

Abstract
Traditional asymmetric PCR uses conventional PCR primers at unequal concentrations to generate single-stranded DNA. This method, however, is difficult to optimize, often inefficient, and tends to promote nonspecific amplification. An alternative approach, Linear-After-The-Exponential (LATE)-PCR, solves these problems by using primer pairs deliberately designed for use at unequal concentrations. The present report systematically examines the primer design parameters that affect the exponential and linear phases of LATE-PCR amplification. In particular, we investigated how altering the concentration-adjusted melting temperature (Tm) of the limiting primer (TmL) relative to that of the excess primer (TmX) affects both amplification efficiency and specificity during the exponential phase of LATE-PCR. The highest reaction efficiency and specificity were observed when TmL - TmX ≥ 5°C. We also investigated how altering TmX relative to the higher Tm of the double-stranded amplicon (TmA) affects the rate and extent of linear amplification. Excess primers with TmX closer to TmA yielded higher rates of linear amplification and stronger signals from a hybridization probe. These design criteria maximize the yield of specific single-stranded DNA products and make LATE-PCR more robust and easier to implement. The conclusions were validated by using primer pairs that amplify sequences within the cystic fibrosis transmembrane regulator (CFTR) gene, mutations of which are responsible for cystic fibrosis.
Keywords: asymmetric PCR, primer melting temperature, quantitative PCR
Asymmetric PCR, as first described by Gyllensten and Erlich (1), can produce single-stranded DNA for sequencing, for use as probes, or for improving detection signals in real-time PCR. Unfortunately, traditional asymmetric PCR is highly variable and often requires extensive optimization to maximize the production of specific single-stranded product and minimize nonspecific amplification (2, 3).
We recently demonstrated (4) that these problems can be understood by considering that the melting temperature (Tm) of any primer decreases when its concentration is lowered (5–7). If the concentration of a primer designed for symmetric PCR is simply lowered for use as a limiting primer, the efficiency of the resulting reaction decreases. A conceivable solution to this problem is to lower the annealing temperature of the reaction, but this, in turn, increases the likelihood of nonspecific amplification due to increased mispriming by the excess primer.
Our preferred strategy, Linear-After-The-Exponential (LATE)-PCR, uses primers that are deliberately designed for use at unequal concentrations, such that the concentration-adjusted Tm of the limiting primer (TmL) is at least as high as the concentration-adjusted melting temperature of the excess primer (TmX) (i.e., TmL - TmX ≥ 0) (4, 8). LATE-PCR begins with an exponential phase in which amplification efficiency is similar to that of symmetric PCR. Once the limiting primer is depleted, the reaction abruptly switches to linear amplification, and the single-stranded product is made for many additional thermal cycles.
Our first goal was to analyze the relationship between TmL and TmX to optimize the efficiency and specificity of the exponential phase of LATE-PCR. Amplification of the sequences of the cystic fibrosis transmembrane regulator (CFTR) gene offers an appropriate challenge, because it encodes a member of the ATP-binding cassette family of transporter proteins with conserved sequence domains (9). Indeed, we have observed high levels of nonspecific amplification when amplification conditions are not stringent (unpublished data). The results presented here demonstrate that high efficiency and specificity are achieved when TmL - TmX ≥ 5°C.
The second goal was to determine how to maximize the rate and amount of single-strand synthesis during the linear amplification phase of LATE-PCR. The accumulating single-stranded products have no complementary strands and are therefore free to hybridize to a probe. These extension products of the excess primer, however, do compete with the excess primer itself for hybridization to the template strand. Therefore, the rate and extent of single-strand synthesis are governed by the relative affinities of the excess primer and the single-stranded product for the same template strand. The experiments presented here demonstrate that modifying the primers to increase TmX and TmL relative to the Tm of the amplicon (TmA) greatly increases the amount of single-stranded DNA that is generated over the course of a reaction and, in turn, increases the sensitivity of LATE-PCR assays.
