

Abstract
Polymerization of actin proteins into dynamic structures is essential to eukaryotic cell life, motivating many in vitro experiments measuring polymerization kinetics of individual filaments. Here, we model these kinetics, accounting for all relevant steps revealed by experiment: polymerization, depolymerization, random ATP hydrolysis, and release of phosphate (Pi). We relate filament growth rates to the dynamics of ATP–actin and ADP–Pi–actin caps that develop at filament ends. At the critical concentration of the barbed end, ccrit, we find a short ATP cap and a long fluctuation-stabilized ADP–Pi cap. We show that growth rates and the critical concentration at the barbed end are intimately related to cap structure and dynamics. Fluctuations in filament lengths are described by the length diffusion coefficient, D. Recently Fujiwara et al. [Fujiwara, I., Takahashi, S., Takaduma, H., Funatsu, T. & Ishiwata, S. (2002) Nat. Cell Biol. 4, 666–673] and Kuhn and Pollard [Kuhn, J. & Pollard, T. D. (2005) Biophys. J. 88, 1387–1402] observed large length fluctuations slightly above ccrit, provoking speculation that growth may proceed by oligomeric rather than monomeric on–off events. For the single-monomer growth process, we find that D exhibits a pronounced peak below ccrit, due to filaments alternating between capped and uncapped states, a mild version of the dynamic instability of microtubules. Fluctuations just above ccrit are enhanced but much smaller than those reported experimentally. Future measurements of D as a function of concentration can help identify the origin of the observed fluctuations.
Keywords: ATP cap, length diffusivity, modeling, critical concentration
The tendency of actin protein to spontaneously polymerize into rapidly growing filaments is fundamental to the life of eukaryotic cells. Cell motility (1, 2), cell division (3), and endocytosis (4) are examples of processes exploiting the dynamic character of actin structures composed of filaments. The regulation of filament growth processes leads to well-defined structures and coordinated function. For example, in combination with branching, capping, and depolymerizing proteins, actin self-assembles into controlled dynamic cross-linked networks forming the dynamic core of lamellipodia (2).
These complex cellular actin-based systems exhibit multiple superposed mechanisms. A large body of in vitro work has sought to unravel these mechanisms and pin down rate constants for the constituent processes in purified systems (5). An important class of experiments entails measuring growth rate at one end by microscopy (6–9) or by bulk spectroscopic methods (10–16) as a function of actin monomer concentration. From these and other in vitro studies using various labeling techniques, the following picture has emerged of filament growth kinetics in the presence of ATP (see Fig. 1). (i) Monomers are added to a growing filament end as ATP–actin. (ii) Rapidly, the ATP is then hydrolyzed to ADP and phosphate (Pi), both remaining bound to the monomer host (ADP–Pi–actin) (10, 14, 17–22). A rate of 0.3 s-1 was reported in ref. 22 in the presence of Mg, assuming random hydrolysis uninfluenced by neighboring monomers. (iii) After a long delay, Pi release into solution occurs, generating ADP–actin (23–25). Reported release rates are in the range of 0.002 to 0.006 s-1 (23–26).
Fig. 1.

Actin cap structure and growth kinetics. (a) Schematic of the threespecies cap at the barbed end of a long actin filament. Near the critical concentration we find a fluctuation-induced cap of Ncap ≈ 25 monomers, with a short ATP–actin component, 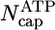 of order one. (b) Mean growth rate and fluctuations: in time t the average number of monomers added to a filament end is jt, with a spread of (2Dt)1/2 about this value.
of order one. (b) Mean growth rate and fluctuations: in time t the average number of monomers added to a filament end is jt, with a spread of (2Dt)1/2 about this value.
A typical filament in a growth rate experiment is thousands of monomer units (mon) in length and thus consists mainly of ADP–actin. Hence, the picture that emerges is of a long ADP–actin filament with a complex three-state “cap” region at the filament end (5) (see Fig. 1). A major goal of this work is to establish the composition and kinetics of the cap and how these determine growth rates and measurable length fluctuations. The monomer composition is important in the context of cellular processes where it is thought to regulate actin-binding proteins in a timely and spatially organized way (2). For example, it has been suggested that rates of branching generated by the Arp2/3 protein complex and/or debranching processes may depend on which of the following three monomer species is involved: ATP–actin, ADP–Pi–actin, or ADP–actin (7, 26, 27). Pi release has been proposed to act as a timer for the action of the depolymerizing/severing protein ADF/cofilin, which preferentially attacks ADP–actin (2).
Our aim in this work is to establish theoretically the quantitative implications of the currently held picture of actin polymerization. Previous theoretical works addressed growth rates before the important process of Pi release was established (28–30). To our knowledge, to date, there has been no theoretical analysis of single filament non-steady-state growth rates rigorously accounting for the processes (i)–(iii) above. A recent theoretical work (31) has addressed steady-state filament compositions.
The cap has important consequences for the growth rate j as a function of ATP–actin concentration, c. Measured j(c) curves, such as those in Fig. 5, are strikingly nonlinear in the region near the concentration where growth rate vanishes (16, 32). These curves become almost linear in excess Pi studies, where presumably the ADP–actin species is no longer involved (16). The complexity of the cap structure and dynamics also underlies the values of the critical concentration ccrit at the fast-growing “barbed” end and slow-growing “pointed” end of the polar actin filament (ccrit denotes the concentration where mean growth rate at one end vanishes). It is well known that in general these critical concentrations are different because detailed balance cannot be invoked for these nonequilibrium polymers (30). Our work explores how these differences are related to cap structure.
Fig. 5.

Growth rate j(c) vs. concentration from data taken from figure 1 of ref. 14 for simultaneous growth at both ends (in KCl and Mg). Solid line indicates numerical results, barbed end (parameters from Table 1), multiplied by a prefactor to fit data that lack absolute scale. Differences between numerical and experimental results may originate from the pointed end contribution or possibly are due to the experimental ionic conditions.
