

Abstract
Influenza poses a significant threat to global health, with the ability to cause severe epidemics and pandemics. The polymerase basic protein 2 (PB2) of the influenza virus plays a crucial role in the viral replication process, making the CAP-binding domain of PB2 an attractive target for antiviral drug development. This study aimed to identify and evaluate potential inhibitors of the influenza polymerase PB2 CAP-binding domain using computational drug discovery methods. We employed a comprehensive computational approach involving virtual screening, molecular docking, and 500 ns molecular dynamics (MD) simulations. Compounds were selected from the Diverse lib database and assessed for their binding affinity and stability in interaction with the PB2 CAP-binding domain. The study utilized the generalized amber force field (GAFF) for MD simulations to further evaluate the dynamic behaviour and stability of the interactions. Among the screened compounds, compounds 1, 3, and 4 showed promising binding affinities. Compound 4 demonstrated the highest binding stability and the most favourable free energy profile, indicating strong and consistent interaction with the target domain. Compound 3 displayed moderate stability with dynamic conformational changes, while Compound 1 maintained robust interactions throughout the simulations. Comparative analyses of these compounds against a control compound highlighted their potential efficacy. Compound 4 emerged as the most promising inhibitor, with substantial stability and strong binding affinity to the PB2 CAP-binding domain. These findings suggest that compound 4, along with compounds 1 and 3, holds the potential for further development into effective antiviral agents against influenza. Future studies should focus on experimental validation of these compounds and exploration of resistance mechanisms to enhance their therapeutic utility.
Keywords: Influenza virus, PB2 CAP-binding domain, Virtual screening, Molecular dynamics simulations, Free binding energy, Free energy landscape
Subject terms: Virtual drug screening, Virtual screening
Introduction
Influenza remains one of the most challenging viral diseases affecting global public health due to its capacity to cause widespread epidemics and occasional pandemics1. The disease mainly results from the influenza virus, which targets the respiratory tract and can cause severe respiratory illness or even death, particularly in high-risk groups such as the elderly, young children, and people with chronic health conditions2. Influenza viruses belong to the Orthomyxoviridae family and are categorized into types A, B, and C, with type A viruses being the most virulent in humans3. One of the key proteins involved in the replication and transcription of the influenza virus is the polymerase basic protein 2 (PB2) of the viral RNA polymerase complex4. The segmented genomes of these viruses facilitate frequent reassortment, leading to antigenic variations and drifts. This genetic shuffling can produce new strains that occasionally evade pre-existing immune responses, resulting in seasonal epidemics and pandemics3. PB2 is crucial for recognizing and binding to the cap structure of pre-mRNAs, a process essential for viral RNA synthesis and transcription5. It is primarily responsible for recognizing and binding to the cap structure of pre-mRNAs, which is crucial for the viral RNA synthesis process5. This cap-snatching mechanism is essential for viral transcription and replication, making the CAP-binding domain of PB2 an attractive target for antiviral drug development6. Historically, influenza outbreaks such as the infamous 1918 Spanish flu, 1957 Asian flu, 1968 Hong Kong flu, and the more recent 2009 H1N1 pandemic have underscored the need for effective antiviral agents7. The 1918 H1N1 pandemic, for instance, caused unprecedented mortality and morbidity, largely due to the lack of effective antiviral treatments and vaccines. This pandemic provided early evidence of the virus’s potential for widespread devastation, thus marking the inception of the urgent need for antiviral research. Subsequent outbreaks have continued to reflect this pattern, where the appearance of novel influenza strains outpaced the effectiveness of existing treatments, compelling the scientific community to innovate and develop new antiviral strategies8. Furthermore, the recent H1N1 outbreak in 2009 illustrated the rapidity with which influenza viruses can disseminate globally, challenging public health systems and antiviral stockpiles. This event emphasized the importance of having a diverse arsenal of antiviral agents that can be deployed promptly in response to new strains7. These events highlight the potential global health crises that can arise from new influenza strains, emphasizing the importance of continued surveillance, vaccine development, and therapeutic interventions9.
Despite the availability of vaccines and existing antiviral drugs like oseltamivir and zanamivir, there is a persistent need for new therapeutic options, particularly those that can target different stages of the viral life cycle or offer treatment alternatives resistant to current therapies10. The PB2 CAP-binding domain, due to its essential role in viral replication, represents a potent target for such interventions11 Currently, the inhibitor landscape for targeting the PB2 CAP-binding domain is still in its developmental stages. The inhibitors aim to disrupt the cap-snatching activity of the PB2 protein, thereby inhibiting the replication of the influenza virus. However, the development of PB2 inhibitors has been challenging due to the need for high specificity and the potential for rapid development of resistance by the virus12. The exploration of potential inhibitors has been significantly advanced by computational approaches, particularly through the use of diverse chemical libraries. The Diverse lib database is an expansive collection of molecular entities that provides a broad range of chemical diversity, enhancing the likelihood of identifying unique structures with potential inhibitory activity against specific viral targets13. Our research leverages computational drug discovery methodologies to screen the Diverse lib database for potential inhibitors of the Influenza Polymerase PB2 CAP-Binding Domain14. By applying a combination of virtual screening with Lipinski rule of five as a filter, followed by re-docking procedures, molecular dynamics (MD) simulations, and free energy calculations, we aim to identify promising candidates for further experimental validation. Additionally, RG-RMSD-based free energy landscapes provide insights into the dynamic stability and binding affinities of the interactions between potential inhibitors and the PB2 CAP-binding domain. This comprehensive approach not only enhances our understanding of the interaction mechanisms but also accelerates the discovery process for efficacious and safe antiviral agents.
Methodology
Data collection and high-throughput virtual screening
In this study, we adopted a computational approach to identify potential inhibitors of the Influenza Polymerase Pb2 CAP-Binding Domain. Initially, protein and ligand datasets were collected. Protein data was obtained from the PDB (ID: 4CB5) and ligand data from the Diverse lib database13,15,16. Protein preparation involved cleaning the structure and optimizing geometry using Chimera's Dock Prep tool17,18. This step involved cleaning the protein structure by removing water molecules and any bound ligands or ions, followed by the addition of hydrogen atoms to correct the protonation state and optimize the overall geometry of the protein.
