

Abstract
SNARE proteins (soluble NSF-attachment protein receptors) are thought to be central components of the exocytotic mechanism in neurosecretory cells, but their precise function remained unclear. Here, we show that each of the vesicle-associated SNARE proteins (v-SNARE) of a chromaffin granule, synaptobrevin II or cellubrevin, is sufficient to support Ca2+-dependent exocytosis and to establish a pool of primed, readily releasable vesicles. In the absence of both proteins, secretion is abolished, without affecting biogenesis or docking of granules indicating that v-SNAREs are absolutely required for granule exocytosis. We find that synaptobrevin II and cellubrevin differentially control the pool of readily releasable vesicles and show that the v-SNARE's amino terminus regulates the vesicle's primed state. We demonstrate that dynamics of fusion pore dilation are regulated by v-SNAREs, indicating their action throughout exocytosis from priming to fusion of vesicles.
Keywords: amperometry, capacitance measurements, cellubrevin, chromaffin cells, synaptobrevin II
Introduction
Neuronal SNAREs (soluble NSF-attachment protein receptors) include synaptobrevin, which resides on vesicles and the plasma membrane proteins, syntaxin and SNAP-25 (Chen and Scheller, 2001; Jahn et al, 2003). At synapses, null mutants of any of the three SNARE proteins exhibit a strong inhibition of action potential-evoked transmitter release (Schulze et al, 1995; Deitcher et al, 1998; Schoch et al, 2001; Washbourne et al, 2002), an observation that agrees well with the finding that these proteins are specifically targeted by clostridial neurotoxins, which are potent inhibitors of neurotransmitter release. Morphological evidence showed that docking of secretory vesicles at the plasma membrane is not impaired after genetic ablation or proteolysis of SNARE proteins, implicating a ‘postdocking' role of these proteins in the exocytotic pathway (Hunt et al, 1994; Broadie et al, 1995). Recent studies on SNAP-25 in chromaffin cells using either single amino-acid substitutions, site-specific antibodies or expression of splice variants in knockout cells suggest that the function of this protein provides the molecular basis for ‘priming' of secretory vesicles, a reaction that transfers morphologically docked vesicles into an exocytosis-competent state (Xu et al, 1999; Wei et al, 2000; Sorensen et al, 2003; Nagy et al, 2004). Still, neither SNAP-25 deficiency nor the lack of synaptobrevin II abolishes spontaneously occurring vesicle fusion or secretion when a long-lasting Ca2+ stimulus is applied, raising the question of whether remaining exocytosis occurs with the help of an alternative SNARE complex or by a SNARE-independent mechanism (Yoshihara et al, 1999; Schoch et al, 2001; Washbourne et al, 2002; Sorensen et al, 2003). Furthermore, data from other systems have indicated that SNAREs may act before fusion and are not directly involved in executing fusion (Tahara et al, 1998; Ungermann et al, 1998).
To gain new insight into the mode of SNARE function in exocytosis, we examined alternative v-SNAREs in the same exocytotic pathway. In following this strategy, we analyzed how structurally homologous v-SNAREs of a chromaffin granule, synaptobrevin II (sybII) and cellubrevin (ceb, also referred to as VAMP 2 and VAMP 3, respectively), contribute to catecholamine secretion from chromaffin cells (Baumert et al, 1989; Elferink et al, 1989; McMahon et al, 1993). Ceb shares a high degree of similarity with sybII, is expressed in a wide variety of tissues and is also a substrate for the proteolytic action of tetanus toxin (McMahon et al, 1993; Galli et al, 1994). Previous studies identified ceb on secretory vesicles of neuroendocrine cells (Chilcote et al, 1995) and glia cells (Bezzi et al, 2004) but also implicated the protein in the fusion of endosome-derived, transferrin receptor-containing vesicles that undergo exocytosis constitutively and in a Ca2+-independent manner (Galli et al, 1994). To study the functional impact of these v-SNARE proteins in Ca2+-triggered exocytosis, we took advantage of recently developed mouse strains, being deficient either for sybII (Schoch et al, 2001) or for ceb (Yang et al, 2001) and, in addition, generated a new double v-SNARE null mouse.
Our experiments demonstrate that secretion is abolished only in the absence of both sybII and ceb, without affecting biogenesis or docking of granules, indicating that one of these v-SNAREs is absolutely required for Ca2+-dependent granule exocytosis. We provide evidence that sybII is the major, if not the only, v-SNARE acting in wild-type cells, but is efficiently substituted by ceb in sybII knockouts (ko), suggesting that v-SNAREs act promiscuously in genetically deficient but specifically in wild-type cells. Furthermore, membrane capacitance measurements and carbon fiber amperometry are used to elucidate how the individual v-SNARE proteins regulate Ca2+-triggered stimulus–secretion coupling and exocytotic membrane merger.
Results
Granule exocytosis in isolated chromaffin cells was synchronized using flash photolysis of caged Ca2+ (NP-EGTA), and changes in the intracellular calcium concentration ([Ca]i) were measured with a combination of calcium indicators (Fura-2 and Furaptra). Photolytic ‘uncaging' of Ca2+ leads to a step-like increase in [Ca]i that stimulates granule exocytosis, which is detected by measuring the membrane capacitance at high time resolution (Figure 1). The secretory response of a wild-type chromaffin cell to such a stimulus can be roughly divided into two kinetically distinct phases (Heinemann et al, 1994; Voets et al, 1999): an initial phase (also referred to as exocytotic burst), reflecting the exocytosis of granules from a release-ready state and a subsequent sustained phase that is thought to represent refilling of the just emptied pool by recruitment of new vesicles into the exocytosis-competent state (‘priming'). At high [Ca]i, priming is immediately followed by fusion producing a sustained increase in membrane capacitance. At resting levels of [Ca]i, the balance between ‘forward-directed' priming and ‘backward-directed' depriming reactions should determine the pool size of release-ready organelles (Voets, 2000; Rettig and Neher, 2002).
Figure 1.
