

Abstract
Epigenetic clocks that quantify rates of aging from DNA methylation patterns across the genome have emerged as a potential biomarker for risk of age-related diseases, like Alzheimer’s disease (AD), and environmental and social stressors. However, methylation clocks have not been validated in genetically diverse cohorts. Here we evaluate a set of methylation clocks in 621 AD patients and matched controls from African American, Hispanic, and white cohorts. The clocks are less accurate at predicting age in genetically admixed individuals, especially those with substantial African ancestry, than in the white cohort. The clocks also do not consistently identify age acceleration in admixed AD cases compared to controls. Methylation QTL (meQTL) commonly influence CpGs in clocks, and these meQTL have significantly higher frequencies in African genetic ancestries. Our results demonstrate that methylation clocks often fail to predict age and AD risk beyond their training populations and suggest avenues for improving their portability.
1. Introduction
Biological aging is the progressive accumulation of cellular damage leading to degeneration and organismal death (Aunan et al., 2016). DNA methylation patterns at CpG sites across the genome correlate strongly with the aging process, an effect that has been quantified using statistical models called “methylation clocks” (Jones, Goodman, and Kobor, 2015). The first-generation of methylation clocks were trained to predict chronological age from methylation levels at selected CpGs from across the genome (Hannum et al., 2013; Horvath, 2013; Zhang et al., 2019). A second-generation of clocks were trained to use methylation levels to predict mortality risk as proxied by a combination of biomarkers of frailty and physiological decline (Morgan E. Levine et al., 2018; Lu et al., 2019). Finally, a third-generation of clocks have been trained to predict the rate of aging based cohorts with longitudinal data on biomarkers of frailty (Belsky et al., 2022).
Greater predicted DNA methylation age compared to an individual’s chronological age, known as epigenetic age acceleration, has been associated with an increased risk of many age-related diseases, including coronary heart disease, white matter hyperintensities, Type 2 diabetes mellitus, Parkinson’s disease, and Alzheimer’s disease (AD) (Hodgson et al., 2017; Horvath, Gurven, et al., 2016; Horvath and Ritz, 2015; Morgan E Levine et al., 2015, 2018; Lu et al., 2019; Raina et al., 2017). As such, methylation clocks show potential as predictive biomarkers of the aging process and age-related health outcomes, and may capture relevant biological signals associated with aging. The clocks are also increasingly being used in social epidemiology research to quantify associations of epigenetic aging with exposure to adverse social and environmental factors that often differ across groups (Aiello et al., 2024; Chiu et al., 2024; Krieger et al., 2024; Non, 2021).
While methylation is shaped by the environment of an individual, it is also strongly influenced by genetic variation (Kader and Ghai, 2017). Millions of methylation quantitative trait loci (meQTLs)—genetic variants that associate with the methylation level of a CpG site across individuals—have been identified (Smith et al., 2014). MeQTL influence methylation levels via many mechanisms, including disruption of CpGs and effects on transcription factor binding, gene expression, and other gene regulatory processes (Banovich et al., 2014; Oliva et al., 2023). Methylation patterns also vary between human groups, and approximately 75% of variance in methylation between human groups associates with genetic ancestry (Galanter et al., 2017). This suggests that meQTLs often vary in frequency in different genetic ancestries.
Despite these factors that lead to differential methylation levels in different genetic ancestries, methylation clocks have been developed and evaluated primarily individuals of European genetic ancestry due to biases in available genomic data. We hypothesized that lack of genetic diversity in the training data of methylation clocks could limit their generalizability across global and admixed populations. Similar factors have posed challenges for the application of polygenic risk scores (PRS) across human groups; PRS models often rapidly decrease in accuracy when applied to individuals not represented in the training set (Martin et al., 2019; Novembre et al., 2022; Privé et al., 2022).
To quantify whether current methylation clocks are generalizable across global populations, we analyzed data from MAGENTA, a diverse AD study which has generated blood methylation and genotyping data for 621 individuals from the Americas, including genetically admixed individuals from African American, Puerto Rican, Cuban, and Peruvian cohorts. We evaluate the accuracy of first-, second-, and third-generation methylation clocks at predicting age in these individuals, and evaluate whether age acceleration metrics from these clocks associate with AD risk, as they do in individuals of European ancestry. We also investigate the impact of genetic diversity on clock CpGs by intersecting clock CpG sites with variants from different human groups from gnomAD, and by comparing the frequencies of meQTL that influence clock CpG sites from three sets of independent meQTLs across different genetic ancestries. Our results highlight obstacles to the application of methylation clocks as biomarkers for precision medicine and epidemiology, but they also identify promising avenues for considering genetic diversity in the development, application, and interpretation of methylation clocks.
2. Results
Our analyses are based on genotyping and blood DNA methylation data collected by the MAGENTA study from 621 individuals from the Americas with AD and non-demented controls. The MAGENTA study is focused on late-onset Alzheimer’s and thus the average age of participants is 76 years old. Reflecting AD prevalence, the study is 68% female. The individuals come from five cohorts, collected from the United States (white, African American, Cuban), Peru, and Puerto Rico (Table 1). As described in detail in the Methods, to facilitate comparisons relevant to understand global differences in our study, we opted to use a combination of geographic and race-based identifiers that are likely to best reflect underlying differences in genetic ancestry and admixture components.
Table 1 :
Demographics of the MAGENTA study cohorts
| Alzheimer’s (N=313) | Control (N=308) | Overall (N=621) | |
|---|---|---|---|
|
| |||
| COHORT | |||
| African American | 98 (31.3%) | 107 (34.7%) | 205 (33.0%) |
| Cuban | 22 (7.0%) | 21 (6.8%) | 43 (6.9%) |
| White | 68 (21.7%) | 65 (21.1%) | 133 (21.4%) |
| Peruvian | 41 (13.1%) | 41 (13.3%) | 82 (13.2%) |
| Puerto Rican | 84 (26.8%) | 74 (24.0%) | 158 (25.4%) |
|
| |||
| SEX | |||
| Female | 206 (65.8%) | 213 (69.2%) | 419 (67.5%) |
| Male | 107 (34.2%) | 95 (30.8%) | 202 (32.5%) |
We apply a range of first-, second-, and third-generation methylation-based epigenetic aging clocks to these individuals. We then evaluate their accuracy in predicting chronological age, quantify whether they identify accelerated aging in individuals with AD, and explore genetic factors that may influence clock performance (Figure 1).