This step involved cleaning the protein structure by removing water molecules and any bound ligands or ions, followed by the addition of hydrogen atoms to correct the protonation state and optimize the overall geometry of the protein. For the virtual screening, we utilized the MtiOpenScreen web server13, which integrates a Lipinski filter to ensure that the screened compounds adhered to basic pharmacokinetic principles19. This screening process resulted in the identification of several candidate compounds, from which four were selected based on superior docking scores indicating potential high-affinity binding to the target domain. Re-docking was performed using the Autodock Vina Chimera plugin20. This step served to validate the initial docking results through a more refined computational model. Both the candidate compounds and a reference molecule were docked into the binding site of the target protein. This re-docking process helped in comparing the binding affinities and conformations of the candidates relative to the known inhibitor, thereby affirming their potential efficacy.
MD simulation
In this study, molecular dynamics (MD) simulations were employed to investigate the dynamic behaviour of Influenza Polymerase Pb2 CAP-Binding Domain in a complex with four potential inhibitors sourced from the Diverse lib database, alongside a control compound. MD simulations were performed using the generalized amber force field (GAFF) in AMBER21. Ligand preparation included assigning force field parameters and charges. Each complex was solvated in TIP3P water, equilibrated, and subjected to a 500 ns production run22. The preparation of the ligand molecules began with the antechamber tool within AMBER23, which facilitated the assignment of force field parameters and atomic charges suitable for non-standard residues in the ligands. This preparation is essential for ensuring accurate interactions within the simulated system. Following the ligand preparation, the complex assembly and system configuration were accomplished using the leap tool from AMBER24. The simulation protocol initiated with a comprehensive energy minimization Energy minimization was performed by PMEMD.cuda module24. This was succeeded by a temperature equilibration phase where each system was heated progressively from 0 to 300 K across a span of 100 ps, utilizing the NVT ensemble (constant volume and temperature). Following temperature equilibration, the systems were further equilibrated under NPT conditions (constant pressure and temperature) for 1 ns to ensure stable density and thermal conditions, setting the stage for the production MD simulations. For each complex, including the control, a 500 ns production run was carried out under constant pressure (1 atm) and temperature (300 K). The Langevin thermostat was employed for temperature regulation25, while pressure was managed using the Berendsen barostat26. Trajectory analysis included RMSD, RMSF, and the number of hydrogen bonds to assess stability and dynamics. The RG-RMSD-based free energy landscape provided insights into the collective movements and binding efficacy. Additionally, free energy calculations were performed to delineate the binding efficacy and identify favorable conformational states, offering a detailed portrayal of the interaction landscape and energetics.
Free binding energy
Entropy effects are crucial in drug–target interactions, yet the contribution of entropy to ligand-binding affinity is frequently overlooked by end-point binding free energy calculation methods, like MM/GBSA27. In this study, the free binding energy of selected inhibitor complexes was quantified to assess their potential efficacy. The analysis was conducted on the last 50 ns of molecular dynamics simulation trajectories. We employed the MMPBSA.py tool from the AMBER software suite to calculate these energies28. The methodology involved three primary steps, simulation preparation, trajectory analysis, and energy calculation. Initially, the system comprising the protein–ligand complex was stabilized using a standard equilibration protocol in AMBER24. This protocol adjusted the system to a suitable temperature and pressure, ensuring realistic physiological conditions. Following stabilization, the production phase of molecular dynamics simulations was executed. During this phase, data on the atomic positions and velocities were recorded, providing detailed insights into the dynamic interactions within the complex. For the analysis, the MMPBSA.py module was utilized to compute the free binding energy from these simulations. This computation was based on the (MM-GBSA) method, a widely recognized approach for this purpose29. The module calculates the energy by considering the difference between the total free energy of the protein–ligand complex and the sum of the free energies of the unbound protein and ligand. This difference represents the binding energy, which is crucial for understanding the binding efficacy of the compounds. This detailed analysis allows for the evaluation of the thermodynamic stability of the inhibitor complexes under investigation, providing valuable insights into their potential as therapeutic agents against influenza.
RG-RMSD-based free energy landscape
The study investigated the dynamic conformational changes and stability of four lead compounds bound to a target protein using the RG-RMSD-based free energy landscape approach30. This method calculates Radius of Gyration (RG) and Root Mean Square Deviation (RMSD) values for each protein–ligand complex to understand their interaction dynamics during molecular dynamics simulations using the Geo-Measure plugin in PyMOL31. The RG measures the compactness of the molecule, while RMSD indicates the average distance between atoms of the complex compared to a control structure over time. By plotting these values, a free energy landscape was created, illustrating energetically favorable conformations and transitional states. This analysis is crucial for predicting the stability and effectiveness of potential inhibitors.
Results
Virtual screening
Virtual screening is a valuable computational technique for discovering bioactive molecules within extensive chemical libraries, aiming to identify those that can bind to specific biological targets. In our research, we employed virtual screening to pinpoint compounds from the Diverse lib database on the MTIopen web server. The selection process for these compounds was guided by their binding energy values obtained during the screening. We carried out a thorough analysis of 1500 compounds from the diverse lib Database on the MTIopen web server13. The binding energies of these molecules ranged from − 10.5 to − 7.8 kcal/mol (Supplementary Table S1). Based on their notable binding energies, four compounds—compound 1 (81066370), compound 2 (24334311), compound 3 (85303482) and compound 4 (85176812) as shown in Table 2 (Table 1).
Table 2.
Intermolecular interaction of four complexes in complex with the protein (a) compound 1, (b) compound 2, (c) compound 3, (d) compound 4 and (e) control compound.
| S no | Complex | H-bond | Hydrophobic | π–π stacking/π–π cation* |
|---|---|---|---|---|
| 1 | Compound 1 | Arg355 | Phe363, Ser337, His432, Ser324, Met431, Asn429, Arg332, Phe404 | Phe323, His357 |
| 2 | Compound 3 | – | Glu361, Arg332, Ser337, Phe363, Phe404, Asn429, Ser324 | Phe323, His357, Phe325 |
| 3 | Compound 4 | His357 | His432, Arg332, Ser337, Glu361, Lys376, Gln406, Ile354, Lys353, Glu341 | Phe323 |
| 4 | Control | Glu361, Ser324 | His432, Phe325, Arg332, Ser337, Phe363, Lys376, Phe404 | Phe323 |
Table 1.
