1–20 of A were prepared from PMA-ionomycin-activated cells. STAT and YY1 binding sites in the probe sequence are boxed; NFAT and Oct motifs are underlined. The competitors (fold molar excess) and supershifting Abs used for each experiment are noted above the corresponding lanes in the gels. Probes are noted below the gels. FP, free probe; a (as in aSTAT), anti.
When we analyzed nuclear factor binding to the −1127/−1097 IL13 promoter region using nuclear extracts from nonpolarized primary CD4+ T cells (in which the T allele was transcribed less actively), Ab supershift analysis again demonstrated the presence of activation-induced complexes containing STAT6 (Fig. 3B, lanes 4 and 10) and STAT1 (Fig. 3B, lanes 5 and 11), that interacted equivalently with the −1112C and T allele (Fig. 3B, lanes 2 and 8). Interestingly, in CD4+ T cell extracts the T allele selectively bound not only constitutively expressed YY1 (Fig. 3B, lanes 7 and 8), but also an additional inducible complex. The latter contained NFAT2, because its formation was inhibited by an anti-NFAT2 Ab (Fig. 3B, lane 9), but not by Abs to STAT6 (Fig. 3B lane 10), STAT1 (Fig. 3B, lane 11) or C/EBPγ (Fig. 3B, lane 12). Moreover, pretreatment of PMA-ionomycin-stimulated CD4+ T cells with cyclosporine A (50 μg/ml) selectively prevented complex formation (data not shown), further supporting the identification of NFAT as a main complex constituent.
Finally, EMSA analysis of nuclear extracts from Jurkat T cells (which, like fresh CD4+ T cells, supported weaker transcription of the −1112T allele) showed strong specific binding of STAT1 to both alleles (Fig. 3C, lanes 3, 4, 8, and 9) and selective binding of YY1 to −1112T (Fig. 3C, lanes 11 and 13). However, no STAT6-containing complex was detected, consistent with deficient STAT6 activity in Jurkat T cells (25). A slowly migrating complex supershifted by an anti-Oct-1 Ab (Fig. 3C, lane 12), but not by Abs to YY1 (Fig. 3C, lane 13), C/EBPγ (Fig. 3C, lane 14), or Oct-2 (data not shown), was also detected in Jurkat T cell extracts using a −1112T allele probe. The same complex was selectively competed by a bona fide Oct consensus oligonucleotide (data not shown), and thus contained Oct-1.
These results show that the higher activity of the IL13-1112T allele in Th2 cells correlated with a unique pattern of DNA-protein interactions marked by the combination of STAT6/STAT1 and YY1, and provide a molecular rationale for the differential transcription of the −1112 alleles in distinct T cell nuclear environments.
IL13-1112T attenuates STAT6-mediated repression of IL13 transcription
YY1 is a ubiquitously expressed nuclear protein that can either activate or repress transcription (26). Because the sequences flanking the YY1 core motif (TCAT) can be quite variable, many promoters contain YY1 sites that overlap those for other transcriptional regulators (26). This topology fosters interactions between YY1 and other factors, the outcome of which depends on the function of the proteins involved. In silico analysis of the IL13 promoter sequence and mutational EMSA analysis by nucleotide transversions (data not shown) demonstrated that the YY1 motif created by −1112T overlaps the 3′ end of a STAT palindrome that is present on both alleles and binds STAT6 or STAT1 in Th2 cells (Fig. 3A, top). In view of the dual role of YY1 in transcriptional regulation, two models may explain the role of YY1 in the increased activity of the IL13-1112T allele in Th2 cells. Both YY1 and STAT proteins may act cooperatively as transcriptional activators, as reported for YY1 and STAT5b binding to the serine protease inhibitor 2.1 gene (27). Alternatively, STAT binding to the IL13-1112 region may repress transcription, as reported for the human IL4 promoter (25), and YY1 may relieve STAT-mediated repression by displacing STATs or recruiting STAT corepressors.
We reasoned that understanding the role played by YY1 in the increased activity of the IL13-1112T allele required the functional characterization of the −1123/−1115 STAT motif overlapping the YY1 site. Therefore, the IL13-1112C and T reporter vectors were mutated in the 5′ half of the STAT motif. EMSA analysis confirmed that the mutation disrupted binding of STAT1/STAT6, but not YY1 (data not shown). Fig. 4 (left) shows that the −1112C STAT mutant construct was markedly more active than either allelic variant when nucleofected into primary human Th2 cells. In contrast, the activity of the minor −1112T allele was only marginally altered by the STAT site mutation. These results demonstrate that the STAT motif upstream of the polymorphism plays a strong negative regulatory role in the context of the −1112C IL13 promoter and indicate that YY1 binding to its newly created site is virtually sufficient to relieve STAT-dependent promoter repression. Strong independent support for both these conclusions was provided by the experiments shown in Fig. 4 (center). Inhibition of STAT6 activation by neutralization of endogenous IL-4 significantly up-regulated transcription of the common −1112C promoter variant in D10.G4.1 Th2 cells, raising the activity of the −1112C allele to the levels achieved by the −1112T allele. In contrast, IL-4 blockade did not affect the activity of the −1112T promoter.
FIGURE 4.
