

Abstract
The ETS-domain transcription factor Elk-1 is a MAP kinase-inducible transcriptional activator protein. However, in the basal state, its activity is repressed by SUMO-dependent histone deacetylase (HDAC) recruitment. Relief of this repression accompanies the activation process. Here, we demonstrate that PIASxα acts to facilitate this derepression process. Members of the PIAS family of proteins can act as E3 enzymes that enhance the sumoylation status of a variety of substrates. However, PIASx-mediated coactivation of Elk-1 occurs in an E3 activity-independent manner. PIASxα binds to Elk-1 in vivo and enhances its transcriptional activity. The coactivating properties of PIASxα require Elk-1 to be modified with SUMO and the integrity of the SUMO binding motif in PIASxα. PIASxα activates Elk-1 through alterations in the HAT/HDAC activities associated with Elk-1. In particular, PIASxα facilitates the loss of the repressive HDAC-2 from sumoylated Elk-1, a key event in the activation of Elk-1 in response to signalling through the ERK MAP kinase pathway. Our data therefore reveal a novel coactivator function for PIASxα through reversing SUMO-mediated repression of transcription factor activity.
Keywords: Elk-1, HDAC-2, PIAS, SUMO, transcriptional coactivator
Introduction
The modification of proteins by SUMO has recently been shown to play an important role in regulating the activities of a variety of transcriptional regulatory proteins (reviewed in Gill, 2003, 2004; Verger et al, 2003). Generally, protein modification by SUMO imparts repressive properties on these target proteins. Recent studies are beginning to unravel the molecular basis to this repression, and in the case of p300 and Elk-1, sumoylation has been shown to be important for the recruitment of the histone deacetylases HDAC-6 and HDAC-2, respectively (Girdwood et al, 2003; Yang and Sharrocks, 2004). Furthermore, histone sumoylation has been associated with histone deacetylase recruitment and transcriptional repression (Shiio and Eisenman, 2003). In other cases such as SP3, sumoylation-dependent changes in subnuclear localisation are thought to contribute to its repressive properties (Ross et al, 2002).
The ETS-domain transcription factor Elk-1 acts together in a complex with the transcription factor SRF and is rapidly activated by ERK MAP kinase-mediated phosphorylation of its transcriptional activation domain (TAD) in response to mitogenic stimulation (reviewed in Sharrocks, 2002; Shaw and Saxton, 2003). This modification correlates with the rapid induction of genes, including c-fos, egr-1, mcl-1 and srf (reviewed in Townsend et al, 1999; Sharrocks, 2002; Shaw and Saxton, 2003; Vickers et al, 2004; Kasza et al, 2005). In addition to a TAD, Elk-1 contains two repression domains, one of which, known as the R-motif contains the sites for SUMO modification (Yang et al, 2003; Salinas et al, 2004). This SUMO modification is lost upon activation of the MAP kinase pathway and this loss contributes to the stimulation of the transactivation potential of Elk-1 caused by this pathway (Yang et al, 2003). Thus, Elk-1 sumoylation plays an important role in maintaining Elk-1 in a repressed state and has also been shown to reduce its rate of nucleocytoplasmic shuttling (Salinas et al, 2004).
Protein sumoylation is mediated by a pathway consisting of an E1 SUMO activating enzyme, SAE1/2, and an E2 SUMO conjugating enzyme, Ubc9 (reviewed in Hay, 2001; Muller et al, 2001). These enzymes are sufficient for substrate sumoylation in vitro. However, recently, a series of proteins have been identified that appear to act to promote substrate sumoylation in an analogous manner to the E3 ligases found in the ubiquitin pathway. These include Pc2 (Kagey et al, 2003), RanBP2 (Kirsh et al, 2002) and members of the PIAS family (reviewed in Schmidt and Muller, 2003). The PIAS family is characterised by the presence of a RING finger motif and includes PIAS1, PIAS3, PIASy, PIASxα/β and hZimp10 (reviewed in Schmidt and Muller, 2003). It is the RING finger motif that imparts E3 ligase activity on the PIAS proteins (Kahyo et al, 2001; Kotaja et al, 2002; Schmidt and Muller, 2002). In addition, PIAS proteins contain a SAP domain (reviewed in Aravind and Koonin, 2000) and a small motif required for noncovalent binding to SUMO (Kotaja et al, 2002; Song et al, 2004). Enhanced SUMO modification mediated by PIAS proteins often leads to further reductions in transcription factor activity as demonstrated for PIASxβ on p53 (Schmidt and Muller, 2002). Furthermore, PIAS proteins were first identified by their ability to inhibit the transcriptional activity of STAT proteins (Chung et al, 1997) and this repression needs the sumoylation activity of the PIAS proteins (Rogers et al, 2003; Ungureanu et al, 2003). However, in some cases, PIAS proteins can activate transcription factors as exemplified by the effects of PIASy on Tcf-4 (Yamamoto et al, 2003) and Zimp10 on the androgen receptor (Sharma et al, 2003). Again, in both cases, activation is thought to be due to the SUMO ligase activity of the PIAS proteins. However, in the case of p53 activation by PIAS1, the SUMO ligase activity is not required (Megidish et al, 2002). In addition, PIAS proteins have been shown to affect transcription factor function through altering their subnuclear localisation as shown for the action of PIASy on LEF1 (Sachdev et al, 2001). The sumoylation activity of PIASy is again important for this process, although the SUMO acceptor sites on LEF1 are not required.
In this study, we have investigated the effect of PIAS proteins on the activity of Elk-1. Instead of the expected role in promoting transcriptional repression through enhanced Elk-1 sumoylation, we demonstrate that PIASxα acts as a coactivator protein. This coactivator activity is independent of its E3 ligase activity but still requires Elk-1 to be SUMO modified. Molecularly, PIASxα activates Elk-1 through altering the HDAC/HAT complexes bound to Elk-1 and facilitating the removal of histone deacetylases.
Results
PIASx proteins upregulate the activity of SUMO-modified Elk-1
Sumoylation of Elk-1 has been shown to repress its transactivation capacity (Yang et al, 2003). To begin to probe the potential role of E3 ligases in establishing the repressive state in Elk-1, we first examined the role of PIASx proteins in regulating Elk-1 activity by depletion using RNA interference (RNAi). Western analysis showed that both HeLa and 293 cells expressed only a single PIASx isoform that ran as a doublet on SDS gels, whose molecular weight was consistent with being the alpha isoform (Figure 1A, and data not shown). siRNA duplexes against PIASxα knocked down the levels of the band corresponding to endogenous PIASxα in HeLa cells (typically 50–90%; Figure 1A, and data not shown) and 293 cells (data not shown), but did not affect the levels of other proteins such as HDAC-2. However, rather than the expected increase in Elk-1-mediated transcriptional activation, which should occur due to decreased sumoylation levels, reductions in PIASxα levels were accompanied by reductions in the transcriptional activity of Elk-1 in both unstimulated and phorbol 12-myristate 13-acetate (PMA)-stimulated cells (Figure 1B). However, similar decreases in activity were not seen upon knocking down PIASxα levels with the non-sumoylatable Elk-1(K2R) mutant (Figure 1C), suggesting an important role of Elk-1 sumoylation in PIASxα-mediated coactivation. In contrast, in comparison to the large decrease in Elk-1 activity observed, the activity of the VP16 TAD was only slightly affected by PIASxα depletion (Figure 1D).
Figure 1.