The major experimental focus has been mean growth rates, j(c). However, equally revealing are fluctuations about the mean whose measurement can expose features of the dynamical processes occurring at filament ends unavailable from j(c). These fluctuations are characterized by a “length diffusivity” D measuring the spread in filament lengths (see Fig. 1b) similarly to simple 1D Fickian diffusion: after time t, the root mean square fluctuation in filament length is (2Dt)1/2 about the mean value j(c)t. By using single-filament microscopy, Fujiwara et al. (8) and Kuhn and Pollard (9) recently measured unexpectedly high values of this diffusivity near steady-state conditions, D ≈ 30 mon2/s. This value should be compared with what would be expected of an equilibrium polymerization involving the measured on/off rates of order 1 mon/s, which would lead to D ≈ 1 mon2/s (8, 30, 33, 34). A number of possible explanations were proposed. (i) Fluctuations arise from “dynamic instability” due to stochastic cap loss episodes. This phenomenon would be a far milder version of the “catastrophes” in microtubule polymerization (8, 35). (ii) Filament polymerization proceeds by addition and subtraction of oligomeric actin segment (8, 35); such kinetics would constitute a radical departure from the accepted picture of filament growth kinetics involving single monomer addition events. (iii) Growth involves extra stochastic events such as short pauses possibly originating in filament–surface attachments (9). (iv) Enhanced fluctuations result from an artifact due to monomer labeling (36). (v) These observed fluctuations result from experimental error in filament length measurements (9). A major focus of this work is to calculate the concentration-dependent length diffusivity, D(c), assuming that the standard monomer-by-monomer addition picture is valid. We will see that large D values are realized below ccrit; just above the critical concentration fluctuations are enhanced, although much less than the experimental values.
We consider the initial condition where long preformed ADP–actin seeds are exposed initially to a buffer of fixed actin concentration c and excess ATP. Thus, for a given c value, a filament consists of a very long ADP–actin core at the end of which lies a complex steady-state (but fluctuating) ATP–actin/ADP–Pi–actin cap. Our analysis emphasizes the barbed end, with the pointed end assumed blocked. Our results apply to very dilute filaments where only ATP–actin is assumed to add to filaments because (i) free monomers bind ATP more strongly than ADP (37) and (ii) depolymerized ADP–actin or ADP–Pi–actin has enough time to exchange its nucleotide for ATP before repolymerization. An important issue is the nature of the ATP hydrolysis mechanism: the experiments of refs. 20 and 21 support a random mechanism, although others have suggested a cooperative vectorial mechanism occurring at the interface between ADP–Pi–actin and ATP–actin with rate 13.6 s-1 (19, 28). In this work, random hydrolysis is assumed throughout.
Parameter Values and Mathematical Methods
One of the major aims of this work is to identify qualitative, but experimentally measurable, features of the growth kinetics that are independent of the precise values of rate constants, because the latter depend on experimental conditions such as ionic strength (38) and the values themselves are often controversial. The parameter values we use are shown in Table 1, in which 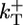 is the polymerization rate constant of ATP–actin, and
is the polymerization rate constant of ATP–actin, and 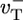 ,
, 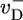 , and
, and 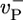 are the depolymerization rates of ATP–actin, ADP–actin, and ADP–Pi–actin, respectively. The rates of ATP hydrolysis and Pi release (both assumed irreversible) are rH and rPi, respectively. In addition, we will explore the effects of changing some of these parameter values. Because the monomer at the tip makes bonds with the two nearest neighbors, each belonging to a different protofilament, one expects that rate constants also may depend on the state of neighbors. Here, however, we study the simplest “one-body” model, assuming that on/off rates depend only on the attaching/detaching species (6) and that hydrolysis and Pi release rates are uniform along the filament. The influence of “many-body” effects will be discussed briefly below.
are the depolymerization rates of ATP–actin, ADP–actin, and ADP–Pi–actin, respectively. The rates of ATP hydrolysis and Pi release (both assumed irreversible) are rH and rPi, respectively. In addition, we will explore the effects of changing some of these parameter values. Because the monomer at the tip makes bonds with the two nearest neighbors, each belonging to a different protofilament, one expects that rate constants also may depend on the state of neighbors. Here, however, we study the simplest “one-body” model, assuming that on/off rates depend only on the attaching/detaching species (6) and that hydrolysis and Pi release rates are uniform along the filament. The influence of “many-body” effects will be discussed briefly below.
Table 1. Values of barbed end rate constants used in this work, appropriate for solutions of 50 mM KCl and 1 mM MgCl2.
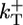
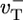
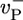
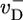
To calculate filament growth kinetics and composition, one is faced with the formidable task of obtaining the steady-state probability distribution of all possible actin monomer sequences along the filament; there are three possible states per monomer, so for filaments of N units long 3N coupled equations must be solved. We have managed, however, to obtain a solution for the mean elongation rate j(c) by projecting the full system of 3N equations onto a set of just 3 exact equations for the return probabilities 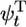 ,
, 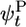 , and
, and 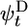 . These are the probabilities that a given monomer that was polymerized at t = 0 is again at the tip at time t as ATP–actin, ADP–Pi–actin, or ADP–actin, respectively.
. These are the probabilities that a given monomer that was polymerized at t = 0 is again at the tip at time t as ATP–actin, ADP–Pi–actin, or ADP–actin, respectively.