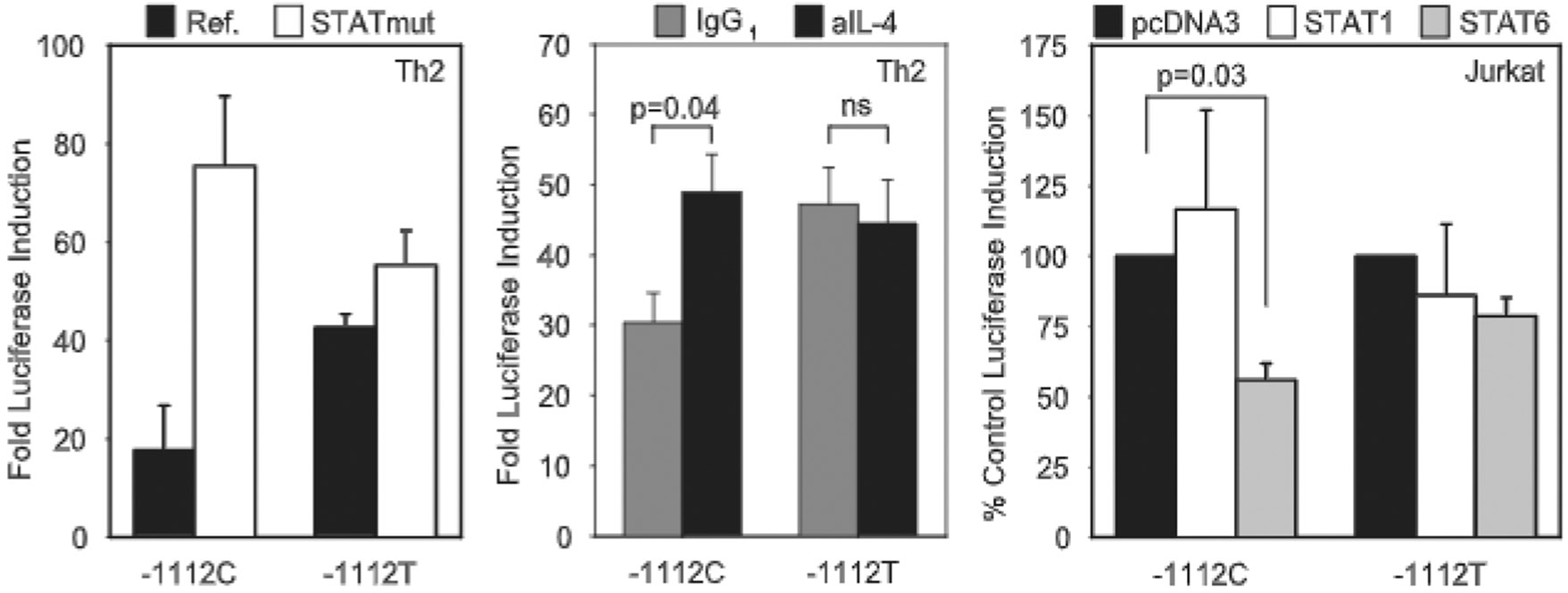
IL13-1112T altcnuatcs STAT6-mediated repression of IL-13 transcriplion. Left, Primary polarized human Th2 cells were transienlly transfected with −1112 allelic variants of the reference 2.7-kh IL13 promoter reporter construct (−1112C/Luc or −1112T/Luc) or equivalent constructs in which the STAT site adjacent to the SNP had been disrupted (−1112C STATmut/Luc and −1112T STATmut/Luc) (n = 2). Cells were left in medium or stimulated with PMA (20 ng/ml) and ionomycin (1 μM) and harvested after 16–18 h. Results are expressed as fold increase in RLA (mean ± SE) after stimulation. Center, Murine D10.G4.1 cells were harvested 6–12 days after antigenic stimulation and nucleofected with pGL3 Basic or IL13-1112C or T promoter reporter variants. Cells were incubated with neutralizing anti (a)-murine IL-4 or control IgG1 Ab for 16 h before harvesting (n = 6). Results are expressed as fold increase in the activity of the IL13 reporter constructs relative to pGL3 Basic. Right, Jurkat T cells were cotransfected with the −1112/Luc IL13 promoter constructs and pcDNA3, STAT1 (n = 4), or a STAT6 (n = 11) expression vectors. Cells were incubated with medium or PMA (20 ng/ml) and ionomycin (1 μM). IL-4 (10 ng/ml) was added to the STAT6 cotransfections. Cells were harvested after 16–18 h. Results arc expressed as percentage of fold luciferase induction in response to stimulation for STAT-transfected cells relative to cells transfected with pcDNA3 (mean ± SE). For all panels, statistical significance was determined using the Wilcoxon two-sample test.
STAT6, not STAT1, was involved in transcriptional attenuation through the −1123/−1115 site adjacent to IL13-1112C>T, because overexpression of STAT6, but not STAT1, significantly decreased −1112C/Luc activity in Jurkat T cells (Fig. 4, right). Interestingly, STAT6 overexpression did not affect transcription from −1112T/Luc, consistent with blockade of the STAT6 site by YY1, which is highly expressed in Jurkat T cells (Fig. 3C). We conclude that binding of YY1 to a motif created by IL13-1112T relieves STAT6-mediated repression, leading to increased activity of the −1112T allele in Th2 cells.
STAT6 and YY1 bind at the IL13-1112 region in vivo
To confirm the role of STAT6 and YY1 as critical determinants of IL13 transcription in vivo, the ability of these factors to bind the endogenous IL13-1112 promoter region in primary human Th2 cells was assessed by ChIP. Fig. 5 (top) shows that the region of interest was readily immunoprecipitated by anti-STAT6 and anti-YY1 Abs but not control IgG, demonstrating STAT6 and YY1 interact with the IL13 promoter in chromatin. STAT6-anti-STAT6 interactions were specific, because the anti-STAT6 Ab failed to immunoprecipitate CNS-1 (Fig. 5, bottom), a regulatory element located in the IL13/IL4 intergenic region, that does not encompass bona fide STAT6 motifs. A weak YY1 signal was detected in CNS-1, in line with the presence of several predicted YY1 sites (J. M. Strempel and D. Vercelli, unpublished observations). Detection of DNA-bound STAT6 in resting Th2 cells, although at variance with our EMSA findings, may reflect the continuous presence of IL-4 in Th2 cell cultures and the sensitivity of ChIP to modifications in chromatin architecture.
FIGURE 5.