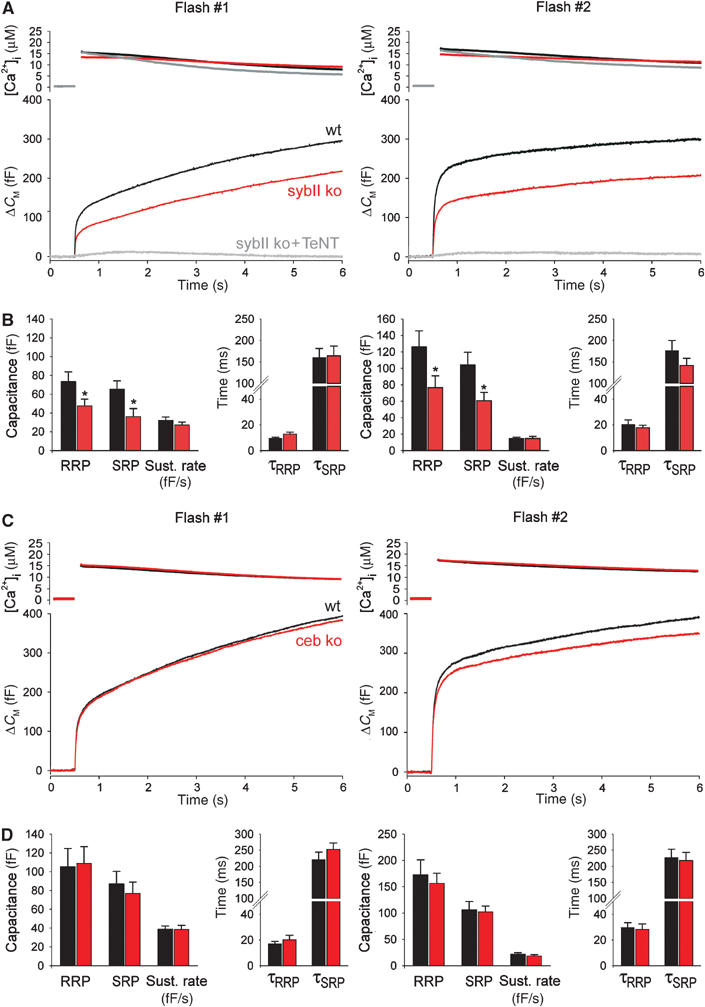
Lack of sybII or ceb differentially affects secretion from chromaffin cells. (A) Averaged flash-induced [Ca2+]i levels (upper panel; flash at t=0.5 s) and corresponding capacitance responses (lower panel) of wild-type (wt; n=22), sybII ko (n=22) and sybII ko cells treated with tetanus toxin light chain (sybII ko+TeNT, n=6). Secretion is diminished in mutant cells but almost completely blocked upon additional poisoning with TeNT. Similar effects are observed for the second flash response given with a 2 min interval (right panels). Preflash [Ca2+]i levels (in μM): first flash: wt 0.31±0.02, sybII ko 0.32±0.01, sybII ko+TeNT 0.38±0.04; second flash: wt 0.43±0.04, sybII ko 0.38±0.02, sybII ko+TeNT 0.49±0.1. (B) Amplitudes of the two exocytotic burst components (RRP, SRP) and the rate of sustained release (fF/s) for control (black bars) and sybII ko cells (red bars). Compared with controls, the average RRP and SRP response of mutant cells is similarly reduced to 65 and 55% for the first flash and to 61 and 58% for the second flash, respectively. No change is observed for the time constants of the individual exocytotic burst components (τRRP, τSRP). *P<0.05 (Student's t-test). (C) Secretion of ceb ko cells is indistinguishable from that of wt cells. Preflash [Ca2+]i was the same for wt and ceb ko cells (in μM): first flash, 0.36±0.01; second flash, 0.45±0.02. (D) Neither the magnitude nor the kinetic properties are significantly changed in ceb ko cells (red bars) compared with controls (black bars).
Synaptobrevin II deficiency versus cellubrevin deficiency
In the absence of synaptobrevin II (sybII ko), Ca2+-triggered secretion from chromaffin cells is only moderately diminished (Figure 1A). This finding was unexpected considering the observation that poisoning of wild-type cells with tetanus toxin (TeNT) leads to a complete loss of exocytosis (Xu et al, 1998). Indeed, expression of TeNT in sybII ko cells abolishes secretion, showing that an alternative toxin substrate, most likely ceb, mediates sybII-independent exocytosis. The analysis of wild-type and mutant cells reveals that loss of sybII diminishes the exocytotic burst size (Figure 1B). Both components of the exocytotic burst, the fast phase (RRP, readily releasable pool) and the slow phase (SRP, slowly releasable pool), are similarly affected, but no corresponding change in the sustained phase of the exocytotic response is observed. Thus, lack of sybII facilitates depriming of the vesicles leading to the observed reduction of RRP and SRP size. In contrast, the kinetic rates of the exocytotic burst (τRRP, τSRP; Figure 1B) and the delay between flash and onset of the response (wt: 4.4±0.77 ms; sybII ko: 5.01±0.5 ms) remain unchanged, showing that the alternative v-SNARE component enables bona fide Ca2+-triggered exocytosis. The same effects are measured for a second flash (given with a 2 min interval), indicating that differences in ‘depriming' of wild-type and sybII ko granules are robust and remain even after strong stimulation of exocytosis.
Astonishingly, lack of cellubrevin (ceb ko) has no profound effect on secretion (Figure 1C and D), neither the first nor the second flash response shows significant alterations compared with controls. Therefore, sybII is either sufficient or can readily compensate for the lack of ceb in the exocytotic mechanism. To address this issue further, we determined the expression levels of sybII and ceb on the background of the different v-SNARE knockout phenotypes in cultured chromaffin cells (Supplementary Figure A and B). The data show that, in the absence of sybII, ceb's expression level is significantly upregulated, on average by 36%. Similarly, the mean immunosignal tlsbfor sybII is increased by about 27% when ceb is absent. These observations are compatible with the view that sybII and ceb are functionally interchangeable and can act in the same exocytotic pathway.
Double v-SNARE deficiency
To test whether cellubrevin mediates secretion in sybII ko cells, we generated a mouse strain that is deficient for both proteins (double v-SNARE null mouse, dko; see Materials and methods). In fact, Ca2+-evoked secretion from dko cells is nearly abolished (Figure 2A). The flash-evoked response from these mutant cells exhibits no significant exocytotic burst (less than 7% of control measured at 0.5 s after stimulation). This result suggests that each of the two v-SNAREs is sufficient to establish a standing pool of release-ready organelles. In close correlation, strong immunoreactive bands for both, sybII and ceb are observed with Western blots of wild-type adrenal gland homogenate prepared at embryonic stage E18.5 (Figure 2C). Furthermore, TI-VAMP (toxin-insensitive VAMP; Coco et al, 1999) but not synaptobrevin I, the other known TeNT substrate, was detectable on immunoblots. Treatment of dko cells with TeNT led to no further reduction of the signal (Figure 2A), implicating TeNT-insensitive VAMPs like TI-VAMP or VAMP 8 (Paumet et al, 2000) in mediating the small remaining response and excluding synaptobrevin I that may be expressed below the detection limits of the immunoblot analysis. Western blot analysis of adrenal gland and of brain homogenate furthermore reveals that double v-SNARE deficiency causes no compensatory changes in the expression levels of other synaptic proteins (Figure 2D and E).
Figure 2.
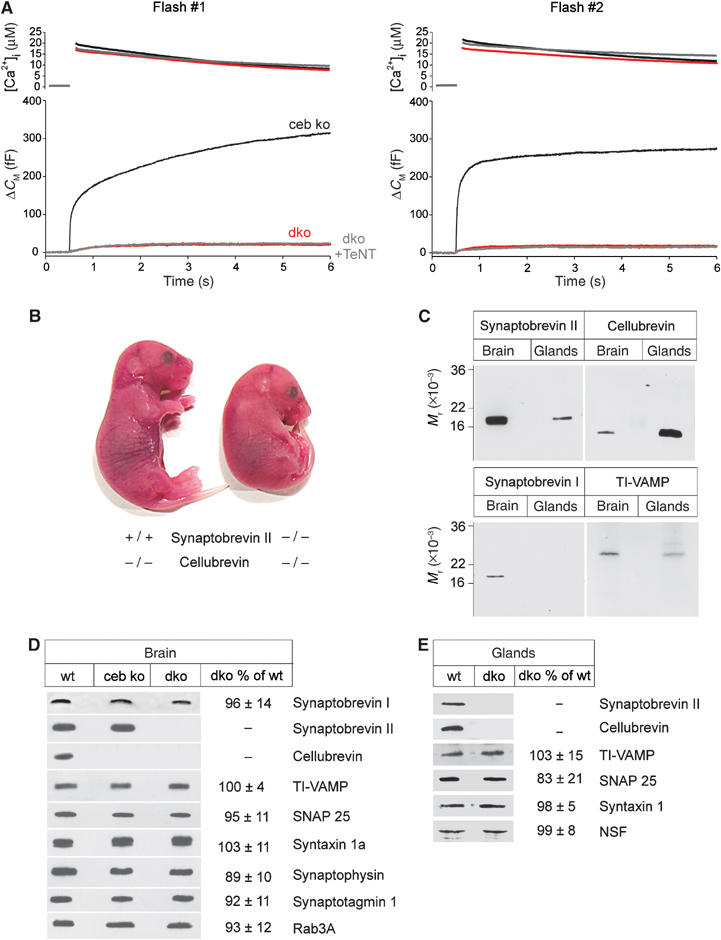
Double v-SNARE deficiency abolishes Ca2+-triggered secretion from chromaffin cells. (A) The flash-evoked capacitance increase is almost abolished in dko cells (n=20). ceb ko cells (n=20) serve as littermate control in these experiments. The residual capacitance response of dko cells is resistant to TeNT poisoning (dko+TeNT, n=12). Preflash [Ca2+]i levels (in μM): first flash: ceb ko 0.34±0.01, dko 0.33±0.01, dko+TeNT 0.39±0.02; second flash: ceb ko 0.43±0.02, dko 0.41±0.02, dko+TeNT 0.54±0.04. (B) Appearance of double v-SNARE-deficient mutant mice at stage E18.5 compared with littermates (ceb ko). (C) Survey on the expression of v-SNARE proteins in embryonic adrenal glands and the brain of wild-type (wt) animals. Each lane is an immunoblot of total homogenate (sybII and ceb, 5 μg/lane; synaptobrevin I, 15 μg/lane; TI-VAMP, 25 μg/lane). Note that ceb, detectable in brain homogenate, likely derives from glia cells (Chilcote et al, 1995). (D) Immunoblot analysis of total brain homogenate from wt and dko mice illustrates no major changes in the expression level of other synaptic proteins. Homogenates were probed with antibodies raised against the indicated proteins. Signals were visualized with enhanced chemiluminescence. (E) Immunoblot of embryonic adrenal glands from wt and dko animals indicates no change in the expression pattern of other synaptic proteins in the absence of sybII and ceb. Quantification of the immunosignals from dko in panels D and E is given as mean percentage±s.d. compared with wt. Data were collected from three independent preparations of brain and gland homogenate.