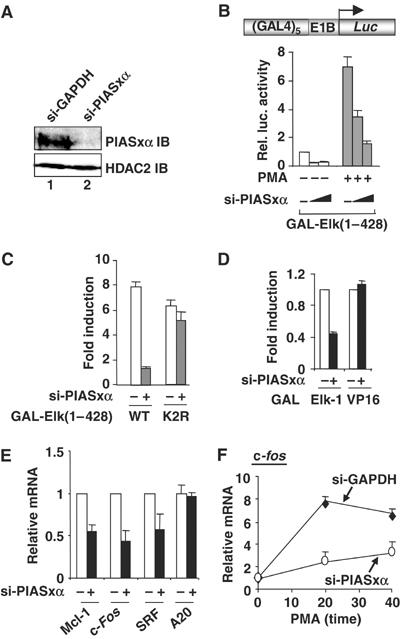
siRNAs show that endogenous PIASxα is a coactivator of Elk-1. (A) Western blot showing a reduction in PIASxα levels in the presence of specific RNAi duplexes. Total lysates from HeLa cells transfected with siRNAs against either GAPDH or PIASxα were probed with PIASxα or HDAC-2 antibodies. (B–D) Reporter gene analyses of the activities of the indicated GAL fusion proteins in 293 cells in the presence of GAPDH (−) or PIASxα RNAi duplexes. (B) The activity of GAL-Elk(1–428) in the presence or absence of PMA stimulation as indicated and siRNAs against either GAPDH (−) (100 pmol) or increasing amounts of PIASxα (50 and 100 pmol). (C) Activities of the wild-type (WT) and mutant (K2R) GAL-Elk(1–428) in the presence of PMA and cells in the presence of GAPDH (−) or PIASxα RNAi duplexes. Data are shown as relative luciferase activity relative to GAL-Elk(1–428) plus GAPDH (−) RNAi duplexes, in the absence of PMA, taken as 1 (B, C). (D) Activity of GAL-Elk(1–428) and GAL-VP16 fusion proteins in the presence of GAPDH (−) or PIASxα RNAi duplexes. Data are shown relative to the activity of each construct in the absence of RNAi duplexes against PIASxα. Luciferase assays are representative of at least two independent experiments (standard errors are shown; n=2). (E, F) Real-time RT–PCR analysis of the indicated genes in serum-starved (E) or PMA-stimulated (F) HeLa cells in the presence or absence of RNAi duplexes to either GAPDH (−) or PIASxα. PMA stimulation was for the indicated times. Data are averages of two experiments performed in duplicate.
The functional consequences of PIASxα knockdown on the activity of several Elk-1-regulated target genes were then tested. Reductions in the basal expression of mcl-1, c-fos and srf were observed upon depletion of PIASxα (Figure 1E). However, this was not a general effect, as the activity of a different gene, A20 (encoding a zinc-finger protein), was unaffected. The basal level of expression of many Elk-1 target genes is low, making substantial further decreases hard to achieve; therefore, we also examined the effect of depleting PIASxα following PMA-mediated gene activation. PMA signals through the ERK pathway to activate c-fos expression through Elk-1 (reviewed in Shaw and Saxton, 2003). Reductions in the level of c-fos transcription were again observed in the presence of siRNA against PIASxα under these conditions, consistent with a role of PIASxα in Elk-1-mediated transcriptional activation in response to mitogenic signalling (Figure 1F).
As the loss of PIASxα leads to decreased Elk-1 activity, overexpression of PIASxα would be expected to cause the opposite effect and enhance Elk-1 activity. We therefore tested PIASxα and the PIASxβ isoform for their ability to promote Elk-1-mediated transcriptional activation. We also tested additional PIAS proteins to exclude nonspecific effects that might occur through perturbation of the SUMO pathway. Both PIASxα and PIASxβ enhanced the transactivation capacity of Elk-1 (Figure 2A). In contrast, neither PIASy nor PIAS1 strongly activated Elk-1, although PIAS1 did show some activating capacity at higher concentrations. PIASxα strongly activated wild-type Elk-1 in a dose-dependent manner but, in comparison, only weakly transactivated a mutant form of Elk-1 that lacks its major SUMO modification sites (Elk-1(K2R); Figure 2B and Supplementary Figure S1). To further probe the requirement for Elk-1 sumoylation for coactivation by PIASxα, we carried out reporter gene assays in the presence of a dominant-negative version of Ubc9 (DN-Ubc9) to inhibit the SUMO pathway. Under these conditions, PIASxα was unable to potentiate Elk-1 transcriptional activity (Figure 2C). PIASxα could also potentiate Elk-1 activity under conditions where the ERK MAP kinase pathway was activated by PMA (data not shown), demonstrating that PIASxα can coactivate both basal and ‘activated' forms of Elk-1. Furthermore, fusion of SUMO to Elk-1 can permit PIASxα to transactivate an Elk-1 derivative, Elk-1(260–428), which lacks the R-motif containing the SUMO modification sites (Figure 2D).
Figure 2.
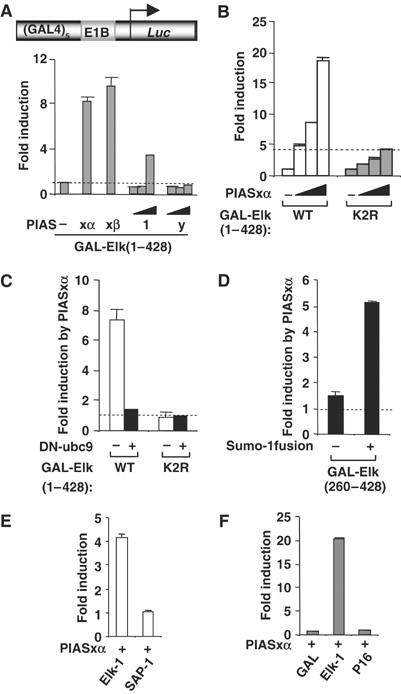
Activation of sumoylated Elk-1 by PIASxα. Reporter gene analysis of the activities of GAL-Elk fusion proteins on the GAL-driven E1b promoter-reporter construct (schematic shown in [A]) in 293 cells is shown. (A) The activity of GAL-Elk(1–428) was tested in the presence and absence of cotransfected PIASxα (0.1 μg), PIASxβ (0.1 μg), PIAS1 (0, 0.1, 0.5 and 1 μg) and PIASy (0, 0.1, 0.5 and 1 μg) as indicated and presented as fold induction of reporter activity relative to the absence of PIAS proteins. (B) The activities of the wild-type (WT) and mutant (K2R) GAL-Elk(1–428) fusion proteins were tested in the presence or absence of increasing amounts of PIASxα (0, 10, 25 and 100 ng) as indicated and presented as fold induction of reporter activity relative to the absence of PIASxα (taken as 1). For alternative depiction of data, see Supplementary Figure S1. (C) The activities of GAL-Elk(1–428) WT and K2R were tested in the presence or absence of PIASxα (0.25 μg) and/or DN-ubc9 (1 μg) as indicated. The data are presented as fold induction by PIASxα of reporter activity and are calculated from the activity of the reporter under each condition relative to the activity in the absence of PIASxα. (D) The activities of GAL-Elk(260–428) and GAL-Sumo-Elk(260–428) were tested in the presence or absence of PIASxα (0.25 μg). The data are presented as fold induction by PIASxα of reporter activity and are relative to the activity of each construct in the absence of PIASxα. (E, F) Activities of GAL-Elk(223–428), GAL-SAP-1(234–431) (E), GAL-DBD, GAL-Elk(1–428) and GAL-VP16 (F) in 293 cells in the presence or absence of transfected PIASxα. Data are shown as fold induction relative to the activity of each GAL fusion in the absence of PIASxα. Luciferase assays are representative of at least two independent experiments (standard errors are shown; n=2).
To investigate the specificity of PIASxα action, we tested its activity against the closely related SAP-1 (Figure 2E) and the unrelated VP16 (Figure 2F) TADs. The activities of both of these transcription factors were unaffected by PIASxα, demonstrating that this is not a general effect on the activity of TADs.
Collectively, these data therefore show that PIASxα can specifically upregulate the activity of Elk-1, but importantly requires Elk-1 to be sumoylated to perform this role. This is fully consistent with the opposite effects mediated by the PIASxα depletion experiments. Thus, loss-of-function and gain-of-function analyses support a role of PIASxα in enhancing the transactivation capacity of SUMO-modified Elk-1 and in promoting target gene activation.