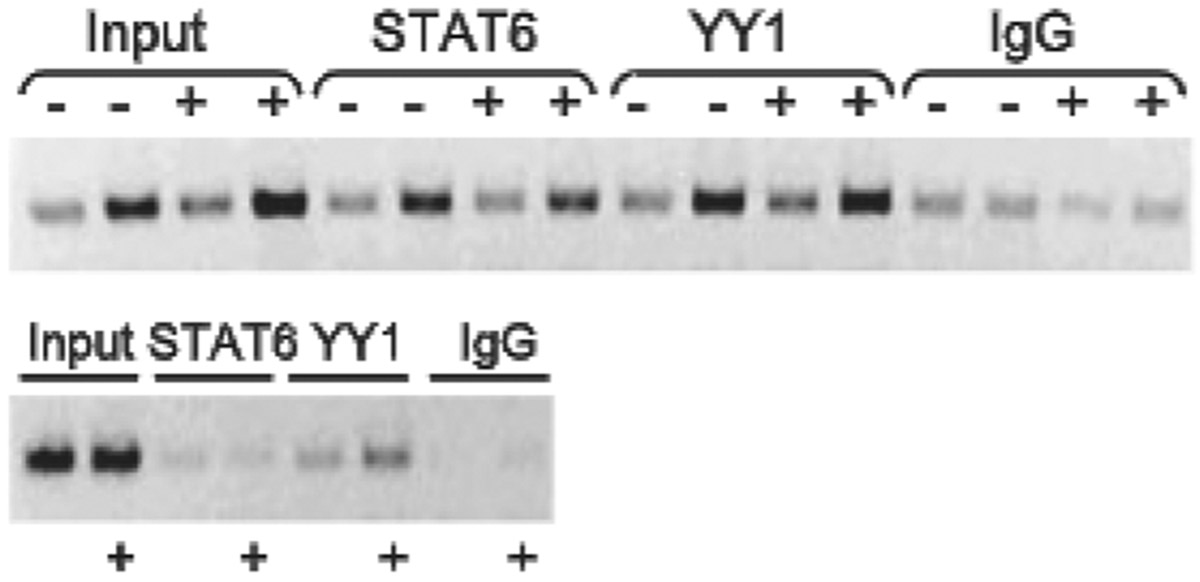
STAT6 and YY1 bind at the IL13–1112 region in vivo. Naive peripheral blood CD4+ T cells from a healthy IL13–1112T heterozygote were differentiated in vitro for 2 wk under Th2 conditions. ChIP assays with an anti-STAT6, anti-YY1, or control Ab (mouse IgG1) were performed on cells (5 × 107 per immunoprecipitation) cultured in the presence (+) or absence (−) of PMA and ionomycin (P/I) for 3 h. Top, Input DNA (5 or 10 ng) or immunoprecipitated DNA (one-twentieth and one-tenth of total) was used as template for PCR amplification of a 202-bp amplicon encompassing IL13-1112. Bottom, Input DNA (10 ng) or immunoprecipitated DNA (one-tenth of total) was used as template for PCR amplification of a 152-bp amplicon corresponding to CNS-1. Results are from one representative experiment of three independent immunoprecipitations.
Given that the DNA fragments generated by sonication were on average 600 bp long but exhibited some expected heterogeneity, and at least 6 YY1 and 9 STAT putative binding motifs are located within 1 kb of IL13-1112C>T (data not shown), ChIP analysis could not formally prove that the docking sites for the immunoprecipitated STAT6- and/or YY1-containing complexes were those immediately adjacent to, or overlapping, IL13-1112C>T. However, our results point to an involvement of both STAT6 and YY1 in the regulation of IL13 promoter activity in the endogenous nuclear environment and as such lend further support to the view that the interplay between these factors is critical for the functional outcome of IL13-1112C>T.
Increased IL-13 secretion in IL13-1112TT homozygotes
The IL13-1112T allele resulted in increased IL13 transcription, (Figs. 2 and 4) and was associated with allergic phenotypes in multiple studies (14), but the existence of a direct correlation between IL13-1112 genotypes and levels of IL-13 production was never tested in a large population. We assessed IL-13 secretion in subjects enrolled in the Tucson Infant Immune Study, a large prospective study of the development of immunological markers of asthma risk (22). We focused on 174 women at the third trimester of pregnancy, unselected for atopy and asthma. Fig. 6 shows that mitogen-activated PBMC from IL13-1112TT homozygotes secreted significantly higher levels of IL-13 compared with −1112CC and CT individuals (ANOVA, p = 0.015). The effect was strengthened after adjusting for ethnicity and IL13-2044 genotype in a multivariate linear regression (−1112TT vs CC, p = 0.007: −1112TT vs CT, p = 0.003). The latter adjustment was required because IL13-2044G>A, a common coding SNP in strong LD with IL13-1112C>T in the Tucson population (12), results in the expression of an IL-13 variant (IL-13 R130Q) with increased biological activity and altered antigenic properties that decrease its ELISA-based detection by a factor of 25.7% (13). Therefore, the data shown in Fig. 6, if anything, would represent under detection of IL-13 in carriers of IL13-2044A. Although the complex patterns of LD in the IL13 locus warrant caution in the interpretation of these data, our findings strongly support the possibility that IL13-1112C>T may contribute to increased IL13 expression in vivo. The low IL-13 levels detected in −1112CT heterozygotes may result from monoallelic IL13 expression, a pattern well documented in mouse strains (28), and preferential expression of the C allele.
FIGURE 6.