Materials and Methods
Primer Design and Tm Calculations. Primers were designed to amplify the sequences at the F508 region of the CFTR gene. All of the limiting primers had the same sequence at their 3′ end to ensure that potential mispriming sites and primer dimer formation would be similar for all tested pairs. Table 1 lists the primers tested and their concentration-adjusted Tm values, as determined by the nearest-neighbor formula (10):
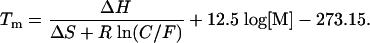 |
Table 1. Primer sequences and melting temperatures.
| Designation | Sequence | Tm |
|---|---|---|
| Series A and B | ||
| Limiting primers | ||
| CF403 S18 | GATTATGCCTGGCACCAT | 51 |
| CF402 S19 | GGATTATGCCTGGCACCAT | 54 |
| CF401 S21 | CTGGATTATGCCTGGCACCAT | 57 |
| CF399 S22 | CCTGGATTATGCCTGGCACCAT | 59 |
| CF398 S23 | TCCTGGATTATGCCTGGCACCAT | 61 |
| CF396 S25 | TTTCCTGGATTATGCCTGGCACCAT | 61 |
| Excess primer | ||
| CF479 A20 | TGATGACGCTTCTGTATCTA | 54 |
| Series C | ||
| Limiting primers | ||
| CF402 S19t | TGATTATGCCTGGCACCAT | 53 |
| CF401 S20 | TGGATTATGCCTGGCACCAT | 56 |
| CF399 S22 | CCTGGATTATGCCTGGCACCAT | 59 |
| CF392 S29 | CAGTTTTCCTGGATTATGCCTGGCACCAT | 64 |
| Excess primers | ||
| CF475 A16 | GACGCTTCTGTATCTA | 47 |
| CF477 A18 | ATGACGCTTCTGTATCTA | 50 |
| CF479 A20 | TGATGACGCTTCTGTATCTA | 54 |
| CF483 A24 | GCTTTGATGACGCTTCTGTATCTA | 59 |
The thermodynamic values ΔH and ΔS are calculated according to Allawi and SantaLucia (11), and the salt correction is that of SantaLucia et al. (6), with M equal to the total concentration of monovalent cations. These calculations are among the most accurate for estimating oligo Tm (7, 12). The Tm calculations can be made by using the melting program available at http://bioweb.pasteur.fr/seqanal/interfaces/melting.html.
For long oligonucleotides, the helix growth steps dominate the helix initiation step, producing a pseudo-first-order equilibrium for which no concentration effect is observed (5). Therefore, good estimates of amplicon Tm are obtained by using a “%GC” formula (13):
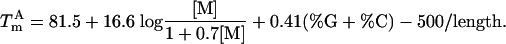 |
The above formulas, although accurate for oligonucleotides in buffers containing monovalent salts, underestimate the Tm in PCR buffers that contain magnesium but provide valuable comparisons for designing amplification reactions.
PCR Conditions. PCR samples included 1,000 nM excess primer, 50 nM limiting primer, 0.4 mM each dNTP, 3.5 mM MgCl2, 0.06 units/μl Platinum TaqDNA Polymerase (Invitrogen), 1× PCR buffer [20 mM Tris·HCl (pH 8.4)/50 mM KCl], 1× Additive Reagent (1 mg/ml BSA/750 mM trehalose/1% Tween-20), 600 pg of human genomic DNA (Coriell Cell Repositories, Camden, NJ), and either 0.2× SYBR green I (Molecular Probes) or 1 μM molecular beacon (8) in a 25-μl volume. Amplification and fluorescence detection were carried out in a Smart Cycler (Cepheid, Sunnyvale, CA).
For samples containing SYBR green, the choice of annealing temperatures was made based on preliminary tests of several LATE-PCR primer pairs (20:1 ratio) with matched Tm. An annealing temperature 2°C below the primer Tm generally yielded the earliest possible detection (lowest CT value) without the generation of nonspecific product detectable on postamplification melting curves (14) (data not shown). Therefore, one series of amplifications was done by using an annealing temperature that was 2°C below TmX, 52°C for all samples, and a second series of amplifications was done by using an annealing temperature that was 2°C below each TmL. An initial denaturation step of 3 min at 95°C was followed by 60 or 75 cycles of 95°C for 5 sec, 52°C (or other specified annealing temperature) for 15 sec, and 72°C for 15 sec with fluorescence acquisition. Product specificity was assessed by electrophoresis through agarose gels or by melting curve analysis (14). Detection threshold for determining CT values was set at 10 standard deviations above background fluorescence. Fluorescence values of different experiments were normalized to the same average final fluorescence to compensate for experimental variation in SYBR green fluorescence intensity.