List of 4 selected compound.
| Compound | Pubchem ID | Formula |
|---|---|---|
| 81066370 | CID 42628216 | (R)-5-(benzo[d][1,3]dioxol-5-yl)-3-(quinolin-6-yl)cyclohex-2-enone |
| 24334311 | CID 3845657 | (1-(3-(4-bromophenyl)-5-(quinolin-6-yl)-4,5-dihydro-1H-pyrazol-1-yl)ethanone) |
| 85303482 | CID 645155 | 1,3-Dimethyl-5-m-tolyl-5,5a-dihydro-1H-indeno[2',1':5,6]pyrido[2,3-d]pyrimidine-2,4,6-trione |
| 85176812 | CID 24817336 | (N-[1-(3,4-dimethylphenyl)-4,5,6,7-tetrahydroindazol-4-yl]-2,5-dimethyl-3-pyrazolecarboxamide) |
| 93G | CID 72206625 | 9-N-(3-CARBOXY-4-HYDROXYPHENYL)KETOMETHYL-7-N-METHYLGUANINE |
For the safely and toxicity prediction of these compound we have also performed the ADMET analysis by using ADMETLAB 2.0 webserver. The ADMET (absorption, distribution, metabolism, excretion, and toxicity) analysis of compound 81066370 highlights its high lipophilicity (LogP: 4.248) and strong inhibition of P-glycoprotein, which can enhance drug retention in cells. It has high plasma protein binding (98.63%) and moderate blood–brain barrier permeability, suggesting potential central nervous system activity. The compound shows significant interactions with CYP ( cytochrome P450) enzymes, indicating robust metabolic stability. Despite its low solubility and absorption, it meets Lipinski’s rule of five and the Golden Triangle rule, demonstrating good drug-likeness. Compound 24334311 exhibits high lipophilicity (LogP: 3.989) and strong P-glycoprotein inhibition, with excellent blood–brain barrier permeability (BBB: 0.989) and significant interactions with CYP enzymes, ensuring metabolic stability. It has high plasma protein binding (94.40%) and a higher fraction of the drug remains unbound in plasma (10.66%), which is favorable for pharmacological activity. This compound also complies with Lipinski's and the Golden Triangle rules, indicating good drug-likeness and potential therapeutic efficacy. Compound 85303482 shows high lipophilicity (LogP: 2.982) with minimal interaction as a P-glycoprotein inhibitor or substrate. It has high plasma protein binding (94.49%) and moderate blood–brain barrier permeability, which may be beneficial for central nervous system applications. The compound demonstrates robust metabolic stability through significant interactions with CYP enzymes. It meets Lipinski's rule of five and the Golden Triangle rule, highlighting its potential as a viable drug candidate as ashown in Table S2.
Redockin and Intermolecular Analysis
Re-docking is a crucial step in drug development, involving a rigorous reevaluation of the interactions between ligand molecules and receptor proteins. In our study, we employed specific parameters for the re-docking process: the docking grid's center was set at coordinates (X = − 50.0, Y = 4.48, Z = − 3.14) with grid dimensions of 20 Å for each axis (X, Y, Z). This approach is vital for validating initial docking results and clarifying key interactions at the receptor's binding site with the ligand. During the re-docking, each ligand was systematically aligned with the target protein and evaluated against a benchmark molecule, generating at least nine distinct poses per ligand-receptor pair. The pose with the most favorable docking energy, indicated by the lowest negative value, was chosen for in-depth analysis. This step was critical for further investigations into the stability of the complex and the affinity between the ligand and the target protein. Simultaneously, a virtual screening was conducted to explore a vast library of medicinal compounds. This screening highlighted four compounds with notably significant binding energies, recorded as − 10.4 kcal/mol, − 10.1 kcal/mol, − 9.9 kcal/mol, and − 9.8 kcal/mol. These compounds, referred to as compounds 1, 2, 3, and 4, showed promising potential and were selected for additional research. A control molecule, “93G”, with a binding energy of -7.8 kcal/mol, was used for comparative purposes. The three-dimensional structures of these molecules, including the control, were visualized using PyMOL software32, and two-dimensional figures were generated by Biodiscovery Studio33, enhancing our understanding of their spatial conformations and interactions within the binding site as illustrated in Fig. 1.
Figure 1.

The 3D and 2D structure of the selected compound in complex with the protein (a, b) compound 1, (c, d) compound 2, (e, f) compound 3, (g, h) compound 4 and (i, j) control.
As shown in Table 2, the current study involved a thorough examination of the molecular interactions that occurred between the selected compound and a target protein Compound 1 exhibited a singular hydrogen bond with Arginine 355 (Arg355) and engaged in eight hydrophobic interactions involving Phenylalanine 404 (Phe404), Arginine 332 (Arg332), Asparagine 429 (Asn429), Methionine 431 (Met431), Serine 324 (Ser324), Histidine 432 (His432), Serine 337 (Ser337), and Phenylalanine 363 (Phe363). Additionally, two π-π stacking interactions were identified with Phenylalanine 323 (Phe323) and Histidine 357 (His357). Compound 2 was formed eight hydrophobic interactions with Serine 337 (Ser337), Glutamate 361 (Glu361), Phenylalanine 363 (Phe363), Phenylalanine 404 (Phe404), Methionine 431 (Met431), Glutamate 341 (Glu341), Asparagine 429 (Asn429), and Arginine 332 (Arg332). This compound also formed two π–π stacking interactions with protein residue Phe323 and His357. Compound 3 formed seven hydrophobic interactions with Glutamate 361 (Glu361), Arginine 332 (Arg332), Serine 337 (Ser337), Phenylalanine 363 (Phe363), Phenylalanine 404 (Phe404), Asparagine 429 (Asn429), and Serine 324 (Ser324). Furthermore, three π-π stacking interactions were observed with Phenylalanine 323 (Phe323), Histidine 357 (His357), and Phenylalanine 325 (Phe325). Compound 4 exhibited one hydrogen bond with Histidine 357 (His357) and engaged in nine hydrophobic interactions with Glutamate 341 (Glu341), Lysine 353 (Lys353), Isoleucine 354 (Ile354), Glutamine 406 (Gln406), Glutamate 361 (Glu361), Lysine 376 (Lys376), Serine 337 (Ser337), Arginine 332 (Arg332), and Histidine 432 (His432). Additionally, a single π-π stacking interaction was noted with Phenylalanine 323 (Phe323). The control compound exhibited two hydrogen bonds with Serine 324 (Ser324) and Glutamate 361 (Glu361), alongside seven hydrophobic interactions with Histidine 432 (His432), Phenylalanine 325 (Phe325), Arginine 332 (Arg332), Serine 337 (Ser337), Phenylalanine 363 (Phe363), Lysine 376 (Lys376), and Phenylalanine 404 (Phe404), as well as a π-π stacking interaction.