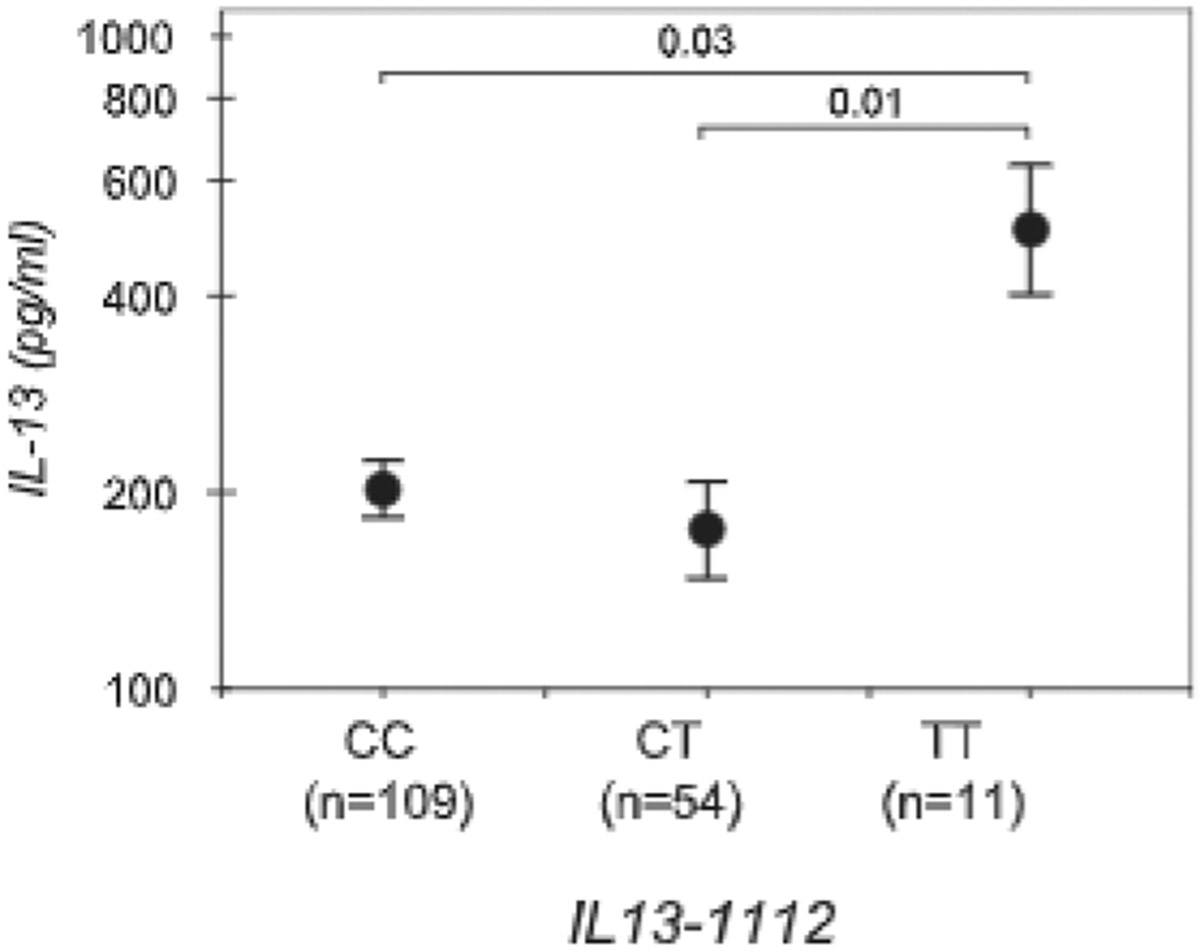
Increased IL-13 secretion in IL13-1112T homozygotes. PBMC from pregnant women enrolled in the Infant Immune Study study and genotyped for IL13-1112C>T and IL13-2044G>A were incubated with Con A (10 μg/ml) and PMA (10 ng/ml) for 18–24 h. IL-13 concentrations in culture supernatants were assessed by ELISA. Results are the mean ± SE of IL-13 concentrations measured in each IL13-1112 genotype group. Statistical significance was assessed by one-way ANOVA for the relation between IL13-1112 genotypes and IL-13 production, and a Bonferroni multiple comparison test for differences in IL-13 levels between individual genotypes.
Discussion
The locus encoding IL-13, a central mediator of allergic inflammation, harbors numerous common polymorphisms in strong LD, and distinct IL13 haplotypes are associated with allergic phenotypes in ethnically distinct populations (1). Because of this genetic complexity, functional studies are necessary to dissect the mechanisms underlying the contribution of natural genetic variation to IL13 dysregulation and susceptibility to allergy. Our study demonstrates IL13-1112C>T is a functional polymorphism that enhances IL13 promoter activity in primary human Th2 lymphocytes, cells programmed for high IL13 expression, but has opposite effects in nonpolarized CD4+ T cells. The nuclear milieu may therefore dictate the functional outcome of genetic variation.
Gene-environment interactions in the nucleus are a phenomenon we previously observed for CD14-159C>T, a SNP which results in different patterns of CD14 promoter activity in monocytes and hepatocytes depending on the Sp1:Sp3 ratio (29). However, our current data are more remarkable in that differential IL13 expression was observed not in distinct cell types but in distinct CD4+ Th cell phenotypes and that they correlated with distinct patterns of transcription factor binding to the IL13 promoter. That the gain-of-function associated with the IL13-1112T allele emerged only in differentiated Th2 cells eloquently shows how subtle the functional impact of genetic variation can be and how essential it is to choose experimental models able to capture it. Furthermore, these results suggest that IL13-1112C>T is likely to influence risk of allergy and/or asthma in the context of an established Th2 response. Thus, this polymorphism may contribute to the maintenance and/or exacerbation of allergic inflammation more than to its inception.