For Series C samples containing the molecular beacon, an initial denaturation step of 3 min at 95°C was followed by 25 cycles of 95°C for 5 sec, TmL - 2°C for 15 sec and 72°C for 15 sec, then an additional 50 cycles of 95°C for 5 sec, 52°C for 15 sec with fluorescence acquisition, and 72°C for 15 sec. Detection threshold for determining CT values was set at 10 standard deviations above background fluorescence.
The experiments testing the quantification of different starting concentrations of genomic DNA were carried out in an Applied Biosystems PRISM 7700 Sequence Detector. Reagents were the same as described above, except that the additive reagent was omitted. Thermal cycling included an initial denaturation step of 2 min at 95°C, followed by 15 cycles of 95°C for 5 sec, 62°C (TmL - 2°C) for 15 sec, and 72°C for 15 sec, then an additional 25 cycles with those three temperature steps plus a detection step at 52°C for 15 sec. The use of a detection step after the extension step enables specific detection of accumulating single-stranded amplicon. Background fluorescence in the initial detection cycle of each sample was normalized to the same average fluorescence. The fluorescence values of all subsequent cycles of a given sample were also multiplied by the same factor (average background divided by sample background) to compensate for technical variations in fluorescence detection. This method of analysis yields more reproducible results than simply subtracting background fluorescence.
Relative Amplification Efficiency Calculations. Amplification efficiencies were compared by using calculations based on the equation Rn = R0 · (1 + E)n, where R0 and Rn are reporter fluorescence at the start of the reaction and after n cycles, and E is the efficiency of the reaction (15). Because R0 and Rn are the same for each of the reactions presented if n is set to the CT value, it follows that (1 + EA)CTA = (1 + EB)CTB for two reactions, A and B. Therefore, EA = 10CTB/CTA Log (1 + EB)) - 1. The relative efficiencies of reactions A and B are determined by setting EB to the maximum value of 1 for cases where CTB < CTA.
Statistical Analysis. Sample CT values for each of the different amplification conditions were compared by using the F test for sample variance. Mean CT values and final fluorescence were compared by using Student's t test.
Results
Effect of Primer Tm Differences on Amplification Efficiency and Specificity. Real-time PCR permits a clear comparison of amplification efficiencies among reactions that contain the same number of starting target molecules but different pairs of primers. Under these conditions, reactions with the higher amplification efficiency generate signals that become detectable in fewer cycles, i.e., have lower CT values. In the first sets of experiments, we focus on the conditions for achieving efficient amplification during the exponential phase of asymmetric PCR. Exponential amplification can be monitored by using the fluorescent dye SYBR green I, which intercalates double-stranded DNA but does not detect the accumulation of single-stranded DNA during the linear phase of LATE-PCR.
We previously demonstrated that amplification is inefficient when TmL is below TmX (4). Fig. 1A confirms that result and shows the relative amplification efficiencies of primer pairs with Tm differences over a wider range. Each curve depicts the mean fluorescence increase for replicate samples at each tested value of TmL - TmX, using one of six different limiting primers (always at 50 nM) with the same excess primer (always at 1,000 nM) (Table 1). For this set of samples (Series A), the annealing temperature was always 52°C, chosen based on preliminary tests with the excess primer (see Materials and Methods). The fluorescence increase occurred latest for samples with TmL - TmX equal to -3 (i.e., limiting primer with lower Tm). Detectable fluorescence increased earlier with TmL - TmX equal to 0 (primers of equal Tm)or +3 (limiting primer with higher Tm) and earliest with that value equal to +5 or +7.
Fig. 1.

Detection of double-stranded DNA by SYBR green fluorescence during asymmetric PCR reveals differences in exponential amplification efficiency with different limiting primers. Curves show the mean fluorescence increase in replicate samples and are colored to indicate the value of TmL - TmX: -3, red; 0, green; +3, orange, +5, blue; +7, purple. The dashed line indicates thresholds for determining CT values. The starting template was 600 pg of human genomic DNA in each sample. (A) Series A amplifications used an annealing temperature of 52°C, which is 2°C below TmX. (B) Series B amplifications used annealing temperature 2°C below TmL, shown in Table 1.