Using electron microscopy, we investigated whether removal of the two v-SNAREs affects number, size or spatial distribution of chromaffin granules (Figure 3A). dko cells exhibit the same density of vesicles as observed in controls (wt: 2.9±0.2 vesicles/μm2; dko: 3.1±0.3 vesicles/μm2), indicating that lack of the v-SNAREs does not interfere with the de novo synthesis of the secretory organelles (Figure 3B). The size distribution of double v-SNARE-deficient vesicles closely resembles that observed for wild-type vesicles (vesicle radius: wt: 65±18 nm; ceb ko: 66±16; dko: 66±15 nm; Figure 3C) and measurement of the granule-to-plasma membrane distance revealed neither significant changes in the number of morphologically docked vesicles (<50 nm) nor in the overall spatial distribution of the granules (Figure 3D). Thus, sybII and ceb are not required for biogenesis and docking of secretory organelles in chromaffin cells.
Figure 3.

Electron microscopy of chromaffin granules lacking sybII and ceb. (A) Exemplary electron micrographs of isolated mouse chromaffin cells from wild-type (wt) and dko animals. Scale, 2 μm. (B) dko cells exhibit the same density of chromaffin granules as found in ceb ko or wt cells. wt cells exhibit on average 156±13 granules/section. Data were collected from 16 wt, 17 ceb ko and 14 dko cells. (C) Size distribution of wt (black line, n=916), ceb ko (gray, n=1122) and dko granules (red, n=992). A Gaussian fitted to each of the distributions (not shown) was used to determine the parameters indicated in the text (mean±s.d.). (D) Subcellular distribution (bin width 50 nm) of chromaffin granules in wt (black), ceb ko (gray) and dko cells (red) measured as granule membrane to plasma membrane distance. Note that in dko cells, there is a slight (but nonsignificant) increase in the number of docked vesicles that may result from the observed blockade in exocytosis causing an accumulation of vesicles in the docked state. Inset: Morphologically docked granule in close proximity to the plasma membrane; intermembrane distance ∼10 nm. Scale, 100 nm.
v-SNARE deficiencies with other stimuli
The distinct functional consequences of the v-SNARE deficiencies are similarly evident for a physiological stimulus of granule exocytosis (Figure 4A–C). Exocytosis triggered by a train of depolarizations is reduced in sybII ko cells, unchanged in ceb ko cells and almost abolished in dko cells. This finding holds for the first capacitance response (within the stimulus train; Figure 4D), which mainly reflects exocytosis of release-ready vesicles (Voets et al, 1999). In contrast, amplitudes of the corresponding depolarization-induced Ca2+ currents are unaffected in all studied genotypes. Thus, granule exocytosis triggered by Ca2+ influx through voltage-gated Ca2+ channels exhibits the same dependence on the different v-SNARE proteins as observed with photolytic ‘uncaging' of Ca2+, putting no constraints on a preferred use of sybII during depolarizing stimuli, as one might expect from the strong inhibition of depolarization-induced release in sybII ko neurons (Schoch et al, 2001).
Figure 4.

v-SNARE dependence of depolarization-induced capacitance increase and catecholamine secretion. (A–C) Average CM responses from sybII ko (n=22), ceb ko (n=26) and dko cells (n=21) compared with the littermate control responses. Cells were stimulated with a train of 100 ms depolarizations (2.5 Hz, voltage protocol, upper panel). Note that wild-type (wt) cells show a stronger increase of the capacitance response than syb ko cells during the stimulus train. This is expected for a reduced SRP component in syb ko cells (as observed in the flash experiments), which occurs less synchronous with such a stimulus. Data were collected from more than 20 cells for each control group. (D) Mean amplitude of the Ca2+ current (ICa2+) and the corresponding CM increase (CM) for the first depolarization. CM was determined at the end of the capacitance recording interval. Data were normalized to the response of controls. The total capacitance increase recorded from dko cells at the end of the stimulus train amounts for less than 10% of the control response. (E–H) Representative amperometric recordings of catecholamine release induced by application of high K+-containing Ringer's solution (80 mM KCl) recorded for the indicated genotypes. Note that the response is reduced in sybII ko cells compared with wt cells and is abolished in dko cells. (I) Plot of the mean flash-evoked capacitance response (measured 5.5 s after stimulation) versus the mean number of amperometric events recorded for the different v-SNARE deficiencies. Data were normalized to the response of the corresponding control group. A linear regression (solid line) approximates the data (r2=0.98).
To study whether changes in the capacitance response observed for the different knockout phenotypes mirror indeed alterations in granule exocytosis, we used amperometry to measure catecholamine secretion directly. Wild-type cells respond to rapid application of high-potassium-containing solution with a strong increase in the frequency of amperometric spikes (48±3 events/application; Figure 4E). For dko cells, 10 out of 16 cells showed no exocytotic event with such a stimulus. On average, only 1±0.5 event/application is observed, corroborating the result that Ca2+-triggered granule exocytosis is abolished in the absence of both v-SNAREs (Figure 4H). A similarly strong block of exocytosis is found upon poisoning of wild-type cells with TeNT (0.8±0.4 events/application, n=10, data not shown). Figure 4I plots the amplitude of the flash-evoked capacitance response versus the mean number of amperometric spikes recorded for the different v-SNARE deficiencies. The combined set of data reveals a linear relationship, suggesting that changes in the Ca2+-triggered capacitance increase can be attributed to alterations in the frequency of granule exocytosis (r2=0.98). In contrast to other SNARE knockouts (Yoshihara et al, 1999; Schoch et al, 2001; Sorensen et al, 2003), a strong impairment of the release machinery in dko cells is also observed when a massive and long-lasting Ca2+ stimulus is used (Supplementary Figure C). In these experiments, cells were perfused with high-Ca2+-containing solution (10 μM free [Ca2+]i) and secretion was examined simultaneously with amperometry and membrane capacitance measurements. dko cells did not show a significant increase in either membrane capacitance (4% of control) or frequency of amperometric signals (6% of control). Taken together, double v-SNARE deficiency produces a nearly complete loss of Ca2+-dependent granule exocytosis.