MD simulation
Molecular dynamics (MD) simulations play a crucial role in elucidating the dynamic stability of protein–ligand complexes, offering detailed insights into temporal molecular interactions34–37. To advance our comprehension of the binding interactions and mechanistic behaviors of four selected compounds with a target protein, we conducted an extensive 500-ns MD simulation. A well-characterized control compound was used for comparative analysis purposes. Throughout the simulation, the trajectories of each molecule were meticulously recorded, enabling the observation of conformational changes during the binding process. Analysis of the simulation data allowed for the assessment of key interaction parameters, including binding affinity, conformational alterations within the protein, and shifts in the energy landscape that impact the stability and functionality of the protein–ligand complex. However, Compound 2 exhibited suboptimal performance in the MD simulation; therefore, subsequent studies focused solely on Compounds 1, 3, and 4, excluding Compound 2 from further analysis. This targeted approach facilitates a more refined investigation into the structural dynamics and interaction efficiency of the remaining compounds. The last pose and first pose before and after the MD simulation is illustrated in Fig. 2.
Figure 2.

The 2D structure of the selected compound in complex with the protein (a, b) compound 1, (c, d) compound 2, (e, f) compound 3, (g, h) compound 4 and (i, j) control.
RMSD analysis
In our molecular dynamics simulations, Compounds 1 and 3 exhibited the protein root-mean-square deviation (RMSD) values below 2 Å, indicative of stable protein conformations. Notably, Compound 3 exhibited a protein RMSD of 2.5 Å during the interval from 150 to 250 ns. In contrast, both Compound 4 and the control compound maintained protein RMSD values below 3 Å throughout the simulation period. Regarding ligand stability, Compound 1 exhibited a ligand RMSD of 2 Å until 210 ns, followed by an increase to less than 4 Å for the remainder of the simulation. Compound 3 showed a ligand RMSD of 3.5 Å, with minor fluctuations observable up to 200 ns, likely due to conformational changes within the ligand. Meanwhile, Compound 4 maintained a ligand RMSD of less than 4 Å throughout the entire 500 ns simulation. Similarly, the control complex exhibited consistent ligand RMSD values below 3 Å during the 500 ns simulation, suggesting a relatively stable interaction between the ligand and the protein over time. These findings provide crucial insights into the dynamic stability and conformational integrity of the ligand–protein interactions for each compound assessed as illustrated in Figs. 3 and 4.
Figure 3.
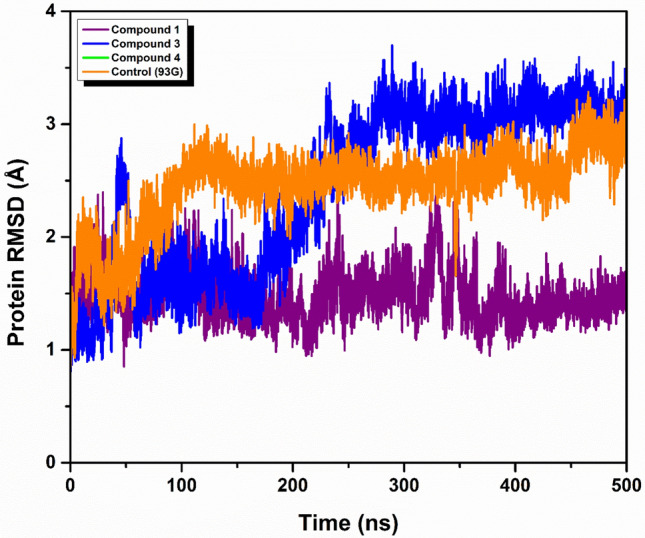
RMSD analysis of protein in complex with three selected compounds protein (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control.
Figure 4.
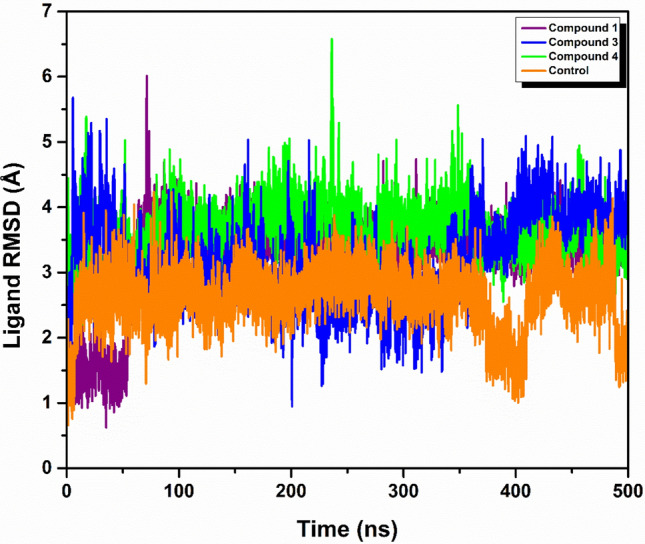
RMSD analysis of three selected ligand (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control in complex with protein.
RMSF analysis
Protein in complex with compound 1 exhibited notable stability in its interaction with the protein, as indicated by a root mean square fluctuation (RMSF) value of less than 3 angstroms (Å) across the amino acid residues numbered 90 to 110. Such low RMSF values suggest minimal deviation in the positions of these residues when bound with Compound 1, indicating a tight and stable interaction within this specific segment of the protein. While Protein in complex with compound 3 exhibited the protein RMSF value below 5 Å for the residues between 90 and 110. While this value is slightly higher than that observed for Protein in complex with compound 1, it still represents a relatively stable interaction within this region of the protein–ligand complex, albeit with slightly more flexibility or movement compared to the protein interaction in compound 1. On other hand protein in complex with compound 4 exhibited a different pattern where the protein RMSF exceeded 3 Å for residues 130 to 140. This indicates a higher degree of fluctuation or movement in this region, suggesting less stability in the protein structure when interacting with Compound 4. The increase in RMSF suggests that these residues experience more dynamic movements, which might affect the efficacy or stability of the compound-protein interaction. For the same residue range of 130 to 140, it was also noted that in control, the protein RMSF was less than 3 Å. The RMSF analysis of all the compounds is illustrated in Fig. 5.
Figure 5.

RMSF analysis of protein in complex with three selected compounds (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control.