Gene-environment interactions in the nucleus may also offer a rationale for the common but disquieting finding that many published associations could not be replicated (30-32). If the functional outcome of genetic variation contributing to disease risk is determined not only by the genetic but also by the biological context, as our data indicate, the conditions under which biological samples are collected for phenotyping may become critically important, and failing to account for gene-environment interactions in the nucleus may hamper detection of susceptibility loci. Interestingly, there are now many examples of established associations with different functional variants within the same gene or with opposite alleles at the same SNP in different populations (1). For example, IgE levels are associated with IL13-1112C>T in some populations (33, 34) and with IL13-2044G>A (12, 33, 35) or IL13-1512A>C (36) in others. Protection from allergy and/or asthma is associated with both the T (9) and the C (37) allele of CD14-159. It is tempting to speculate that these seemingly contradictory results may often represent an outcome of gene-environment interactions in the nucleus.
Increased transcription of IL13-1112T was associated with the creation of a binding site for YY1 that overlapped a STAT motif and attenuated STAT6-mediated repression. Although necessary for Th2 cell differentiation and expression of Th2 cytokine genes, including IL13 (38), STAT6 can also act as a negative transcriptional regulator. STAT6 overexpression was shown to suppress the activity of an IL4 reporter construct in both Jurkat (25) and polarized murine Th2 (39) cells. Furthermore, comparative mRNA profiling of IL-4-stimulated B cells from wild-type and STAT6−/− mice demonstrated that more than one-half of STAT6-controlled genes were down-regulated by this transcription factor in vivo (40). Last but not least, STAT6 inhibited IL4 promoter activity in differentiated D10.G4.1 murine Th2 cells (39). Our work further extended these findings by showing that STAT6 negatively affects IL13 expression in Th2 cells, and furthermore identified the STAT site adjacent to −1112C>T as being critical for STAT6-dependent suppression of IL13 transcription. Indeed, the replacement of −1112C with a T, and the concomitant creation of a YY1 site overlapping the STAT motif, was sufficient to fully counteract STAT6-dependent promoter inhibition. STAT6 may be part of a complex feedback circuit, acting initially as a permissive factor essential for Th2 cytokine locus remodeling (24) leading to IL4 and IL13 expression in Th2 cells. Partnering with a different set of factors, STAT6 may then become an inhibitor that fine tunes or even limits IL13 expression in fully differentiated Th2 effector cells. Our data suggest that the dynamic transcription factor context of differentiating Th2 cells plays a major role in modulating the regulatory effects of IL13-1112C>T. Whereas STAT6 and YY1 docking on the −1112T allele was detected in both nonpolarized CD4+ T cells (in which the T allele was less active) and Th2 cells (in which the T allele was more active), other factors (NFAT, Oct-1) were available for binding only in nonpolarized cells. In view of the topology of the relevant binding sites in the polymorphic IL13 promoter region, the latter factors may impair the ability of YY1 to interfere with STAT6-mediated promoter repression (Fig. 7). The molecular interactions underlying these effects remain to be determined.
FIGURE 7.
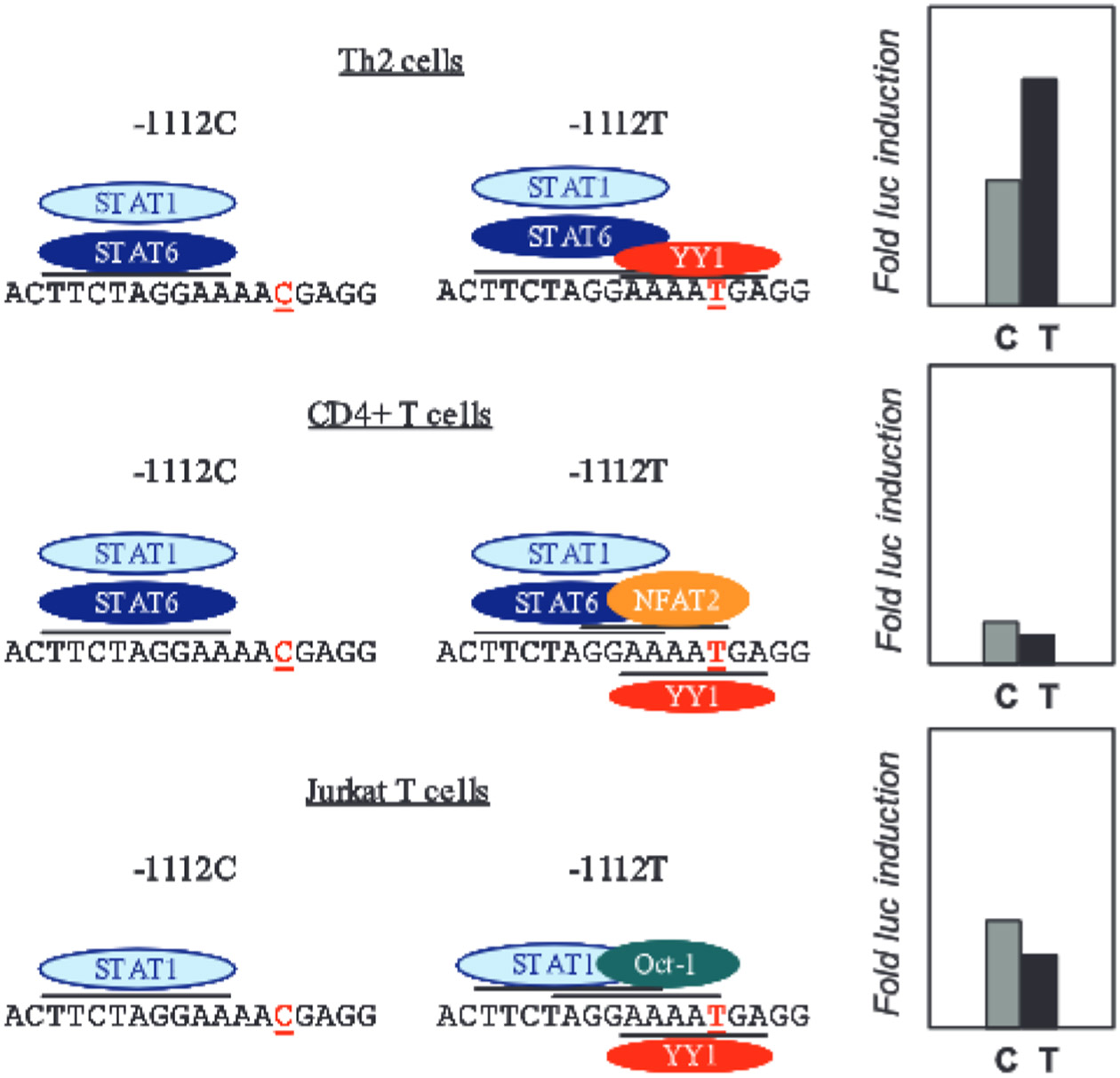
Model of gene-environment interactions in the nucleus at IL13-1112C>T. The results of the DNA/protein interaction analysis are summarized on the left. The corresponding IL13-1112C>T transcription data are presented on the right.
