

Abstract
Background
Depression is common in primary care and is associated with marked personal, social and economic morbidity, thus creating significant demands on service providers. The antidepressant fluoxetine has been studied in many randomised controlled trials (RCTs) in comparison with other conventional and unconventional antidepressants. However, these studies have produced conflicting findings. Other systematic reviews have considered selective serotonin reuptake inhibitor (SSRIs) as a group which limits the applicability of the findings for fluoxetine alone. Therefore, this review intends to provide specific and clinically useful information regarding the effects of fluoxetine for depression compared with tricyclics (TCAs), SSRIs, serotonin‐noradrenaline reuptake inhibitors (SNRIs), monoamine oxidase inhibitors (MAOIs) and newer agents, and other conventional and unconventional agents.
Objectives
To assess the effects of fluoxetine in comparison with all other antidepressive agents for depression in adult individuals with unipolar major depressive disorder.
Search methods
We searched the Cochrane Collaboration Depression, Anxiety and Neurosis Review Group Controlled Trials Register (CCDANCTR) to 11 May 2012. This register includes relevant RCTs from the Cochrane Central Register of Controlled Trials (CENTRAL) (all years), MEDLINE (1950 to date), EMBASE (1974 to date) and PsycINFO (1967 to date). No language restriction was applied. Reference lists of relevant papers and previous systematic reviews were handsearched. The pharmaceutical company marketing fluoxetine and experts in this field were contacted for supplemental data.
Selection criteria
All RCTs comparing fluoxetine with any other AD (including non‐conventional agents such as hypericum) for patients with unipolar major depressive disorder (regardless of the diagnostic criteria used) were included. For trials that had a cross‐over design only results from the first randomisation period were considered.
Data collection and analysis
Data were independently extracted by two review authors using a standard form. Responders to treatment were calculated on an intention‐to‐treat basis: dropouts were always included in this analysis. When data on dropouts were carried forward and included in the efficacy evaluation, they were analysed according to the primary studies; when dropouts were excluded from any assessment in the primary studies, they were considered as treatment failures. Scores from continuous outcomes were analysed by including patients with a final assessment or with the last observation carried forward. Tolerability data were analysed by calculating the proportion of patients who failed to complete the study due to any causes and due to side effects or inefficacy. For dichotomous data, odds ratios (ORs) were calculated with 95% confidence intervals (CI) using the random‐effects model. Continuous data were analysed using standardised mean differences (SMD) with 95% CI.
Main results
A total of 171 studies were included in the analysis (24,868 participants). The included studies were undertaken between 1984 and 2012. Studies had homogenous characteristics in terms of design, intervention and outcome measures. The assessment of quality with the risk of bias tool revealed that the great majority of them failed to report methodological details, like the method of random sequence generation, the allocation concealment and blinding. Moreover, most of the included studies were sponsored by drug companies, so the potential for overestimation of treatment effect due to sponsorship bias should be considered in interpreting the results. Fluoxetine was as effective as the TCAs when considered as a group both on a dichotomous outcome (reduction of at least 50% on the Hamilton Depression Scale) (OR 0.97, 95% CI 0.77 to 1.22, 24 RCTs, 2124 participants) and a continuous outcome (mean scores at the end of the trial or change score on depression measures) (SMD 0.03, 95% CI ‐0.07 to 0.14, 50 RCTs, 3393 participants). On a dichotomous outcome, fluoxetine was less effective than dothiepin or dosulepin (OR 2.13, 95% CI 1.08 to 4.20; number needed to treat (NNT) = 6, 95% CI 3 to 50, 2 RCTs, 144 participants), sertraline (OR 1.37, 95% CI 1.08 to 1.74; NNT = 13, 95% CI 7 to 58, 6 RCTs, 1188 participants), mirtazapine (OR 1.46, 95% CI 1.04 to 2.04; NNT = 12, 95% CI 6 to 134, 4 RCTs, 600 participants) and venlafaxine (OR 1.29, 95% CI 1.10 to 1.51; NNT = 11, 95% CI 8 to 16, 12 RCTs, 3387 participants). On a continuous outcome, fluoxetine was more effective than ABT‐200 (SMD ‐1.85, 95% CI ‐2.25 to ‐1.45, 1 RCT, 141 participants) and milnacipran (SMD ‐0.36, 95% CI ‐0.63 to ‐0.08, 2 RCTs, 213 participants); conversely, it was less effective than venlafaxine (SMD 0.10, 95% CI 0 to 0.19, 13 RCTs, 3097 participants). Fluoxetine was better tolerated than TCAs considered as a group (total dropout OR 0.79, 95% CI 0.65 to 0.96; NNT = 20, 95% CI 13 to 48, 49 RCTs, 4194 participants) and was better tolerated in comparison with individual ADs, in particular amitriptyline (total dropout OR 0.62, 95% CI 0.46 to 0.85; NNT = 13, 95% CI 8 to 39, 18 RCTs, 1089 participants), and among the newer ADs ABT‐200 (total dropout OR 0.18, 95% CI 0.08 to 0.39; NNT = 3, 95% CI 2 to 5, 1 RCT, 144 participants), pramipexole (total dropout OR 0.12, 95% CI 0.03 to 0.42, NNT = 3, 95% CI 2 to 5, 1 RCT, 105 participants), and reboxetine (total dropout OR 0.60, 95% CI 0.44 to 0.82, NNT = 9, 95% CI 6 to 24, 4 RCTs, 764 participants).
Authors' conclusions
The present study detected differences in terms of efficacy and tolerability between fluoxetine and certain ADs, but the clinical meaning of these differences is uncertain. Moreover, the assessment of quality with the risk of bias tool showed that the great majority of included studies failed to report details on methodological procedures. Of consequence, no definitive implications can be drawn from the studies' results. The better efficacy profile of sertraline and venlafaxine (and possibly other ADs) over fluoxetine may be clinically meaningful, as already suggested by other systematic reviews. In addition to efficacy data, treatment decisions should also be based on considerations of drug toxicity, patient acceptability and cost.
Keywords: Humans; Antidepressive Agents; Antidepressive Agents/therapeutic use; Antidepressive Agents, Second‐Generation; Antidepressive Agents, Second‐Generation/therapeutic use; Antidepressive Agents, Tricyclic; Antidepressive Agents, Tricyclic/therapeutic use; Depression; Depression/drug therapy; Fluoxetine; Fluoxetine/therapeutic use; Randomized Controlled Trials as Topic; Selective Serotonin Reuptake Inhibitors; Selective Serotonin Reuptake Inhibitors/therapeutic use
Plain language summary
Fluoxetine compared with other antidepressants for depression in adults
Depression is a severe mental illness characterised by a persistent low mood and loss of all interest and pleasure, usually accompanied by a range of symptoms such as appetite change, sleep disturbance and poor concentration. The predominant treatment options for depression are drugs and psychological therapies, but antidepressant drugs are the most common treatment for moderate to severe depression. Fluoxetine, one of the first new generation antidepressants, is an extremely popular drug treatment for depression. However, findings from studies comparing fluoxetine with other antidepressants are controversial. In this systematic review, the efficacy and tolerability of fluoxetine was compared with other antidepressants for the acute treatment of depression.