Radius of gyration analysis
The radius of gyration (RG) serves as a quantitative measure of the compactness of a protein's tertiary structure and provides insight into its structural stability within biological systems38. An elevated RG value signifies a looser packing configuration, which may correlate with reduced stability due to increased vulnerability to environmental perturbations. Conversely, a lower RG is indicative of a tightly packed, more stable conformation. This relationship is visually elucidated in Figure S1, where varying RG values are correlated with distinct structural transformations in proteins, thereby underscoring their consequential effects on protein stability.
Hydrogen bond analysis
In the initial stages of the simulation, Compound 1 formed one to two hydrogen bonds. However, as the simulation progressed, the stability of these hydrogen bonds varied, with bonding becoming confined to specific areas. This observation suggests a possible alteration in the molecular conformation of its binding sites. Compound 3 demonstrated a more stable yet minimal hydrogen bonding pattern. It started the simulation with a single hydrogen bond, which remained consistent throughout the duration of the simulation. The persistence of only one hydrogen bond suggests a rigid molecular structure or a constrained environment, potentially limiting further bonding opportunities. Compound 4 displayed a consistent hydrogen bonding pattern, maintaining one to two hydrogen bonds throughout the simulation. This consistent bonding indicates a balanced and flexible molecular configuration, which allows for stable interactions without significant changes in bonding patterns. This flexibility could be crucial for its functionality in specific environments or applications. The control compound exhibited a more complex and sustained hydrogen bonding pattern, consistently forming two to three hydrogen bonds up to the 500 ns mark of the simulation. This robust bonding behavior underscores a strong and stable interaction capability, with multiple bonding sites actively engaged over an extended period. This could reflect a complex molecular structure designed to engage more extensively with its surroundings, potentially making it a useful standard for comparison in studies of molecular interaction dynamics as illustrated in Fig. 6.
Figure 6.

Hydrogen bond analysis of three compounds in complex with protein (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control.
Binding free energy analysis
The binding free energies of multiple complexes were quantitatively evaluated using the molecular mechanics/generalized Born surface area (MM/GBSA) approach. This technique facilitates the breakdown of the overall binding energy (ΔG) into its constituent energy components, thereby enabling a detailed analysis of the energetic contributions from the interactions between the ligand and the target protein, as delineated in Table 3. Specifically, Compound 1 exhibited a ΔG of − 48.46 ± 8.48 kcal/mol, while Compound 3 and Compound 4 exhibited ΔGs of − 52.95 ± 8.14 kcal/mol and − 53.30 ± 11.57 kcal/mol, respectively. The control complex showed a ΔG of − 52.14 ± 45.08 kcal/mol. Notably, the ΔG values of these complexes aligned closely with those of the control complex, indicating consistent and robust binding interactions with the target protein. This high degree of agreement with the control complex highlights the effective and stable binding dynamics of these compounds. Additionally, the small standard deviations associated with these values suggest a high level of reliability and reproducibility in the binding energy estimations. Collectively, these data suggest that the compounds under study exhibit a strong affinity for the target protein, classifying them as robust and effective ligands with the potential to modulate protein activity. These results emphasize the potential of these molecules as potent inhibitors within their respective biological pathways.
Table 3.
MMGBSA analysis of three compounds in complex with protein (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control.
| Energy components/complexes | Compound 1 | Compound 3 | Compound 4 | Control |
|---|---|---|---|---|
| Van der Waal energy (ΔVDWAALS) | − 37.79 ± 2.14 | − 41.54 ± 2.39 | − 42.90 ± 3.19 | − 38.93 ± 3.20 |
| Electrostatic energy(ΔEEL) | − 6.24 ± 2.78 | − 6.98 ± 1.95 | − 14.18 ± 3.28 | − 53.06 ± 20.73 |
| Polar solvation energy (ΔEGB) | 16.85 ± 2.39 | 18.02 ± 2.23 | 25.34 ± 3.05 | 56.15 ± 20.31 |
| Non-polar solvation energy (ΔESURF) | − 21.27 ± 1.15 | − 22.44 ± 1.55 | − 21.56 ± 2.04 | − 10.12 ± 0.82 |
| Net gas phase energy (ΔGGAS) | − 44.03 ± 4.93 | − 48.53 ± 4.35 | − 57.08 ± 6.47 | 14.12 ± 23.94 |
| Net solvation energy (ΔGSOLV) | − 4.42 ± 3.55 | − 4.42 ± 3.79 | 3.78 ± 5.09 | 66.27 ± 21.14 |
| ΔGtotal | − 48.46 ± 8.48 | − 52.95 ± 8.14 | − 53.30 ± 11.57 | − 52.14 ± 45.08 |
RG-RMSD and PCA based free energy landscape
The free energy landscape is a fundamental concept in molecular dynamics simulations, offering insights into the energy distribution within biomolecular systems and their conformational behaviour. This landscape is crucial for understanding the thermodynamic and kinetic properties that influence the actions of complex biomolecular structures. It outlines the energy barriers, stable states, and transition pathways among different molecular conformations. Gaining a deep knowledge of thermodynamics and kinetics over this landscape is vital for understanding the complex interactions, structural changes, and stability of biomolecular complexes. The radius of gyration (RG) analysis provides information on the protein folding process, indicating how much the protein compacts or expands as it folds. On the other hand, root-mean-square deviation (RMSD) analysis gives insights into the dynamic stability of the complex by monitoring deviations from a reference conformation over time. To illustrate the 2D (Fig. 7) and 3D (Figure S2) free energy landscapes, graphical representations were created using the geo-measure plugin in pymol, mapping RG against RMSD values. These visuals help in understanding the energy distribution and the structural transitions of the biomolecular complex. For the comparative study we have also performed the PCA based FEL through PCA. The PCA-based free energy landscapes in compound (a), (b), (c), and (d) provide insights into the system's conformational stability. Compound (a) displays a distinct low Gibbs free energy basin, indicating highly stable conformations, while higher energy regions suggest less stable states. Compound (b) similarly highlights stable conformations but also reveals additional metastable states, suggesting the system can occupy several less stable configurations. Compound (c) extends the range of PC1, broadening the low energy basin and indicating greater conformational flexibility and overall stability. Compound (d) adjusts the scale for PC2, reinforcing the stability of the central basin and highlighting energy barriers around it, which represent transitions to less stable states. These landscapes collectively emphasize the stability of the system's conformations and provide a detailed view of its thermodynamic properties as shown in Figure S6.
Figure 7.

RG-RMSD based free energy landscape analysis of three compounds in complex with protein (a) compound 1, (b) compound 3, (c) compound 4 and the (d) control.