The outline of our method is as follows. For j < 0 the growth rate is related to the return probabilities by 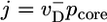 , where
, where 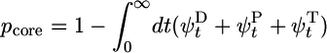 is the probability of exposure of the ADP–actin core at the tip. For j > 0, the relation is
is the probability of exposure of the ADP–actin core at the tip. For j > 0, the relation is 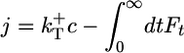 , where
, where 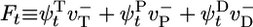 is the mean depolymerization rate at time t of a monomer that added to the tip at t = 0. The integral of Ft is the total depolymerization rate of added monomers. In Supporting Material, which is published as supporting information on the PNAS web site, we present the dynamical equations obeyed by the return probabilities, from which we obtained a closed recursion relation for the Laplace transform of Ft, namely fE. This recursion relation relates fE to fE+rH and fE+rPi. With boundary condition fE → 0 as E → ∞, we started from large E values and evolved this equation numerically toward E = 0 to obtain
is the mean depolymerization rate at time t of a monomer that added to the tip at t = 0. The integral of Ft is the total depolymerization rate of added monomers. In Supporting Material, which is published as supporting information on the PNAS web site, we present the dynamical equations obeyed by the return probabilities, from which we obtained a closed recursion relation for the Laplace transform of Ft, namely fE. This recursion relation relates fE to fE+rH and fE+rPi. With boundary condition fE → 0 as E → ∞, we started from large E values and evolved this equation numerically toward E = 0 to obtain 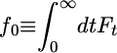 . Given f0, the time integrals of the return probabilities then were obtained directly from the dynamical equations, and j was thereby determined.
. Given f0, the time integrals of the return probabilities then were obtained directly from the dynamical equations, and j was thereby determined.
The above analytically based method does not generate cap sizes and length diffusivities. To calculate these quantities and also to test the validity of the analytical method, we have simulated the stochastic tip dynamics employing the kinetic Monte Carlo (MC) method known as the BKL (39) or Gillespie (40) algorithm to evolve the state of a filament tip in time and to calculate its mean growth rate. Each step of the algorithm entails updating time by an amount depending on the rate and number of possible future events, namely polymerization/depolymerization, hydrolysis, and Pi release. Excellent agreement is found between MC results and the numerical solutions of our closed equations for the growth rate (see Fig. 3 Inset).
Fig. 3.

Dependence of growth rate on concentration: influence of 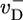 (indicated in s-1 next to each curve). Other parameters are as in Table 1. MC and exact numerical solution results are indistinguishable. The spread in ccrit values for the three curves is 5%. (Left Inset) Blow-up of critical region showing the agreement between MC (squares, error bars are standard deviation of mean) and numerical method (solid line). (Right Inset) Influence of many-body effects; the value shown in s-1 next to curves is the depolymerization rate of ATP–actin next to ADP–actin,
(indicated in s-1 next to each curve). Other parameters are as in Table 1. MC and exact numerical solution results are indistinguishable. The spread in ccrit values for the three curves is 5%. (Left Inset) Blow-up of critical region showing the agreement between MC (squares, error bars are standard deviation of mean) and numerical method (solid line). (Right Inset) Influence of many-body effects; the value shown in s-1 next to curves is the depolymerization rate of ATP–actin next to ADP–actin, 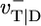 .
.
Our analytical method is exact and avoids preaveraging, an approximation where the joint probability of a given filament nucleotide sequence is approximated as a product of probabilities for individual actin subunits. This approximation neglects correlations between units. To assess the accuracy of this scheme, we compared our results for cap size and growth rate with those obtained by using preaveraging (see Supporting Material for details). Preaveraging has been used in other theoretical studies of actin polymerization such as ref. 31 to study steady state and ref. 32 to study growth rates.
Cap Structure and the Importance of Fluctuations
By using the parameters of Table 1, in Fig. 2 we present MC results for (i) the total cap size, Ncap, namely the mean total number of ATP–actin and ADP–Pi–actin subunits at the barbed end, as a function of concentration, and (ii) the number of ATP–actin cap subunits, 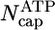 . Fig. 2 shows that both caps become large for large concentrations. This behavior is easy to understand. Consider, for example, the ATP cap: when polymerization rates exceed both the hydrolysis rate rH and the depolymerization rates, the interface between ADP–Pi–actin and ATP–actin follows the growing tip with a lag of j(c)/rH monomers. Thus,
. Fig. 2 shows that both caps become large for large concentrations. This behavior is easy to understand. Consider, for example, the ATP cap: when polymerization rates exceed both the hydrolysis rate rH and the depolymerization rates, the interface between ADP–Pi–actin and ATP–actin follows the growing tip with a lag of j(c)/rH monomers. Thus,
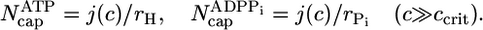 |
[1] |
Here, the number of ADP–Pi subunits, 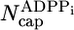 , is found by using similar reasoning as for
, is found by using similar reasoning as for 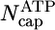 . The validity of Eq. 1 for large concentrations is verified against MC data in Fig. 2.
. The validity of Eq. 1 for large concentrations is verified against MC data in Fig. 2.
Fig. 2.

Total cap length Ncap(c) and ATP–actin cap length 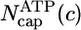 at barbed end. Parameters are from Table 1. Squares indicate MC results. Dashed lines refer to Eq. 1. Solid lines indicate preaveraging approximation. Vertical dashed line indicates ccrit = 0.119 μM.
at barbed end. Parameters are from Table 1. Squares indicate MC results. Dashed lines refer to Eq. 1. Solid lines indicate preaveraging approximation. Vertical dashed line indicates ccrit = 0.119 μM.
The striking feature of Fig. 2 is that the total cap remains long even below the critical concentration of the barbed end, being 25 units at ccrit and remaining larger than unity down to c ≈ ccrit/2. One might naively have guessed that below ccrit there would be no cap at all, because the filament is shrinking into its ADP core. (Indeed, the absence of a cap would also be suggested by Eq. 1 if one were to extend its validity down to ccrit where j = 0.) This reasoning is, however, invalid because it neglects fluctuations due to randomness of monomer addition/subtraction.