Expression of synaptobrevin II and cellubrevin restores secretion in dko cells
To test whether the observed effects are specific for the absence of the individual v-SNAREs, we acutely overexpressed the proteins in dko cells using the Semliki forest virus system (Liljestrom and Garoff, 1991). Expression of sybII completely restores the flash-evoked response of dko cells to the level found in ceb ko cells, the littermate control (Figure 5A). Thus, except for the lack of the v-SNARE proteins, the release machinery remains intact in mutant cells, agreeing well with our biochemical and morphological results (Figures 2 and 3). A large amount of secretory activity is also regained by expression of ceb in dko cells resembling the sybII ko phenotype (Figure 1A). Evidently, the alternating expression of the individual v-SNAREs in dko cells creates a mirror image of the single-knockout phenotypes.
Figure 5.
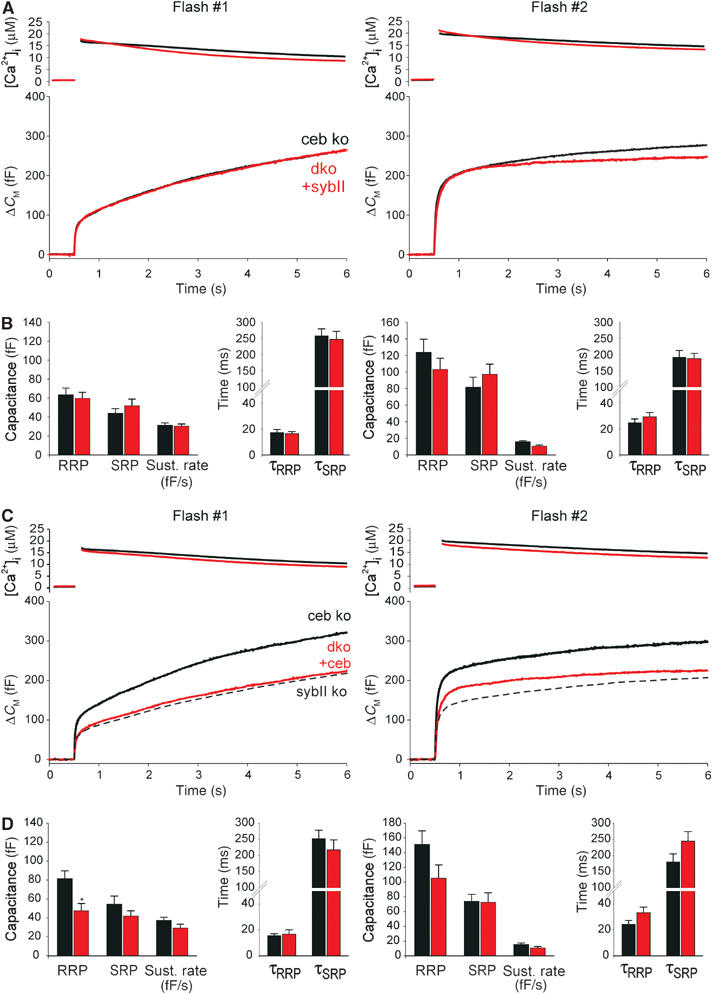
Expression of either v-SNARE, sybII or ceb, ‘rescues' secretion in dko cells. (A) Mean flash-evoked capacitance response of dko cells expressing sybII (dko+sybII, n=28) compared with controls (ceb ko, n=30). Preflash [Ca2+]i levels (in μM): first flash: ceb ko 0.40±0.01, dko+sybII 0.41±0.01; second flash: ceb ko 0.51±0.02, dko+sybII 0.61±0.03. Notably, secretion from dko cells is rescued within 4–6 h after start of virus-driven protein expression. This may be accomplished either by direct sorting of new v-SNAREs to pre-existing granules or by rapid de novo synthesis of granules. The latter scenario requires preferential recruitment of newly formed vesicles for exocytosis as observed by Duncan et al (2003). (B) Expression of sybII in dko cells (red bars) restores magnitude and kinetics of RRP and SRP as well as the sustained rate of secretion, control (black bars). (C) Average flash-evoked capacitance response of dko cells expressing ceb (dko+ceb, n=19) compared with controls (ceb ko, n=20). Preflash [Ca2+]i levels (in μM): first flash: ceb ko 0.35±0.01, dko+ceb 0.34±0.01; second flash: ceb ko 0.44±0.02, dko+ceb 0.47±0.02. The dashed line represents sybII ko measurement (from Figure 1A) indicating near identity with the dko+ceb phenotype. Note that the slight, but nonsignificant difference in the exocytotic burst size for the second flash response of dko+ceb and sybII ko cells is likely due to higher preflash [Ca2+]i levels in virus-transfected cells (0.47 μM) compared with nontransfected sybII ko cells (0.38 μM). (D) Expression of ceb in dko cells (red bars) partially restores secretion compared with controls (black bars). RRP and SRP sizes are ‘rescued' to 58 and 70% for the first flash and to 77 and 98% for the second flash, respectively. *P<0.05 compared with controls (Student's t-test).
By measuring the change in protein levels upon overexpression in dko cells, we found that sybII and ceb levels increase on average more than 14-fold compared with endogenous levels in wild-type cells (Supplementary Figure A and B), indicating that molecular differences rather than different expression levels of the proteins determine the phenotypes. Consistent with this result, secretion from cells heterozygous for sybII, analyzed either with amperometry or with capacitance measurements, was indistinguishable from wild-type responses (data not shown).
The cytoplasmic domains of sybII and ceb are nearly identical but exhibit one amino-acid exchange A37N and a nonconserved amino terminus (aa 1–32, numbers refer to sybII; Figure 6A). To test whether these structural determinants regulate the vesicle's priming stability, we exchanged the amino-terminal domains (aa 1–37) of the v-SNAREs. Indeed, expression of the chimera protein Syb-N (sybII N-terminus+ceb) in dko cells (Figure 6B and C) supports a larger secretory response and a larger exocytotic burst than the Ceb-N chimera (ceb N-terminus+sybII). Notably, Syb-N-mediated exocytosis closely resembles the ceb ko response (Figure 6B, gray line). For Ceb-N, the RRP and the SRP components are significantly reduced, whereas the kinetic rates of the exocytotic burst are unchanged. These results indicate that the amino-terminal domain of the v-SNARE protein controls the priming stability of chromaffin granules.
Figure 6.
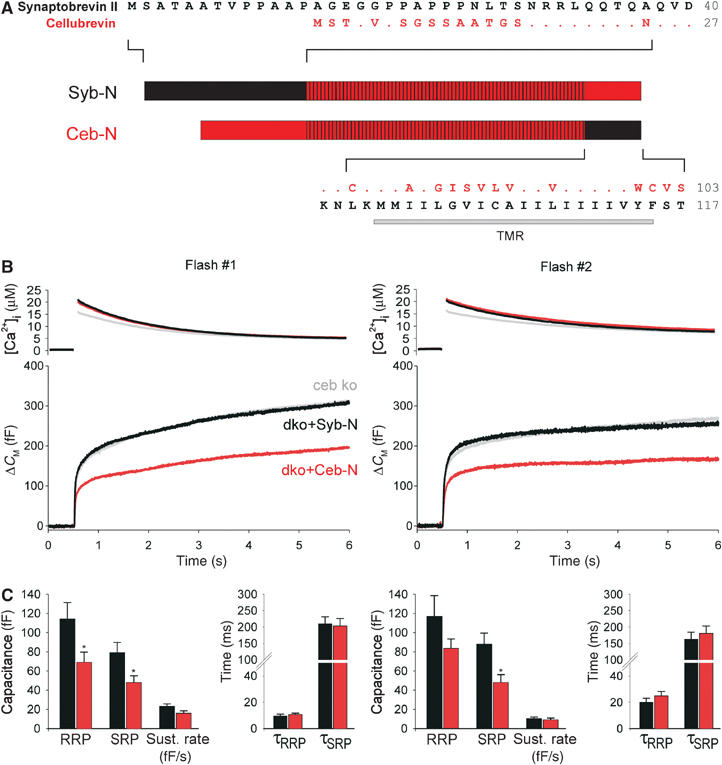
The amino terminus of the v-SNARE protein determines the vesicle's priming stability. (A) Schematic presentation of sybII/ceb chimeras: Syb-N (aa 1–37 sybII+aa 25–103 ceb); Ceb-N (aa 1–24 ceb+aa 38–117 sybII). The hatched area illustrates the conserved region of the proteins. Dots indicate amino acids of ceb (red) that are identical to those of sybII (black). TMR, transmembrane region. (B) Mean flash-evoked capacitance response of ceb ko cells (gray, n=37), dko cells expressing the chimera syb-N (black, n=24) and those expressing the chimera ceb-N (red, n=24). Preflash [Ca2+]i levels (in μM): first flash: ceb ko 0.4±0.1, syb-N 0.44±0.02, ceb-N 0.42±0.02; second flash: ceb ko 0.54±0.03, syb-N 0.58±0.03, ceb-N 0.56±0.05. (C) Syb-N expression in dko cells (black bars) supports a larger pool of exocytosis-competent vesicles than Ceb-N expression (red bars). *P<0.05 (Student's t-test).