Figure 1: Schematic of the workflow of the study.

We analyzed genome-wide methylation and genotyping data from blood samples from 621 AD and non-demented control individuals from the MAGENTA study. We applied a set of first-, second-, and third-generation methylation clocks to the individuals and estimated their genetic ancestry. This enabled us to explore the performance of methylation clocks in individuals with different genetic ancestries.
2.1. Methylation clock accuracy is lower in cohorts with substantial African ancestry
To test whether current methylation clocks are able to predict age accurately in diverse, genetically admixed groups, we evaluated age predictions for the control individuals in the MAGENTA study. We first analyzed the widely used Horvath clock, which was trained on data from several tissues and cell types to accurately predict age across the lifespan using methylation levels at 353 CpG sites.
Age predicted from DNA methylation (DNAm age) using the Horvath clock was strongly correlated with chronological age (Pearson r = 0.72) (Figure 2A). While this correlation is lower than reported in the original study (>0.9), it is consistent with previous studies of older individuals (Horvath, 2013; Marioni et al., 2015).
Figure 2: Methylation clock accuracy is lower in cohorts with substantial African genetic ancestry.

A: Pearson correlation between chronological age and DNAm age predicted by the Horvath clock for controls in the white MAGENTA cohort. The correlation of 0.72 is similar to previous studies of older cohorts. B: Pearson correlation between chronological age and DNAm age predicted by the Horvath clock for the genetically admixed cohorts in MAGENTA. The right plot in each pair shows the proportion of European (CEU), African (YRI), and American (PEL) global ancestry for each individual in each cohort. The two cohorts with substantial African ancestry—African Americans and Puerto Ricans—have significantly lower correlations than the other cohorts. C: Difference in correlation between chronological and predicted DNAm age for controls in each cohort compared to the white cohort controls for four methylation clocks. The baseline correlation for the white cohort controls is given in each panel; asterisks indicate a statistically significant difference from the baseline. * p < 0.05.
In contrast to the white cohort, the correlation between DNAm age and chronological age were significantly lower for Puerto Ricans (r = 0.45, p = 0.007) and African Americans (r = 0.51, p = 0.016) (Figure 2B). The correlations for Cubans (r = 0.68, p = 0.385) and Peruvians (r = 0.72, p = 0.52) were similar in magnitude to the white cohort.
We noticed that the two cohorts with low correlation come from regions where individuals often have substantial amounts of African ancestry. To explore how admixture levels associated with the accuracy of the Horvath clock in predicting age, we estimated the global proportions of African (YRI), European (CEU), and American (PEL) ancestries in each individual from the MAGENTA cohort using reference groups from the 1000 Genomes Project (The 1000 Genomes Project Consortium et al., 2015).
Methylation clock accuracy was lowest for the cohorts with substantial African ancestry: African Americans (median 85% African) and Puerto Ricans (median 15% African). In contrast, the clocks performed similarly to the white cohort in groups lacking substantial African ancestry: Cubans (6% African) and Peruvians (2% African).
2.2. Accuracy of age prediction on admixed individuals varies across methylation clocks
To investigate the performance of other methylation clocks at predicting chronological age in admixed individuals, we selected several additional publicly available open-source clocks. We considered two other “first-generation” clocks that were trained to predict chronological age: the Hannum clock (Hannum et al., 2013) and a model developed by Zhang et al., 2019 that used large datasets for training and achieved substantially higher performance than previous age predictors. We hereafter refer to this elastic net model as “Zhang2019_EN”.
Both models achieved higher correlations with chronological age than the Horvath clock across the cohorts in the MAGENTA study. For example, Hannum has a correlation of 0.74 in the white cohort, and consistent with previous evaluations, Zhang2019_EN has a correlation of 0.88. These relative performance trends held across cohorts, but again, the African Americans and Puerto Ricans, the cohorts with substantial African ancestry, had the lowest age correlations for each clock (Figure 2C).
Next, we evaluated the PhenoAge clock, a “second-generation” clock that is trained on biomarkers of frailty and physiological deterioration (Morgan E. Levine et al., 2018). The correlation between DNAm age and chronological age was lower for this clock in comparison to the other methylation clocks (r = 0.53 in the white cohort). This is likely due to the fact that this clock was not trained to predict age directly, but rather markers of aging. This clock did not show as substantial a difference in performance between cohorts as seen for the Horvath clock, but the African American and Puerto Rican populations again had the lowest correlation of all cohorts.
Overall, these results demonstrate that current methylation clocks vary in the correlation of their predicted DNAm age with chronological age in genetically admixed cohorts. The clocks are also consistently the least accurate in predicting age cohorts with substantial proportions of African ancestry.
2.3. Most methylation clocks do not identify accelerated aging in admixed Alzheimer’s cohorts
DNAm age has been proposed as a promising biomarker and predictive tool for age-related disease risk, particularly because of associations between accelerated DNAm age (compared to chronological age) and the presence of diseases such as coronary heart disease, Parkinson’s disease, and AD. However, these results have largely been observed in European-ancestry cohorts.
To evaluate the ability of methylation clocks to identify accelerated aging and risk for age-related disease in diverse, genetically admixed individuals, we quantified the association of methylation age acceleration with AD status in cohorts from the MAGENTA study. In addition to the clocks tested in the previous section, we also included a “third generation” clock, DunedinPACE, that aims to predict the pace of aging as measured by change in biomarkers over time from methylation data, rather than age itself (Belsky et al., 2022).
The cell type composition in blood is known to change with age, which if not accounted for, can confound age acceleration estimates (Jaiswal and Ebert, 2019). Thus, we focused on intrinsic age acceleration estimates computed using established algorithms to correct for cell type composition.
To establish a baseline for this analysis, we tested whether individuals with AD in the white cohort show significantly greater age acceleration than non-demented controls. As expected from previous studies (Morgan E Levine et al., 2015, 2018), AD cases have modest but significantly greater age acceleration as measured by the Horvath clock than controls (Supplementary Figure 1; median 1.7 vs. 1.5 years, p = 0.041). For each of the other clocks, AD cases had higher median age acceleration than controls (Figure 3B), though due to the relatively small sample size, the differences only reached statistical significance for the DunedinPACE clock (median 1.09 vs. 1.07, p = 0.044).
Figure 3: Methylation clocks rarely identify accelerated aging in admixed Alzheimer’s cohorts.