The relatively modest impact of IL13-1112C>T on transcription reflects the nature of single nucleotide variations, subtle differences that alter fine tuning or sensitivity thresholds of promoters and regulatory elements rather than impose the drastic effects of loss- or gain-of-function mutations seen in Mendelian disorders. Indeed, the magnitude of the effect was similar to other regulatory polymorphisms such as the SNP in SLC22A associated with rheumatoid arthritis and loss of transcriptional activity (41) and the variant CD14 and TGF-β promoters (29, 42). Because the functional effects of individual polymorphisms may be small, risk for complex diseases is substantially increased by synergism between multiple SNPs arrayed along a single regulatory pathway. Indeed, asthma risk was 5-fold higher in carriers of IL13-1112T and IL4RA 478Ser (43), compared with individuals with both nonrisk genotypes, and the effects of the IL13-1112TT genotype on risk for food sensitization were modified by IL4RA S478P and IL4RA Q155R (17). Risk for high IgE and asthma was increased even further by the combination of IL13-1112C>T, IL4-589C>T, IL4RA-148A>G and STAT6 IL4RA-2892C>T (44).
Comparative analysis of the IL13 promoter showed the IL13-1112T allele that increases risk for allergic disease is the ancestral allele, whereas the derived −1112C allele is protective. Furthermore, this analysis revealed the topology of STAT6 and YY1 motifs resulting in increased IL13 promoter activity has been fully conserved through at least 30 million years of evolutionary history, and all the replacements found in the STAT motif in Old World and New World monkeys occurred within the 3N spacer, not in the TTC/GAA palindrome critical for DNA-protein interactions (Fig. 1, bottom). These findings and their relevance to common diseases are best interpreted in the framework of the ancestral susceptibility model (45), according to which ancestral alleles reflect ancient adaptations to the lifestyle of ancient human populations. In that context, derived alleles were deleterious. With the shift in environment and lifestyle that has occurred in modern populations, ancestral alleles can increase the risk of common diseases, as exemplified by variants involved in energy metabolism and sodium homeostasis (45). An equivalent role of IL13-1112C>T among immunity genes is suggested by its current associations with allergy and asthma susceptibility in Western environments (1) in the face of strong associations between IL13-1112T and protection from Schistosoma hematobium in Africa (46) and severe malaria in Thailand (47). Although it is unclear why IL13-1112C rose abruptly in frequency to become the common allele in most human populations, the IL13 locus shows signatures of a recent selective sweep in the Caucasian and Chinese populations (48). We speculate that a genetically determined propensity for high IL13 expression may have become detrimental through deleterious effects on reproduction. Indeed, endometriosis, which increases the risk of infertility, has been associated with elevated IL13 mRNA and protein expression within the ectopic endometrium (49). IL13 may therefore be the first immunity gene that conforms to the ancestral susceptibility model.
Acknowledgments
This work was supported by National Institutes of Health Grants HL66391 (to D.V.) and AI42268 (to A.L.W.), a fellowship from the Canadian Institutes of Health Research (to L.C.), and an Interest Section Award from the American Academy of Allergy, Asthma and Immunology (to L.C.).
Footnotes
Abbreviations used in this paper: SNP, single nucleotide polymorphism; ChIP, chromatin immunoprecipitation; LD, linkage disequilibrium; RLA, relative luciferasc activity; YY1, Yin Yang 1; DT, divergence threshold; rh, recombinant human.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Obcr C, and Hoffjan S. 2006. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 7: 95–100. [DOI] [PubMed] [Google Scholar]
- 2.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, and Donaldson DD. 1998. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261. [DOI] [PubMed] [Google Scholar]
- 3.Grünig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, Sheppard D, Mohrs M, Donaldson DD, Locksley RM, and Corry DB. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282: 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghaffar O, Laberge S, Jacobson MR, Lowhagen O, Rak S, Durham SR, and Hamid Q. 1997. IL-13 mRNA and immunoreactivity in allergen-induced rhinitis: comparison with IL-4 expression and modulation by topical glucocorticoid therapy. Am. J. Respir. Cell. Mol. Biol 17: 17–24. [DOI] [PubMed] [Google Scholar]
- 5.Lordan JL, Bucchieri F, Richter A, Konstantinidis A, Holloway JW, Thornber M, Puddicombe SM, Buchanan D, Wilson SJ, Djukanovic R, Holgate ST, and Davies DE. 2002. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. J. Immunol 169: 407–414. [DOI] [PubMed] [Google Scholar]
- 6.Wynn TA 2003. IL-13 effector functions. Annu. Rev. Immunol 21: 425–456. [DOI] [PubMed] [Google Scholar]