Mapping the regions required for PIASxα-mediated coactivation of Elk-1
To establish the regions of Elk-1 required for coactivation by PIASxα, we first tested a series of truncated Elk-1 derivatives (Figure 3A). The R-motif was included in all these constructs as it is essential for PIASxα-mediated coactivation (Figure 2B and D). PIASxα was unable to potentiate the activity of the N-terminal part of Elk-1, but the coactivation function of PIASxα was retained with the C-terminal part of Elk-1 (Elk-1(205–428)) (Figure 3B). The investigation of further C-terminal truncations within the context of Elk-1(205–428) demonstrated that upon deletion of the region encompassing the Elk-1 TAD (amino acids 375–399), PIASxα-mediated Elk-1 activation was lost (Figure 3C). Thus, it appears that the intrinsic activity of the Elk-1 TAD must be intact for PIASxα-mediated coactivation. To further probe this, we investigated a series of Elk-1 point mutants, which had diminished transactivation capacity. The mutant Elk-1 proteins F378A/W379A and I376A/H377A exhibit greatly reduced transactivation capacity when activated by the ERK pathway (Price et al, 1995; data not shown) and both show greatly reduced responses to PIASxα-mediated transactivation (Figure 3D). In contrast, the more active mutants, V365A/L366A and V412A/D413A, exhibit similar responses to the wild-type protein. Thus, PIASxα appears to modify the transcriptional output from the Elk-1 TAD rather than providing an additional TAD.
Figure 3.
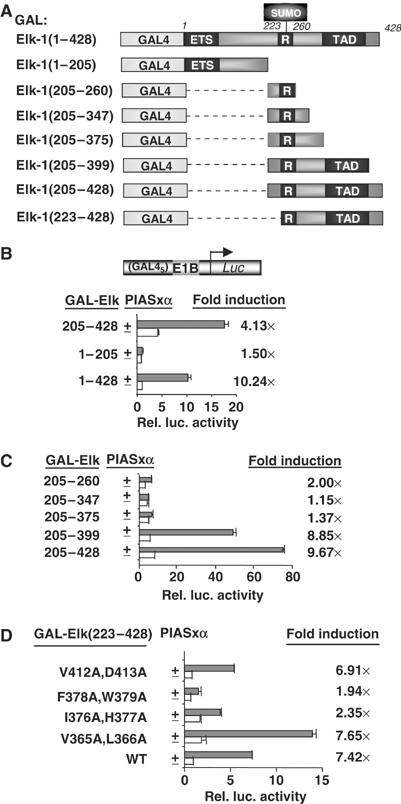
Mapping the determinants in Elk-1 for PIASxα coactivation. (A) Schematic of the GAL fusion proteins used in this figure. The Elk-1 ETS domain, R-motif and TAD are indicated in dark grey boxes. (B–D) Reporter gene analysis of activities of indicated GAL-Elk fusion proteins on the GAL-driven E1b promoter-reporter construct in 293 cells. The activities of each construct were tested in the presence or absence of transfected PIASxα and presented as relative luciferase activities (relative to activities of GAL-Elk(1–428) (B), GAL-Elk(205–428) (C) and GAL-Elk(223–428) (D) in the absence of PIASxα) and n-fold induction by PIASxα. Luciferase assays are representative of at least two independent experiments (standard errors are shown; n=2).
Elk-1 and PIASxα interact in vivo
The coactivating properties of PIASxα suggest that it interacts with Elk-1. To investigate this possibility, we carried out co-immunoprecipitation assays from 293 cells transfected with Elk-1 and PIASxα (Figure 4A). Efficient PIASxα precipitation was only observed when both proteins were overexpressed (Figure 4A, lane 4), although a weaker band can be detected with endogenous levels of Elk-1 (Figure 4A, lane 2). To demonstrate that endogenous Elk-1 and PIASxα interact, co-immunoprecipitation experiments were performed in HeLa cells. While PIASxα was efficiently precipitated with an Elk-1 antibody, only background levels were observed with normal rabbit IgG (Figure 4B). As Elk-1 sumoylation is required for PIASxα-mediated transactivation, we tested whether the SUMO binding motif on PIASxα or the SUMO modification site on Elk-1 is required for PIASxα–Elk-1 interactions. However, neither PIASxα(Δ467–487) (lacking the SUMO binding motif) (Figure 4C) nor the non-sumoylatable Elk-1(K2R) mutant (Figure 4D) was deficient in complex formation. Furthermore, cotransfection of the SUMO-specific protease Ssp3 or stimulation with PMA had no effect on complex formation (data not shown). Thus, binding of PIASxα to Elk-1 is not simply through interactions with the SUMO moiety.
Figure 4.
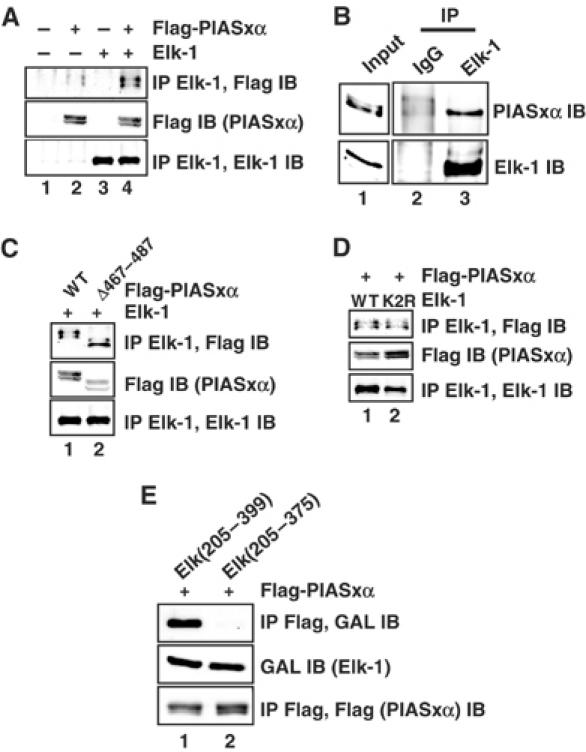
Elk-1 and PIASxα interact in vivo. (A) Co-immunoprecipitation (IP) of the indicated PIASxα proteins with Elk-1 from 293 cells. Cells were cotransfected with 2 μg of wild-type (WT) or mutant (K2R) pcDNA-Elk-1 and 3 μg of WT or mutant (Δ467–487) pFlag-PIASxα as indicated. Elk-1 proteins were immunoprecipitated with an anti-Elk-1 antibody, and co-precipitated PIASxα proteins were subsequently detected with a Flag antibody (top panel). (B) Co-immunoprecipitation of endogenous Elk-1 and PIASxα from HeLa cells. Immunoprecipitation (IP) was carried out using Elk-1 or control IgG antibodies. (C, D) Co-immunoprecipitation (IP) of the indicated PIASxα proteins with Elk-1 was carried out as in (A), except that 2 μg of mutant (K2R) pcDNA-Elk-1 and 3 μg of mutant (Δ467–487) pFlag-PIASxα were also tested as indicated. (E) Co-immunoprecipitation (IP) of the indicated GAL-Elk fusion proteins with PIASxα from 293 cells. Cells were cotransfected with 2 μg of each GAL-Elk fusion protein and 3 μg of pFlag-PIASxα. PIASxα was immunoprecipitated with an anti-Flag antibody, and co-precipitated GAL-Elk fusion proteins were subsequently detected with a GAL antibody (top panel). Immunoprecipitates (bottom panel) and total cell extracts (middle panel) were also analysed by immunoblot with the indicated antibodies to detect total levels of proteins in the immunoprecipitate and total cell lysates, respectively.
To localise the interaction surface on Elk-1, we tested two C-terminal Elk-1 deletion derivatives for PIASxα binding that contained either the intact TAD (Elk(205–399)) or a truncation thereof (Elk(205–375)). Complexes could readily be detected with Elk(205–399) but, in comparison, interactions were much weaker for Elk(205–375) (Figure 4E), demonstrating an important role of residues in the TAD (375–399) in complex formation with PIASxα. This is consistent with the reporter gene analysis, which identified this region as necessary for PIASx-mediated coactivation (Figure 3). Thus, PIASxα interacts with Elk-1 and this interaction requires the TAD of Elk-1.