The Series A differences in real-time kinetics were quantified by calculating their mean CT values (Table 2). Each increase in TmL - TmX yielded a statistically significant decrease in mean CT value (P < 0.05), with the exception of the 0 and +3 primer combinations. The difference between the samples with negative values and those with the highest positive values of TmL - TmX was almost four cycles, a difference in amplification efficiency of ≈15%. Melting analysis confirmed that the CFTR-specific amplicon was the major product in each sample, including those in which TmL - TmX was equal to +5 or +7. Substantial levels of nonspecific products were present in only 4 of the 49 total samples, including 1 of 15 samples in the +5 and +7 groups.
Table 2. Effect of TmL - TmX differences on double-strand DNA amplification efficiency.
| Series A
|
Series B
|
|||||
|---|---|---|---|---|---|---|
| Primer TmL - TmX | n | CT | E | n | CT | E |
| -3 | 9 | 35.6 ± 1.1 | 0.85 | 13 | 34.7 ± 0.5 | 0.93 |
| 0 | 19 | 34.6 ± 0.9 | 0.89 | 19 | 34.6 ± 0.9 | 0.94 |
| +3 | 6 | 34.4 ± 0.6 | 0.89 | 6 | 34.9 ± 0.9 | 0.93 |
| +5 | 9 | 32.4 ± 0.5 | 0.97 | 13 | 33.2 ± 0.3 | 0.99 |
| +7 | 6 | 31.7 ± 0.3 | 1 | 6 | 33.0 ± 0.4 | 1 |
CT is expressed as mean ± standard deviation. Series A anneals at 2 degrees below TmX; Series B anneals at 2 degrees below TmL. Data for TmL - TmX = 0 are the same for each series. E, relative amplification efficiency.
Series B (Fig. 1B) used the same primer pairs as Series A but annealing temperatures that were always 2°C below the TmL of the particular limiting primer in the reaction. As in Series A, the mean CT values when TmL - TmX was +5 or +7 were significantly lower than when TmL - TmX equaled +3, 0, or -3 (P < 0.01). Moreover, melting analysis revealed that samples with TmL - TmX equal to -3 included varying amounts of nonspecific products that contributed to the fluorescence increase and masked a relatively low amplification efficiency.
The variance in CT values among replicate reactions decreased as TmL - TmX increased. In Series B, the variance for samples in which TmL - TmX = +5 was significantly smaller than for groups having lower TmL - TmX values (P < 0.01, P < 0.001, and P < 0.05 for comparisons with sample groups having TmL - TmX values of +3, 0, and -3, respectively). These differences in the reliability of replicate reactions suggest that quantification of the amount of starting DNA will be more accurate when TmL values are relatively high.
Gel electrophoresis of the reaction products of Series B confirmed that the higher annealing temperature for samples with a TmL - TmX value of +5 enhanced amplification specificity (Fig. 2). In contrast, nonspecific products were common in samples with TmL - TmX values of -3 and annealing temperature of 49°C. Even in samples with a 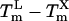 value of 0, one sample contained high levels of nonspecific product, and others showed low levels.
value of 0, one sample contained high levels of nonspecific product, and others showed low levels.
Fig. 2.

Amplification products following electrophoresis through a 3% agarose gel and ethidium bromide staining. Numbers above the bars indicate TmL - TmX values for each set of replicate samples. Annealing temperature in each case was 2°C below TmL, and starting template was 600 pg of human genomic DNA. Specific CFTR product is the lowest band in each lane, 77–81 nucleotides. Double- and single-stranded products were not resolved on this gel. S, 50-bp DNA ladder size standards. Lengths are shown at left.