A: Comparison of the distributions of Horvath intrinsic age acceleration for AD patients and non-demented controls for each of the admixed cohorts in MAGENTA. AD patients do not show significantly higher age acceleration in any of the admixed cohorts. In contrast, the AD cases had significantly greater acceleration than controls in the white cohort (Supplementary Figure 1). NS = Not significant. B: Median differences in intrinsic age acceleration between AD patients and non-demented controls for five methylation clocks for each cohort in MAGENTA. The clocks do not consistently identfy accelerated aging in AD across cohorts, and the results also vary within cohorts. * p < 0.05.
Having established that previously reported age acceleration in AD was detectable in our framework, we evaluated whether the clocks found accelerated aging in the admixed AD cohorts. Focusing first on the Horvath clock, we observed inconsistent relationships between age acceleration and AD status. None of the admixed cohorts showed a significant difference, and controls even had higher median age acceleration in Peruvians and Cubans (Figure 3A). Across the other clocks, none consistently identified greater age acceleration in AD cases across all populations (Figure 3B). Among the first- and second-generation clocks, only PhenoAge demonstrated a significant ability to differentiate AD cases from controls in any of the non-European ancestry groups, specifically in African American individuals (p = 0.008), which were included in its training set. Cubans consistently showed greater age acceleration in controls rather than cases, while none of the other admixed cohorts even had consistent directions of effect across methods.
DunedinPACE stood out in the evaluation, as it identified significantly greater aging in AD cases compared to controls in the white (p = 0.044), African American (p = 0.0019), and Puerto Rican (p = 0.0090) cohorts using its “pace of aging” metric. However, no significant differences were found for Cubans (p = 0.26) or Peruvians (p = 0.81).
2.4. Combining predictions across methylation clocks does not improve their performance
Inspired by recent work on the ensembling of PRS to better predict disease risk from genetic variation across populations (Monti et al., 2024), we evaluated whether combining age predictions would lead to greater accuracy in age prediction and AD risk prediction in the admixed cohorts. To investigate this, we averaged the age predictions for each individual in the MAGENTA study across five methylation clocks: Horvath, Hannum, Zhang2019_EN, Zhang2019 BLUP, and PhenoAge clocks. (The Zheng2019_BLUP is a variation of the Zhang2019_EN clock that does not use strong regularization.)
The ensemble method’s DNAm age prediction is more strongly correlated with chronological age in comparison to the Horvath and PhenoAge clocks, but it could not improve upon the best predictors (Zhang2019 clocks) across populations (Supplementary Figure 2A).
We next evaluated whether the ensemble intrinsic age acceleration estimates would associate more strongly with AD disease status relative to the standalone methylation clocks. Following the same evaluation framework as for the individual clocks, we found only one significant difference in age acceleration. Cuban control individuals had significantly lower age acceleration than AD cases (Supplementary Figure 2B). Thus, a simple ensemble does not lead to stronger performance at either task in admixed cohorts.
2.5. Many methylation clock CpGs are disrupted by genetic variants, but the variants are extremely low frequency
Our results so far demonstrate that existing methylation clocks do not perform consistently across genetically admixed individuals with ancestries under-represented in clock training data. Thus, we sought to investigate potential mechanisms underlying the decreased performance of some clocks. We first quantified how often a genetic variant disrupted a CpG site included in a clock. This scenario could lead to inaccuracies across cohorts given the loss of potential for methylation and the ability of the site to contribute to the age prediction.
Of the 353 CpG sites considered in the Horvath clock, 245 (69%) have at least one disruptive genetic variant observed in at least one individual in the gnomAD database of variants identified in a cohort of 76,156, including thousands of individuals of non-European ancestry. However, these variants are extremely rare (Figure 4); the average frequency is 0.0001, with the most common case being a variant observed in just one individual. Only one clock CpG disrupting variant had a frequency greater than 1%. Thus, genetic variation in clock CpG sites themselves is unlikely to be the main cause of lack of generalization of the Horvath clock.
Figure 4: Genetic variants that disrupt methylation clock CpG sites appear at extremely low frequencies.
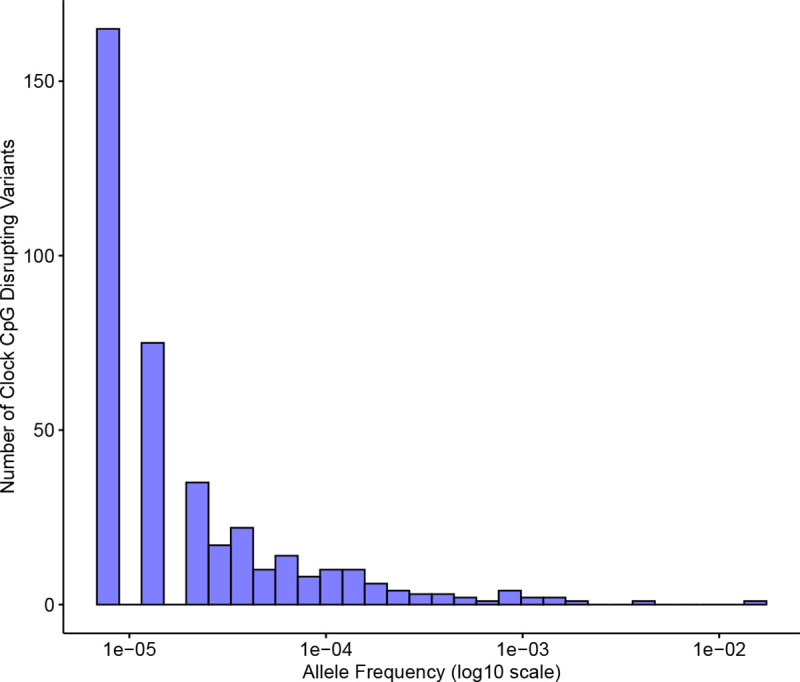
The allele frequency distribution for single nucletide variants that disrupt one of the 353 CpG sites considered by the Horvath clock. Allele frequencies were computed across 76,156 individuals from large-scale sequencing studies harmonized in gnomAD (version 3.0).
2.6. Common methylation QTL influence clock CpGs
We next assessed the prevalence of meQTLs that affect clock CpG sites, another modifier of methylation levels that could lead to spurious DNAm age predictions across individuals. We gathered three sets of meQTLs from Europeans, South Asians, and African Americans. We intersected the affected CpGs for the meQTLs with the Horvath clock CpG sites.