In May 2012 we searched, in a wide‐ranging way, for all the useful studies (randomised controlled trials, or RCTs) we could find which compared fluoxetine with any other antidepressant in treating people with depression. One hundred and seventy‐one RCTs were included, with 24,868 people in the analyses. Combining the results from all the trials, fluoxetine was similarly effective, but better tolerated, than older generation (tricyclic) antidepressants. In comparison with other new generation antidepressants, important differences in efficacy and in tolerability were found between fluoxetine and some of the antidepressants, for example, fluoxetine was less effective than sertraline and mirtazapine but better tolerated than reboxetine. These differences might have a clinical impact in everyday practice. However, when interpreting these differences it is important to bear in mind that the studies were short in duration (eight weeks or less) and that the average size of each trial was small (each included around 100 people). Moreover, most of the included studies were sponsored by drug companies, which could potentially have led to an overestimation of treatment effect. As a consequence, it is difficult to draw clear, clinically meaningful conclusions. More reliable information is needed about the respective safety profiles of antidepressants.
Summary of findings
Summary of findings for the main comparison. Fluoxetine compared to TCAs.
| Fluoxetine compared to TCAs | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: TCAs | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| TCAs | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
471 per 1000 | 463 per 1000 (406 to 520) | OR 0.97 (0.77 to 1.22) | 2124 (24 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.03 higher (0.07 lower to 0.14 higher) | 3393 (50 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 335 per 1000 | 284 per 1000 (246 to 326) | OR 0.79 (0.65 to 0.96) | 4194 (49 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 193 per 1000 | 116 per 1000 (87 to 152) | OR 0.55 (0.40 to 0.75) | 3647 (40 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 68 per 1000 | 87 per 1000 (66 to 112) | OR 1.29 (0.96 to 1.72) | 2911 (33 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in study design: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 2. Fluoxetine compared to ABT‐200.
| Fluoxetine compared to ABT 200 | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: ABT 200 | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ABT 200 | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 1.85 standard deviations lower (2.25 to 1.45 lower) | 141 (1 study) | ⊕⊕⊝⊝ low1 | This corresponds to a large effect according to conventions proposed by Cohen 1992. However, only one study contributed to this analysis | ||
| Failure to complete ‐ total ‐ | 528 per 1000 | 167 per 1000 (82 to 304) | OR 0.18 (0.08 to 0.39) | 144 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ inefficacy ‐ | 56 per 1000 | 14 per 1000 (2 to 115) | OR 0.24 (0.03 to 2.20) | 144 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ side effects ‐ | 361 per 1000 | 43 per 1000 (11 to 132) | OR 0.08 (0.02 to 0.27) | 144 (1 study) | ⊕⊕⊝⊝ low1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in study design: no details on randomisation procedures and allocation concealment. Blinding stated but not tested. Only one study included in the analysis.
Summary of findings 3. Fluoxetine compared to agomelatine.
| Fluoxetine compared to agomelatine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: agomelatine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Agomelatine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
282 per 1000 | 361 per 1000 (280 to 450) | OR 1.44 (0.99 to 2.09) | 515 (1 study) | ⊕⊕⊝⊝ low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.02 standard deviations higher (0.18 lower to 0.23 higher) | 1213 (3 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 135 per 1000 | 170 per 1000 (122 to 233) | OR 1.31 (0.89 to 1.94) | 785 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 55 per 1000 | 59 per 1000 (23 to 142) | OR 1.08 (0.41 to 2.88) | 785 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 34 per 1000 | 50 per 1000 (25 to 97) | OR 1.51 (0.74 to 3.07) | 785 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in this analysis.
Summary of findings 4. Fluoxetine compared to amineptine.
| Fluoxetine compared to amineptine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: amineptine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Amineptine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
719 per 1000 | 486 per 1000 (249 to 727) | OR 0.37 (0.13 to 1.04) | 63 (1 study) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 210 per 1000 | 140 per 1000 (43 to 370) | OR 0.61 (0.17 to 2.21) | 232 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 94 per 1000 | 97 per 1000 (19 to 366) | OR 1.04 (0.19 to 5.57) | 63 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ (Copy) | 84 per 1000 | 46 per 1000 (3 to 418) | OR 0.52 (0.03 to 7.82) | 232 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 5. Fluoxetine compared to amisulpride.
| Fluoxetine compared to amisulpride | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: amisulpride | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Amisulpride | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.17 standard deviations higher (0.07 lower to 0.41 higher) | 268 (1 study) | ⊕⊕⊝⊝ low1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 225 per 1000 | 288 per 1000 (191 to 409) | OR 1.39 (0.81 to 2.38) | 281 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ inefficacy ‐ | 56 per 1000 | 65 per 1000 (25 to 156) | OR 1.16 (0.43 to 3.10) | 281 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ side effects ‐ | 92 per 1000 | 72 per 1000 (32 to 155) | OR 0.77 (0.33 to 1.82) | 281 (1 study) | ⊕⊕⊝⊝ low1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested. Only one study included in the analysis.
Summary of findings 6. Fluoxetine compared to bupropion.
| Fluoxetine compared to bupropion | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: bupropion | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Bupropion | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
493 per 1000 | 447 per 1000 (318 to 582) | OR 0.83 (0.48 to 1.43) | 436 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 356 per 1000 | 356 per 1000 (270 to 450) | OR 1.00 (0.67 to 1.48) | 436 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 23 per 1000 | 0 per 1000 (0 to 87) | OR 1.16 (0.33 to 4.10) | 436 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 59 per 1000 | 60 per 1000 (28 to 124) | OR 1.01 (0.45 to 2.25) | 436 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 7. Fluoxetine compared to citalopram.
| Fluoxetine compared to citalopram | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: citalopram | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Citalopram | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
379 per 1000 | 268 per 1000 (109 to 522) | OR 0.60 (0.20 to 1.79) | 59 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.06 standard deviations higher (0.10 lower to 0.21 higher) | 661 (3 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 211 per 1000 | 189 per 1000 (138 to 254) | OR 0.87 (0.60 to 1.27) | 732 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 75 per 1000 | 66 per 1000 (37 to 112) | OR 0.87 (0.48 to 1.56) | 732 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 75 per 1000 | 49 per 1000 (27 to 89) | OR 0.64 (0.34 to 1.20) | 732 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis and less than 100 patients.
Summary of findings 8. Fluoxetine compared to Crocus sativus.
| Fluoxetine compared to Crocus sativus | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison:Crocus sativus | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Crocus sativus | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
250 per 1000 | 150 per 1000 (35 to 464) | OR 0.53 (0.11 to 2.60) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 50 per 1000 | 50 per 1000 (3 to 475) | OR 1.00 (0.06 to 17.18) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | |
| Failure to complete ‐ inefficacy ‐ | 0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
| Failure to complete ‐ side effects ‐ | 0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested. Only one study included in the analysis and less than 50 patients.
Summary of findings 9. Fluoxetine compared to duloxetine.
| Fluoxetine compared to for duloxetine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: duloxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Fluoxetine | ||||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
400 per 1000 | 485 per 1000 (289 to 684) | OR 1.41 (0.61 to 3.25) | 103 (1 study) | ⊕⊕⊝⊝ low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 281 per 1000 | 260 per 1000 (171 to 372) | OR 0.90 (0.53 to 1.52) | 532 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 15 per 1000 | 47 per 1000 (14 to 152) | OR 3.33 (0.93 to 12.11) | 432 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 66 per 1000 | 19 per 1000 (5 to 80) | OR 0.28 (0.07 to 1.23) | 532 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis.