To understand why fluctuations lead to long caps, consider the length changes of the cap only, excluding changes in the ADP–actin core length. Just below the critical concentration, the tip of a typical long cap has a net shrinkage rate (33, 34), vcap(c). This value is a weighted average of rates, summed over all possible states of the short ATP–actin segment on top of the long ADP–Pi–actin segment. Because vcap is a smooth function of c, it can be Taylor-expanded near the critical concentration and expressed as 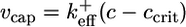 , where
, where 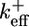 is an effective on rate constant, different from
is an effective on rate constant, different from 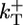 . Now superposed on this average shrinkage, the cap tip also performs a random walk in cap length space, described by a diffusivity Dcap(c) (8, 33, 34), also an average over the states of the short ATP cap. (Dcap is in fact the short-time diffusivity of the entire filament; see discussion below.) For small times, diffusivity dominates and of order (2Dcapt)1/2 units add to or subtract from the cap. For times less than the cap turnover time tcap, this number is much bigger than the number of units wiped out by coherent shrinkage, vcapt. The cap lifetime tcap is the time when the shrinkage just catches up, vcaptcap ≈ (2Dcaptcap)1/2. Hence, the approximate dependence of cap length on concentration is
. Now superposed on this average shrinkage, the cap tip also performs a random walk in cap length space, described by a diffusivity Dcap(c) (8, 33, 34), also an average over the states of the short ATP cap. (Dcap is in fact the short-time diffusivity of the entire filament; see discussion below.) For small times, diffusivity dominates and of order (2Dcapt)1/2 units add to or subtract from the cap. For times less than the cap turnover time tcap, this number is much bigger than the number of units wiped out by coherent shrinkage, vcapt. The cap lifetime tcap is the time when the shrinkage just catches up, vcaptcap ≈ (2Dcaptcap)1/2. Hence, the approximate dependence of cap length on concentration is
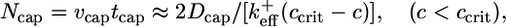 |
[2] |
which indeed becomes large as ccrit is approached from below.
In summary, even though on average below ccrit no ATP–actin monomers are being added to the tip, fluctuations in addition/subtraction rates allow a cap to grow to length (2Dcaptcap)1/2 because the cap length diffusivity is dominant for times less than tcap. Now because Pi release is very slow, for simplicity in deriving Eq. 2 we assumed the release rate was zero, rPi = 0. However, the result of Eq. 2 is valid even for a nonzero rPi except for concentrations so close to ccrit that the cap turnover time exceeds the Pi release time. In this inner region, diffusion is only able to grow the cap for a time of order 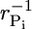 before Pi release intervenes. The maximum possible cap length, attained very close to ccrit, is thus,
before Pi release intervenes. The maximum possible cap length, attained very close to ccrit, is thus,
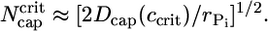 |
[3] |
Eq. 2 is valid until Ncap reaches this bound.
These arguments explain the origin of the long caps below ccrit. To make a quantitative comparison of Eqs. 2 and 3 to the numerics of Fig. 2, the values of Dcap and vcap must be determined. Now because for our parameter set 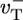 and
and 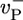 have similar values (see Table 1), an estimate can be obtained by considering the special case where
have similar values (see Table 1), an estimate can be obtained by considering the special case where 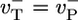 (identical ATP–actin and ADP–Pi–actin). This case is convenient because Dcap and vcap can be calculated exactly; the cap has just one monomer species, so
(identical ATP–actin and ADP–Pi–actin). This case is convenient because Dcap and vcap can be calculated exactly; the cap has just one monomer species, so 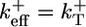 and
and 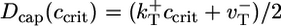 (8, 33, 34). By using the values of Table 1 in these expressions and in Eq. 3 gives
(8, 33, 34). By using the values of Table 1 in these expressions and in Eq. 3 gives 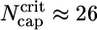 , of the same order as the numerics of Fig. 2.
, of the same order as the numerics of Fig. 2.
Finally, note that the preaveraging method shown in Fig. 2 is an excellent approximation in regions where fluctuations are unimportant (very large or very small c), producing almost identical results to MC. However, below ccrit it considerably underestimates cap lengths. This error results from the preaveraged treatment of fluctuations.
Mean Growth Rate, j(c)
How is the behavior of the average rate of growth j(c) correlated to cap structure and dynamics? The lowest curve of Fig. 3 shows numerical results for barbed end growth, using identical parameters to those of Fig. 2. A noticeable feature is that the slopes are very different above and below the critical concentration of the barbed end. This difference directly reflects the cap structure just discussed, as follows. For c ≫ ccrit the ATP–actin segment is long and hides the remaining ADP–Pi–actin portion of the cap, so j ≈ 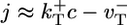 has simple linear form and slope
has simple linear form and slope 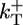 , approximately behaving as if ATP–actin were the only species involved. In the region where c < ccrit, the slope of j(c) is large because the cap length is changing rapidly as concentration increases (see Fig. 2). Filament length change is now generated by capless episodes, when the ADP–actin core is exposed, and the filament shrinks with velocity
, approximately behaving as if ATP–actin were the only species involved. In the region where c < ccrit, the slope of j(c) is large because the cap length is changing rapidly as concentration increases (see Fig. 2). Filament length change is now generated by capless episodes, when the ADP–actin core is exposed, and the filament shrinks with velocity 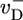 (the steady-state cap has fixed mean length and does not on average contribute). Thus,
(the steady-state cap has fixed mean length and does not on average contribute). Thus, 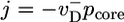 where pcore ≈ 1/Ncap is the probability the cap length vanishes, assuming a broad distribution of cap lengths with mean Ncap. By using Eq. 2, this expression gives
where pcore ≈ 1/Ncap is the probability the cap length vanishes, assuming a broad distribution of cap lengths with mean Ncap. By using Eq. 2, this expression gives 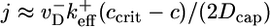 in the region where Eq. 2 is valid. Because
in the region where Eq. 2 is valid. Because 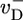 is large, this is a much larger slope than for concentrations above ccrit.
is large, this is a much larger slope than for concentrations above ccrit.