Out of the 353 CpGs included in the Horvath clock, 271 (77%) had at least one meQTL. Overall, a total of 29,033 unique variants associated with methylation levels at clock CpGs. The meQTL had an average allele frequency of 0.19, and 26,500 were common (≥1%; Figure 5A).
Figure 5: Common meQTLs affect most Horvath clock CpGs and vary in frequency across ancestries.
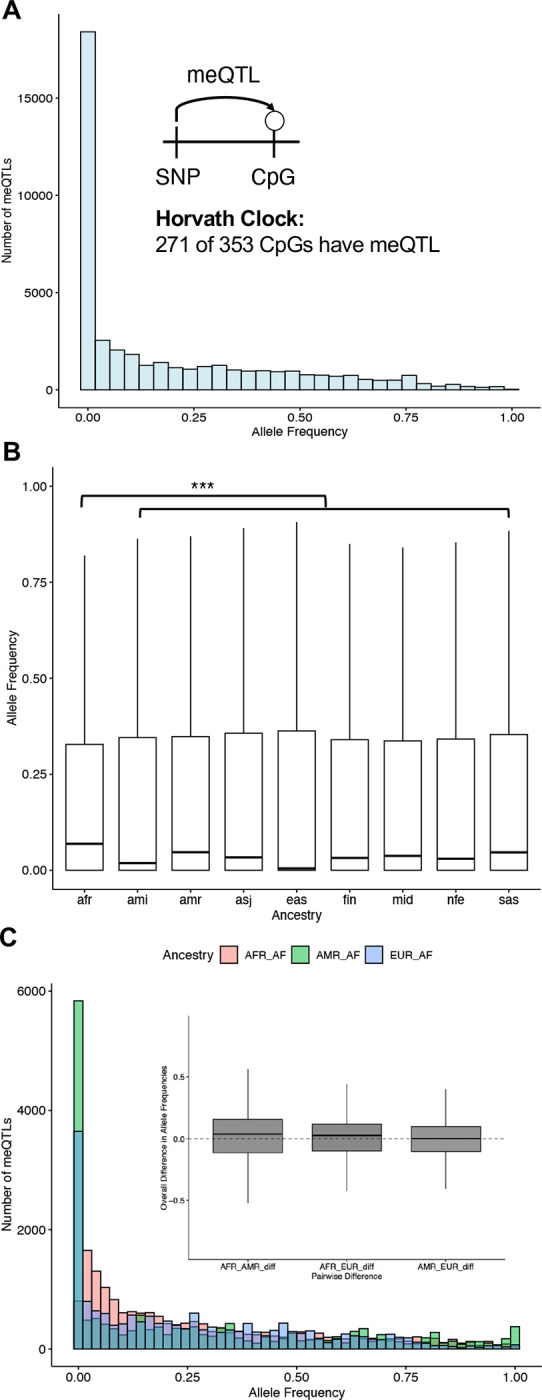
A: The allele frequency distribution of the 29,033 unique variants associated with methylation levels at Horvath clock CpGs. Allele frequencies were computed over the 76,215 genomes in gnomAD version 4.1. Inset: Out of the 353 CpGs in the Horvath clock, 271 (77%) have at least one meQTL, i.e., a genetic variant that is associated with methylation level. B: Clock meQTL have significantly higher alelle frequency in individuals with African genetic ancestry from gnomAD than all other ancestry groups (median 0.068 for African vs. 0.004–0.046; p < 3.85×10−25). C: Clock meQTL have significantly higher allele frequency on African local ancestry genomic segments in 7,612 Latino admixed individuals with varying proportions of European, American, and African ancestry from gnomAD. Inset: The distribution of difference in frequencies for each meQTL for each pair of populations.
2.7. Clock CpG methylation QTLs vary in frequency across ancestries
Differences in the presence or frequency of meQTL that influence clock CpGs between genetic ancestries could lead to decreased DNAm age prediction accuracy (and therefore weaker associations with disease) in diverse and admixed cohorts. For example, if a clock is trained on a cohort without an meQTL, the learned weights for the CpG will not have accounted for the effects of the meQTL.
To quantify whether differences in meQTL across genetic ancestries could potentially influence methylation clocks, we analyzed the gnomAD allele frequencies for the 29,033 Horvath clock meQTL tag variants in multiple global populations (Figure 5B). The meQTLs are at significantly higher frequencies in African ancestry populations (median 0.068) than in each of the eight other population groups considered (0.004–0.046; p < 3.85 × 10−25). There were also 2,328 meQTL that were only observed in African populations.
To connect these results to individuals with recent admixture, like many in the MAGENTA study, we also tested whether the Horvath clock meQTL differed in frequency in local ancestry blocks of different origins in genetically admixed individuals from gnomAD. We used pre-computed local ancestry calls for 7,612 Latino/Admixed American individuals to compare allele frequencies for each meQTL in three ancestral backgrounds: African, Amerindigenous, and European. The clock CpG-affecting meQTLs were at higher frequencies in African local ancestry backgrounds (Figure 5C), consistent with our finding that these meQTLs are most frequent in African ancestry individuals at the global population level.
2.8. Susceptibility to meQTL varies across methylation clocks
Given the strong potential for meQTL to influence the Horvath clock (76% of its CpGs have an meQTL), we expanded this analysis and quantified the number and proportion of clock CpGs that are affected by meQTL for all clocks considered here (Figure 6). The other first-generation clocks have lower proportions of their CpG sites affected by meQTLs (Hannum: 8%, Zhang2019_EN: 1%). PhenoAge is similarly low, with 7% of its CpGs affected by at least one meQTL. Finally, DunedinPACE had no meQTLs affecting its 173 clock CpG sites.
Figure 6: Methylation clocks vary in the number and proportion of CpGs affected by meQTLs.

The proportion of clock CpGs for each clock that have at least one known meQTL. The number of unique clock CpGs affected by meQTLs for each clock is given on top of each bar. The meQTL were taken from three genome-wide studies in Europeans, South Asians, and African Americans.
Thus, the clock with the largest decrease in performance in admixed cohorts (in terms of predicting chronological age and identifying age acceleration in AD) has the most and largest fraction of meQTLs influencing its CpGs. On the opposite side of the spectrum, DunedinPACE, the best performing clock at identifying AD cases in the MAGENTA study, had no meQTLs. The three other clocks with intermediate performance in the admixed cohorts, all have meQTL for some CpGs, but much lower fraction than the Horvath clock.