Summary of findings 10. Fluoxetine compared to escitalopram.
| Fluoxetine compared to escitalopram | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: escitalopram | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Escitalopram | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
236 per 1000 | 239 per 1000 (147 to 363) | OR 1.02 (0.56 to 1.85) | 240 (1 study) | ⊕⊕⊝⊝ low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.07 standard deviations higher (0.19 lower to 0.33 higher) | 231 (1 study) | ⊕⊕⊝⊝ low1,2 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 148 per 1000 | 210 per 1000 (148 to 292) | OR 1.53 (1.00 to 2.37) | 578 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 13 per 1000 | 23 per 1000 (6 to 82) | OR 1.74 (0.46 to 6.53) | 578 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 77 per 1000 | 89 per 1000 (51 to 151) | OR 1.17 (0.64 to 2.12) | 578 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis.
Summary of findings 11. Fluoxetine compared to fluvoxamine.
| Fluoxetine compared to fluvoxamine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: fluvoxamine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Fluvoxamine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
605 per 1000 | 592 per 1000 (443 to 727) | OR 0.95 (0.52 to 1.74) | 177 (1 study) | ⊕⊕⊝⊝ low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 170 per 1000 | 936 per 1000 (69 to 219) | OR 071 (0.36 to 1.37) | 284 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
| Failure to complete ‐ side effects ‐ | 39 per 1000 | 41 per 1000 (6 to 239) | OR 1.04 (0.14 to 7.71) | 100 (1 study) | ⊕⊕⊝⊝ low1,2 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis.
Summary of findings 12. Fluoxetine compared to hypericum.
| Fluoxetine compared to hypericum | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: hypericum | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Hypericum | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
490 per 1000 | 485 per 1000 (346 to 625) | OR 0.98 (0.55 to 1.73) | 717 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.13 standard deviations higher (0.02 lower to 0.29 higher) | 648 (5 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 129 per 1000 | 133 per 1000 (88 to 189) | OR 1.04 (0.65 to 1. 68) | 679 (5 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | OR 4.70 (0.22 to 99.39) | 401 (2 studies) | ⊕⊕⊕⊝ moderate1 | |||
| Failure to complete ‐ side effects ‐ | 35 per 1000 | 42 per 1000 (20 to 88) | OR 1.21 (0.56 to 2.64) | 679 (5 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 13. Fluoxetine compared to maprotiline.
| Fluoxetine compared to maprotiline | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: maprotiline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Maprotiline | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
398 per 1000 | 563 per 1000 (984 to 734) | OR 1.95 (0.91 to 4.18) | 163 (2 studies) | ⊕⊕⊕⊝ moderate | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.04 standard deviations higher (0.15 lower to 0.23 higher) | 433 (5 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 92 per 1000 | 151 per 1000 (84 to 257) | OR 1.75 (0.90 to 3.41) | 351 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 67 per 1000 | 36 per 1000 (11 to 121) | OR 0.53 (0.15 to 1.93) | 209 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 19 per 1000 | 47 per 1000 (6 to 279) | OR 2.54 (0.33 to 19.9) | 209 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 14. Fluoxetine compared to mianserin.
| Fluoxetine compared to mianserin | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: mianserin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Mianserin | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
593 per 1000 | 538 per 1000 (282 to 776) | OR 0.80 (0.27 to 2.38) | 53 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.43 standard deviations higher (0.38 lower to 1.23 higher) | 128 (3 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 362 per 1000 | 263 per 1000 (93 to 560) | OR 0.63 (0.18 to 2.25) | 93 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 74 per 1000 | 154 per 1000 (30 to 522) | OR 2.27 (0.38 to 13.63) | 53 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
| Failure to complete ‐ side effects ‐ | 148 per 1000 | 154 per 1000 (38 to 450) | OR 1.05 (0.23 to 4.70) | 53 (1 study) | ⊕⊝⊝⊝ very low1,2 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis and less than 100 patients.
Summary of findings 15. Fluoxetine compared to milnacipran.
| Fluoxetine compared to milnacipran | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: milnacipran | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Milnacipran | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
473 per 1000 | 518 per 1000 (412 to 623) | OR 1.20 (0.78 to 1.84) | 370 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.36 standard deviations lower (0.63 to 0.08 lower) | 213 (2 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 411 per 1000 | 406 per 1000 (322 to 497) | OR 0.98 (0.68 to 1.42) | 560 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 137 per 1000 | 165 per 1000 (97 to 267) | OR 1.25 (0.68 to 2.30) | 560 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 71 per 1000 | 103 per 1000 (59 to 175) | OR 1.50 (0.81 to 2.76) | 560 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 16. Fluoxetine compared to mirtazapine.
| Fluoxetine compared to mirtazapine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: Fluoxetine Comparison: mirtazapine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Mirtazapine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
354 per 1000 | 444 per 1000 (363 to 527) | OR 1.46 (1.04 to 2.04) | 600 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.57 standard deviations higher (0.15 lower to 1.29 higher) | 31 (1 study) | ⊕⊝⊝⊝ very low1,2 | This corresponds to a medium effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 327 per 1000 | 304 per 1000 (211 to 416) | OR 0.90 (0.55 to 1.47) | 301 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 44 per 1000 | 62 per 1000 (31 to 119) | OR 1.45 (0.71 to 2.96) | 600 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 98 per 1000 | 93 per 1000 (56 to 151) | OR 0.95 (0.55 to 1.64) | 600 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
2 Only one study included in the analysis and less than 100 patients.
Summary of findings 17. Fluoxetine compared to moclobemide.
| Fluoxetine compared to moclobemide | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: moclobemide | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Moclobemide | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
436 per 1000 | 496 per 1000 (416 to 575) | OR 1.27 (0.92 to 1.75) | 721 (7 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.13 standard deviations higher (0.04 lower to 0.30 higher) | 540 (6 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 207 per 1000 | 209 per 1000 (155 to 275) | OR 1.01 (0.70 to 1.45) | 721 (7 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 62 per 1000 | 44 per 1000 (21 to 93) | OR 0.70 (0.32 to 1.56) | 679 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 86 per 1000 | 91 per 1000 (57 to 144) | OR 1.07 (0.64 to 1.80) | 721 (7 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 18. Fluoxetine compared to nefazodone.
| Fluoxetine compared to nefazodone | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: nefazodone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Nefazodone | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.06 standard deviations lower (0.30 lower to 0.18 higher) | 271 (4 studies) | ⊕⊕⊕⊝ moderate1 | This effects approaches zero | ||
| Failure to complete ‐ total ‐ | 220 per 1000 | 132 per 1000 (58 to 269) | OR 0.54 (0.22 to 1.31) | 161 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 24 per 1000 | 17 per 1000 (1 to 211) | OR 0.71 (0.05 to 10.71) | 161 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 96 per 1000 | 75 per 1000 (33 to 161) | OR 0.76 (0.32 to 1.81) | 286 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 19. Fluoxetine compared to paroxetine.