The region very close to ccrit, where Eq. 3 takes over, is an interesting one. (i) Here the total cap becomes long, of length approximately 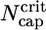 , implying that ADP–actin is rarely exposed at the tip. It follows that the depolymerization rate of ADP–actin will have only a small influence on the value of ccrit. This effect is verified in Fig. 3 where we display j(c) curves for
, implying that ADP–actin is rarely exposed at the tip. It follows that the depolymerization rate of ADP–actin will have only a small influence on the value of ccrit. This effect is verified in Fig. 3 where we display j(c) curves for 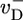 values ranging from 2.2 to 7.2 s-1. These changes produce only a very small shift in ccrit, even though j(c) changes significantly for c < ccrit. (ii) The mean ATP-cap length is small (of order unity), and because the tip composition and cap length are constantly fluctuating, both ATP–actin and ADP–Pi–actin are frequently exposed at the tip. Thus, we expect a dependence of ccrit on the value of
values ranging from 2.2 to 7.2 s-1. These changes produce only a very small shift in ccrit, even though j(c) changes significantly for c < ccrit. (ii) The mean ATP-cap length is small (of order unity), and because the tip composition and cap length are constantly fluctuating, both ATP–actin and ADP–Pi–actin are frequently exposed at the tip. Thus, we expect a dependence of ccrit on the value of 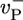 . This dependence is verified in Fig. 4, where we display how the growth rate and ccrit change with the value of
. This dependence is verified in Fig. 4, where we display how the growth rate and ccrit change with the value of 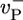 . The magnitude of the shift is influenced by the assumed rate of ATP hydrolysis: if one uses, for example, a hydrolysis rate 10 times smaller, the change in growth remains substantial but is considerably reduced (see Fig. 4 Inset).
. The magnitude of the shift is influenced by the assumed rate of ATP hydrolysis: if one uses, for example, a hydrolysis rate 10 times smaller, the change in growth remains substantial but is considerably reduced (see Fig. 4 Inset).
Fig. 4.

Growth rate: influence of the value of 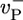 (shown in s-1). Other values are as in Table 1. Solid lines indicate numerical solutions and MC simulations (indistinguishable). Dashed line indicates preaveraging approximation for
(shown in s-1). Other values are as in Table 1. Solid lines indicate numerical solutions and MC simulations (indistinguishable). Dashed line indicates preaveraging approximation for 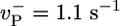 . (Inset) Same but with rH = 0.03 s-1.
. (Inset) Same but with rH = 0.03 s-1.
Note also that preaveraging estimates the growth rate very accurately (see Fig. 4). Even in the fluctuation-dominated region just below ccrit, where cap size is substantially underestimated, it remains accurate although slightly less so than elsewhere.
An important question is the effect of many-body interactions between actin subunits, so far neglected in this work. We have found that the shape of the mean growth rate near and below the critical concentration is sensitive to these interactions. As an example, Fig. 3 Inset shows the dependence of j(c) on the depolymerization rate of ATP–actin when its nearest neighbor is ADP–actin 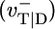 , with all other rates as in Table 1. Other types of many-body interactions can lead also to shifts in ccrit (data not shown). Including many-body interactions rapidly increases the number of rate constants. Because these constants are unknown and presumably hard to measure, the uniqueness with which growth rate curves can be modeled near ccrit is limited. We stress, however, that the central qualitative conclusions, namely the existence of a long cap at ccrit and the associated change of slope of the growth rate, are general. An example of fitting experimental j(c) curves with a one-body model is shown in Fig. 5.
, with all other rates as in Table 1. Other types of many-body interactions can lead also to shifts in ccrit (data not shown). Including many-body interactions rapidly increases the number of rate constants. Because these constants are unknown and presumably hard to measure, the uniqueness with which growth rate curves can be modeled near ccrit is limited. We stress, however, that the central qualitative conclusions, namely the existence of a long cap at ccrit and the associated change of slope of the growth rate, are general. An example of fitting experimental j(c) curves with a one-body model is shown in Fig. 5.
Fluctuations in Growth Rate
Turning now to fluctuations in growth rates, we find these behave dramatically around the critical concentration, reflecting a mild version of the dynamic instability exhibited by microtubules (30, 41). In Fig. 6 Inset, we used MC to evaluate the length diffusivity, D(t) ≡ (〈L2〉 - 〈L〉2)/(2t), where L is the number of subunits added/subtracted after time t, starting from filaments with steady-state caps at t = 0. For c = 0.15 μM (above ccrit), we find D is essentially independent of time. Its magnitude is of order 1 mon2/s, as would be expected for a growth process of identical subunits that add/subtract with rates of order 1 s-1 (8, 30, 33, 34). However, for c = 0.1 μM (below ccrit), D is increasing with time, reaching a large asymptotic value D∞ after several hundred seconds. Fig. 6 shows the dependence of D∞ on concentration; it exhibits a sharp peak below ccrit and then drops rapidly.
Fig. 6.

Long time length diffusion coefficient, D∞(c). Squares indicate MC results, using parameters from Table 1. Vertical dashed line indicates ccrit. Solid line indicates prediction of simple model: 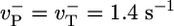 , rPi = 0, other values from Table 1. (Inset) Time-dependence of diffusivity at two concentrations.
, rPi = 0, other values from Table 1. (Inset) Time-dependence of diffusivity at two concentrations.