3. Discussion
Methylation clocks are promising biomarkers of aging and social stress, and as tools for mechanistic studies of diseases related to the aging process. Despite their widespread use in these applications, methylation clocks have not been comprehensively evaluated in diverse human groups. These groups are underrepresented in genetic and genomic databases and underserved in biomedical research in terms of access to healthcare and quality of healthcare.
In this study, we sought to fill this gap by evaluating commonly used methylation clocks in genetically admixed individuals from the Americas. We found that most clocks did not predict age as accurately in admixed control individuals as in the white cohort, especially for individuals with substantial African genetic ancestry. We next found that most methylation clocks could not discern between AD patients and non-demented controls based on age acceleration metrics in cohorts with genetic ancestries distinct from their training populations.
To evaluate potential genetic factors that could contribute to this decrease in performance, we hypothesized that two types of variants could reduce clock accuracy: 1) variants that disrupt clock CpG sites prevent methylation, and 2) meQTLs that influence clock CpG site methylation. Both scenarios could lead to over or under estimations of age, and therefore spurious associations with age-related disease, if they differ in frequency across genetic ancestries. While we discovered that 245 of the 353 CpG sites used by the Horvath clock are disrupted in at least one individual in gnomAD, these variants are extremely rare and thus unlikely to be a major driver of differences between the cohorts. In contrast, among meQTLs from multiple global populations, we found 29,033 unique variants that affected the Horvath clock CpG sites. Many of these variants are common, and they are most frequent in African ancestry individuals from gnomAD. We also showed that they are most frequent in African local ancestry blocks in admixed Latino individuals from the Americas.
Our findings demonstrate that methylation clocks—a widespread tool in aging, genomics, and social epidemiology research—perform inconsistently across individuals of different genetic ancestries. These results further underline the need for more diversity in the development and evaluation of genomic tools for precision medicine.
We hope that these results also encourage researchers using these tools to study diseases of aging or social stressors in diverse groups to exercise caution when interpreting differences in age acceleration. We have shown that many methylation clocks differ significantly in their accuracy at predicting age between cohorts. Thus, what might appear to be a faster pace of aging, could simply be the result of a difference in genetic ancestry from the training cohort. Applying existing methylation clocks to diverse individuals could lead to grave consequences and exacerbate existing disparities in access to quality healthcare, as well as provide spurious conclusions about an individual’s health. Due to the increased potential for false positives and false negatives when applying the clocks as predictive biomarkers, individuals at risk might not receive the medical attention they need, and additional stress could unncessarily be placed on individuals in good health. These challenges must be addressed before methylation clock are adopted as biomarkers for precision medicine.
The challenges we identify here for methylation clocks mirror the limitations of PRS, wherein phenotype prediction models decrease in accuracy on individuals genetically distant from the training population. While there are substantial biological differences in the processes modeled by methylation clocks and PRS, we are optimistic that recent progress on building PRS that are more portable across cohorts will provide strategies for improving methylation clocks.
Our results suggest two promising approaches for building more robust clocks. First, given the large number of meQTL in the human genome and their differences in frequency across human populations, we suggest training clocks only on CpG sites that do not have known meQTL. Given that the biological signatures driving methylation clock performance appear to influence large fractions of the genome, we do not anticipate that this will substantially limit overall performance. The strong and relatively consistent performance across cohorts of the DunedinPACE clock, which lacks CpGs with meQTL, supports this approach. It also suggests that methylation clocks that predict the pace of aging (rather than age itself) may be more robust, but further work is needed to validate this hypothesis. Second, we encourage including individuals from multiple genetic ancestries in the training cohorts. The ability of the PhenoAge clock, which included African Americans in the training cohort, to detect significant age acceleration in the African American AD cases suggests this may improve generalizability.
There are caveats and limitations to our study that we hope future work will address. First, the impact of environment on methylation clock accuracy and differences in environmental factors for global populations that might lead to decreases in methylation clock accuracy are difficult to study with the data at hand. Given this, we focused on genetic influences on CpG sites that could lead to spurious associations in diverse populations. However, work is needed to investigate other factors that might cause methylation clocks to not generalize across individuals, such as the methods that account for cell type composition heterogeneity in blood that might not be as accurate across individuals of different populations. Second, while we attempted to evaluate a representative set of first-, second-, and third-generation clocks, we were not able to evaluate all methylation clocks. In particular, some with closed source that were only available as a web server could not be used due to data privacy restrictions for the MAGENTA samples. Another limitation is the use of blood samples to generate methylation data for the study of a neurodegenerative disease focused on the central nervous system. However, we note that all methylation clocks tested in the present study were developed using blood samples, either exclusively (Hannum, PhenoAge, Zheng2019_EN, Zheng2019_BLUP, and DunedinPACE) or as part of the tissues used in training (Horvath). In addition, these clocks and their association with age-related diseases such as AD have all been validated in multiple tissues, including blood. Multiple studies in the AD literature point to changes in blood such as gene expression changes, immune cell type composition, and disruption of the blood brain barrier in AD patients relative to non-demented controls, such that signals related to AD pathology can be identified from this peripheral tissue (Griswold et al., 2020; Shigemizu et al., 2022) and through epigenetic age acceleration (Hodgson et al., 2017; Marioni et al., 2015; Raina et al., 2017). Finally, while the MAGENTA study is an excellent resource for exploring methylation clocks and AD in admixed individuals, it is not representative of all genetic ancestries and combinations. Moreover, the sample sizes vary between the MAGENTA cohorts, with the Cuban and Peruvian cohorts being particularly small relative to the others. This limits our ability to find differences in the age accelerations between AD patients and controls as measured by the methylation clocks. However, we are reassured by the complementary findings in African cohorts on the decreased performance of methylation clocks at age prediction and the role of meQTL (Meeks et al., 2024).
In conclusion, our results show that many existing methylation clocks have inconsistent performance and limited portability across genetically admixed cohorts. We encourage future efforts in the development of methylation clocks and other genomics-based aging biomarkers to be genetics- and ancestry-aware to ensure the accuracy of these tools for all individuals, regardless of their genetic background.
4. Methods
4.1. MAGENTA study
Cohort Selection
All participants in the MAGENTA study were recruited through previous studies of AD, including Feliciano-Astacio et al., 2019, Marca-Ysabel et al., 2021, and Griswold et al., 2020. Blood samples were taken for all individuals ascertained and processed at the following sites: the University of Miami Miller School of Medicine (Miami, FL, US), Wake Forest University (Winston-Salem, NC, US), Case Western Reserve University (Cleveland, OH, US), Universidad Central Del Caribe (Bayamón, PR), and the Instituto Nacional de Ciencias Neurologicas (Lima, PE). Ascertainment protocols were consistent across sites and capture cognitive function, family history of AD/related dementias, sociodemographic factors, and dementia staging. All diagnoses were assigned by clinical experts following criteria for diagnosis and staging from the National Institute on Aging Alzheimer’s Association (NIA-AA).