Synaptobrevin II promotes faster fusion pore expansion than cellubrevin
To determine whether the functional differences between sybII and ceb extend to the level of membrane merger, we took advantage of the high-time resolution of amperometry (Figure 7A). For each amperometric spike, evoked by high-K+ depolarization in wild-type and single-knockout cells, quantal size, amplitude and kinetic parameters were determined. As shown in Figure 7B and C, neither the amount of transmitter released from a granule nor the peak amplitude of the oxidative current spike changes in the absence of one of the v-SNARE proteins. Exocytosis starts with the formation of a fusion pore that restricts initial transmitter efflux from the vesicle generating a ‘foot' signal, which precedes the amperometric spike (Chow et al, 1992; Bruns and Jahn, 1995; Albillos et al, 1997; Figure 7A). This initial phase of release is significantly altered in the absence of sybII. Fusion events, recorded in sybII ko cells, exhibit a much longer ‘foot' duration (Figure 7F–H) leading to an increase in charge and in maximum current amplitude of the ‘foot' signal as expected for prolonged fusion pore expansion (Table I, mean of median values determined for each cell). In contrast, the kinetic properties of the main phase of transmitter discharge are unaffected (Figure 7D and E), rendering the possibility unlikely that experimental inconsistencies, such as variable cell-electrode spacing, are responsible for the prolongation of the fusion pore signal. Thus, lack of sybII specifically delays the moment when the slow expansion of the fusion pore proceeds to a more rapid expansion mode (spike phase). In contrast, ‘foot' signals recorded from ceb-deficient cells are identical to those of wild-type cells (Figure 7F–H), indicating that chromaffin granules equipped only with ceb fuse more slowly with the plasma membrane than sybII-carrying granules. Expression of sybII in sybII ko cells fully restores the wild-type phenotype (Figure 7G and H), whereas expression of ceb failed to accelerate fusion pore expansion. Therefore, not the number of v-SNARE proteins, but their molecular properties determine the fusion pore dynamics. We conclude that v-SNAREs act at a late stage of exocytosis regulating the timing of fusion pore dilation.
Figure 7.
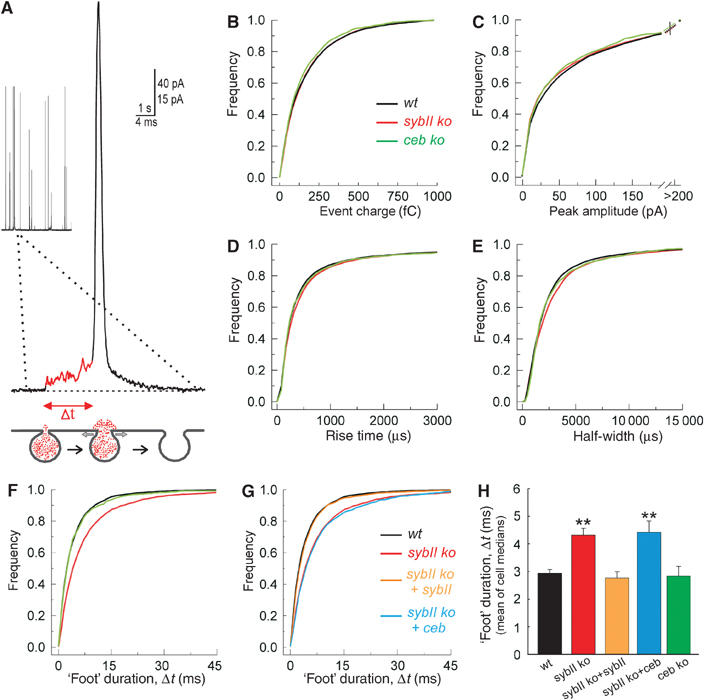
Lack of sybII prolongs the expansion time of the exocytotic fusion pore. (A) Exemplary amperometric event recorded with a carbon fiber in direct contact with the plasma membrane of a chromaffin cell. A ‘foot' signal (red line) precedes the main amperometric spike indicative for restricted transmitter efflux through a narrow fusion pore that slowly expands (t=‘foot' duration, see scheme) before bulk release, coinciding with more rapid pore dilation. (B–E) Characteristics of single spikes recorded from wild-type (wt; black), sybII ko (red) and ceb ko cells (green) displayed as cumulative frequency distributions for the indicated parameters. The properties of the amperometric signals from the v-SNARE-deficient cells are very similar compared with controls and curves partially overlap (values are given in Table I). (F) The ‘foot' duration of amperometric spikes is significantly longer in sybII ko (red) than in wt (black) or ceb ko cells (green). No change in the frequency of ‘foot' signals is observed (see Table I). (G) Expression of sybII (orange), but not of ceb (blue), in sybII ko cells (red) ‘rescues' the ‘foot' duration to wt levels (black). (H) Mean value of cell medians of ‘foot' duration for all studied conditions. *P<0.05 compared with wt cells.
Table 1.