Effect of PIASxα on Elk-1 sumoylation and phosphorylation status
Elk-1 transcriptional activity can be enhanced through phosphorylation of its TAD by members of the MAP kinase family (reviewed in Sharrocks, 2002; Shaw and Saxton, 2003) or by interfering with the SUMO modification pathway and loss of SUMO from the R-motif repression domain (Yang et al, 2003). We therefore investigated whether PIASxα works through either of these mechanisms. First, we examined the sumoylation status of Elk-1 in the presence of cotransfected PIASxα. However, no decreases in sumoylation levels were observed as would be expected during Elk-1 activation (Figure 5A). Next, we tested whether Elk-1 phosphorylation at the critical Ser383 residue is enhanced by PIASxα. However, PIASxα had little effect on Elk-1 phosphorylation (Figure 5B). Furthermore, PIASxα-mediated Elk-1 activation was not affected by the ERK pathway inhibitor U01260, demonstrating that PIASxα does not increase Elk-1-mediated transcription by activating the ERK pathway (Figure 5C).
Figure 5.
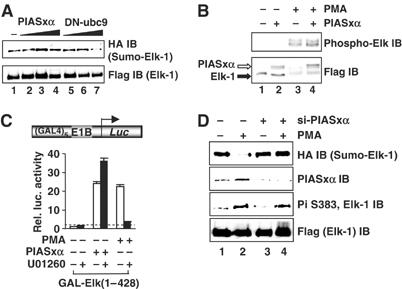
PIASxα effects on Elk-1 sumoylation and phosphorylation status. (A) In vivo sumoylation of Elk-1 in COS7 cells. The indicated His-tagged Elk-1- and HA-tagged SUMO-1-encoding constructs were cotransfected in the presence or absence of increasing amounts of PIASxα (0.25, 1 and 2 μg) or DN-ubc9 (0.25, 1 and 2 μg) plasmids. Elk-1 was isolated by nickel affinity chromatography, and SUMO-modified Elk-1 was detected with anti-HA antibody. Total Elk-1 was detected by anti-Flag antibody. (B) Western blots of extracts from cells transfected with Elk-1 and/or PIASxα in the presence or absence of PMA, probed with an anti-phospho-Ser383 Elk-1 antibody (top panel) and an anti-Flag antibody (bottom panel). (C) Reporter gene analysis of the activity of GAL-Elk(1–428) in the presence or absence of PIASxα and/or PMA and/or MEK inhibitor (U01260) on the GAL-driven E1B promoter-reporter construct in 293 cells. The data are shown relative to GAL-Elk(1–428) in the absence of PMA and PIASxα. (D) In vivo sumoylation of Elk-1 in HeLa cells. His-tagged Elk-1 and HA-tagged SUMO-1 were cotransfected in the absence and presence of PIASxα siRNAs. Cells were either serum-starved or treated with PMA for 30 min as indicated. Sumoylation and phosphorylation status were determined as in (A, B).
To establish whether PIASxα affects the modification status of activated Elk-1, we depleted PIASxα in the presence of PMA stimulation. Activation of the ERK pathway by PMA leads to enhanced Elk-1 phosphorylation and a decrease in Elk-1 sumoylation (Figure 5D, lanes 1 and 2; Yang et al, 2003). However, upon depletion of PIASxα, while increases in Elk-1 phosphorylation were unperturbed, the loss of sumoylation was inhibited (Figure 5D, lanes 3 and 4). Thus, PIASxα is unimportant for sumoylation and ERK-inducible phosphorylation of Elk-1 but plays an important role in the loss of Elk-1 sumoylation following activation of the ERK pathway. A role in promoting SUMO loss, an activating event, is consistent with our observation that PIASxα is a coactivator of Elk-1.
Mapping the regions of PIASxα involved in coactivation of Elk-1
Next, we determined which regions of PIASxα are important for coactivation of Elk-1. First we studied the activity of two PIASxα mutants, PIASxα(Δ467–487), which lacks a SUMO binding motif, and PIASxα(W383A), which contains a mutation in the RING finger domain that inhibits its E3 SUMO ligase activity (Figure 6A; Kotaja et al, 2002). Of these mutants, only PIASxα(Δ467–487) showed a reduced ability to activate Elk-1 (Figure 6B). This inability to activate Elk-1 was not due to reduced expression, as PIASxα(Δ467–487) was expressed to higher levels than the wild-type protein (Figure 6C). To further probe a potential defect with PIASxα(W383A), we performed a dose–response experiment with lower levels of PIASxα. However, even at lower concentrations, this mutant was still as active as the wild-type protein (Supplementary Figure S2). Thus, importantly, the activity of PIASxα(W383A) was unperturbed, indicating that the E3 ligase activity associated with the RING finger is not required for PIASxα-mediated Elk-1 activation.
Figure 6.
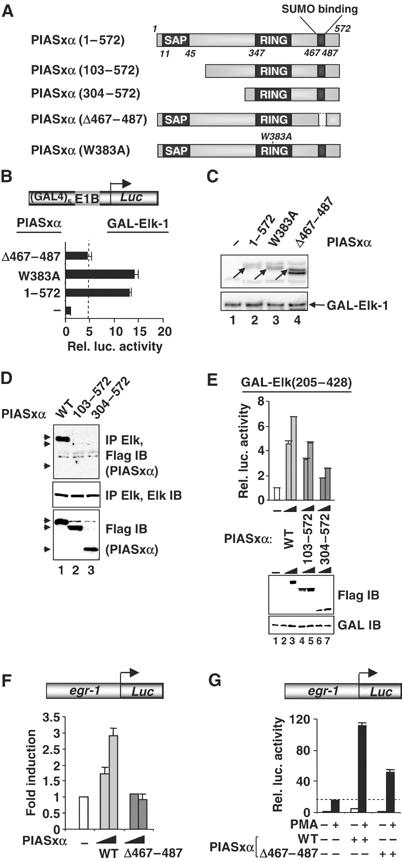
Mapping the determinants in PIASxα for coactivation of Elk-1. (A) Schematic of PIASxα proteins used in this figure. The known PIAS domains (SAP, RING and SUMO binding) are indicated in dark grey boxes. (B) Reporter gene analysis of activities of GAL-Elk(1–428) in the presence or absence of the indicated PIASxα proteins on the GAL-driven E1b promoter-reporter construct in 293 cells. The data are shown relative to GAL-Elk(1–428) in the absence of PIASxα. (C) Western blots probed with Flag (PIASxα) and Gal4 (GAL-Elk) antibodies. Arrows indicate the band corresponding to the protein with the correct size. (D) Co-immunoprecipitation analysis of the indicated PIASxα deletion mutants and cotransfected Elk-1 using an anti-Elk-1 antibody. Arrows represent the locations of the truncated PIASxα proteins. (E) Reporter gene analysis of increasing amounts of the indicated PIASxα proteins (wild type (WT), 125 and 250 ng; 103–572, 500 and 1000 ng; 304–572, 750 and 1500 ng) and the Elk-1 derivative GAL-Elk(205–428) on the GAL-driven E1b promoter-reporter construct in 293 cells. Expression levels of the mutants are shown in the accompanying Western blot. (F, G) Reporter gene analysis of activities of egr-1 promoter-driven luciferase reporter construct in 293 cells. (F) Reporter activity in the absence (white bar) or presence of increasing amounts of WT (light grey bars) or mutant (Δ467–487) (0.1 and 1 μg) (dark grey bars) PIASxα proteins (relative to the basal level of the reporter construct). (G) Reporter activities in the absence (white bar) or presence of WT or mutant (Δ467–487) (dark grey bars) PIASxα proteins in the presence or absence of PMA. The data are shown relative to the basal level of the reporter construct (taken as 1). Luciferase assays are representative of at least two independent experiments (standard errors are shown; n=2).