| Fluoxetine compared to paroxetine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: paroxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Paroxetine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
426 per 1000 | 477 per 1000 (408 to 550) | OR 1.23 (0.93 to 1.65) | 1574 (9 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.01 standard deviations lower (0.25 lower to 0.24 higher) | 2061 (11 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 317 per 1000 | 313 per 1000 (273 to 358) | OR 0.98 (0.81 to 1.20) | 1848 (10 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 52 per 1000 | 39 per 1000 (22 to 71) | OR 0.75 (0.41 to 1.39) | 1005 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 133 per 1000 | 115 per 1000 (87 to 151) | OR 0.85 (0.62 to 1.16) | 1509 (9 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 20. Fluoxetine compared to phenelzine.
| Fluoxetine compared to phenelzine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: phenelzine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenelzine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
150 per 1000 | 200 per 1000 (45 to 564) | OR 1.42 (0.27 to 7.34) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.05 standard deviations lower (0.67 lower to 0.57 higher) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 100 per 1000 | 20 per 1000 (1 to 308) | OR 0.18 (0.01 to 4.01) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | |
| Failure to complete ‐ inefficacy ‐ | 0 per 1000 (0 to 0) | Not estimable | 0 (0) | |||
| Failure to complete ‐ side effects ‐ | 50 per 1000 | 17 per 1000 (1 to 303) | OR 0.32 (0.01 to 8.26) | 40 (1 study) | ⊕⊝⊝⊝ very low1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested. Only one study included in the analysis and less than 50 patients.
Summary of findings 21. Fluoxetine compared to pramipexole.
| Fluoxetine compared to pramipexole | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: pramipexole | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Pramipexole | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
657 per 1000 | 513 per 1000 (315 to 707) | OR 0.55 (0.24 to 1.26) | 105 (1 study) | ⊕⊕⊝⊝ low1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0 standard deviations higher (0 to 0 higher) | 0 (0) | No data available on this outcome | |||
| Failure to complete ‐ total ‐ | 443 per 1000 | 87 per 1000 (23 to 250) | OR 0.12 (0.03 to 0.42) | 105 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ inefficacy ‐ | 57 per 1000 | 29 per 1000 (3 to 215) | OR 0.49 (0.05 to 4.51) | 105 (1 study) | ⊕⊕⊝⊝ low1 | |
| Failure to complete ‐ side effects ‐ | 314 per 1000 | 27 per 1000 (5 to 186) | OR 0.06 (0.01 to 0.50) | 105 (1 study) | ⊕⊕⊝⊝ low1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested. Only one study included in the analysis.
Summary of findings 22. Fluoxetine compared to reboxetine.
| Fluoxetine compared to reboxetine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: reboxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Reboxetine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
566 per 1000 | 501 per 1000 (418 to 589) | OR 0.77 (0.55 to 1.10) | 721 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.04 standard deviations higher (0.31 lower to 0.40 higher) | 205 (2 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero | ||
| Failure to complete ‐ total ‐ | 361 per 1000 | 253 per 1000 (199 to 316) | OR 0.60 (0.44 to 0.82) | 764 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 88 per 1000 | 82 per 1000 (43 to 146) | OR 0.92 (0.47 to 1.77) | 464 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 129 per 1000 | 57 per 1000 (22 to 139) | OR 0.41 (0.15 to 1.09) | 211 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 23. Fluoxetine compared to sertraline.
| Fluoxetine compared to sertraline | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: sertraline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Sertraline | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
416 per 1000 | 494 per 1000 (435 to 554) | OR 1.37 (1.08 to 1.74) | 1188 (6 studies) |
⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.09 standard deviations higher (0.03 lower to 0.20 higher) | 1160 (7 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 229 per 1000 | 258 per 1000 (217 to 307) | OR 1.17 (0.93 to 1.49) | 1591 (9 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 70 per 1000 | 76 per 1000 (49 to 118) | OR 1.09 (0.68 to 1.77) | 1056 (5 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 110 per 1000 | 134 per 1000 (102 to 174) | OR 1.25 (0.92 to 1.70) | 1591 (9 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 24. Fluoxetine compared to tianeptine.
| Fluoxetine compared to tianeptine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: tianeptine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Tianeptine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
534 per 1000 | 562 per 1000 (462 to 657) | OR 1.12 (0.75 to 1.67) | 387 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.15 standard deviations lower (0.40 lower to 0.10 higher) | 730 (3 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 225 per 1000 | 218 per 1000 (167 to 279) | OR 0.96 (0.69 to 1.33) | 830 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 47 per 1000 | 39 per 1000 (13 to 110) | OR 0.82 (0.27 to 2.53) | 830 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 91 per 1000 | 101 per 1000 (66 to 152) | OR 1.13 (0.71 to 1.80) | 830 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 25. Fluoxetine compared to trazodone.
| Fluoxetine compared to trazodone | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: trazodone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Trazodone | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
642 per 1000 | 467 per 1000 (189 to 769) | OR 0.49 (0.13 to 1.86) | 110 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.25 standard deviations lower (0.76 lower to 0.26 higher) | 203 (4 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 250 per 1000 | 145 per 1000 (71 to 274) | OR 0.51 (0.23 to 1.13) | 230 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 147 per 1000 | 38 per 1000 (7 to 207) | OR 0.23 (0.04 to 1.51) | 70 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 151 per 1000 | 105 per 1000 (34 to 280) | OR 0.66 (0.20 to 2.19) | 110 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Summary of findings 26. Fluoxetine compared to venlafaxine.
| Fluoxetine compared to venlafaxine | ||||||
| Patient or population: patients with depression Settings: in‐ and outpatients Intervention: fluoxetine Comparison: venlafaxine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Venlafaxine | Fluoxetine | |||||
|
Failure to respond (reduction ≥ 50% on HDRS) |
341 per 1000 | 400 per 1000 (363 to 439) | OR 1.29 (1.10 to 1.51) | 3387 (12 studies) | ⊕⊕⊕⊝ moderate1 | |
|
Endpoint score (HDRS or MADRS) |
The mean endpoint score in the intervention groups was 0.10 standard deviations higher (0.0 to 0.19 higher) | 3097 (13 studies) | ⊕⊕⊕⊝ moderate1 | This corresponds to a very small effect according to conventions proposed by Cohen 1992 | ||
| Failure to complete ‐ total ‐ | 256 per 1000 | 234 per 1000 (203 to 267) | OR 0.89 (0.74 to 1.06) | 2683 (14 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ inefficacy ‐ | 43 per 1000 | 56 per 1000 (40 to 79) | OR 1.31 (0.91 to 1.89) | 2640 (13 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to complete ‐ side effects ‐ | 116 per 1000 | 87 per 1000 (69 to 110) | OR 0.72 (0.56 to 0.94) | 2640 (13 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Limitations in studies designs: no details on randomisation procedures and allocation concealment. Blinding stated but not tested.