We employed the methodical framework to analyze ligand-bound complexes along with a control group. The employment of graphical representations enhances the comprehension of energy consequences arising from conformational alterations within the chemical systems studied. Moreover, two-dimensional (Fig. 7) and three-dimensional (Figure S2) visualizations effectively illustrate the dynamic shifts in conformation occurring throughout the simulation process. This visual approach facilitated the identification of a low-energy conformation, unveiling an emergent structural form that provides essential insights into the energetically favorable states of the chemical system. The observation of a deep blue region within the extensive free energy landscape signifies the presence of localized energy minima, consistently observed as the protein structures transitioned into their lowest energy configurations. These localized minima, distinctly indicated by dark blue zones, elucidate the assembly of chemical group structures within minimal energy states. It was noted repeatedly that these complexes preserved localized energy minima within a broader free energy framework, as denoted by the dark blue areas. Further examination of the free energy landscape, in both two and three dimensions, unveils the thermodynamic properties of the studied chemical complexes, including a control compound. These analyses furnish comprehensive insights into their molecular dynamics and stability profiles. A notable characteristic of this landscape is the consistent presence of a relative maximal energy state among these complexes, sustained within an energy range of 14 to 16 kJ/mol. This uniformity across the complexes suggests a potential homogeneity in structural or functional traits, such as molecular interactions or stability. Additionally, all complexes demonstrated a stable conformation at energy levels below 2 kJ/mol, as depicted by the dark blue area on the landscape, corroborating their thermodynamic stability.
In this study, we focused on extracting three distinct poses from each biomolecular complex identified within lower energy states, which are typically associated with more stable and energetically favorable conformations. These poses were superimposed on the initial pose of their respective complex, which served as a control to facilitate a direct comparison of structural deviations that occurred during the simulation as illustrated in the Fig. 8. The initial pose acts as a reference point, providing a baseline against which the structural integrity and deviations of subsequent poses can be assessed. To quantify these deviations, we calculated the root-mean-square deviation (RMSD) for each superimposed structure. RMSD is a statistical measure used extensively in structural biology to calculate the average distance between the backbone atoms of superimposed proteins. It is an indicator of the conformational differences between compared structures, providing insights into the degree of similarity or variance from the initial pose. The Compound 1 exhibited an RMSD of 1.179 Å, indicating minimal deviation from the control and suggesting high structural stability under the simulation conditions. Compound 3 had an RMSD of 1.364 Å, pointing to more significant conformational changes and suggesting a potentially more dynamic behavior within the simulation environment. Compound 4 showed the smallest RMSD of 1.067 Å, implying the greatest conformational stability among the compounds tested. Lastly, the control complex, which is crucial for comparative analysis, showed an RMSD of 1.206 Å. This value establishes a baseline for understanding the degree of structural fluctuations that might be considered normal within this specific experimental setup. These RMSD values are crucial in providing a clear, quantitative view of how each compound's conformation varies from the initial structure, offering essential insights into the dynamic stability and conformational behaviours of the complexes under study. Through this analysis, we can better understand the structural dynamics that underpin the behaviour of these complexes in their respective lower energy states.
Figure 8.

Superimposed representation of three selected compounds in complex with the target protein (a) compound 1 (b) compound 3 (c) compound 4 and (d) control.
Discussion
This study has rigorously evaluated the potential of specific compounds as inhibitors of the influenza polymerase PB2 CAP-binding domain, a critical target for antiviral strategies against the influenza virus. The comprehensive use of virtual screening, molecular docking, and molecular dynamics (MD) simulations provided a detailed comparative analysis of compounds 1, 3, and 4, alongside a control compound39,40. Our findings offer valuable insights into their respective binding affinities, dynamic stability, and molecular interactions, facilitating a nuanced understanding of their potential as therapeutic agents41. Each compound demonstrated unique properties in terms of binding affinity and stability within the binding domain of PB2, as revealed through our computational analyses42. Compound 1 consistently showed a high binding affinity, underscored by its low RMSD values throughout the simulation, indicating a stable interaction with the target. It was noted for engaging in extensive hydrophobic interactions and crucial π–π stacking interactions, contributing significantly to its strong binding profile. Compound 3 exhibited moderate RMSD values, indicating relatively less stability compared to Compounds 1 and 4. The dynamic nature observed through the simulation suggests that while it is an effective binder, its fluctuating conformation could impact its functional efficacy under varying physiological conditions. Compound 4 stood out with the lowest RMSD values among the compounds tested, implying superior stability and potentially the most effective inhibition of the PB2 domain. It also demonstrated a robust interaction profile similar to Compound 1, characterized by multiple effective hydrophobic and π–π stacking interactions. Another work conducted on the PB2 domain also discovered some common interactions, such as Phe363, Lys376, Arg332, Asn429, Glu361, and Phe404which support our findings43. Simmilarly research conducted by lei zhao in 2020 is also supports are finding. The control compound provided a standard for comparison, exhibiting RMSD values that help benchmark the novel compounds. Its performance was instrumental in establishing a context for evaluating the novel compounds' relative effectiveness and stability. Through MD simulations, we gained deeper insights into the structural dynamics of the interactions34,39, Both Compound 1 and Compound 4 displayed substantial stability, suggesting that they maintain consistent interactions with the PB2 domain over time. Compound 3, while effective, showed potential variability in its binding, which could be explored further to understand any implications for its therapeutic use. The binding free energies calculated for the compounds provided a quantitative measure of their binding efficacy. Compound 4 exhibited the most favourable free energy profile, indicating the strongest and most stable binding among the compounds tested. Compound 3 and Compound 1 followed, with Compound 3 slightly outperforming Compound 1 in terms of energy favorability, although with less stability as noted in the RMSD and dynamic observations44.