To understand the physics underlying this behavior, consider the simple model where ATP–actin and ADP–Pi–actin are identical 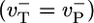 and Pi release very slow (rPi → 0). Now D describes the random walk performed by the filament tip; if the tip makes a random forwards or backwards step of L monomer units every time interval T, then one can write D = L2/T. Just above the critical concentration, where on and off rates are approximately equal, the tip randomly adds or subtracts one ATP–actin (L = 1) in a mean time
and Pi release very slow (rPi → 0). Now D describes the random walk performed by the filament tip; if the tip makes a random forwards or backwards step of L monomer units every time interval T, then one can write D = L2/T. Just above the critical concentration, where on and off rates are approximately equal, the tip randomly adds or subtracts one ATP–actin (L = 1) in a mean time 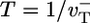 , giving
, giving 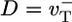 . Just below the critical concentration, however, we know there is a long steady-state cap. Because most filaments are capped, at short times D is determined by length changes of the cap, and its value is thus close to the cap diffusivity, Dcap. As time increases, more and more uncapping episodes occur, each episode now contributing to filament length change. Such events are correlated on the timescale of the cap lifetime,
. Just below the critical concentration, however, we know there is a long steady-state cap. Because most filaments are capped, at short times D is determined by length changes of the cap, and its value is thus close to the cap diffusivity, Dcap. As time increases, more and more uncapping episodes occur, each episode now contributing to filament length change. Such events are correlated on the timescale of the cap lifetime, 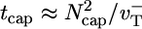 [we used
[we used 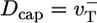 for the simple model (33, 34)]. This fact explains why D(t) changes with time up to the cap lifetime (see Fig. 6 Inset). Thus, to determine D∞, one must take T = tcap. By using a well known result from the theory of 1D random walks (42), the number of uncapping events during the time tcap is approximately (Dcaptcap)1/2 ≈ Ncap. Because the number of core monomers lost during each uncapping episode before a polymerizing monomer arrives is of order
for the simple model (33, 34)]. This fact explains why D(t) changes with time up to the cap lifetime (see Fig. 6 Inset). Thus, to determine D∞, one must take T = tcap. By using a well known result from the theory of 1D random walks (42), the number of uncapping events during the time tcap is approximately (Dcaptcap)1/2 ≈ Ncap. Because the number of core monomers lost during each uncapping episode before a polymerizing monomer arrives is of order 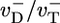 , thus
, thus 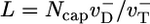 . Thus, one obtains a very different expression for the diffusivity,
. Thus, one obtains a very different expression for the diffusivity, 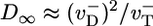 ; there is a discontinuity in diffusivity at ccrit of magnitude
; there is a discontinuity in diffusivity at ccrit of magnitude
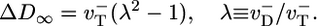 |
[4] |
At the barbed end the instability parameter λ ≈ 5.1 and fluctuations at the critical concentration are very large, with a pronounced discontinuous drop in D∞ as one passes to higher c. A rigorous derivation of Eq. 4 is shown in Supporting Material where in addition we obtain the full sawtooth curve shown in Fig. 6; evidently, the simple model captures many features of the actual D∞(c) profile. The effect of Pi release and ATP–actin/ADP–Pi–actin differences is to shift ccrit and to smooth the sharp peak and shift it to somewhat below ccrit.
How do the results of Fig. 6 compare with the large fluctuations observed by Fujiwara et al. (8) and Kuhn and Pollard (9) and also suggested by the findings of ref. 43? Fig. 6 shows a peak value of D∞ ≈ 34 mon2·s-1, dropping to D∞ ≈ 5 mon2·s-1 at ccrit. The experimentally reported value was ≈30 mon2·s-1; however, these measurements were performed at (8) or close to (9) a treadmilling steady state, i.e., at a concentration slightly above ccrit for the barbed end and well below that for the pointed end. At this concentration, Fig. 6 shows a diffusivity of <5 mon2·s-1. Thus, both theory and experiment exhibit large fluctuations near ccrit but at different concentrations. Further experimental measurements of the full D∞(c) profile are needed to establish the relationship, if any, between these.
Our work leads also to the following prediction: Because Pi will bind to ADP–actin and eliminate the effect of a large instability parameter, thus fluctuations and D at the barbed end will be suppressed in the presence of excess Pi.
Discussion
Pointed End j(c): Why Is ccrit So Different? In this work, we emphasized the barbed end, but our methods are also applicable to the pointed end, provided the same mechanisms of uniform random hydrolysis and slow Pi release remain valid. Making this assumption, let us now discuss why ccrit (for ATP–actin) at the pointed end is almost six times the value at the barbed end (6). Now an important issue is how different the ATP–actin and ADP–Pi–actin species are, in terms of on and off rate constants. That they are similar is suggested by the observation that excess Pi reduces the critical concentration in a pure ADP–actin polymerization to a value rather close to the barbed end ccrit in ATP (16, 44–46). However, the assumption that the two species are similar and that the same basic mechanisms apply at the pointed end is inconsistent with the very different ccrit values. This inconsistency is due to the cap structure we have established here: the cap includes a long ADP–Pi segment essentially hiding the ADP–actin core, which is thus rarely seen at the filament tip (see Fig. 1a). For the barbed end (Fig. 2) Ncap ≈ 25 at ccrit, and we find a large value for the pointed end at its ccrit, although smaller than the barbed end (data not shown). Thus, ADP–actin on/off rates are almost irrelevant to ccrit (see Fig. 3), and hence differences between ATP–actin and ADP–actin cannot account for the large ccrit differences. Thus, the origin must be different ATP–actin/ADP–Pi–actin compositions at the pointed and barbed ends; because the ATP–actin segment is short, both species are regularly exposed at filament ends, and substantially different ccrit values will result, provided the two species have different rate constants. Were these identical, ccrit at both ends would be very similar, because the on/off rates at the filament ends would then be very close to the values for an all ATP–actin filament; for such a filament, detailed balance dictates that the ratio of on/off rates at each end are identical (30). [However, in apparent contradiction to this conclusion are the findings of ref. 6. where different on/off ratios were reported at each end, under conditions where long ATP–actin caps are expected. A conceivable explanation is possibility (ii); see below.] Many-body effects will further affect ccrit.