The MAGENTA study is based on pre-existing sample collections which vary in terms of the demographic information collected for each participant. Because the original ascertainment of MAGENTA study participants was international, different population descriptors were used across different ascertainment sites/cohorts. To facilitate comparisons relevant to understand the global differences noted in our study, we use a combination of geographic and race-based identifiers that are likely to best reflect underlying differences in genetic ancestry and admixture components. The label “white” is applied to legacy samples from North Carolina, Tennessee, and South Florida where participants either self-identified with this descriptor or were (in some legacy instances) administratively assigned as White race. The label “African American” is applied to samples collected via ascertainment in North Carolina and South Florida using population descriptors that specifically targeted enrollment of self-identified Black/African American participants. “Puerto Rican”, “Cuban”, and “Peruvian” labels are applied to samples collected as part of ascertainment efforts in these geographic areas. While more precise descriptors of self-identity are preferred, the advanced age of study participants and the older dates of some sample collections make recontact to collect these data impossible. All participants or their consenting proxy provided written informed consent as part of the study protocols approved by the site-specific Institutional Review Boards.
Genetic data and ancestry analysis
Genome-wide SNP genotyping was previously performed as previously described for the MAGENTA study cohorts. Briefly, samples were genotyped on the Illumina Infinium Global Screening Array using standard quality control filters on call rate, quality, missingness, and Hardy-Weinberg equilibrium.
For our analyses, local ancestry calls were generated using the FLARE software (S. R. Browning, Waples, and B. L. Browning, 2023) with three reference panels from the 1000 Genomes Project: Utah residents with Northern and Western European ancestry (CEU), Peruvians in Lima (PEL), and Yoruba in Ibadan, Nigeria (YRI). To estimate global ancestry proportions, we summed the haplotype lengths for each ancestry in each individual and divided by the total number of sites considered.
Methylation profiles
DNA methylation was quantified using the Illumina HumanMethylation EPICv2.0 according to the manufacturer’s instructions. All quality control and data normalization were performed using the the openSeSAMe pipeline from the SeSAMe (Wanding Zhou, 2018) tools for analyzing Illumina Infinium DNA methylation arrays. Probes of poor design were removed from the analysis as well as probes with signal detection P-value >0.05 in more than 5% of the samples. Non-CG probes and probes located on the X, Y, and mitochondrial chromosomes were also removed. Samples with incomplete bisulfite conversion (GCT score >1.5) and principal component analysis outliers were excluded. Noob normalization was performed with SeSAMe, using a nonlinear dye-bias correction.
4.2. Estimating epigenetic age and its correlation with chronological age in the MAGENTA study
We applied multiple commonly used first-, second-, and third-generation methylation clocks to all individuals in the MAGENTA study with genome-wide methylation data. We used established implementations of the Horvath, Hannum, Zhang2019_EN, Zhang2019 BLUP, and PhenoAge clocks from the methylClock R library (Pelegí-Sisó et al., 2021). We also applied DunedinPACE, a third-generation clock separately, because it was not included in the methylclock library (Belsky et al., 2022). Because this clock does not explicitly predict age, it is not included in the analyses of correlation with biological age. Unless otherwise specified, default options were used for all clocks.
The methylation clocks considered analyze different numbers of CpG sites. For each clock, the sites considered were taken from the methylclock library or the original publication. In the case of missing data, the methylClock library imputes methylation status using the mpute.knn function from the impute R library. The MAGENTA cohort had low proportions of missing data for clock CpGs. Specifically, there were 3.7% missing for the Horvath clock, 12.7% for the Hannum clock, 4.5% for the Zhang2019_EN clock, 3.7% for the PhenoAge clock, and 17.9% for the DunedinPACE clock.
We computed the Pearson correlation of estimated epigenetic age and chronological age for the controls in each cohort. To compare the strength of correlation between cohorts, we computed p-values for the observed differences using Fisher’s z test and the Zou method for computing confidence intervals as implemented in the cocor library (Diedenhofen and Musch, 2015).
4.3. Computing epigenetic age acceleration in Alzheimer’s disease patients and controls
In order to quantify epigenetic age acceleration from blood methylation data, we estimated raw, intrinsic, and extrinsic age acceleration for all clocks, except DunedinPACE, from the methylClock library. Blood cell type composition differs between individuals and over the lifespan; thus, we report results in the main text using intrinsic age acceleration estimates, which capture age acceleration independently of blood cell proportions.
Using the age acceleration estimates, we compared epigenetic age association between AD cases and matched controls using a Mann-Whitney U test, as implemented in the stats library in R. We analyzed the study as a whole and stratified by cohort.
4.4. Evaluating ensembles of age predictors
We tested the performance of a simple ensemble of age prediction methods at both estimating chronological age and distinguishing AD cases and controls. The ensemble was computed as the average of the estimate of each method on each individual. The resulting predictions were evaluated as described for each clock itself.
4.5. Analysis of genetic variation at clock CpG sites
To test whether the CpG sites included in the methylation clocks are variable between individuals, we intersected all the clock CpGs with variants identified in version 3.0 of gnomAD (Karczewski et al., 2020). This database covers 76,156 individuals with whole genome sequencing harmonized from many large-scale sequencing studies. The intersection was performed using bedtools in hg38 coordinates (Quinlan and Hall, 2010).
4.6. Analysis of meQTL affecting clock CpGs
Identification of meQTL affecting clock CpGs
We leveraged meQTL sets identified from blood samples by three independent studies. The first study identified 11,165,559 meQTLs from 3,799 Europeans and 3,195 South Asians (Hawe et al., 2022). The second study generated 4,565,687 meQTLs from 961 African Americans (Shang et al., 2023). The final study (EPIGEN) identified 249,710 meQTLs from 2,358 UK individuals (Villicaña et al., 2023). We filtered these sets separately on at a false discovery rate threshold of 0.05, correcting for multiple tests using the Benjamini-Hochberg method. These meQTL studies published their results in hg19 coordinates. To integrate with genetic variation and clock CpG data, we mapped the meQTL positions to hg38 using the UCSC liftOver tool (Hinrichs, 2006). For each meQTL set, we intersected the target CpG site with the CpGs considered in each clock and then combined across meQTL sets to generate a set of clock CpGs with evidence of meQTL.