In addition the excluded compound 2 demonstrated a protein root-mean-square deviation (RMSD) value of 10 Å, indicating the degree of structural deviation from the initial conformation over the course of the molecular dynamics (MD) simulation. For the ligand associated with Compound 2, the RMSD value remained below 7.5 Å during the initial 100 ns (ns) of the simulation, suggesting a relatively stable interaction and minimal deviation from its original binding position. However, as the simulation progressed from 100 to 500 ns, the ligand RMSD increased to values less than 20 Å (Figure S3 (a)). This rise in RMSD indicates greater fluctuations and possibly a less stable interaction between the ligand and the protein over this period. The protein's root-mean-square fluctuation (RMSF) analysis showed a value of 2.5 Å for the region encompassing amino acid residues 100 to 110 (Figure S3 (b)). RMSF values reflect the flexibility of specific regions of the protein, and a value of 2.5 Å in this segment suggests moderate flexibility in this region. Throughout the MD simulation, the formation of two hydrogen bonds was detected shortly after the simulation began. Hydrogen bond formation is crucial for maintaining the stability of the protein–ligand complex and influences the binding affinity (Figure S3 (e)). Furthermore, radius of gyration (Rg) and RMSD-based analysis of the free energy landscape (FEL) revealed a less stable energy transition (Figure S3 (c-d)) and PCA based FEL (Figure S4). This analysis provides insights into the stability and conformational states of the protein–ligand complex, suggesting that the system experiences transitions between less stable states over the course of the simulation. The examination of the superimposed structural poses, obtained by aligning the structures at different time points, indicated an overall RMSD value of 1.705 Å as shown in (Figure S5). This value represents the average deviation of the backbone atoms from the reference structure, reflecting the overall structural stability and conformational changes of the protein–ligand complex throughout the simulation.
Our approach resonates with contemporary antiviral research methodologies, particularly those targeting essential viral proteins. Research methodologies similar to ours have been pivotal in advancing inhibitors for other critical viral targets, such as the HIV reverse transcriptase and hepatitis C virus polymerase. These studies emphasize the importance of understanding molecular interactions and dynamics, which are crucial for optimizing compound efficacy and developing effective therapeutic agents.
Conclusion
This study systematically explored the inhibitory potential of compounds targeting the influenza polymerase PB2 CAP-binding domain, utilizing a combination of virtual screening, molecular docking, and molecular dynamics simulations. Our findings provide a detailed comparative analysis of three novel compounds—compounds 1, 3, and 4—against a control, revealing critical insights into their binding affinities, stability, and interaction dynamics. Compound 4 emerged as the most promising candidate, demonstrating superior binding stability and the lowest root-mean-square deviation (RMSD) values, suggesting it maintains the most consistent interaction with the target protein. This compound also exhibited the most favorable free energy profile, indicating strong and stable binding, which is crucial for effective inhibition. Compound 3, while displaying slightly higher RMSD values, also showed potential with its considerable binding affinity, although its dynamic fluctuations suggest further investigation is needed to fully understand its therapeutic potential. Compound 1, though slightly less effective in binding energy compared to Compound 4, still maintained robust interactions and stability, marking it as a viable candidate. These computational insights lay a strong foundation for future experimental validation. The next steps should involve in vitro and in vivo studies to confirm the inhibitory effects observed in silico and to assess the pharmacological properties of these compounds. Additionally, exploring resistance mechanisms and potential combination therapies could enhance the therapeutic utility and lifespan of these inhibitors.
Supplementary Information
Author contributions
A.S.A., S.A., K.M.Y., S.M.A., D.M.A., R.K.A., B.H., and A.M.E. contributed towards conceptualization. A.M.E. contributed towards supervision. A.S.A., S.A., K.M.Y., S.M.A., D.M.A., R.K.A. and A.M.E. contributed towards the methodology and data analysis. A.S.A. and A.M.E. contributed towards writing—original draft preparation. A.S.A., S.A., K.M.Y., S.M.A., D.M.A., R.K.A., B.H., and A.M.E. contributed towards Writing—review & editing.
Funding
This research has been funded by the Scientific Research Deanship at the University of Ha’il-Saudi Arabia through project number RG-23 204.
Data availability
The datasets generated and/or analysed during the current study are available upon request from the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this Article was revised: In the original version of this Article, Amr S. Abouzied was incorrectly affiliated with ‘Department of Pharmaceutical Chemistry, Egyptian Drug Authority, Giza, Egypt’. The correct affiliation is listed here. Department of Pharmaceutical Chemistry, College of Pharmacy, University of Hail, 81442, Hail, Saudi Arabia.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
1/29/2025
A Correction to this paper has been published: 10.1038/s41598-025-87888-7
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-69816-3.
References
- 1.WHO, Influenza (Seasonal) (2023). https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal)?gad_source=1&gclid=Cj0KCQjw8pKxBhD_ARIsAPrG45kJYli0_XBYBwBTsKBiZcz-jcBg8HkgBZp2x4Ji4hhOm4Y43TF0cI4aAq9xEALw_wcB (accessed April 21, 2024).
- 2.PAHO, Influenza, SARS-CoV-2, RSV and other respiratory viruses, (n.d.). https://www.paho.org/en/topics/influenza-sars-cov-2-rsv-and-other-respiratory-viruses (accessed April 21, 2024).
- 3.Russell, C. J. Orthomyxoviruses: Structure of Antigens☆. In Reference Module in Biomedical Sciences (Elsevier, 2016). 10.1016/B978-0-12-801238-3.95721-0
- 4.te Velthuis, A. J. W. & Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol.14, 479–493. 10.1038/nrmicro.2016.87 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graef, K. M. et al. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol.84, 8433–8445. 10.1128/JVI.00879-10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Severin, C. et al. The cap-binding site of influenza virus protein PB2 as a drug target. Acta Crystallogr. D Struct. Biol.72, 245–253. 10.1107/S2059798316000085 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Al Hajjar, S. & McIntosh, K. The first influenza pandemic of the 21st century. Ann. Saudi Med.30, 1–10. 10.4103/0256-4947.59365 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jester, B., Uyeki, T. & Jernigan, D. Readiness for responding to a severe pandemic 100 years After 1918. Am. J. Epidemiol.187, 2596–2602. 10.1093/aje/kwy165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tosh, P. K., Jacobson, R. M. & Poland, G. A. Influenza vaccines: From surveillance through production to protection. Mayo Clin. Proc.85, 257–273. 10.4065/mcp.2009.0615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meseko, C., Sanicas, M., Asha, K., Sulaiman, L. & Kumar, B. Antiviral options and therapeutics against influenza: History, latest developments and future prospects. Front Cell Infect. Microbiol.13, 1269344. 10.3389/fcimb.2023.1269344 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevaert, A. & Naesens, L. The influenza virus polymerase complex: An update on its structure, functions, and significance for antiviral drug design. Med. Res. Rev.36, 1127–1173. 10.1002/med.21401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massari, S., Desantis, J., Nizi, M. G., Cecchetti, V. & Tabarrini, O. Inhibition of influenza virus polymerase by interfering with its protein-protein interactions. ACS Infect. Dis.7, 1332–1350. 10.1021/acsinfecdis.0c00552 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labbé, C. M. et al. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res.43, W448–W454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahamad, S., Hema, K. & Gupta, D. Identification of novel tau-tubulin kinase 2 inhibitors using computational approaches, PubMed (2023). https://pubmed.ncbi.nlm.nih.gov/37065061/ (accessed June 23, 2024). [DOI] [PMC free article] [PubMed]
- 15.Berman, H. M. et al. The protein data bank. Nucleic Acids Res.28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pautus, S. et al. New 7-methylguanine derivatives targeting the influenza polymerase PB2 cap-binding domain. J. Med. Chem.56, 8915–8930. 10.1021/jm401369y (2013). [DOI] [PubMed] [Google Scholar]
- 17.Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem.25, 1605–1612. 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Yadav, S., Ahamad, S., Gupta, D. & Mathur, P. Lead optimization, pharmacophore development and scaffold design of protein kinase CK2 inhibitors as potential COVID-19 therapeutics, PubMed (n.d.). https://pubmed.ncbi.nlm.nih.gov/35014595/ (accessed June 23, 2024). [DOI] [PubMed]
- 19.Lipinski’s Rule of Five—An overview | ScienceDirect Topics, (n.d.). https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/lipinskis-rule-of-five (accessed January 2, 2024).