- 7.Punnonen J, Aversa G, Cocks BJ, Mckcnzie ANJ, Menon S, Zurawski G, de Waal Malefytqq R, and de Vries JE. 1993. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc. Natl. Acad. Sci. USA 90: 3730–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosentino G, Soprana E, Thienes CP, Siccardi AG, Viale G, and Vercelli D. 1995. IL-13 downregulates CD14 expression and TNF-α secretion in human monocytes. J. Immunol 155: 3145–3151. [PubMed] [Google Scholar]
- 9.Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, and Martinez FD. 1999. A polymorphism in the 5′ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum IgE. Am. J. Respir. Cell Mol. Biol 20: 976–983. [DOI] [PubMed] [Google Scholar]
- 10.Dealtry GB, Clark DE, Sharkey A, Charnock-Jones DS, and Smith SK. 1998. Expression and localization of the Th2-type cytokine interleukin-13 and its receptor in the placenta during human pregnancy. Am. J. Reprod. Immunol 40: 283–290. [DOI] [PubMed] [Google Scholar]
- 11.Ribeiro-do-Couto LM, Boeije LC, Kroon JS, Hooibrink B, Breur-Vriesendorp BS, Aarden LA, and Boog CJ. 2001. High IL-13 production by human neonatal T cells: neonate immune system regulator? Eur. J. Immunol 31: 3394–3402. [DOI] [PubMed] [Google Scholar]
- 12.Graves PE, Kabcsch M, Halonen M, Holberg CJ, Baldini M, Fritzsch C, Weiland S, Erickson RP, von Mutius E, and Martinez FD. 2000. A cluster of seven tightly linked polymorphisms in the IL-13 gene is associated with total serum IgE levels in three populations of white children. J. Allergy Clin. Immunol 105: 506–513. [DOI] [PubMed] [Google Scholar]
- 13.Vladich FD, Brazille SM, Stern D, Peck ML, Ghittoni R, and Vercelli D. 2005. IL-13 R130Q, a common variant associated with allergy and asthma, enhances effector mechanisms essential for human allergic inflammation. J. Clin. Invest 115: 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffjan S, Nicolae D, and Ober C. 2003. Association studies for asthma and atopic diseases: a comprehensive review of the literature. Respir. Res 4: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howard TD, Whittaker PA, Zaiman AL, Koppelman GH, Xu J, Hanley MT, Meyers DA, Postma DS, and Bleecker ER. 2001. Identification and association of polymorphisms in the interleukin-13 gene with asthma and atopy in a Dutch population. Am. J. Respir. Cell. Mol. Biol 25: 377–384. [DOI] [PubMed] [Google Scholar]
- 16.Hummelshoj T, Bodtger U, Datta P, Malling HJ, Oturai A, Poulsen LK, Ryder LP, Sorensen PS, Svejgaard E, and Svejgaard A. 2003. Association between an interleukin-13 promoter polymorphism and atopy. Eur. J. Immunogenet 30: 355–359. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Beaty TH, Deindl P, Huang SK, Lau S, Sommerfeld C, Fallin MD, Kao WH, Wahn U, and Nickel R. 2004. Associations between specific serum IgE response and 6 variants within the genes IL4, IL13, and IL4RA in German children: the German Multicenter Atopy Study. J. Allergy Clin. Immunol 113:489–495. [DOI] [PubMed] [Google Scholar]
- 18.van der Pouw Kraan TCTM, van Veen A, Boeije LCM, van Tuyl SAP, de Groot ER, Stapel SO, Bakker A, Verweij CL, Arden LA, and van der Zee JS. 1999. An IL-13 promoter polymorphism associated with increased risk of allergic asthma. Genes Immun. 1: 61–65. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Beaty T, Deindl P, Huang S, Lau S, Sommerfeld C, Fallin M, Kao W, Wahn U, and Nickel R. 2003. Associations between total serum IgE levels and the 6 potentially functional variants within the genes IL4, IL13, and IL4RA in German children: the German Multicenter Atopy Study. J. Allergy Clin. Immunol 112: 382–388. [DOI] [PubMed] [Google Scholar]
- 20.Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, and Rubin EM. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394. [DOI] [PubMed] [Google Scholar]
- 21.Ovcharenko I, Boffelli D, and Loots GG. 2004. eShadow: a tool for comparing closely related sequences. Genome Res. 14: 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oddy WH, Halonen M, Martinez FD, Lohman IC, Stern DA, Kurzius-Spencer M, Guerra S, and Wright AL. 2003. TGF-β in human milk is associated with wheeze in infancy. J. Allergy Clin. Immunol 112: 723–728. [DOI] [PubMed] [Google Scholar]
- 23.Boffelli D, Nobrega MA, and Rubin EM. 2004. Comparative genomics at the vertebrate extremes. Nat. Rev. Genet 5: 456–465. [DOI] [PubMed] [Google Scholar]
- 24.Agarwal S, and Rao A. 1998. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 9: 765–775. [DOI] [PubMed] [Google Scholar]
- 25.Georas SN, Cumberland JE, Burke TF, Chen R, Schindler U, and Casolaro V. 1998. Stat6 inhibits human interleukin-4 promoter activity in T cells. Blood 92: 4529–4538. [PubMed] [Google Scholar]
- 26.Gordon S, Akopyan G, Garban H, and Bonavida B. 2006. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene 25: 1125–1142. [DOI] [PubMed] [Google Scholar]
- 27.Bergad PL, Towle HC, and Berry SA. 2000. Yin-Yang 1 and glucocorticoid receptor participate in the Stat5-mediated growth hormone response of the serine protease inhibitor 2.1 gene. J. Biol. Chem 275: 8114–8120. [DOI] [PubMed] [Google Scholar]
- 28.Guo L, Hu-Li J, and Paul WE. 2005. Probabilistic regulation in TH2 cells accounts for monoallelic expression of IL-4 and IL-13. Immunity 23: 89–99. [DOI] [PubMed] [Google Scholar]