Population-level allele frequencies of meQTL affecting clock CpGs
We analyzed the frequency of clock CpG-affecting meQTLs within two different versions of gnomAD. We used version 4.1 to quantify the allele frequencies of these meQTLs in the following global populations: African, Middle Eastern, Admixed American, European (non-Finnish), South Asian, Ashkenazi Jewish, East Asian, European (Finnish), Amish, and a “Remaining” group defined by gnomAD as individuals that did not unambiguously cluster within these previous groups in a principal component analysis. We then used gnomAD version 3.1 to gather allele frequencies for local ancestry blocks identified in 7,612 Latino admixed individuals with varying proportions of European, Amerindigenous, and African ancestry.
Supplementary Material
4.9. Acknowledgements
First, we acknowledge the individuals who participated in the MAGENTA study. We are also grateful to members of the Capra Lab, Param Priya Singh, Gillian Meeks, Brenna Henn, and Shyamalika Gopalan for helpful feedback on the project.
This work was supported by the National Institutes of Health (NIH) awards R35GM127087, R01AG070935, U01AG076482, U01AG072579, AG070864, AG072547, and AG074865. It was also supported by a Genentech-UCSF School of Pharmacy Diversity Fellowship to SCG.
This work was conducted in part using the resources of the Wynton High Performance Compute cluster at the University of California, San Francisco.
Footnotes
Competing interestsre
The authors declare no competing interests.
Code availability
The publicly available code for analysis are available in the following repository: https://github.com/seba2550/methyl-clocks-admixture
4.7. Data availability
Raw and normalized beta matrices, along with genotyping data used in this study will be made available at time of publication in the NIAGADS platform. Age predictions for all clocks mentioned in this article will be made available in tab-delimited format in the same Github repository in which all code used for these analyses is available.
References
- Aiello Allison E. et al. (July 2024). “Familial Loss of a Loved One and Biological Aging: NIMHD Social Epigenomics Program”. en. In: JAMA Network Open 7.7, e2421869. issn: 2574–3805. doi: 10.1001/jamanetworkopen.2024.21869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunan J R et al. (Jan. 2016). “Molecular and biological hallmarks of ageing”. en. In: British Journal of Surgery 103.2, e29–e46. issn: 0007–1323, 1365–2168. doi: 10.1002/bjs.10053. [DOI] [PubMed] [Google Scholar]
- Banovich Nicholas E. et al. (Sept. 2014). “Methylation QTLs Are Associated with Coordinated Changes in Transcription Factor Binding, Histone Modifications, and Gene Expression Levels”. en. In: PLoS Genetics 10.9. Ed. by Reddy Timothy E., e1004663. issn: 1553–7404. doi: 10.1371/journal.pgen.1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky Daniel W et al. (Jan. 2022). “DunedinPACE, a DNA methylation biomarker of the pace of aging”. In: eLife 11. Ed. by Deelen Joris et al. Publisher: eLife Sciences Publications, Ltd, e73420. issn: 2050–084X. doi: 10.7554/eLife.73420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning Sharon R., Waples Ryan K., and Browning Brian L. (Feb. 2023). “Fast, accurate local ancestry inference with FLARE”. en. In: The American Journal of Human Genetics 110.2, pp. 326–335. issn: 00029297. doi: 10.1016/j.ajhg.2022.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Dorothy T. et al. (July 2024). “Essential Nutrients, Added Sugar Intake, and Epigenetic Age in Midlife Black and White Women: NIMHD Social Epigenomics Program”. en. In: JAMA Network Open 7.7, e2422749. issn: 2574–3805. doi: 10.1001/jamanetworkopen.2024.22749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedenhofen Birk and Musch Jochen (Apr. 2015). “cocor: A Comprehensive Solution for the Statistical Comparison of Correlations”. en. In: PLOS ONE 10.4. Ed. by Olivier Jake, e0121945. issn: 1932–6203. doi: 10.1371/journal.pone.0121945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshua M Galanter et al. (Jan. 2017). “Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures”. en. In: eLife 6, e20532. issn: 2050–084X. doi: 10.7554/eLife.20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold Anthony J. et al. (Aug. 2020). “Immune and Inflammatory Pathways Implicated by Whole Blood Transcriptomic Analysis in a Diverse Ancestry Alzheimer’s Disease Cohort”. In: Journal of Alzheimer’s Disease 76.3, pp. 1047–1060. issn: 13872877, 18758908. doi: 10.3233/JAD-190855. [DOI] [PubMed] [Google Scholar]
- Hannum Gregory et al. (Jan. 2013). “Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates”. en. In: Molecular Cell 49.2, pp. 359–367. issn: 10972765. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawe Johann S. et al. (Jan. 2022). “Genetic variation influencing DNA methylation provides insights into molecular mechanisms regulating genomic function”. en. In: Nature Genetics 54.1, pp. 18–29. issn: 1061–4036, 1546–1718. doi: 10.1038/s41588-021-00969-x. [DOI] [PubMed] [Google Scholar]
- Hinrichs A. S. (Jan. 2006). “The UCSC Genome Browser Database: update 2006”. en. In: Nucleic Acids Research 34.90001, pp. D590–D598. issn: 0305–1048, 1362–4962. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson Karen et al. (May 2017). “Epigenetic Age Acceleration Assessed with Human White-Matter Images”. en. In: The Journal of Neuroscience 37.18, pp. 4735–4743. issn: 0270–6474, 1529–2401. doi: 10.1523/JNEUROSCI.0177-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath Steve (2013). “DNA methylation age of human tissues and cell types”. en. In: Genome Biology 14.10, R115. issn: 1465–6906. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath Steve, Gurven Michael, et al. (Dec. 2016). “An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease”. en. In: Genome Biology 17.1, p. 171. issn: 1474–760X. doi: 10.1186/s13059-016-1030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath Steve and Ritz Beate R (Dec. 2015). “Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients”. en. In: Aging 7.12, pp. 1130–1142. issn: 1945–4589. doi: 10.18632/aging.100859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal Siddhartha and Ebert Benjamin L. (Nov. 2019). “Clonal hematopoiesis in human aging and disease”. en. In: Science 366.6465, eaan4673. issn: 0036–8075, 1095–9203. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones Meaghan J., Goodman Sarah J., and Kobor Michael S. (Dec. 2015). “¡span style=”font-variant:small-caps;”¿DNA¡/span¿ methylation and healthy human aging”. en. In: Aging Cell 14.6, pp. 924–932. issn: 1474–9718, 1474–9726. doi: 10.1111/acel.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kader Farzeen and Ghai Meenu (Feb. 2017). “DNA methylation-based variation between human populations”. en. In: Molecular Genetics and Genomics 292.1, pp. 5–35. issn: 1617–4615, 1617–4623. doi: 10.1007/s00438-016-1264-2. [DOI] [PubMed] [Google Scholar]