We are driven to the following two possibilities: (i) ATP–actin and ADP–Pi–actin are substantially different, or (ii) different mechanisms operate during pointed end growth. Certain workers (47, 48) have proposed possibility (i), based on the irreversibility of hydrolysis (47), which suggests a large energetic change, possibly a structural change of the filament. Possibility (i) may in fact be consistent with the experiments of refs. 16 and 44–46, which did not probe individual on/off rate constants of ADP–Pi–actin and that may have involved significant ADP–actin polymerization (45). We are unaware of any crystallographic (49) or electron microscopic (50) experiments examining ATP/ADP–Pi differences for filamentous actin.
If we adhere to the assumption that the growth mechanisms as previously outlined apply to both ends, we then are led to the following prediction: the values of ccrit for ATP–actin at both ends will be only weakly affected by the presence of excess Pi (provided ionic conditions are strictly unchanged). This prediction follows because the binding of Pi to ADP–actin segments is almost irrelevant because these are rarely exposed at the tip due to long caps at ccrit. Indeed, for the barbed end no significant shift has been observed in the presence of Pi (16, 44–46). For the pointed end, however, a reduction of ccrit has been reported in the presence of Pi and barbed end capping proteins (16, 44–46). This observation cannot be explained within the present framework and suggests possibility (ii). Future experiments will hopefully settle this important issue.
Conclusions
In this work, filament growth rates j(c) and their fluctuations, as measured by the diffusivity D(c), were calculated as functions of ATP–actin concentration c. This work presents a rigorous calculation of these quantities accounting for all known basic mechanisms. Pantaloni et al. (28, 29) studied j(c) at the barbed end in a work before the mechanism of Pi release was discovered. Infinitely fast Pi release and vectorial hydrolysis were assumed. Given the data available at that time, to explain the sharp change in slope of j(c) at ccrit (see, e.g., Fig. 5), they further assumed (i) strong three-body ATP–actin/ADP–actin interactions that lead to stable short ATP–actin caps, and (ii) zero hydrolysis rate of the nucleotide bound to the terminal monomer. In our work, the origin of the sharp change in slope is precisely the fact that Pi release is slow, similar to an earlier model of microtubule polymerization (51).
Recently, Bindschadler et al. (31) studied the composition of actin filaments accounting for all three actin species at steady state. We have examined the preaveraging approximation used in their work and showed that it leads to very accurate j(c) curves, but the cap lengths are underestimated below ccrit.
Here, we have addressed random ATP hydrolysis only. Further work is needed to analyze the implications of the vectorial hydrolysis suggested by refs. 19 and 28. We showed that for random hydrolysis j(c) is linear far above the critical concentration. Growth rate experiments for both ends together in the absence of KCl have exhibited nonlinearities up to c = 10 μM, far above the critical concentration of the barbed end, which is 1 μM under these conditions (10, 11). In refs. 10 and 28, this observation was attributed to vectorial hydrolysis at the barbed end, whereas in ref. 6 this behavior was assigned to the nonlinear contribution of the pointed end whose critical concentration is ≈5 μM under the same conditions.
Perhaps our most interesting finding is that the long time diffusivity D∞ has a large peak below the critical concentration ccrit of the barbed end, followed by a sharp drop in a narrow range above ccrit. This conclusion is quite general, and its origin is the smallness of the Pi release rate and the large value of the off rate of ADP–actin at the barbed end. Future measurements of length diffusivities over a range of concentrations promise to provide new information and insight on the fundamentals of actin polymerization.
Supplementary Material
Acknowledgments
We thank Ikuko Fujiwara, Jeffrey Kuhn, and Thomas Pollard for stimulating discussions. This work was supported by Petroleum Research Fund Grant 33944-AC7 and National Science Foundation Grant CHE-00-91460.
Author contributions: B.O., D.V., and Q.Y. performed research and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MC, Monte Carlo; mon, monomers.
References
- 1.Bray, D. (2000) Cell Movements. From Molecules to Motility (Garland, New York), 2nd Ed.
- 2.Pollard, T. D. & Borisy, G. G. (2003) Cell 112, 453-465. [DOI] [PubMed] [Google Scholar]
- 3.Pelham, R. J. & Chang, F. (2002) Nature 419, 82-86. [DOI] [PubMed] [Google Scholar]
- 4.Young, M. E., Cooper, J. A. & Bridgman, P. C. (2004) J. Cell. Biol. 166, 629-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korn, E. D., Carlier, M.-F. & Pantaloni, D. (1987) Science 238, 638-643. [DOI] [PubMed] [Google Scholar]