Background
Description of the condition
Major depression is generally diagnosed when a persistent and unreactive low mood or loss of interest and pleasure, or both, are accompanied by a range of symptoms including appetite loss, insomnia, fatigue, loss of energy, poor concentration, psychomotor symptoms, inappropriate guilt and morbid thoughts of death (APA 1994). It was the third leading cause of burden among all diseases in the year 2004 and it is expected to be the greatest cause in 2030 (WHO 2006). This condition is associated with marked personal, social and economic morbidity, loss of functioning and productivity, and creates significant demands on service providers in terms of workload (APA 2000; NICE 2010). Although pharmacological and psychological interventions are both effective for major depression, in primary and secondary care settings antidepressant (AD) drugs remain the mainstay of treatment in moderate to severe major depression (APA 2006; NICE 2010). Amongst ADs many different agents are available, including tricyclics (TCAs); monoamine oxidase inhibitors (MAOIs); selective serotonin reuptake inhibitors (SSRIs); serotonin‐noradrenaline reuptake inhibitors (SNRIs) such as venlafaxine, duloxetine and milnacipran; and other agents (mirtazapine, reboxetine, bupropion). Over the last 20 years prescribing ADs has dramatically risen in Western countries, mainly because of the increasing number of prescriptions for SSRIs, which have progressively become the most commonly prescribed ADs (Ciuna 2004). The selective action of SSRIs is purported to be the rationale for potential advantages over other existing therapies. Rather than a breakthrough in pharmacology, the development of SSRIs may be seen as a process of refining the action of existing and commonly used alternatives and this process may be clinically important (Freemantle 2000). SSRIs are generally more acceptable than TCAs, and there is evidence of similar efficacy (NICE 2010). However, head‐to‐head comparisons have provided contrasting findings (Cipriani 2006a).
Description of the intervention
Fluoxetine hydrochloride (3‐(p‐trifluoromethylphenoxy)‐N‐methyl‐3‐phenylpropylamine HCl; Lilly (LY) 110140) was first described in a scientific journal in 1974 as a selective serotonin (5‐hydroxytryptamine or 5‐HT)‐uptake inhibitor (Wong 2005). It was marketed as an AD in December 1987 and went off patent in August 2001. From its marketing fluoxetine quickly became the most prescribed AD in the United States (Marshall 2009) and, despite the availability of newer agents, it remains extremely popular in the pharmacological treatment of major depression and in the treatment of several anxiety disorders.
How the intervention might work
Fluoxetine’s presumed mechanism of action is through inhibiting the reuptake of serotonin. It is not clear, however, if reuptake inhibition correlates with clinical effect, either between patients or over time.
The bioavailability of fluoxetine is relatively high, and peak plasma concentrations are reached in six to eight hours. It is highly bound to plasma proteins, mostly albumin. Fluoxetine is metabolised in the liver by isoenzymes of the cytochrome P450 system, including CYP2D6. Only one metabolite of fluoxetine, norfluoxetine (N‐demethylated fluoxetine), is biologically active. The extremely slow elimination of fluoxetine and its active metabolite norfluoxetine from the body distinguishes it from other ADs. With time, fluoxetine and norfluoxetine inhibit their own metabolism so the fluoxetine elimination half‐life changes from one to three days after a single dose to four to six days after long‐term use.
Why it is important to do this review
In 2000 Geddes and colleagues (Geddes 2000) completed a Cochrane systematic review comparing the group of SSRIs with all other ADs and concluded that there were no large differences between the AD drug classes; however it was suggested that differences may emerge when single, head‐to‐head drug comparisons were considered. Starting from this consideration, and with the aim to shed light on the field of AD trials and treatment of major depression, a group of researchers agreed to join forces under the rubric of the Meta‐Analyses of New Generation Antidepressants Study Group (MANGA Study Group) to systematically review all available evidence for each specific newer AD. We have up to now completed individual reviews on sertraline (Cipriani 2009a), escitalopram (Cipriani 2009b), milnacipran (Nagakawa 2009), fluvoxamine (Omori 2010), mirtazapine (Watanabe 2011), duloxetine (Cipriani 2012a) and citalopram (Cipriani 2012b), and a number of other reviews are now underway. A systematic review comparing fluoxetine with TCAs, heterocyclics, MAOIs, SSRIs, SNRIs and other antidepressants was first published in 2005 (Cipriani 2005) but since then new randomised evidence has been produced. We therefore sought to update that review with the aim of providing the ‘best available’ and most up‐to‐date evidence on the efficacy and acceptability of fluoxetine in individuals with unipolar major depression.
Objectives
To assess the effects of fluoxetine in comparison with all other antidepressive agents for depression in adult individuals with unipolar major depressive disorder. Specifically:
To determine the efficacy of fluoxetine in comparison with other ADs in alleviating the acute symptoms of unipolar major depressive disorder in adults; and
Review the acceptability of treatment with fluoxetine in comparison with other ADs.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) comparing fluoxetine with all other active ADs as monotherapy in the acute phase treatment of unipolar depression were included. We included RCTs with a cross‐over design but only used the results from the first randomisation period.
We excluded quasi‐randomised trials, such as those allocating participants by using alternate days of the week.
Types of participants
The review included participants 18 years or older, of both sexes, with a primary diagnosis of unipolar major depression according to standardised criteria, DSM‐III, DSM‐III‐R, DSM‐IV (APA 2000), ICD‐10 (WHO 1992), Feighner criteria (Feighner 1972) or Research Diagnostic Criteria (Spitzer 1972). Studies using ICD‐9 were excluded as it only lists disease names and does not have diagnostic criteria.
We included participants with the following subtypes of depression: chronic, with catatonic features, with melancholic features, with atypical features, with postpartum onset, and with a seasonal pattern. We included studies in which up to 20% of participants presented with depressive episodes in bipolar affective disorder. We also included participants with a concurrent secondary diagnosis of another psychiatric disorder.
We excluded participants with a concurrent primary diagnosis of Axis I or II disorders and participants with a serious concomitant medical illness.
Types of interventions
We examined fluoxetine in comparison with conventional pharmacological treatments for acute depression. We also examined fluoxetine in comparison with non‐conventional ADs (hypericum or other non‐conventional ADs). We excluded trials in which fluoxetine was compared to another type of psychopharmacological agent (that is anxiolytics, anticonvulsants, antipsychotics or mood‐stabilisers) and trials in which fluoxetine was used as an augmentation strategy.
Experimental intervention
Fluoxetine (as monotherapy). No restrictions on dose, frequency, intensity and duration were applied.
Comparator interventions
Conventional antidepressive agents:
tricyclics (TCAs);
heterocyclics;
SSRIs;
SNRIs;
MAOIs or newer ADs; and
other conventional psychotropic drugs.
Non‐conventional antidepressive agents:
hypericum; and
other non‐conventional antidepressive agents (e.g. Crocus sativus).
No restrictions on dose, frequency, intensity and duration were applied.
Types of outcome measures
Primary outcomes
Efficacy
Efficacy was evaluated using the following outcome measures.
(1) Dichotomous outcome
Number of participants who responded to treatment at the end of the trial by showing a reduction of at least 50% on the Hamilton Depression Scale (HDRS) (Hamilton 1960) out of the total number of randomised participants (intention‐to‐treat analysis).
(2) Continuous outcome
Group mean scores at the end of the trial or change scores on HDRS, or Montgomery‐Asberg Depression Scale (MADRS) (Montgomery 1979), or any other depression scale. If both endpoint and change scores were available, we considered endpoint scores.