Effect of Amplicon and Excess Primer Tm Difference on the Rate and Extent of Linear Amplification. A separate set of experiments (Series C) was carried out to test how the difference between TmA and TmX affects the rate and extent of single-strand synthesis during the linear amplification phase of LATE-PCR. Reactions included a molecular beacon to monitor CFTR-specific single-stranded DNA synthesis. Primer pairs were designed so that TmL - TmX = +5° to +6°C and are listed in Table 1. Fig. 3 shows the molecular beacon fluorescence for samples with TmA - TmX ranging from +13 to +23. Mean CT values and rates of signal increase (line slopes) are presented in Table 3. The greatest fluorescence increase was generated by samples with TmA - TmX = 13. The rate and extent of fluorescence increase were progressively lower as the value of TmA - TmX increased. Final fluorescence levels were significantly different for each of the primer pairs tested (P < 0.01). Three pairs of primers yielded similar CT values, suggesting that exponential amplification efficiency was similar in each case, and that the observed differences in subsequent fluorescence increase were almost exclusively due to differences in the rate of linear amplification. Thus, the rate of linear amplification increases as the difference between TmA and TmX decreases, so long as the difference between TmL and TmX is maintained.
Fig. 3.

Detection of CFTR-specific product during the linear amplification phase of LATE-PCR. Curves show increases in molecular beacon fluorescence in individual samples with TmA - TmX values of 13 (green), 17 (blue), 20 (red), or 23 (orange). Starting template was 600 pg of human genomic DNA in each sample.
Table 3. Effect of TmA - TmX differences on CFTR molecular beacon signals.
| Limiting primer | TmL | Excess primer | TmX | TmA | TmA - TmX | CT | Early slope (cycles 45–60) | Late slope (cycles 60–75) |
|---|---|---|---|---|---|---|---|---|
| CF402 S19t | 53 | CF475 A16 | 47 | 70 | 23 | 43.9 ± 2.7 | 2.3 | 3.6 |
| CF401 S20 | 56 | CF477 A18 | 50 | 70 | 20 | 39.2 ± 0.7 | 4.2 | 3.8 |
| CF399 S22 | 59 | CF479 A20 | 54 | 71 | 17 | 39.1 ± 0.3 | 7.6 | 5.7 |
| CF392 S29 | 64 | CF483 A24 | 59 | 72 | 13 | 38.4 ± 0.4 | 11.3 | 7.6 |
Quantifying Starting Template Concentration Using Optimized LATE-PCR. Real-time symmetric PCR is commonly used to measure the initial template concentration in a sample through the comparison of CT values. Fig. 4A shows that real-time LATE-PCR conveys the same quantitative information with low variation among replicate samples. Samples containing 10–100,000 copies of human genomic DNA plus the best primer pair from Series C generated fluorescent signals that reached threshold in relation to that starting concentration. Each 10-fold decrease in starting template concentration delayed detection ≈3.3 cycles, as expected for efficient exponential amplification. In contrast to symmetric PCR, plots from LATE-PCR samples neither diverged nor plateaued at different fluorescent values. Instead, fluorescence increased linearly and uniformly over several cycles. Thus, fluorescent values at later cycles continued to convey quantitative information. Fig. 4B shows a standard curve for starting template concentration based on the fluorescent values at cycle 40. It should be noted that the analysis did not require setting a baseline over several cycles, as is typical for standard real-time analysis. Instead, background fluorescence in the different samples was normalized by using values from the first detection cycle. Thus, where real-time detection is unavailable, it may still be possible to quantify the starting template by measuring the final amplicon concentration in LATE-PCR reactions using fluorescent probes or other detection methods.
Fig. 4.

Detection of CFTR-specific product in samples containing different initial concentrations of DNA. (A) Optimized LATE-PCR was carried out by using 100,000 (red), 10,000 (green), 1,000 (orange), 100 (blue), and 10 (purple) copies of human genomic DNA. Curves show molecular beacon fluorescence increase in eight replicate samples at each starting template concentration. (B) Plots of initial DNA concentration vs. cycle 40 fluorescence demonstrates the quantitative nature of these endpoint values. R2 = 0.974
Discussion
The efficiency and specificity of double-stranded DNA amplification during the exponential phase of asymmetric PCR depend on the relative Tm of the limiting and excess primers and the annealing temperature of the reaction. This report confirms that reactions in which TmL is below TmX and the annealing temperature is set in relation to TmX, as is generally the case for traditional asymmetric PCR primers, have lower efficiencies than reactions in which TmL equals TmX (4). The present findings also demonstrate that increasing TmL above TmX can increase efficiency and specificity even further. The improved specificity is of particular importance for applications intended to generate useful quantities of single-stranded DNA.