- 6.Pollard, T. D. (1986) J. Cell Biol. 103, 2747-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amann, K. J. & Pollard, T. D. (2001) Proc. Natl. Acad. Sci. USA 98, 15009-15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujiwara, I., Takahashi, S., Takaduma, H., Funatsu, T. & Ishiwata, S. (2002) Nat. Cell Biol. 4, 666-673. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn, J. & Pollard, T. D. (2005) Biophys. J. 88, 1387-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlier, M.-F., Pantaloni, D. & Korn, E. D. (1984) J. Biol. Chem. 259, 9983-9986. [PubMed] [Google Scholar]
- 11.Carlier, M.-F., Pantaloni, D. & Korn, E. D. (1985) J. Biol. Chem. 260, 6565-6571. [PubMed] [Google Scholar]
- 12.Coué, M. & Korn, E. D. (1986) J. Biol. Chem. 261, 1588-1593. [PubMed] [Google Scholar]
- 13.Carlier, M.-F., Criquet, P., Pantaloni, D. & Korn, E. D. (1986) J. Biol. Chem. 261, 2041-2050. [PubMed] [Google Scholar]
- 14.Carlier, M.-F., Pantaloni, D. & Korn, E. D. (1986) J. Biol. Chem. 261, 10785-10792. [PubMed] [Google Scholar]
- 15.Weber, A., Northrop, J., Bishop, M. F., Ferrone, F. A. & Mooseker, M. S. (1987) Biochemistry 26, 2537-2544. [DOI] [PubMed] [Google Scholar]
- 16.Carlier, M.-F. & Pantaloni, D. (1988) J. Biol. Chem. 263, 817-825. [PubMed] [Google Scholar]
- 17.Pardee, J. D. & Spudich, J. A. (1982) J. Cell Biol. 93, 648-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pollard, T. D. & Weeds, A. G. (1984) FEBS Lett. 170, 94-98. [DOI] [PubMed] [Google Scholar]
- 19.Carlier, M.-F., Pantaloni, D. & Korn, E. D. (1987) J. Biol. Chem. 262, 3052-3059. [PubMed] [Google Scholar]
- 20.Ohm, T. & Wegner, A. (1994) Biochim. Biophys. Acta 1208, 8-14. [DOI] [PubMed] [Google Scholar]
- 21.Pieper, U. & Wegner, A. (1996) Biochemistry 35, 4396-4402. [DOI] [PubMed] [Google Scholar]
- 22.Blanchoin, L. & Pollard, T. D. (2002) Biochemistry 41, 597-602. [DOI] [PubMed] [Google Scholar]
- 23.Carlier, M.-F. & Panataloni, D. (1986) Biochemistry 35, 7789-7792. [DOI] [PubMed] [Google Scholar]
- 24.Carlier, M.-F. (1987) Biochem. Biophys. Res. Commun. 143, 1069-1075. [DOI] [PubMed] [Google Scholar]
- 25.Melki, R., Fievez, S. & Carlier, M.-F. (1996) Biochemistry 35, 12038-12045. [DOI] [PubMed] [Google Scholar]
- 26.Blanchoin, L., Pollard, T. D. & Mullins, R. D. (2000) Curr. Biol. 10, 1273-1282. [DOI] [PubMed] [Google Scholar]
- 27.Ichetovkin, I., Grant, W. & Condeelis, J. (2002) Curr. Biol. 12, 79-84. [DOI] [PubMed] [Google Scholar]
- 28.Pantaloni, D., Hill, T. L., Carlier, M. F. & Korn, E. D. (1985) Proc. Natl. Acad. Sci. USA 82, 7207-7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill, T. L. (1986) Biophys. J. 49, 981-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill, T. L. (1987) Linear Aggregation Theory in Cell Biology (Springer, New York).
- 31.Bindschadler, M., Osborn, E. A., Dewey, C. F. J. & McGrath, J. L. (2004) Biophys. J. 86, 2720-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keiser, T., Schiller, A. & Wegner, A. (1986) Biochemistry 25, 4899-4906. [DOI] [PubMed] [Google Scholar]
- 33.O'Shaughnessy, B. & Vavylonis, D. (2003) Phys. Rev. Lett. 90, 118301. [DOI] [PubMed] [Google Scholar]
- 34.O'Shaughnessy, B. & Vavylonis, D. (2003) Eur. Phys. J. E 12, 481-496. [DOI] [PubMed] [Google Scholar]
- 35.Littlefield, R. & Fowler, V. M. (2002) Nat. Cell Biol. 4, E209-E210. [DOI] [PubMed] [Google Scholar]
- 36.Kudryasov, D. S., Phillips, M. & Reisler, E. (2004) Biophys. J. 87, 1136-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinosian, H. J., Selden, L. A., Estes, J. E. & Gershman, L. C. (1993) J. Biol. Chem. 268, 8683-8691. [PubMed] [Google Scholar]
- 38.Drenckhahn, D. & Pollard, T. D. (1986) J. Biol. Chem. 261, 12754-12758. [PubMed] [Google Scholar]
- 39.Bortz, A. B., Kalos, M. H. & Lebowitz, J. L. (1975) J. Comput. Phys. 17, 10-18. [Google Scholar]
- 40.Gillespie, D. T. (1977) J. Phys. Chem. 81, 2340-2361. [Google Scholar]
- 41.Hill, T. L. & Chen, Y.-D. (1984) Proc. Natl. Acad. Sci. USA 81, 5772-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fisher, M. E. (1984) J. Stat. Phys. 34, 667-729. [Google Scholar]
- 43.Brenner, S. L. & Korn, E. D. (1983) J. Biol. Chem. 258, 5013-5020. [PubMed] [Google Scholar]
- 44.Rickard, J. E. & Sheterline, P. (1986) J. Mol. Biol. 191, 273-280. [DOI] [PubMed] [Google Scholar]
- 45.Wanger, M. & Wegner, A. (1987) Biochim. Biophys. Acta 915, 105-113. [DOI] [PubMed] [Google Scholar]
- 46.Weber, A., Pennise, C. R. & Fowler, V. M. (1999) J. Biol. Chem. 274, 34637-34645. [DOI] [PubMed] [Google Scholar]
- 47.Carlier, M.-F., Pantaloni, D., Evans, J. A., Lambooy, P. K., Korn, E. D. & Webb, M. R. (1988) FEBS Lett. 235, 211-214. [DOI] [PubMed] [Google Scholar]
- 48.Dickinson, R. B., Caro, L. & Purich, D. L. (2004) Biophys. J. 87, 2838-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otterbein, L. R., Graceffa, P. & Dominguez, R. (2001) Science 293, 708-711. [DOI] [PubMed] [Google Scholar]
- 50.Belmont, L. D., Orlova, A., Drubin, D. G. & Egelman, E. H. (1999) Proc. Natl. Acad. Sci. USA 96, 29-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill, T. L. & Carlier, M.-F. (1983) Proc. Natl. Acad. Sci. USA 80, 7234-7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.