Secondary outcomes
Acceptability
(3) Failure to complete due to any reason
Number of participants who dropped out during the trial as a proportion of the total number of randomised participants.
(4) Failure to complete due to inefficacy
Number of participants who dropped out during the trial due to inefficacy as a proportion of the total number of randomised participants.
(5) Failure to complete due to side effects
Number of participants who dropped out during the trial due to side effects as a proportion of the total number of randomised participants.
Search methods for identification of studies
CCDAN's Specialized Register (CCDANCTR) The Cochrane Depression, Anxiety and Neurosis Group (CCDAN) maintains two clinical trials registers at their editorial base in Bristol, UK, a references register and a studies based register. The CCDANCTR‐References Register contains over 31,500 reports of randomised controlled trials in depression, anxiety and neurosis. Approximately 65% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual. Please contact the CCDAN Trials Search Coordinator for further details. Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950‐), EMBASE (1974‐) and PsycINFO (1967‐); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review specific searches of additional databases. Reports of trials are also sourced from international trials registers c/o the World Health Organization’s trials portal (ICTRP), ClinicalTrials.gov, drug companies, the hand‐searching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCDAN's generic search strategies can be found on the Group's website.
Electronic searches
The CCDANCTR‐Studies Register was searched by the Trials Search Co‐ordinator (TSC) using the following search strategy: Diagnosis = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder*" or "Affective Symptoms" and Intervention = Fluoxetine
The CCDANCTR‐References Register was searched using similar terms to identify additional untagged/uncoded references: Keyword = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder" or "Affective Symptoms" and Free‐Text = (Fluoxetin* or Prozac)
Searches were conducted to 11 May 2012. No language restrictions were applied.
Other trial registers
The CCDAN TSC also searched Clinicalstudyresults.org to December 2011 (before this website was phased out) together with ClinicalTrials.gov and the WHO ICTRP to 16 July 2012 for additional published, unpublished or ongoing studies.
We searched trial databases of the following drug‐approving agencies: the Food and Drug Administration (FDA) in the USA, the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK, the European Medicines Agency (EMA) in the EU, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan and the Therapeutic Goods Administration (TGA) in Australia. We also searched ongoing trial registers: ClinicalTrials.gov in the USA, Controlled‐Trials.com (ISRCTN) in the UK, the Nederland's Trial Register, the European Union Drug Regulating Authorities Clinical Trials register (EudraCT), UMIN‐CTR in Japan and the Australian New Zealand Clinical Trials Registry (ACTRN). These searches were undertaken in November 2010.
Searching other resources
Handsearches
Appropriate journals and conference proceedings relating to fluoxetine treatment for depression have already been handsearched and incorporated into the CCDANCTR databases.
Personal communication
Pharmaceutical companies and experts in this field were asked if they knew of any study that met the inclusion criteria of this review.
Reference checking
Reference lists of the included studies, previous systematic reviews and major textbooks of affective disorder that were written in English were checked for published reports and citations of unpublished research (Trespidi 2011).
Data collection and analysis
Selection of studies
Two independent review authors (LRM, MP) checked to ensure that studies relating to fluoxetine generated by the search of the CCDANCTR‐References Register and the other complementary searches met the rough inclusion criteria, firstly based on the titles and abstracts. All studies that were rated as possible candidates by either of the two review authors were added to a preliminary list, and the full text articles were then retrieved. LRM, CG, MP and AC assessed the full text articles to see if they met the strict inclusion criteria. If the raters disagreed, the final rating was made by consensus with the involvement of another member of the review group (CB). Considerable care was taken to group multiple publications to the main study to which they related.
Data extraction and management
Two review authors, working independently and in duplicate (LRM and MP), extracted data from the included studies. Again, any disagreement was discussed with other authors, and decisions were documented. If necessary, we contacted authors of studies for clarification. We extracted the following data from the included studies:
(i) participant characteristics (age, depression diagnosis, comorbidity, depression severity, antidepressant treatment history for the index episode, study setting); (ii) intervention details (intended dosage range, mean daily dosage actually prescribed, sponsorship); and (iii) outcome measures of interest.
The results were compared with those in the completed reviews of individual antidepressants in The Cochrane Library.
We considered the following comparisons:
fluoxetine versus TCAs;
fluoxetine versus the heterocyclics maprotiline, mianserin;
fluoxetine versus the SSRIs citalopram, escitalopram, fluvoxamine, paroxetine, sertraline;
fluoxetine versus the SNRIs duloxetine, milnacipran, venlafaxine;
fluoxetine versus the MAOIs or newer ADs agomelatine, mirtazapine, moclobemide, phenelzine, reboxetine;
fluoxetine versus other conventional psychotropic drugs amineptine, bupropion, pramipexole, tianeptine, trazodone; and
fluoxetine versus the other non‐conventional AD agents Crocus sativus, hypericum.
In the analysis TCAs were pooled as data have shown that drugs belonging to the TCA group are similar in terms of efficacy and tolerability, while drugs belonging to the other classes have rather different efficacy and tolerability profiles (Cipriani 2011).
Assessment of risk of bias in included studies
Two independent review authors independently assessed trial quality in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). This set of criteria is based on evidence of associations between effect overestimation and a high risk of bias in an article, such as sequence generation, allocation concealment, blinding (of participants and personnel, outcome assessment), incomplete outcome data, selective reporting and other source of bias. The categories are defined as:
low risk of bias;
high risk of bias; and
unclear risk of bias.
If the raters disagreed, the final rating was made by consensus with the involvement of another member of the review group.
Non‐congruence in quality assessment was reported as percentage disagreement. The ratings were also compared with those in the completed reviews of individual antidepressants in The Cochrane Library. If there were any discrepancies, they were fed back to the authors of the completed reviews.
Measures of treatment effect
All comparisons were performed between fluoxetine and comparator ADs as individual ADs. Additionally, fluoxetine was compared with TCAs considered as a class.
Skewed data and non‐quantitative data were presented descriptively. An outcome was considered skewed when the mean was smaller than twice the SD. In terms of change score, data were difficult to depict as skewed or not as the possibility existed for negative values; therefore, we entered all of the results of the outcome into the meta‐analysis.
Dichotomous data
For dichotomous, or event‐like, data, odds ratios (ORs) were calculated with their 95% confidence intervals (CI). For statistically significant results, we calculated the number needed to treat to provide benefit (NNT).
Continuous data
For continuous data we calculated the standardised mean differences (SMD) with 95% CI.
Unit of analysis issues
Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (for example pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state, even with a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in major depression, we only used data from the first phase of the cross‐over studies.
Cluster‐randomised trials
Studies increasingly employ 'cluster randomisation’ (such as randomisation by clinician or practice) but analysis and pooling of clustered data pose problems (Barbui 2011b). They are commonly analysed as if the randomisation was performed on the individuals rather than the clusters. In this case, approximately correct analyses were performed by dividing the binary data (the number of participants and the number experiencing the event) as presented in a report by a 'design effect’ (Higgins 2011). This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC): design effect = 1 + (m‐1) *ICC (Higgins 2011). If the ICC was not reported it was assumed to be 0.1. For continuous data only the sample size was reduced; means and standard deviations remained unchanged.