A priori, it seemed possible that increasing TmL well above the annealing temperature might reduce amplification specificity. Our results demonstrate, however, that setting TmL at 7°C or 9°C above the annealing temperature did not increase the amount of nonspecific product, and amplification efficiency was improved by the use of those limiting primers, as evidenced by a decrease in mean CT values. In contrast, a large amount of nonspecific product was generated when TmX was only 5°C above the annealing temperature. Thus, one advantage of having TmL > TmX is that the true optimal annealing temperature for both primers can be used, one sufficiently low relative to TmL to allow for efficient utilization of the limiting primer but sufficiently high relative to TmX to limit amplification of nonspecific products by the excess primer.
The use of primer pairs with TmL above TmX is likely to improve amplification efficiency for many target sequences, although the optimal Tm difference between limiting and excess primers may vary with the target and primer ratio. Similar tests amplifying the β-hexosaminidase gene (mutations of which are responsible for Tay-Sachs disease) with primer ratios of 40:1 show that increasing TmL - TmX from 0 to +4 increases amplification efficiency while maintaining specificity (unpublished data). We have incorporated the use of positive TmL - TmX values as a general strategy for LATE-PCR primer design.
Improving the rate and extent of single-stranded DNA synthesis can be accomplished by reducing the difference between TmA and TmX. Presumably, this relates to the ability of the excess primer molecules to hybridize to their target strand in the presence of an increasing concentration of competing single-stranded product. CFTR excess primers with calculated TmX within 20°C of TmA generated reasonable rates of single-stranded synthesis, with the best rates obtained when that difference was 13°C. We have also obtained good results amplifying sequences within the β-hexosaminidase and β-globin genes when these limits are maintained (ref. 4 and unpublished results). Those amplicons have higher TmA than the CFTR amplicon generated here and thus require primers with higher TmX and TmL, suggesting this rule can be generally applied to other asymmetric amplifications. It may be difficult, however, to design primers that meet this criterion for amplicons that are long or GC-rich. In such cases, achieving good yields may require altering the primers or amplicon in other ways, for example, by incorporating nucleotide analogs that lower TmA. It is also important to recognize that other factors besides primer Tm may influence the extent of linear amplification. For instance, subtle changes in the 5′ end of the limiting or excess primer can have a profound effect on the rate of single-strand production, even when those changes have minor effects on primer Tm.
One of the significant advantages of LATE-PCR over symmetric PCR is the increased intensity and reproducibility of real-time signals. In LATE-PCR, the concentration of the limiting primer is chosen such that it becomes depleted just as the product becomes detectable with a labeled probe. In contrast to symmetric PCR, the probe signals in LATE-PCR continue to increase at similar rates in all samples, and the highly variable reaction plateau is avoided. Like symmetric PCR, LATE-PCR is able to quantify the initial targets in a sample using CT values, something that would be difficult or impossible using traditional methods of asymmetric PCR. In fact, there are few publications in which hybridization probes have been used with unequal concentrations of primers; most used low primer ratios insufficient to sustain linear amplification (16–19).
The highly reproducible linear increase in single-stranded product means that LATE-PCR end-point analysis can be used to quantify starting template concentrations. Quantification is possible for at least 5 orders of magnitude in starting template but depends on primer concentrations, reaction optimization, and number of cycles. For some applications, end-point analysis may provide an alternative to expensive real-time detection equipment and licensing fees.
Another advantage of LATE-PCR is the flexibility that it provides for detection methods. It is worth noting that the same molecular beacon probe was used for all samples, despite the use of primers with Tm over a wide range. This is not possible with symmetric PCR, because probes must be designed with a relatively high Tm to ensure that probe hybridization during the annealing step occurs before primer hybridization and extension. In contrast, LATE-PCR provides single-stranded product that is freely available to hybridize with the probe at whatever temperature is appropriate for that probe. This permits the use of hybridization probes with Tm below the primer Tm. Advantages of using probes with relatively low Tm include simplified design, lowered background signal, and increased allele discrimination (4). These improvements are accrued by using any type of probe that emits a signal upon hybridization.
Conclusion
LATE-PCR establishes straightforward primer design criteria for generating high yields of single-stranded DNA products without extensive reaction optimization. LATE-PCR effectively eliminates the need for multiple rounds of amplification or laborious steps for purifying single-stranded DNA. When combined with real-time or end-point detection using low-Tm probes, this method offers unique potential in forensics, bioweapons detection, and other fields where assay sensitivity and consistency are essential.