Properties of individual fusion events in v-SNARE-deficient chromaffin cells
| ‘Foot' parameters |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype (no. of cells) | Events/applic. | Charge (fC) | Amplitude (pA) | Rise time (ms) | Half-width (ms) | % of events with ‘foot' | Amplitude (pA) | Duration (ms) | Charge (fC) |
| Wild type (51) | 48±3.0 | 100±5 | 30±2 | 0.28±0.01 | 1.7±0.07 | 56±3 | 6.5±0.3 | 2.9±0.1 | 9.2±0.5 |
| sybII ko (49) | 37±2.5** | 110±6 | 29±3 | 0.31±0.02 | 1.9±0.11 | 56±4 | 7.8±0.4** | 4.3±0.2** | 14±0.9** |
| sybII ko+sybII (21) | 45±4.5 | 105±8 | 26±3 | 0.32±0.02 | 1.9±0.17 | 57±5 | 7.2±0.5 | 2.8±0.2 | 10.7±1.2 |
| sybII ko+ceb (18) | 37±3.7** | 106±9 | 30±3 | 0.27±0.02 | 1.6±0.15 | 57±6 | 9.1±0.8** | 4.4±0.2** | 17.7±2.0** |
| ceb ko (13) | 42±3.5 | 96±9 | 28±4 | 0.28±0.02 | 1.8±0.14 | 57±5 | 6.4±0.8 | 2.8±0.3 | 9.4±1.1 |
| v-SNARE dko (16) |
1±0.5** |
— |
— |
— |
— |
— |
— |
— |
— |
| Events (>4 pA) were characterized with respect to their charge, peak amplitude and kinetic parameters (50–90% rise time, half-width) and concerning the properties of the ‘foot' signal (max. amplitude, duration and charge of the ‘foot'). The parameter ‘events per applic.' refers to the mean number of events stimulated by two applications of high-potassium solution (each stimulation 20 s, interval 60 s). Note the control group for ceb ko cells had a release frequency of 40±5.5 events/application (n=17). The parameter ‘% of events with foot' indicates the mean percentage of amperometric spikes preceded by a ‘foot' signal. Other values are given as mean±s.e.m. of the median determined from the parameter's frequency distribution for each cell. Total number of events analyzed per genotype is as follows: wt 4896, sybII ko 3620, sybII ko+sybII 1899 sybII ko+ceb 1332, ceb ko 1089. No significant changes were observed in mice heterozygous for sybII (n=32 cells) or for ceb (n=16 cells) compared with controls. **P<0.05 compared with wild-type cells. | |||||||||
Discussion
Functional redundancy of v-SNAREs in Ca2+-dependent granule exocytosis
In this study, we show that the presence of one of the v-SNARE proteins, sybII or ceb, suffices chromaffin granules to perform bona fide Ca2+-dependent exocytosis. The fidelity by which ceb promotes stimulus–secretion coupling was unexpected, considering that this protein is virtually undetectable in neurons, but it is in line with the high degree of sequence homology to sybII within the SNARE domain, which mediates the interaction with other SNARE partners (Sutton et al, 1998). This result, together with recent studies in yeast and Drosophila (Bhattacharya et al, 2002; Liu and Barlowe, 2002; Vilinsky et al, 2002), suggests functional redundancy among SNARE proteins as a more widespread phenomenon than expected from the hypothesis that these proteins encode for compartmental specificity in intracellular membrane transport (McNew et al, 2000). For example, experiments in yeast have shown that Ykt6p, a v-SNARE protein functioning at late stages of the secretory pathway, compensates for the lack of Sec22p, which is required for ER–Golgi transport (Liu and Barlowe, 2002). The ubiquitously expressed Drosophila v-SNARE syb, which might be comparable with ceb, is able to substitute, at least upon strong overexpression, for the neuronal isoform (n-Syb) in n-Syb nulls by supporting some evoked exocytosis at the neuromuscular junction (Bhattacharya et al, 2002). Our experiments in chromaffin cells, using high-resolution membrane capacitance measurements together with a well-defined Ca2+ stimulus, extend these findings. Particularly, ceb's property to support a standing pool of release-ready organelles with unchanged stimulus–secretion coupling identifies this protein as a true functionally redundant v-SNARE for rapid Ca2+-triggered exocytosis. Still, our results also provide evidence that sybII is the preferred, if not the only, v-SNARE acting in the secretion of wild-type chromaffin cells. First, lack of sybII attenuates secretion and, furthermore, its overexpression (but not of ceb) in double v-SNARE-deficient cells rescues the exocytotic response to wild-type levels showing that sybII is required and functionally sufficient as cognate v-SNARE. Second, ceb supports exocytosis only with reduced efficiency, but its removal causes no increase in secretion, as expected if both proteins would compete in the exocytotic mechanism. One might speculate that secretion has reached an upper limit under wild-type conditions preventing any further increase of the response upon removal of ceb. However, overexpression of ceb in wild-type cells produces no reduction of the secretory response (Supplementary Figure E), reinforcing the view that ceb does not compete with sybII in the exocytosis of wild-type cells. Taken together, these results suggest that ceb plays a minor, if any, role in granule exocytosis of wild-type cells, but efficiently substitutes for sybII in its absence. The different contribution of ceb to secretion from sybII ko and wild-type cells suggests a precise control that functionally segregates the individual v-SNARE components in the full context of the protein machinery but fails under genetically deficient conditions. Most likely, structures outside of the highly homologous SNARE domain encode for the preferential use of sybII. Its long, nonconserved amino terminus is well suited to confer specificity and to direct initial contact upon membrane-bridging SNARE interactions. Taken together, v-SNAREs fulfill an essential role in exocytosis in which they are promiscuous and exhibit independently regulatory elements, which contribute to specificity of v-SNARE action in wild-type cells. Interestingly, the nearly complete loss of secretion, even with long-lasting Ca2+ stimuli, clearly distinguishes double v-SNARE deficiency from other SNARE knockout phenotypes (Yoshihara et al, 1999; Schoch et al, 2001; Sorensen et al, 2003), likely because it removes the majority of promiscuously acting SNAREs in the exocytotic pathway.
v-SNARE specifics provide insight into the mode of SNARE function in exocytosis
Two distinct properties of granule exocytosis are affected by v-SNARE proteins—the number of granules that readily undergo exocytosis and the mode of fusion pore expansion once exocytosis has been initiated. In neurons, sybII deficiency strongly reduces the response to hypertonic sucrose stimulation that measures the pool of readily releasable vesicles (Schoch et al, 2001). Both, the single-knockout phenotypes and the rescue experiments in dko cells show that sybII supports a larger exocytotic burst than ceb. Since the sustained rate of secretion (reflecting ‘priming') remains unchanged, the increased pool size in the presence of sybII is best explained by reduced ‘depriming' of release-ready granules. Importantly, the effects observed for the two cognate v-SNAREs are remarkably similar to those found for different t-SNARE variants of SNAP-25, SNAP-25A and SNAP-25B (Sorensen et al, 2003). This suggests that the assembled (or assembling) SNARE complexes control the pool of release-ready organelles and by this may provide the platform for auxiliary proteins to regulate exocytosis competence either by controlling the availability of SNAREs or their direct association. We find that structural components within the amino-terminal domain (aa 1–37) of the v-SNARE protein comprise key factors that regulate the priming stability of chromaffin granules. It remains to be shown whether the single amino-acid difference (A37N) at the very N-terminal end of the SNARE domain or the neighboring, nonconserved amino terminus (aa 1–32) is responsible for the differential regulation of the primed vesicle pool by the two v-SNAREs. In any case, our results extend recent indications that SNAREs may partially assemble at their amino-terminal ends in the exocytosis-competent state, as judged from a differential sensitivity of neurotransmitter release to poisoning with TeNT and BoNT-B (Hua and Charlton, 1999).
Previous studies led to opposing views on the mode of SNARE action in exocytosis, suggesting that these proteins are either dispensable before fusion (Tahara et al, 1998; Ungermann et al, 1998) or have an executive role (Weber et al, 1998; McNew et al, 2000). In the latter studies, using a lipid dequenching assay, SNAREs reconstituted in liposomes have been shown to be essential and sufficient for fusion on the time scale of minutes to hours, leaving the question unanswered whether SNARE action directly links lipid mixing to full membrane merger and fusion pore opening or simply generates a hemifused state from which liposomes may fuse in a SNARE-independent fashion. We show that sybII and ceb differentially regulate the dynamics of the exocytotic fusion pore on a millisecond timescale. Thus, v-SNARE function remains visible after initiation of exocytosis and is therefore intimately tied to the mechanism that makes vesicles fuse. Recent models hypothesize that SNARE proteins ‘zip' from their membrane distant amino-terminal ends toward the membrane proximal carboxy termini (for review, see Chen and Scheller, 2001). While differences in the amino-terminal sequence of the v-SNAREs alter the priming stability, their nonconserved carboxy-terminal transmembrane domains (TMDs, amino-acid identity 44%) may be responsible for the fusion pore effect. The TMDs of sybII and syntaxin directly interact via contiguous areas of interfacial residues (Margittai et al, 1999; Laage et al, 2000) that are only partially conserved within the TMD of ceb (Figure 6A). Since fusion of membranes likely involves the transition of a trans-SNARE complex to its cis-orientation (all proteins within the same membrane), alterations of SNARE assembly between their carboxy-terminal TMDs can perturb local membrane curvature and fusion pore dilation.