To further probe the interaction between Elk-1 and PIASxα and its functional consequences, we tested a series of N-terminally deleted PIASxα mutants (Figure 6A) for their ability to bind Elk-1 and to activate Elk-1-mediated transcription. In contrast to wild-type PIASxα, binding was much reduced with PIASxα(101–572) (Figure 6D, lane 2). The residual binding observed with PIASxα(101–572) was reduced below detectable levels upon further truncation in PIASxα(304–572) (Figure 6D, lane 3). To examine the functional consequences of this loss of binding, the activities of the wild-type and truncated versions of PIASxα were tested in a reporter assay using GAL-Elk-1 fusion proteins. In comparison to wild-type PIASxα, PIASxα(101–572) showed reduced transactivation capacity, which was further reduced in PIASxα(304–572) (Figure 6E). This reduction in activity was not attributable to differences in expression levels in the reporter assay. Thus, a correlation exists between the ability of Elk-1 and PIASxα to interact and the ability of PIASxα to function as an Elk-1 coactivator.
We also tested whether the loss of the SUMO binding motif in PIASxα(Δ467–487) affected its ability to activate the promoter of an Elk-1-regulated target gene, egr-1. While wild-type PIASxα was able to activate the egr-1 promoter, PIASxα(Δ467–487) was unable to do so (Figure 6F). Furthermore, in the presence of PMA, PIASxα cooperatively enhanced transactivation by Elk-1, while PIASxα(Δ467–487) was much less effective (Figure 6G). Thus, the SUMO binding motif is an important determinant of the coactivating capacity of PIASxα on Elk-1.
PIASxα potentiates p300-mediated coactivation of Elk-1
Transcriptional activation by Elk-1 is mediated, at least in part, through the coactivators p300/CBP (Janknecht and Nordheim, 1996; Li et al, 2003). We therefore tested whether PIASxα also affected the activity of p300. Increasing concentrations of PIASxα greatly enhanced the activity of p300, but this enhancement was much reduced upon deletion of the CRD1 repression domain in p300ΔR (Figure 7A). The CRD1-motif has previously been shown to be sumoylated (Girdwood et al, 2003), so this effect is consistent with the role of PIASxα in regulating the activity of other sumoylated transcription factors. Next, we tested which regions of PIASxα are essential for activating p300. The E3 ligase-defective mutant, PIASxα(W383A), activated p300 to similar levels as the wild-type protein (Figure 7B). However, deletion of either the SAP domain in PIASxα(103–572) or the SUMO binding motif in PIASxα(Δ467–487) resulted in reduced levels of activation (Figure 7B). The defects observed with PIASxα(103–572) towards p300 were greater than those observed with Elk-1, suggesting that the mode of action is not identical (data not shown).
Figure 7.
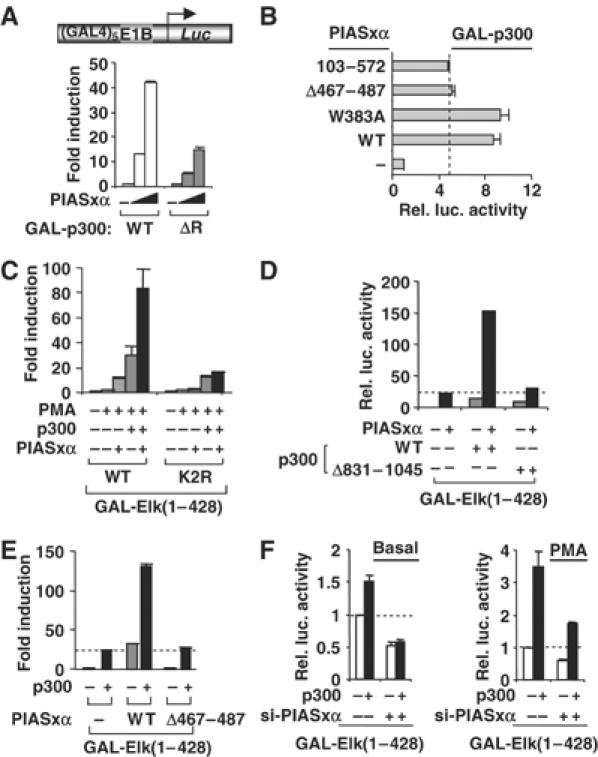
PIASxα upregulates p300 activity. (A, B) Reporter gene analysis of activities of indicated GAL-p300 fusion proteins on the GAL-driven E1b promoter-reporter construct in 293 cells. (A) The activities of the indicated GAL-p300 constructs were tested in the presence or absence of increasing amounts of transfected PIASxα (10, 25 and 100 ng). Data are presented as fold activation of each construct in the absence of PIASxα. (B) Reporter gene analysis of activities of WT GAL-p300 in the presence and absence of PIASxα. The data are shown relative to GAL-p300 in the absence of PIASxα. (C–E) Reporter gene analysis of activities of GAL-Elk(1–428) WT and K2R in the presence and absence of the indicated p300 constructs and/or PIASxα proteins and/or PMA on the GAL-driven E1B promoter-reporter construct in COS7 cells. Data are shown relative to the activity of GAL-Elk(1–428) alone (taken as 1). All samples in panels D and E were stimulated with PMA. (F) Reporter assays were carried out in 293 cells in the presence and absence of siRNA duplexes against PIASxα in serum-free (basal) or in stimulated (PMA) conditions. Luciferase assays are representative of at least two independent experiments (standard errors are shown; n=2).
As PIASxα activates both p300 and Elk-1, we asked whether functional synergy could be observed between the coactivation activities of p300 and PIASxα. Individually, PIASxα and p300 activate Elk-1 five- and 12-fold, respectively (Figure 7C). However, together, a synergistic 35-fold activation of Elk-1 is observed. Importantly, this synergism is lost with the non-sumoylatable Elk-1(K2R) mutant that is refractory to PIASxα-mediated coactivation (Figure 7C) and is also lost in the presence of the non-sumoylatable p300 mutant p300(Δ831–1045) (Figure 7D). Moreover, a reduction in synergism between PIASxα and p300 is also observed with the PIASxα(Δ467–487) mutant (Figure 7E).
To establish a role of endogenous PIASxα in p300-mediated Elk-1 activation, we compared the coactivating properties of p300 in the absence and presence of siRNAs against PIASxα. The increases in Elk-1 activity elicited by p300 in both the presence and absence of ERK pathway activation were compromised upon depletion of PIASxα (Figure 7F).
Collectively, these data therefore demonstrate that PIASxα can also act through p300 to affect its coactivator activity and that PIASxα and p300 synergise in coactivation of Elk-1. This synergism is dependent on the sumoylation sites in both p300 and Elk-1 and the SUMO binding site in PIASxα.
PIASxα reduces HDAC activity and promotes increased acetylation at Elk-1 target promoters
The transcriptional activity of Elk-1 has previously been shown to be repressed under basal conditions by the recruitment of HDAC-2 through the repressive R-motif in a SUMO-dependent manner (Yang and Sharrocks, 2004). Furthermore, our results demonstrate that PIASxα coactivation needs Elk-1 to be sumoylated and is required to facilitate SUMO removal from Elk-1 following ERK pathway activation. Thus, a possible molecular mechanism to explain the role of PIASxα would be to promote the loss of HDAC-2 and hence derepression of Elk-1 activity.
To establish whether PIASxα affects the HDAC activity associated with Elk-1, we first investigated the sensitivity of Elk-1 to the HDAC inhibitor trichostatin A (TSA) in the presence and absence of PIASxα. In the presence of PIASxα, Elk-1 became less sensitive to TSA treatment (Figure 8A), suggesting a reduced involvement of HDACs in repressing Elk-1 activity. We then tested whether overexpression of a panel of different HDACs was able to reverse PIASxα-mediated activation (Figure 8B). Of the HDACs tested, only HDAC-2, and to a lesser extent HDAC-6, caused a reduction in PIAS-mediated Elk-1 activation. No such effect by HDAC-2 was seen on the Elk-1(K2R) mutant, although a small but reproducible reduction was still observed with HDAC-6 (Figure 8B). To further probe a specific role of HDAC-2 in the PIASxα response, we depleted endogenous HDAC-2 by siRNA to investigate whether this sensitised Elk-1 to PIASxα-mediated transactivation. Depletion of HDAC-2, but not HDAC-3, potentiated the effect of PIASxα on Elk-1 (Figure 8C). Again, this effect was not seen on the Elk-1(K2R) mutant.