- Karczewski Konrad J. et al. (May 2020). “The mutational constraint spectrum quantified from variation in 141,456 humans”. en. In: Nature 581.7809, pp. 434–443. issn: 0028–0836, 1476–4687. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger Nancy et al. (July 2024). “Epigenetic Aging and Racialized, Economic, and Environmental Injustice: NIMHD Social Epigenomics Program”. en. In: JAMA Network Open 7.7, e2421832. issn: 2574–3805. doi: 10.1001/jamanetworkopen.2024.21832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine Morgan E et al. (Dec. 2015). “Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning”. en. In: Aging 7.12, pp. 1198–1211. issn: 1945–4589. doi: 10.18632/aging.100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine Morgan E. et al. (Apr. 2018). “An epigenetic biomarker of aging for lifespan and healthspan”. en. In: Aging 10.4, pp. 573–591. issn: 1945–4589. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Ake T. et al. (Jan. 2019). “DNA methylation GrimAge strongly predicts lifespan and healthspan”. In: Aging (Albany NY) 11.2, pp. 303–327. issn: 1945–4589. doi: 10.18632/aging.101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni Riccardo E. et al. (Jan. 2015). “DNA methylation age of blood predicts all-cause mortality in later life”. In: Genome Biology 16.1, p. 25. issn: 1465–6906. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin Alicia R. et al. (Apr. 2019). “Clinical use of current polygenic risk scores may exacerbate health disparities”. en. In: Nature Genetics 51.4. Number: 4 Publisher: Nature Publishing Group, pp. 584–591. issn: 1546–1718. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks Gillian L. et al. (2024). “Common DNA sequence variation influences epigenetic aging in African populations”. In: bioRxiv. Publisher: Cold Spring Harbor Laboratory; eprint: https://www.biorxiv.org/content/early/2024/08/26/2024.08.26.608843.full.pdf. doi: 10.1101/2024.08.26.608843. [DOI] [Google Scholar]
- Monti Remo et al. (July 2024). “Evaluation of polygenic scoring methods in five biobanks shows larger variation between biobanks than methods and finds benefits of ensemble learning”. en. In: The American Journal of Human Genetics 111.7, pp. 1431–1447. issn: 00029297. doi: 10.1016/j.ajhg.2024.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Non Amy L (Nov. 2021). “Social Epigenomics: Are we at an Impasse?” en. In: Epigenomics 13.21, pp. 1747–1759. issn: 1750–1911, 1750–192X. doi: 10.2217/epi-2020-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre John et al. (Dec. 2022). “Addressing the challenges of polygenic scores in human genetic research”. English. In: The American Journal of Human Genetics 109.12. Publisher: Elsevier, pp. 2095–2100. issn: 0002–9297, 1537–6605. doi: 10.1016/j.ajhg.2022.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva Meritxell et al. (Jan. 2023). “DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits”. en. In: Nature Genetics 55.1, pp. 112–122. issn: 1061–4036, 1546–1718. doi: 10.1038/s41588-022-01248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegí-Sisó Dolors et al. (June 2021). “methylclock: a Bioconductor package to estimate DNA methylation age”. In: Bioinformatics 37.12, pp. 1759–1760. issn: 1367–4803. doi: 10.1093/bioinformatics/btaa825. [DOI] [PubMed] [Google Scholar]
- Privé Florian et al. (Jan. 2022). “Portability of 245 polygenic scores when derived from the UK Biobank and applied to 9 ancestry groups from the same cohort”. en. In: The American Journal of Human Genetics 109.1, pp. 12–23. issn: 0002–9297. doi: 10.1016/j.ajhg.2021.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan Aaron R. and Hall Ira M. (Mar. 2010). “BEDTools: a flexible suite of utilities for comparing genomic features”. en. In: Bioinformatics 26.6, pp. 841–842. issn: 1367–4811, 1367–4803. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina Abhay et al. (Dec. 2017). “Cerebral white matter hyperintensities on MRI and acceleration of epigenetic aging: the atherosclerosis risk in communities study”. en. In: Clinical Epigenetics 9.1, p. 21. issn: 1868–7075, 1868–7083. doi: 10.1186/s13148-016-0302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Lulu et al. (May 2023). “meQTL mapping in the GENOA study reveals genetic determinants of DNA methylation in African Americans”. en. In: Nature Communications 14.1, p. 2711. issn: 2041–1723. doi: 10.1038/s41467-023-37961-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemizu Daichi et al. (Nov. 2022). “Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through immune landscape analysis”. en. In: npj Aging 8.1. Number: 1 Publisher: Nature Publishing Group, pp. 1–9. issn: 2731–6068. doi: 10.1038/s41514-022-00096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Alicia K et al. (2014). “Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type”. en. In: BMC Genomics 15.1, p. 145. issn: 1471–2164. doi: 10.1186/1471-2164-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium et al. (Oct. 2015). “A global reference for human genetic variation”. en. In: Nature 526.7571, pp. 68–74. issn: 0028–0836, 1476–4687. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villicaña Sergio et al. (July 2023). “Genetic impacts on DNA methylation help elucidate regulatory genomic processes”. en. In: Genome Biology 24.1, p. 176. issn: 1474–760X. doi: 10.1186/s13059-023-03011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Wanding, Shen Hui (2018). sesame. doi: 10.18129/B9.BIOC.SESAME. [DOI] [Google Scholar]
- Zhang Qian et al. (Aug. 2019). “Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing”. In: Genome Medicine 11.1, p. 54. issn: 1756–994X. doi: 10.1186/s13073-019-0667-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and normalized beta matrices, along with genotyping data used in this study will be made available at time of publication in the NIAGADS platform. Age predictions for all clocks mentioned in this article will be made available in tab-delimited format in the same Github repository in which all code used for these analyses is available.