Acknowledgments
We thank Dr. Fred Kramer for thought-provoking discussions and encouragement. Brandeis University provided financial support for this research.
Author contributions: K.E.P., J.A.S., J.E.R., and L.J.W. designed research; K.E.P. performed research; K.E.P., J.A.S., J.E.R., and L.J.W. analyzed data; and K.E.P. and L.J.W. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LATE-PCR, Linear-After-The-Exponential PCR; Tm, melting temperature; TmL, limiting primer Tm; TmX, excess primer Tm; TmA, amplicon Tm;CT, threshold cycle; CFTR, cystic fibrosis transmembrane regulator.
References
- 1.Gyllensten, U. B. & Erlich, H. A. (1988) Proc. Natl. Acad. Sci. USA 85, 7652-7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCabe, P. C. (1990) in PCR Protocols: A Guide to Methods and Applications, eds. Innis, M. A., Gelfand, D. H., Sninsky, J. J. & White, T. J. (Academic, New York), pp. 76-83.
- 3.Gyllensten, U. B. & Allen, M. (1993) Methods Enzymol. 218, 3-16. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez, J. A., Pierce, K. E., Rice, J. E. & Wangh, L. J. (2004) Proc. Natl. Acad. Sci. USA 101, 1933-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breslauer, K. J. (1986) in Thermodynamic Data for Biochemistry and Biotechnology, ed. Hinz, H. (Springer, New York), pp. 402-427.
- 6.SantaLucia, J., Allawi, H. T. & Seneviratne, P. A. (1996) Biochemistry 35, 3555-3562. [DOI] [PubMed] [Google Scholar]
- 7.SantaLucia, J. (1998) Proc. Natl. Acad. Sci. USA 95, 1460-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pierce, K. E., Rice, J. E., Sanchez, J. A. & Wangh, L. J. (2003) Mol. Hum. Reprod. 9, 815-820. [DOI] [PubMed] [Google Scholar]
- 9.Welsh, M. J., Tsui. L-P., Boat, T. F. & Beaudet, A. L. (1995) in The Metabolic and Molecular Basis of Inherited Disease, eds. Scriver, C. R., Beaudet, A. L., Sly, W. S. & Valle, D. (McGraw–Hill, New York), Vol. III, pp. 3799-3876. [Google Scholar]
- 10.Le Novère, N. (2001) Bioinformatics 17, 1226-1227. [DOI] [PubMed] [Google Scholar]
- 11.Allawi, H. T. & SantaLucia, J. (1997) Biochemistry 36, 10581-10594. [DOI] [PubMed] [Google Scholar]
- 12.Owczarzy, R., Vallone, P. M., Gallo, F. J., Paner, T. M., Lane, M. J. & Benight, A. S. (1998) Biopolymers 44, 217-239. [DOI] [PubMed] [Google Scholar]
- 13.Wetmur, J. G. (1991) Crit. Rev. Biochem. Mol. Biol. 26, 227-259. [DOI] [PubMed] [Google Scholar]
- 14.Ririe, K. M., Rasmussen, R. P. & Wittwer, C. T. (1997) Anal. Biochem. 245, 154-160. [DOI] [PubMed] [Google Scholar]
- 15.Liu, W. & Saint, D. A. (2002) Anal. Biochem. 302, 52-59. [DOI] [PubMed] [Google Scholar]
- 16.Lay, M. J. & Wittwer, C. T. (1997) Clin. Chem. 43, 2262-2267. [PubMed] [Google Scholar]
- 17.Nurmi, J., Ylikoski, A., Soukka, T., Karp, M. & Lovgren, T. (2000) Nucleic Acids Res. 28, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Millward, H., Samowitz, W., Wittwer, C. T. & Bernard, P. S. (2002) Clin. Chem. 48, 1321-1328. [PubMed] [Google Scholar]
- 19.Afonina, I. A., Reed, M. W., Lusby, E., Shishkina, I. G. & Belousov, Y. S. (2002) BioTechniques 32, 940-949. [DOI] [PubMed] [Google Scholar]