Figure 8.
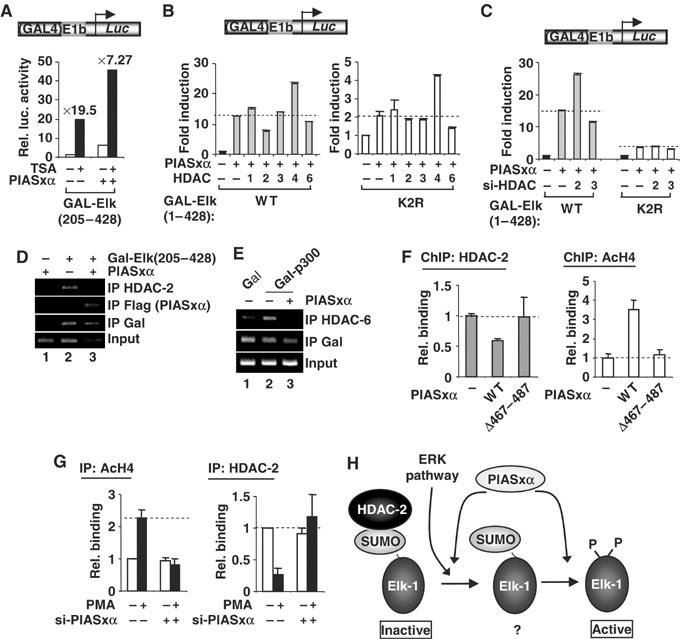
PIASxα affects HDAC-2 binding and histone acetylation levels at Elk-1-regulated promoters. (A–C) Reporter gene analysis of the activities of GAL-Elk-1 constructs on a GAL-driven E1B promoter-reporter construct in 293 cells. (A) The activity of GAL-Elk(205–428) in the presence or absence of PIASxα and/or TSA. Data are shown as luciferase assays relative to GAL-Elk(205–428) alone and as fold induction by TSA. (B) The activities of wild-type (WT) and mutant (K2R) GAL-Elk(1–428) in the presence and absence of PIASxα and/or indicated HDACs. Data are shown as fold induction to the reporter activity relative to each of GAL fusion proteins in the absence of PIASxα (taken as 1). Note the different scales on the axes for WT and K2R versions of Elk-1. (C) The activities of GAL-Elk(1–428) WT and K2R in the presence or absence of RNAi duplexes against GAPDH (−) or the indicated HDACs. Data are shown as fold induction to the reporter activity relative to each of GAL fusion proteins in the presence of GAPDH RNAi duplexes. (D, E) ChIP analysis of HDAC-2, PIASxα (D) and HDAC-6 (E) recruitment to a GAL-driven E1b promoter-reporter construct in 293 cells. The recruitment is monitored following immunoprecipitation (IP) with the indicated antibodies in the presence of the indicated combinations of Gal4 DBD, Gal-Elk(205–428) or Gal-p300 and PIASxα. (F, G) ChIP analysis of HDAC-2 and acetyl-H4 levels on the c-fos promoter. (F) Assays were carried out in 293 cells in the presence of transfected Elk-1 and in the presence or absence of the indicated PIASxα proteins using antisera specific for the indicated proteins. Following immunoprecipitation (IP) of crosslinked lysates, real-time PCR analysis of eluted DNA was performed. All quantitative PCRs were normalised to the input control. Data are shown as relative enrichment for each individual antibody used. (G) ChIP analysis of HeLa cells containing transfected RNAi duplexes against PIASxα using antisera specific for the indicated proteins. (H) Model for the action of PIASxα in facilitating HDAC-2 removal from sumoylated Elk-1. The ERK pathway causes HDAC-2 release and Elk-1 phosphorylation and desumoylation. PIASxα promotes HDAC-2 release and is required for desumoylation. A poised ‘intermediate' sumoylated form of Elk-1 (indicated by ‘?'), which precedes the appearance of a phosphorylated, desumoylated fully active form, is indicated to reflect the molecular role of PIASxα.
Together, these results point to a role of PIASxα in specifically affecting HDAC-2 activity associated with Elk-1 and suggest that PIASxα might affect HDAC-2 occupancy at Elk-1-regulated promoters. To test this possibility, we first used chromatin immunoprecipitation (ChIP) analysis to investigate whether PIASxα was recruited to a reporter gene by Elk-1. In the absence of Elk-1, little recruitment of PIASxα could be observed, but was readily detectable in the presence of Elk-1 (Figure 8D, lanes 1 and 3). However, while Elk-1 promoted HDAC-2 recruitment, the presence of PIASxα led to a reduction in this recruitment (Figure 8D, lanes 2 and 3). To probe the generality of this phenomenon, we also tested the effect of PIASxα on the recruitment of HDAC-6 to p300-regulated promoters. As observed with Elk-1, a loss of HDAC-6 recruitment to a p300-regulated reporter construct was observed upon overexpression of PIASxα (Figure 8E). This is consistent with the observation that PIASxα also activates p300-mediated transcriptional activation (Figure 7) and demonstrates a wider role of PIASxα in promoting HDAC exclusion from different transcription factor complexes.
To confirm that the PIASxα-mediated HDAC dissociation occurs on endogenous Elk-1-regulated target genes, we examined the presence of HDAC-2 at the c-fos promoter by ChIP analysis (Figure 8F). In the presence of PIASxα, HDAC-2 promoter occupancy decreased. Conversely, an increase in histone H4 acetylation was observed, consistent with HDAC loss and promoter activation. Importantly, the PIASxα(Δ467–487) mutant that cannot activate Elk-1 does not cause a reduction in HDAC-2 occupancy or a change in promoter acetylation status (Figure 8F). Finally, we investigated the consequences of depleting endogenous PIASxα on HDAC occupancy and acetylation status of the c-fos promoter. In the absence of siRNAs directed against PIASxα, PMA stimulation enhanced acetylation and decreased HDAC-2 occupancy at the c-fos promoter (Figure 8G). In contrast, upon depletion of PIASxα, these PMA-mediated increases in histone acetylation and decreases in HDAC-2 occupancy were blocked. This is consistent with the reductions in c-fos activation observed upon PIASxα depletion (Figure 1F).
Collectively, these results therefore demonstrate that a major function of PIASxα is to facilitate HDAC-2 removal from Elk-1 and thereby promote derepression and alter the histone acetylation status of Elk-1-regulated promoters.
Discussion
Recent studies suggest that the modification of transcriptional regulatory proteins by SUMO is a common event and that SUMO modification often imparts transcriptional regulatory properties on the target protein (reviewed in Gill, 2003, 2004; Verger et al, 2003). However, the molecular mechanisms regulating protein sumoylation and the mechanistic actions of SUMO modification are still unclear. Here, we have probed the role of PIAS proteins in Elk-1 regulation, and uncovered an unexpected mode of action. Rather than acting as an E3 ligase to promote transcriptional repression, PIASxα acts as an E3 activity-independent coactivator that facilitates derepression of Elk-1 activity.
Molecularly, PIASxα promotes a switch in the HDAC/HAT activities associated with Elk-1, and hence transcriptional activation. One major point of action is in promoting the loss of HDAC-2 activity associated with Elk-1 (Figure 8H). HDAC-2 has previously been shown to play a major role in downregulating the transcriptional activity of Elk-1 (Yang and Sharrocks, 2004). However, in addition, PIASxα can also upregulate the activity of the Elk-1 coactivator protein p300, thereby increasing the HAT activity associated with Elk-1. This change might also contribute to the change in the acetylation equilibrium mediated by PIASxα. Mechanistically, it appears that PIASxα also works on p300 by HDAC removal, although in this case, its point of action is on HDAC-6, which is specifically recruited by p300 (Girdwood et al, 2003). This mechanism of action is consistent with the observation that HDAC-6 can partially reverse PIASxα-mediated Elk-1 activation and that this occurs irrespective of the Elk-1 sumoylation status. The overall change in the HDAC/HAT equilibrium is reflected by the changes seen in histone acetylation observed in Elk-1-regulated promoters upon overexpression or depletion of PIASxα and the subsequent gene expression changes observed.