- 20.Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem.31, 455–461. 10.1002/jcc.21334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem.25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A105, 9954–9960. 10.1021/jp003020w (2001). [Google Scholar]
- 23.Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Modell.25, 247–260 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput. Chem.26, 1668–1688. 10.1002/jcc.20290 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farago, O. Langevin thermostat for robust configurational and kinetic sampling. Phys. A Stat. Mech. Appl.534, 122210. 10.1016/j.physa.2019.122210 (2019). [Google Scholar]
- 26.Lin, Y., Pan, D., Li, J., Zhang, L. & Shao, X. Application of Berendsen barostat in dissipative particle dynamics for nonequilibrium dynamic simulation. J. Chem. Phys.146, 12 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Sun, H. et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys.10.1039/C7CP07623A (2018). [DOI] [PubMed] [Google Scholar]
- 28.Miller, B. R. et al. MMPBSA.py : An efficient program for end-state free energy calculations. J. Chem. Theory Comput.8, 3314–3321. 10.1021/ct300418h (2012). [DOI] [PubMed] [Google Scholar]
- 29.Homeyer, N. & Gohlke, H. Free energy calculations by the molecular mechanics Poisson−Boltzmann surface area method. Mol. Inf.31, 114–122 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Duan, L. et al. Accelerated molecular dynamics simulation for helical proteins folding in explicit water. Front. Chem.7, 540. 10.3389/fchem.2019.00540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagami, L. P., Das Neves, G. M., Timmers, L. F. S. M., Caceres, R. A. & Eifler-Lima, V. L. Geo-measures: A PyMOL plugin for protein structure ensembles analysis. Comput. Biol. Chem.87, 107322. 10.1016/j.compbiolchem.2020.107322 (2020). [DOI] [PubMed] [Google Scholar]
- 32.DeLano, W. L. Pymol: An open-source molecular graphics tool, CCP4 Newsl. Protein Crystallogr.40, 82–92 (2002). [Google Scholar]
- 33.Studio, D. Discovery studio, Accelrys [2.1] (2008).
- 34.Bowers, K., Chow, E., Xu, H., Dror, R., Eastwood, M., Gregersen, B., Klepeis, J., Kolossváry, I., Moraes, M., Sacerdoti, F., Salmon, J., Shan, Y. & Shaw, D. Molecular dynamics—Scalable algorithms for molecular dynamics simulations on commodity clusters 84 (2006). 10.1145/1188455.1188544
- 35.Ahamad, S., Kanipakam, H., Kumar, V. & Gupta, D. A molecular journey to check the conformational dynamics of tau tubulin kinase 2 mutations associated with Alzheimer’s disease, PubMed (2021). https://pubmed.ncbi.nlm.nih.gov/35424125/ (accessed June 23, 2024). [DOI] [PMC free article] [PubMed]
- 36.Ahamad, S., Hema, K., Kumar, V. & Gupta, D. The structural, functional, and dynamic effect of Tau tubulin kinase1 upon a mutation: A neuro-degenerative hotspot, PubMed (n.d.). https://pubmed.ncbi.nlm.nih.gov/34297427/ (accessed June 23, 2024). [DOI] [PubMed]
- 37.Alsukaibi, A. K. D., Alenezi, K. M., Haque, A., Ahmad, I., Saeed, M., Verma, M. & Hsieh, M. F. Chemical, biological and in silico assessment of date (P. dactylifera L.) fruits grown in Ha’il region, PubMed (2023). https://pubmed.ncbi.nlm.nih.gov/36936534/ (accessed June 23, 2024). [DOI] [PMC free article] [PubMed]
- 38.Ali, S. et al. Identification and evaluation of inhibitors of lipase from malassezia restricta using virtual high-throughput screening and molecular dynamics studies. IJMS20, 884. 10.3390/ijms20040884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers, K. J., Chow, E., Xu, H., Dror, R. O., Eastwood, M. P., Gregersen, B. A., Klepeis, J. L., Kolossvary, I., Moraes, M. A. & Sacerdoti, F. D. Scalable algorithms for molecular dynamics simulations on commodity clusters 84-es (2006).
- 40.Alexey, R. et al. Structure-based virtual screening and biological evaluation of novel inhibitors of mycobacterium Z-ring formation. J. Cell. Biochem.123, 852–862 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Pantsar, T. & Poso, A. Binding affinity via docking: Fact and fiction. Molecules23, 1899. 10.3390/molecules23081899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yunta, M. J. R. It is important to compute intramolecular hydrogen bonding in drug design?. AJMO5, 24–57. 10.12691/ajmo-5-1-3 (2017). [Google Scholar]
- 43.Zhao, L. et al. Identification of novel influenza polymerase PB2 inhibitors using a cascade docking virtual screening approach. Molecules25, 5291. 10.3390/molecules25225291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fatriansyah, J. F., Rizqillah, R. K., Yandi, M. Y. & Sahlan, M. Molecular docking and dynamics studies on propolis sulabiroin-A as a potential inhibitor of SARS-CoV-2. J. King Saud. Univ. Sci.34, 101707. 10.1016/j.jksus.2021.101707 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available upon request from the corresponding author.