Studies with multiple treatment groups
Studies that compared more than two intervention groups were included in the meta‐analysis by combining all relevant experimental intervention groups of the study into a single group, and all relevant control intervention groups into a single control group, as recommended in section 16.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Dealing with missing data
Dichotomous data
Responders and remitters to treatment were calculated on a strict intention‐to‐treat (ITT) basis: dropouts were included in this analysis. Where participants had been excluded from the trial before the endpoint, we assumed that they experienced a negative outcome by the end of the trial (for example failure to respond to treatment). We examined the validity of this decision in the sensitivity analyses by applying worst‐ and best‐case scenarios.
When dichotomous outcomes were not reported but the baseline mean, endpoint mean and their SDs of the HRSD (or other depression scale) were provided, we converted continuous outcome data expressed as mean and SD into the number of responding and remitted patients, according to the validated imputation method (Furukawa 2006). We examined the validity of this imputation in the sensitivity analyses (Altman 1996; Furukawa 2006).
Continuous data
We applied the loose ITT analyses for continuous variables whereby all the patients with at least one post‐baseline measurement were represented by their last observations carried forward (LOCF), with due consideration of the potential bias and uncertainty introduced. Data from trials not using a LOCF approach were extracted and analysed as reported by the authors. Where SDs were not reported, authors were asked to supply the data. When only the standard error (SE) or t‐statistics or P values were reported, SDs were calculated according to Altman (Altman 1996). In the absence of data from the authors, we substituted SDs by those reported in other studies in the review (Furukawa 2006).
Assessment of heterogeneity
Heterogeneity between studies was investigated by visual inspection of the forest plots and using the I2 statistic (Higgins 2003). According to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), the following thresholds for the interpretation of I2 were used: 0% to 40%, might not be important; 30% to 60%, may represent moderate heterogeneity; 50% to 90%, may represent substantial heterogeneity; 75% to 100%, considerable heterogeneity. Moreover, we considered the sample size, and the magnitude and the direction of the treatment effects.
Assessment of reporting biases
Data from included studies were entered into a funnel plot (trial effect against trial variance) to investigate small‐study effects (Sterne 2000). We used the tests for funnel plot asymmetry only when there were at least 10 studies included in the meta‐analysis, and results were interpreted cautiously, with visual inspection of the funnel plots. We followed the Cochrane Handbook for Systematic Reviews of Interventions methodology (Higgins 2011). When evidence of small‐study effects was identified, possible reasons for funnel plot asymmetry, including publication bias, were investigated.
Data synthesis
The primary analysis used a random‐effects model (odds ratio (OR)), which had the highest generalisability in our empirical examination of summary effect measures for meta‐analyses (Furukawa 2002). The robustness of this summary measure was routinely examined by checking the fixed‐effect model OR and the random‐effects model risk ratio (RR). Material differences between the models were reported. A P value of less than 0.05 and a 95% confidence interval (CI) not including 1 (for the dichotomous outcomes) were considered statistically significant.
Fixed‐effect model analyses were performed routinely for the continuous outcomes as well, to investigate the effect of the choice of method on the estimates. Material differences between the models were reported.
Subgroup analysis and investigation of heterogeneity
We undertook a subgroup analysis for the duration of follow up. We considered the following categories: (1) less than 6 weeks, (2) 6 to 16 weeks, and (3) more than 16 weeks. Moreover, with the exception of the TCA group, stratification by each control agent was performed to ascertain whether there were treatment differences between fluoxetine and AD drugs belonging to the same pharmacological class.
Sensitivity analysis
Sensitivity analyses (worst‐case scenario; best‐case scenario; excluding ORs imputed based on continuous data; fixed‐effect rather than random‐effects models; RR rather than OR; excluding trials using LOCF; excluding trials with substituted SD) were not performed in this version of the review. However, we will conduct these analyses in the next update. If cluster‐randomised or cross‐over trials are included in the next update, we will undertake a sensitivy analysis excluding studies with these study designs.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification
Results of the search
The original searches yielded 883 studies; after reading the abstracts, 364 papers were considered potentially relevant for this review. Of these, 219 were excluded because they were not randomised trials or for other reasons. The remaining 145 were retrieved for more detailed evaluation and 132 RCTs meeting the inclusion criteria were included in the 2005 version (see Figure 1).
1.
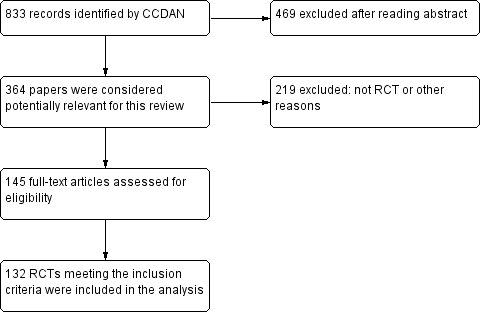
Study flow diagram, 2005 version.
In July 2012 a new search was conducted to update the review. This new search yielded 524 new records of RCTs published between 2005 and 2012, 12 unpublished RCTs, 10 RCTs defined as 'awaiting assessment' in the previous 2005 version, and five studies still ongoing at the time of inclusion. Of these, 536 were considered potentially relevant for this review. After reading the abstracts, 99 references were considered eligible for possible inclusion and the corresponding full papers were retrieved for a detailed evaluation; 55 trials were excluded or are awaiting assessment for wrong study design (not RCT) or other reasons and five are still ongoing. We included a total of 39 RCTs in the qualitative synthesis and 33 RCTs in the quantitative synthesis (meta‐analysis) (see Figure 2).
2.
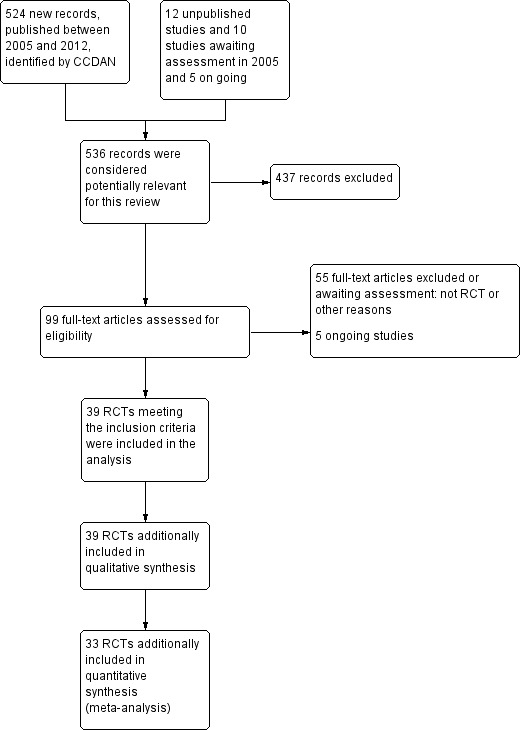
Study flow diagram, 2012 version.
Included studies
See: Characteristics of included studies
Overall, a total of 171 studies were included in the present systematic review (24,868 participants). Attempts to contact authors for additional information were unsuccessful in 22 cases and successful in two cases with additional data provided by the authors.
Design
The great majority of included studies were reported to be double‐blind (161 out of 171 RCTs, that is 95%). The participants were followed up for six weeks (range four to 24 weeks) in a majority of the trials (80 trials).