PIAS proteins are characterised as E3 ligases due to the presence of their RING finger domain. Functionally, this has been shown to be important in E3 ligase activity of several PIAS proteins against different substrates (Kahyo et al, 2001; Kotaja et al, 2002; Schmidt and Muller, 2002). However, the E3 ligase activity of PIASxα is not required for its coactivating activity towards Elk-1 and it does not appear to influence the basal levels of Elk-1 sumoylation. Nevertheless, the action of PIASxα on Elk-1 and p300 requires both substrate sumoylation to occur and also the presence of the SUMO binding motif in PIASxα. This requirement does not reflect a simple need of SUMO binding for recruitment but rather appears to be linked to the major point of action of PIASxα in controlling HDAC binding to Elk-1. As sumoylation is essential for HDAC-2 recruitment to Elk-1 (Yang and Sharrocks, 2004), this provides an explanation as to why PIASxα-mediated coactivation needs prior Elk-1 sumoylation. In common with many transcription factor targets, Elk-1 is sumoylated at low stoichiometry (1–5%; Yang et al, 2003; Salinas et al, 2004), yet large functional effects are seen upon disruption of this sumoylation, on both its transactivation capacity and its rate of nucleocytoplasmic shuttling. It has been proposed that this low stoichiometry reflects sumoylation being either a transient trigger or a modification of a transcriptionally relevant subpopulation (Hay, 2005). The large sumoylation-dependent effects we observe on Elk-1 activity due to changes in PIASxα levels further emphasise the importance of this SUMO-modified subpopulation. Furthermore, siRNA-mediated depletion experiments demonstrate the importance of PIASxα for SUMO removal from Elk-1. However, it is currently unclear what the temporal order of SUMO and HDAC-2 removal is and whether PIASxα first promotes HDAC-2 removal or whether both occur simultaneously. A possible mode of action of PIASxα would be to affect HDAC-2 or HDAC-6 sumoylation, although to date, neither protein has been shown to be SUMO modified and we have been unable to demonstrate any PIASxα-mediated changes in their sumoylation status (data not shown).
Overall, our results therefore provide a novel molecular role of PIAS proteins in regulating transcription factor activity through mechanisms other than promoting direct substrate sumoylation. Indeed, other studies have shown that PIAS proteins can act in a manner independent of substrate sumoylation, although none have been shown to function molecularly through corepressor/HDAC dissociation. For example, the E3 ligase activity of PIAS1 is not needed for its ability to coactivate p53 (Megidish et al, 2002). Moreover, the repression of GATA-1 activity by PIASy does not need the SUMO sites in GATA-1 (Collavin et al, 2004). In addition, the subnuclear relocalisation of LEF1 by PIASy does not require LEF1 sumoylation, although in this case, the sumoylation activity of PIASy is required (Sachdev et al, 2001). Thus, PIAS proteins have the potential to regulate the activities of transcription factors in a variety of SUMO-dependent and -independent ways.
Elk-1 is activated through phosphorylation in its TAD by the ERK MAP kinase pathway (reviewed in Sharrocks, 2002; Shaw and Saxton, 2003). Phosphorylation permits recruitment and activation of coactivators like Sur2/Med23 (Stevens et al, 2002) and p300 (Li et al, 2003), but also promotes the loss of SUMO and the loss of associated HDAC-2 (Yang and Sharrocks, 2004). PIASxα appears to play an integral role in this process, as overexpression and depletion of PIASxα cause increases and decreases in gene activation mediated by the ERK pathway. These changes in gene activation are accompanied by changes in promoter acetylation. One model to explain the role of PIASxα in this process is that it facilitates HDAC-2 loss during the induction process, which would be followed by SUMO loss (Figure 8H). How PIASxα achieves this is unclear, but the requirement for its SUMO binding motif suggests that it might act by engaging the SUMO moiety and displacing components of associated corepressor complexes. The trigger for this might be the conformational change initiated in Elk-1 following its phosphorylation (Yang et al, 1999), or by a direct effect of the ERK pathway on the activity of PIASxα. Future studies will probe these possibilities.
In summary, our work demonstrates a novel facet of PIAS protein action as a coactivator protein. In the case of Elk-1, PIASxα acts through dynamically regulating the recruitment of HDAC-2. A similar effect is elicited on p300 through HDAC-6 displacement. It will be important to probe the potential roles of PIAS proteins against other substrates to see if the regulation of SUMO site-dependent corepressor complex recruitment is a more widespread phenomenon.
Materials and methods
Plasmid constructs and oligonucelotide primers
For details of plasmids and primers used, see Supplementary data.
Tissue culture, cell transfection, reporter gene assays and RNA interference
293, COS7 and HeLa cells were grown and transfection experiments were carried out as described previously (Yang et al, 2001). For PMA stimulation, cells were serum-starved for 18 h and subsequently treated with 10 nM PMA for a further 6 h for reporter analysis or for 30 min for ChIP analysis, before harvesting. For TSA experiments, cells were serum-starved for 8 h and subsequently treated with 330 nM of TSA for a further 12 h for reporter analysis before harvesting. Where indicated, 10 μM of the MEK inhibitor, U01260, was added to cells. For Gal4 fusion-driven luciferase reporter gene assays, 0.5 μg of reporter plasmid and 0.25 μg of pCH110 were cotransfected with 0.1 μg of Gal4 fusion expression plasmids. Cell extracts were prepared and luciferase and β-galactosidase assays were carried out as described previously (Yang et al, 1998).
siRNAs, including the GAPDH control, were constructed by the Silencer™ siRNA construction kit (Ambion). The following were used: HDAC-2 target siRNA sequence: 5′-AAGCATCAGGATTCTGTTACG-3′; HDAC-3 target siRNA sequence: 5′-AAGATGCTGAACCATGCACCT-3′; human PIASxα-H1 target siRNA sequence: 5′-AAGATACTAAGCCCACATTTG-3′.
To carry out RNAi, a two-step transfection protocol was carried out in 12-well plates as described previously (Yang et al, 2003).
SUMO assays in vivo
In vivo sumoylation assays were performed as described previously (Yang et al, 2003).
Immunoprecipitation
For immunoprecipitations, the antibody matrix was prepared by binding the antibodies to Dynabeads protein A (rabbit IgG) or protein G (mouse IgG) (Dynal Biotech.). Lysates were prepared from 6 cm dishes in 300 μl of RIPA-1 buffer containing protease inhibitors, 10 mM NEM and E64, as described previously (Kotaja et al, 2002). The antibody matrix was incubated with whole-cell extracts with rotation overnight at 4°C. Complexes were washed three times with RIPA-1, boiled in sample loading buffer and subjected to SDS–PAGE followed by Western blot analysis.
Western blot analysis
Western blotting was carried out with primary antibodies—Flag (Sigma), Elk-1 and Gal4(DBD) (Santa Cruz Biotech.), HA antibody, phospho-Elk (NEB) and PIASxα (kindly provided by J Palvimo)—and HRP-conjugated secondary antibodies (Transduction Lab.) followed by detection with SuperSignal west dura extended duration substrate (Pierce) and visualised using Quantity-One (Bio-Rad).
ChIP assays and quantitative real-time PCR ChIP assay
ChIP assays using antisera specific to acetylated histone H4 (Upstate Biotechnology), Elk-1 and HDAC-2 (Santa Cruz Biotech.), HDAC-6 (Abcam) and Flag (Sigma) were performed as described previously (Yang and Sharrocks, 2004) except that crosslinking was performed with 0.5% formaldehyde at room temperature for 2 min and Dynabeads protein A or protein G was used.
Quantitative real-time PCR was performed as specified by the manufacturer using QuantiTect™ SYBR Green PCR reagents (QIAGEN). Data were analysed by Opticon2 (MJ Research).