- 29.LeVan TD, Bloom JW, Bailey TJ, Karp CL, Halonen M, Martinez FD, and Vercelli D 2001. A common single nucleotide polymorphism in the CD14 promoter decreases the affinity of Sp protein binding and enhances transcriptional activity. J. Immunol 167: 5838–5844. [DOI] [PubMed] [Google Scholar]
- 30.Lohmueller KE, Pearce CL, Pike M, Lander ES, and Hirschhorn JN. 2003. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet 33: 177–182. [DOI] [PubMed] [Google Scholar]
- 31.Editorial. 2005. Framework for a fully powered risk engine. Nat. Genet 37: 1153. [DOI] [PubMed] [Google Scholar]
- 32.Hall IP, and Blakcy JD. 2005. Genetic association studies in thorax. Thorax 60: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinzmann A, Jerkic SP, Ganter K, Kurz T, Blattmann S, Schuchmann L, Gerhold K, Berner R, and Deichmann KA. 2003. Association study of the IL13 variant Arg110Gln in atopic diseases and juvenile idiopathic arthritis. J. Allergy Clin. Immunol 112: 735–739. [DOI] [PubMed] [Google Scholar]
- 34.Hoffjan S, Ostrovnaja I, Nicolac D, Newman DL, Nicolae R, Gangnon R, Steiner L, Walker K, Reynolds R, Greene D, et al. 2004. Genetic variation in immunoregulatory pathways and atopic phenotypes in infancy. J. Allergy Clin. Immunol. 113: 511–518. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Nickel R, Beyer K, Wahn U, Ehrlich E, Freidhoff LR, Bjorksten B, Beaty TH, and Huang SK. 2000. An IL13 coding region variant is associated with a high total serum IgE level and atopic dermatitis in the German multicenter atopy study (MAS-90). J. Allergy Clin. Immunol 106: 167–170. [DOI] [PubMed] [Google Scholar]
- 36.Maier LM, Howson JMM, Walker N, Spickett GP, Jones RW, Ring SM, McArdle WL, Lowe CE, Bailey R, Payne F, Todd JA, and Strachan DP. 2006. Association of IL13 with total IgE: evidence against an inverse association of atopy and diabetes. J. Allergy Clin. Immunol 117: 1306–1313. [DOI] [PubMed] [Google Scholar]
- 37.Ober C, Leavitt SA, Tsalenko A, Howard TD, Hoki DM, Daniel R, Newman DL, Wu X, Parry R, Lester LA, et al. 2000. Variation in the interleukin 4-receptor β gene confers susceptibility to asthma and atopy in ethnically diverse populations. Am. J. Hum. Genet 66: 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaplan MH, Schindler U, Smiley ST, and Grusby MJ. 1996. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity 4: 313–319. [DOI] [PubMed] [Google Scholar]
- 39.Dorado B, Jerez MJ, Flores N, Martin-Saavedra FM, Duran C, and Ballester S. 2002. Autocrine IL-4 gene regulation at late phases of TCR activation in differentiated Th2 cells. J. Immunol 169: 3030–3037. [DOI] [PubMed] [Google Scholar]
- 40.Schroder AJ, Pavlidis P, Arimura A, Capece D, and Rothman PB. 2002. STAT6 serves as a positive and negative regulator of gene expression in IL-4-stimulated B lymphocytes. J. Immunol 168: 996–1000. [DOI] [PubMed] [Google Scholar]
- 41.Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y, Sawada T, Suzuki M, Nagasaki M, Ohtsuki M, Ono M, et al. 2003. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet 35: 341–348. [DOI] [PubMed] [Google Scholar]
- 42.Silverman ES, Palmer LJ, Subramaniam V, Hallock A, Mathew S, Vallone J, Faffe DS, Shikanai T, Raby BA, Weiss ST, and Shore SA. 2004. Transforming growth factor-β1 promoter polymorphism C–509T is associated with asthma. Am. J. Respir. Crit. Care Med 169: 214–219. [DOI] [PubMed] [Google Scholar]
- 43.Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, and Bleecker ER. 2002. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am. J. Hum. Genet 70: 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabesch M, Schedel M, Carr D, Woitsch B, Fritzsch C, Weiland SK, and von Mutius E. 2006. IL-4/IL-I3 pathway genetics strongly influence serum IgE levels and childhood asthma. J. Allergy Clin. Immunol 117: 269–274. [DOI] [PubMed] [Google Scholar]
- 45.Di Ricnzo A, and Hudson RR. 2005. An evolutionary framework for common diseases: the ancestral susceptibility model. Trends Genet. 21:596–601. [DOI] [PubMed] [Google Scholar]
- 46.Kouriba B, Chevillard C, Bream J, Argiro L, Dessein H, Arnaud V, Sangare L, Dabo A, Beavogui A, Arama C, et al. 2005. Analysis of the 5q31-q33 locus shows an association between IL13–1055C/T IL-13–591A/G polymorphisms and Schistosoma haematobium infections. J. Immunol 174: 6274–6281. [DOI] [PubMed] [Google Scholar]
- 47.Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, and Tokunaga K. 2003. A single-nucleotide substitution from C to T at position–1055 in the IL-13 promoter is associated with protection from severe malaria in Thailand. Genes Immun. 4: 528–531. [DOI] [PubMed] [Google Scholar]
- 48.Zhou G, Zhai Y, Dong X, Zhang X, He F, Zhou K, Zhu Y, Wei H, Yao Z, Zhong S, et al. 2004. Haplotype structure and evidence for positive selection at the human IL13 locus. Mol. Biol. Evol 21: 29–35. [DOI] [PubMed] [Google Scholar]
- 49.Chegini N, Roberts M, and Ripps B. 2003. Differential expression of interleukins (IL)-13 and IL-15 in ectopic and eutopic endometrium of women with endometriosis and normal fertile women. Am. J. Reprod. Immunol 49: 75–83. [DOI] [PubMed] [Google Scholar]