In summary, the comparative analysis of different v-SNAREs favors a model of sequential v-SNARE action in rapid Ca2+-triggered exocytosis, in which the amino terminus regulates v-SNARE engagement and priming stability of the vesicles, the highly conserved SNARE motif ensures unperturbed initiation of exocytosis and the TMD controls the organelle's fusogenic properties.
Materials and methods
Mutant mice and cell culture
SybII and ceb mutant mice were maintained as heterozygotes by continuous crossbreeding with C57BL/6. ceb ko animals are viable and fertile. Thus, crossbreeding of the v-SNARE-deficient mouse strains allowed us to obtain animals that are heterozygous for sybII and homozygous knockouts for ceb. From the offspring of this generation, dko mutants were recovered at the expected Mendelian ratio (1.0:1.82:0.96; 109 animals, 19 preparations). These animals die at birth and exhibit a phenotype that compares well with the sybII ko phenotype (Schoch et al, 2001), having a tucked posture with neither spontaneous movements nor sensorimotor reflexes (Figure 2B). All analyses were performed on littermates at E17.5–18.5 derived from the heterozygote matings. Preparation and culturing of embryonic mouse chromaffin cells was performed as described (Sorensen et al, 2003). Recordings were performed at room temperature on days 1–3 of culture.
Whole-cell capacitance measurements and caged Ca2+ photolysis
Recordings were performed with patch pipettes and an EPC-9 amplifier (HEKA, Lambrecht, Germany). The Ringer's solution contained (in mM) 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 48 glucose, pH 7.3 with NaOH. For measuring depolarization-induced secretion, the pipette solution contained (in mM) 120 Cs-glutamate, 8 NaCl, 0.18 CaCl2, 0.28 BAPTA, 1 MgCl2, 2 Mg-ATP, 0.5 Na2GTP, 10 HEPES–CsOH, pH 7.3, 350 nM calculated free [Ca2+]i. Voltage-dependent Ca2+ currents were isolated by blocking K+ and Na+ currents with TEA-Cl (20 mM) and TTX (0.5 μM) added to the extracellular solution.
For ratiometric measurements of [Ca2+]i, a mixture of indicator dyes FURA-2 and Furaptra (Molecular Probes, Eugene, OR) was excited at 340/380 nm with a monochromator (Polychrome IV, TILL Photonics, Planegg, Germany). Whole-cell recordings with test solutions of defined calcium concentrations were used for in vivo calibration of the ratiometric Ca2+ signals. NP-EGTA (supplied by G Ellis-Davies, MCP Hahnemann University, Philadelphia, PA) was photolysed by a flash of ultraviolet light (xenon flash lamp, Rapp OptoElectronics, Hamburg, Germany) focused through a Zeiss objective (× 40, Fluar, 1.3) of an inverted microscope (Axiovert 200, Zeiss, Germany). The monochromator light was used to adjust [Ca2+]i after the flash by photolysing small amounts of NP-EGTA. The pipette solution for flash experiments contained (in mM) 90 Cs-aspartate, 10 NaCl, 4.63 CaCl2, 5 NP-EGTA, 0.2 FURA-2, 0.3 Furaptra, 2 Mg-ATP, 0.3 Na2GTP, 40 HEPES, 17.5 D-glucose, pH 7.3. For Ca2+ infusion of cells (10 μM free Ca2+), the pipette solution contained (in mM) 90 Cs-aspartate, 10 NaCl, 10 DPTA, 6.8 CaCl2, 0.2 FURA-2, 0.3 Furaptra, 2 Mg-ATP, 0.3 Na2GTP, 40 HEPES, 17.5 D-glucose, pH 7.3. Data were acquired with the Pulse software (HEKA, Lambrecht, Germany) and capacitance measurements were performed according to the Lindau–Neher technique (sine wave stimulus: 1000 Hz, 35 mV peak-to-peak amplitude, DC-holding potential −70 mV). Current signals were digitized at 20 kHz and membrane capacitance was analyzed with customized IgorPro routines (Wavemetrics, Lake Oswego, OR). The flash-evoked capacitance response was approximated with the following function: f(x)=A0+A1(1−exp(−(t)/τ1))+A2(1−exp(−(t)/τ2))+k(t), where A0 represents the cell capacitance before the flash. The parameters A1, τ1 and A2, τ2 represent the amplitudes and time constants of RRP and SRP, respectively.
Amperometry
Carbon fiber electrodes (Pan-T650, 5 μm diameter, Amoco, Greenville, SC) were prepared as described (Bruns, 2004). Amperometric currents were recorded with EPC-7 amplifier (HEKA, Lambrecht, Germany, electrode voltage +800 mV), filtered at 3 kHz (eight-pole Bessel) and digitized gap-free (25 kHz). For data collection and evaluation, the programs pClamp6 (Axon instruments, Foster City, CA) and AutesW (NPI Electronics, Tamm, Germany) were used. Signals were again digitally filtered at 3 kHz and analyzed with a customized event detection routine (Bruns et al, 2000). The analysis was restricted to events with a peak amplitude >4 pA and a total charge ranging from 10 to 1000 fC. The start of the ‘foot' signal is defined as the time point where the current amplitude exceeds two times the standard deviation of the average baseline noise and it ends at the inflection point between the slowly increasing ‘foot' signal and the more rapidly increasing spike current (Figure 7A). External solutions were delivered from a multichannel perfusion pipette (Bruns et al, 2000). Data were collected from release events stimulated by two applications of high K+-containing Ringer's solution (in mM: 50 NaCl, 80 KCl, 2 CaCl2, 1 MgCl2, 48 glucose, 10 HEPES–NaOH, pH 7.3) given with an interval of 1 min. Stimulation periods were bracketed by superfusion of cells with Ringer's solution.
Electron microscopy
Chromaffin cells (plated on Thermanox coverslips, Nunc, Rochester, NY) were fixed with 3% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4), osmicated with 2% OsO4 in 0.1 M cacodylate buffer (pH 7.4) for 1 h at 4°C and serially dehydrated in ethanol followed by infiltration with Embed-812 (EMS, München, Germany; polymerization 60°C, 48 h). Ultrathin sections (gray/silver interference color) were counterstained (2% uranyl acetate and lead citrate, 0.4 mg/ml, pH 12.0) and analyzed with a Philips Tecnai12 Biotwin electron microscope. The morphometric analyses were performed on randomly selected cells from three wild-type, three ceb ko and four dko animals. Images acquired with a MegaView III CCD camera (Soft Imaging System, Münster, Germany) were analyzed with the Metamorph software (Universal Imaging, West Chester, PA). The number and spatial distribution of chromaffin granules were evaluated on micrographs (× 30 000 magnification, digitally zoomed to × 122 500) covering the entire cell's cytoplasmic region. The granule diameter was measured on micrographs (magnification × 49 000, digitally zoomed to × 205 000) as described (Bruns et al, 2000).
Viral constructs
cDNAs encoding for sybII, ceb and TeNT light chain were subcloned into the viral plasmid pSFV1 (Invitrogen, San Diego, CA) upstream of an internal ribosome entry site that is followed by an open reading frame encoding for the enhanced green fluorescent protein (EGFP). EGFP labeling was used to identify infected cells. Preparation of virus particles was performed as described (Ashery et al, 1999).
Western blotting and immunocytochemistry
Whole mouse brain or adrenal gland extracts from embryos (E17.5–18) were prepared by homogenization in solution containing (in mM) 100 NaCl, 10 HEPES, 2 EDTA, 1 DTT, 2% Triton X-100, pH 7.3. Homogenates were incubated on ice for 30 min and insoluble material was removed by centrifugation (Beckmann SS34; 13 000 r.p.m., 15 min, 4°C). Samples were separated by SDS–PAGE, analyzed by immunoblotting and immunoreactive bands were visualized with enhanced chemiluminescence. Mouse monoclonal antibodies against synaptobrevin II (69.1), syntaxin (HPC-1), SNAP-25 (71.2), synaptotagmin (41.1), synaptophysin, synaptotagmin 1 and NSF as well as rabbit sera raised against synaptobrevin I were kindly provided by R Jahn (Göttingen, Germany). The monoclonal antibody against TI-VAMP and the rabbit serum against ceb were kindly provided by T Galli (Paris, France). ECL-processed films were scanned and protein bands were densitometrically quantified using Metamorph software. Antibodies directed against sybII and ceb were affinity-purified.