Real-time RT–PCR
Cells were harvested and total RNA extracted using the RNeasy kit (Qiagen). A 40 ng portion of total RNA from each sample was used for QuantiTect™ SYBR Green RT–PCR kit (QIAGEN) according to the supplier's protocol. Data were analysed by Opticon2 (MJ Research).
Supplementary Material
Supplementary Methods
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
We thank Anne Clancy for excellent technical assistance; Hilary Ashe, Stefan Roberts, Alan Whitmarsh and members of our laboratory for comments on the manuscript and stimulating discussions; Jorma Palvimo, Frances Fuller-Pace, Tony Kouzarides, Ron Hay and Neil Perkins for reagents; and Amanda O'Donnell for help with the real-time PCR. This work was supported by grants from the Wellcome Trust.
References
- Aravind L, Koonin EV (2000) SAP—a putative DNA-binding motif involved in chromosomal organization. Trends Biochem Sci 25: 112–114 [DOI] [PubMed] [Google Scholar]
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K (1997) Specific inhibition of Stat3 signal transduction by PIAS3. Science 278: 1803–1805 [DOI] [PubMed] [Google Scholar]
- Collavin L, Gostissa M, Avolio F, Secco P, Ronchi A, Santoro C, Del Sal G (2004) Modification of the erythroid transcription factor GATA-1 by SUMO-1. Proc Natl Acad Sci USA 101: 8870–8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill G (2003) Post-translational modification by the small ubiquitin-related modifier SUMO has big effects on transcription factor activity. Curr Opin Genet Dev 13: 108–113 [DOI] [PubMed] [Google Scholar]
- Gill G (2004) SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev 18: 2046–2059 [DOI] [PubMed] [Google Scholar]
- Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia-Wilson E, Perkins ND, Hay RT (2003) p300 transcriptional repression is mediated by SUMO modification. Mol Cell 11: 1043–1054 [DOI] [PubMed] [Google Scholar]
- Hay RT (2001) Protein modification by SUMO. Trends Biochem Sci 26: 332–333 [DOI] [PubMed] [Google Scholar]
- Hay RT (2005) SUMO: a history of modification. Mol Cell 18: 1–12 [DOI] [PubMed] [Google Scholar]
- Janknecht R, Nordheim A (1996) MAP kinase-dependent transcriptional coactivation by Elk-1 and its cofactor CBP. Biochem Biophys Res Commun 228: 831–837 [DOI] [PubMed] [Google Scholar]
- Kagey MH, Melhuish TA, Wotton D (2003) The Polycomb protein Pc2 is a SUMO E3. Cell 113: 127–137 [DOI] [PubMed] [Google Scholar]
- Kahyo T, Nishida T, Yasuda H (2001) Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell 8: 713–718 [DOI] [PubMed] [Google Scholar]
- Kasza A, O'Donnell A, Gascoigne K, Zeef LAH, Hayes A, Sharrocks AD (2005) The ETS-domain transcription factor Elk-1 regulates the expression of its partner protein, SRF. J Biol Chem 280: 1149–1155 [DOI] [PubMed] [Google Scholar]
- Kirsh O, Seeler JS, Pichler A, Gast A, Muller S, Miska E, Mathieu M, Harel-Bellan A, Kouzarides T, Melchior F, Dejean A (2002) The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J 21: 2682–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaja N, Karvonen U, Janne OA, Palvimo JJ (2002) PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol Cell Biol 22: 5222–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q-J, Yang S-H, Maeda Y, Sladek FM, Sharrocks AD, Martins-Green M (2003) MAP kinase phosphorylation-dependent activation of Elk-1 leads to activation of the coactivator p300. EMBO J 22: 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megidish T, Xu JH, Xu CW (2002) Activation of p53 by protein inhibitor of activated Stat1 (PIAS1). J Biol Chem 277: 8255–8259 [DOI] [PubMed] [Google Scholar]
- Muller S, Hoege C, Pyrowolakis G, Jentsch S (2001) SUMO, ubiquitin's mysterious cousin. Nat Rev Mol Cell Biol 2: 202–210 [DOI] [PubMed] [Google Scholar]
- Price MA, Rogers AE, Treisman R (1995) Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2 (ERP/NET). EMBO J 14: 2589–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RS, Horvath CM, Matunis MJ (2003) SUMO modification of STAT1 and its role in PIAS-mediated inhibition of gene activation. J Biol Chem 278: 30091–30097 [DOI] [PubMed] [Google Scholar]
- Ross S, Best JL, Zon LI, Gill G (2002) SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol Cell 10: 831–842 [DOI] [PubMed] [Google Scholar]
- Sachdev S, Bruhn L, Sieber H, Pichler A, Melchior F, Grosschedl R (2001) PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev 15: 3088–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Briancon-Marjollet A, Bossis G, Lopez MA, Piechaczyk M, Jariel-Encontre I, Debant A, Hipskind RA (2004) SUMOylation regulates nucleo-cytoplasmic shuttling of Elk-1. J Cell Biol 165: 767–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Muller S (2002) Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci USA 99: 2872–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Muller S (2003) PIAS/SUMO: new partners in transcriptional regulation. Cell Mol Life Sci 60: 2561–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Li X, Wang Y, Zarnegar M, Huang CY, Palvimo JJ, Lim B, Sun Z (2003) hZimp10 is an androgen receptor co-activator and forms a complex with SUMO-1 at replication foci. EMBO J 22: 6101–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD (2002) Complexities in ETS-domain transcription factor function and regulation; lessons from the TCF subfamily. Biochem Soc Trans 30: 1–9 [DOI] [PubMed] [Google Scholar]
- Shaw PE, Saxton J (2003) Ternary complex factors: prime nuclear targets for mitogen-activated protein kinases. Int J Biochem Cell Biol 35: 1210–1226 [DOI] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA 100: 13225–13230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci USA 101: 14373–14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JL, Cantin GT, Wang G, Shevchenko A, Shevchenko A, Berk AJ (2002) Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 296: 755–758 [DOI] [PubMed] [Google Scholar]
- Townsend KJ, Zhou P, Qian L, Bieszczad CK, Lowrey CH, Yen A, Craig RW (1999) Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J Biol Chem 274: 1801–1813 [DOI] [PubMed] [Google Scholar]
- Ungureanu D, Vanhatupa S, Kotaja N, Yang J, Aittomaki S, Janne OA, Palvimo JJ, Silvennoinen O (2003) PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 102: 3311–3313 [DOI] [PubMed] [Google Scholar]
- Verger A, Perdomo J, Crossley M (2003) Modification with SUMO: a role in transcriptional regulation. EMBO Rep 4: 137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers ER, Kasza A, Aksan-Kurnaz I, Seifert A, Zeef LA, O'Donnell A, Hayes A, Sharrocks AD (2004) Ternary complex factor–serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol 24: 10340–10351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Ihara M, Matsuura Y, Kikuchi A (2003) Sumoylation is involved in beta-catenin-dependent activation of Tcf-4. EMBO J 22: 2047–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Jaffray E, Hay RT, Sharrocks AD (2003) Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol Cell 12: 63–74 [DOI] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD (2004) SUMO promotes HDAC-mediated transcriptional repression. Mol Cell 13: 611–617 [DOI] [PubMed] [Google Scholar]
- Yang SH, Shore P, Willingham N, Lakey JH, Sharrocks AD (1999) The mechanism of phosphorylation-inducible activation of the ETS-domain transcription factor Elk-1. EMBO J 18: 5666–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Vickers E, Brehm A, Kouzarides T, Sharrocks AD (2001) Temporal recruitment of the mSin3A–histone deacetylase corepressor complex to the ETS Domain transcription factor Elk-1. Mol Cell Biol 21: 2802–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S-H, Yates PR, Whitmarsh AJ, Davis RJ, Sharrocks AD (1998) The Elk-1 ETS-domain transcription factor contains a MAP kinase targeting motif. Mol Cell Biol 18: 710–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figure S1
Supplementary Figure S2