To determine changes in protein levels of sybII or ceb, chromaffin cells were processed for immunolabeling as described (Hannah et al, 1998). Epifluorescence pictures were acquired with an Imago-CCD camera (TILL Photonics, Martinsried, Germany) and were analyzed using the Metamorph software. For quantification of the immunosignal, the average intensity of the fluorescent immunolabel was determined within an area of interest that comprises the outer cell perimeter.
Supplementary Material
Supplementary Information
Acknowledgments
We express our gratitude to Drs E Neher and D Stevens for valuable discussions. We thank J Pessin (Stony Brook, NY) for providing the ceb ko animals and Drs G Nagy and J Sorensen for helping us to set up the mouse chromaffin cell culture. We are grateful to K Klingler, J Metzger, D Weber, M Wirth, K Wissel and C Bick for excellent technical assistance. The work was supported by grants from the DFG (SFB 530) and from the European Commission (QLG3-CT-2001-02430) to DB and by HOMFOR.
References
- Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389: 509–512 [DOI] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J (1999) An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol 78: 525–532 [DOI] [PubMed] [Google Scholar]
- Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R (1989) Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J 8: 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifer G, Steinhauser C, Pilati E, Volterra A (2004) Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci 7: 613–620 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Stewart BA, Niemeyer BA, Burgess RW, McCabe BD, Lin P, Boulianne G, O'Kane CJ, Schwarz TL (2002) Members of the synaptobrevin/vesicle-associated membrane protein (VAMP) family in Drosophila are functionally interchangeable in vivo for neurotransmitter release and cell viability. Proc Natl Acad Sci USA 99: 13867–13872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST (1995) Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron 15: 663–673 [DOI] [PubMed] [Google Scholar]
- Bruns D (2004) Detection of transmitter release with carbon fiber electrodes. Methods 33: 312–321 [DOI] [PubMed] [Google Scholar]
- Bruns D, Jahn R (1995) Real-time measurement of transmitter release from single synaptic vesicles. Nature 377: 62–65 [DOI] [PubMed] [Google Scholar]
- Bruns D, Riedel D, Klingauf J, Jahn R (2000) Quantal release of serotonin. Neuron 28: 205–220 [DOI] [PubMed] [Google Scholar]
- Chen YA, Scheller RH (2001) SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2: 98–106 [DOI] [PubMed] [Google Scholar]
- Chilcote TJ, Galli T, Mundigl O, Edelmann L, McPherson PS, Takei K, De Camilli P (1995) Cellubrevin and synaptobrevins: similar subcellular localization and biochemical properties in PC12 cells. J Cell Biol 129: 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow RH, von Ruden L, Neher E (1992) Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 356: 60–63 [DOI] [PubMed] [Google Scholar]
- Coco S, Raposo G, Martinez S, Fontaine JJ, Takamori S, Zahraoui A, Jahn R, Matteoli M, Louvard D, Galli T (1999) Subcellular localization of tetanus neurotoxin-insensitive vesicle-associated membrane protein (VAMP)/VAMP7 in neuronal cells: evidence for a novel membrane compartment. J Neurosci 19: 9803–9812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL (1998) Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci 18: 2028–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Greaves J, Wiegand UK, Matskevich I, Bodammer G, Apps DK, Shipston MJ, Chow RH (2003) Functional and spatial segregation of secretory vesicle pools according to vesicle age. Nature 422: 176–180 [DOI] [PubMed] [Google Scholar]
- Elferink LA, Trimble WS, Scheller RH (1989) Two vesicle-associated membrane protein genes are differentially expressed in the rat central nervous system. J Biol Chem 264: 11061–11064 [PubMed] [Google Scholar]
- Galli T, Chilcote T, Mundigl O, Binz T, Niemann H, De Camilli P (1994) Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J Cell Biol 125: 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah MJ, Weiss U, Huttner WB (1998) Differential extraction of proteins from paraformaldehyde-fixed cells: lessons from synaptophysin and other membrane proteins. Methods 16: 170–181 [DOI] [PubMed] [Google Scholar]
- Heinemann C, Chow RH, Neher E, Zucker RS (1994) Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+. Biophys J 67: 2546–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua SY, Charlton MP (1999) Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat Neurosci 2: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H (1994) A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron 12: 1269–1279 [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112: 519–533 [DOI] [PubMed] [Google Scholar]
- Laage R, Rohde J, Brosig B, Langosch D (2000) A conserved membrane-spanning amino acid motif drives homomeric and supports heteromeric assembly of presynaptic SNARE proteins. J Biol Chem 275: 17481–17487 [DOI] [PubMed] [Google Scholar]
- Liljestrom P, Garoff H (1991) A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology (NY) 9: 1356–1361 [DOI] [PubMed] [Google Scholar]
- Liu Y, Barlowe C (2002) Analysis of Sec22p in endoplasmic reticulum/Golgi transport reveals cellular redundancy in SNARE protein function. Mol Biol Cell 13: 3314–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margittai M, Otto H, Jahn R (1999) A stable interaction between syntaxin 1a and synaptobrevin 2 mediated by their transmembrane domains. FEBS Lett 446: 40–44 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof TC (1993) Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature 364: 346–349 [DOI] [PubMed] [Google Scholar]
- McNew JA, Weber T, Parlati F, Johnston RJ, Melia TJ, Sollner TH, Rothman JE (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol 150: 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB (2004) Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 41: 417–429 [DOI] [PubMed] [Google Scholar]
- Paumet F, Le Mao J, Martin S, Galli T, David B, Blank U, Roa M (2000) Soluble NSF attachment protein receptors (SNAREs) in RBL-2H3 mast cells: functional role of syntaxin 4 in exocytosis and identification of a vesicle-associated membrane protein 8-containing secretory compartment. J Immunol 164: 5850–5857 [DOI] [PubMed] [Google Scholar]
- Rettig J, Neher E (2002) Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science 298: 781–785 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122 [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ (1995) Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell 80: 311–320 [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75–86 [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 395: 347–353 [DOI] [PubMed] [Google Scholar]
- Tahara M, Coorssen JR, Timmers K, Blank PS, Whalley T, Scheller R, Zimmerberg J (1998) Calcium can disrupt the SNARE protein complex on sea urchin egg secretory vesicles without irreversibly blocking fusion. J Biol Chem 273: 33667–33673 [DOI] [PubMed] [Google Scholar]
- Ungermann C, Nichols BJ, Pelham HR, Wickner W (1998) A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol 140: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilinsky I, Stewart BA, Drummond J, Robinson I, Deitcher DL (2002) A Drosophila SNAP-25 null mutant reveals context-dependent redundancy with SNAP-24 in neurotransmission. Genetics 162: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T (2000) Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28: 537–545 [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T (1999) Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron 23: 607–615 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26 [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Wei S, Xu T, Ashery U, Kollewe A, Matti U, Antonin W, Rettig J, Neher E (2000) Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J 19: 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E (1998) Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci 1: 192–200 [DOI] [PubMed] [Google Scholar]
- Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R (1999) Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99: 713–722 [DOI] [PubMed] [Google Scholar]
- Yang C, Mora S, Ryder JW, Coker KJ, Hansen P, Allen LA, Pessin JE (2001) VAMP3 null mice display normal constitutive, insulin- and exercise-regulated vesicle trafficking. Mol Cell Biol 21: 1573–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y (1999) Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J Neurosci 19: 2432–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information