Sample sizes
The mean number of participants per study was 135.2, with a minimum sample size of 16 (O'Keane 1992) and a maximum of 1096 (Keller 2007).
Setting
A total of 105 trials enrolled only outpatients, 14 trials enrolled only inpatients, and both inpatients and outpatients were enrolled in the remaining trials. Forty‐one per cent of the included studies were undertaken in Europe, 20% in USA, 5% in Canada and in Central or South America, 4% of the trials were conducted in Iran and for the remaining 12% the geographic area was unclear. Three per cent of the included studies were multicentric international trials: 2% were conducted in Africa (South Africa and Zimbabwe); 2% in China, Australia and New Zealand; less than 1% in Israel and Turkey.
Participants
The majority of included trials (163 RCTs) enrolled patients with a diagnosis of major depression based on DSM‐III (34 studies), DSM‐III‐R (64 studies), DSM‐IV or ICD 10 criteria (67 studies). Seventy‐eight trials excluded patients over 65 years, while 14 trials included only elderly patients. We also included a minority of studies in which up to 20% of patients presented with depressive episodes in bipolar disorder.
Intervention and comparators
In 74 studies fluoxetine was compared with TCAs (22 studies versus amitriptyline, 15 versus imipramine, 6 versus dothiepin or dosulepin, 5 versus maprotiline, 5 versus clomipramine, 6 versus nortriptyline, 4 versus desipramine, 4 versus doxepine, 3 versus mianserin, 2 versus trimipramine, 1 versus lofepramine, 1 versus nimofensine). Thirty RCTs compared fluoxetine with other SSRIs (13 versus paroxetine, 12 versus sertraline, 3 versus citalopram, 2 versus escitalopram), 20 versus SNRIs (15 versus venlafaxine, 3 versus milnacipran, 2 versus duloxetine), and 20 studies with MAOIs or newer agents (7 versus moclobemide, 4 versus reboxetine, 5 versus mirtazapine, 3 versus agomelatine, 1 versus phenelzine). Moreover, in 19 studies fluoxetine was compared with other conventional agents (4 versus trazodone, 4 versus nefazodone, 4 versus tianeptine, 2 versus amineptine, 2 versus bupropion, 1 versus pramipexole, 1 versus amisulpride, 1 versus ABT‐200). Finally, eight studies compared fluoxetine with non‐conventional agents (6 versus hypericum and 2 versus Crocus sativus). A fixed dose regimen for fluoxetine was employed in 72 studies.
Outcomes
At the end of the reviewing process, 165 RCTs were included in the meta‐analysis. For efficacy outcomes, 121 RCTs provided continuous data and 91 dichotomous data. For acceptability outcomes, 139 RCTs provided data on total dropouts, 104 on dropouts due to inefficacy and 125 on dropouts due to side effects. In the majority of trials (130 out of 143, 90%) the 17‐ or 21‐ item HDRS was used for reporting outcomes.
Overall, 13,619 patients were included in the efficacy analysis dichotomous outcome (6441 participants randomised to fluoxetine and 7178 randomised to another antidepressant) and 15,870 were included in the efficacy analysis continuous outcome (7625 participants randomised to fluoxetine and 8245 randomised to another antidepressant). A total of 18,756 patients were included in the acceptability analysis (9009 partIcipants randomised to fluoxetine and 9747 randomised to another antidepressant).
Excluded studies
See: Characteristics of excluded studies; Characteristics of studies awaiting classification
Forty‐four articles that were initially selected did not meet our inclusion criteria and were excluded because of one of the following reasons: wrong design (24 articles), review or pooled analysis (two articles), wrong comparison (nine articles), wrong intervention (one article) and wrong diagnosis or population (eight articles). A total of 34 records were classified as 'awaiting classification'. Of these, 28 were study reports written in Chinese.
Risk of bias in included studies
See: Included studies; Figure 3; Figure 4
3.
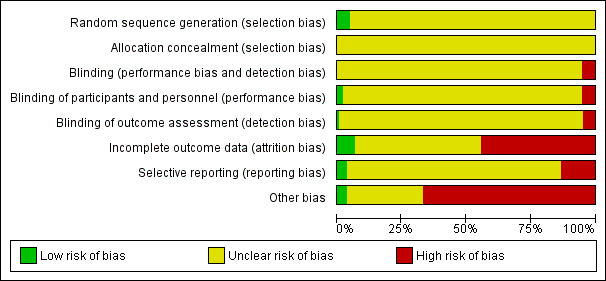
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Our judgement about the overall risk of bias in the individual studies is illustrated in Figure 3 and Figure 4. The methodological quality of many included studies was judged as poor, although judging articles from some time ago by today’s standard might be problematic (Begg 1996). Moreover, many articles failed to report methodologically relevant information on study procedure (in these cases the judgement was defined as 'unclear') and the overall reporting of studies was not good. This type of reporting has been associated with an overestimate of the estimate of effect (Schulz 1995) and it should be considered when interpreting the results. However, in general it is not possible to predict the direction or magnitude of bias associated with a lack of adequate sequence generation or adequate allocation concealment (Odgaard‐Jensen 2011).
Allocation
Random sequence generation
The majority of studies (161) did not report the methods of generating random sequence, while eight studies (Akhondzadeh Basti 2007; Alves 1999; Byerley 1988; Harrer 1999; Hosak 2000; Noorbala 2005; Rudolph 1999; Sheehan 2009) specified this information and they were classified as 'low risk'.
Allocation concealment
All trials failed to report details on allocation concealment and were classified as 'unclear risk'.
Blinding
One hundred and sixty‐three trials were undertaken on a double‐blind basis, four trials employed a single‐blind design and five an open design. None of the double‐blind trials specified if blindness was maintained during the study. Nine studies were classified as 'high risk' of performance and detection bias while the remaining 163 were classified as 'unclear'. For the item on blinding of participants and personnel (performance bias), three RCTs were classified as 'low risk' of bias, nine RCTs as 'high risk' of bias and the remaining 160 as 'unclear'.
Incomplete outcome data
Only 11 trials (Alby 1993; Bennie 1995; Berlanga 1997; Chouinard 1999; Corrigan 2000; Dierick 1996; Fava 2005; Guelfi 1999; O'Keane 1992; Sheehan 2009; Suleman 1997) were rated as adequate in terms of addressing incomplete outcome data, while the majority (83 studies) were classified as 'unclear risk' and 77 as 'high risk'.
Selective reporting
The study protocol was not available for almost all studies so it was difficult to make a judgement on the possibility of outcome reporting bias. However, in six studies (Chouinard 1999; Fava 2005; Guelfi 1999; Hale 2010; O'Keane 1992; Wehmeier 2005) results were consistent with what was stated in the study protocols. Twenty‐three studies were classified as 'high risk' of bias, five studies as 'low risk' of bias and the remaining 144 were classified as 'unclear'.
Other potential sources of bias
Most of the included studies (115 RCTs) were funded by pharmaceutical industry (and classified as 'high risk' of bias) and five studies were independent of industry (and classified as 'low risk' of bias). The remaining studies did not specify the source of funding and were defined as 'unclear'.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15; Table 16; Table 17; Table 18; Table 19; Table 20; Table 21; Table 22; Table 23; Table 24; Table 25; Table 26
Comparison 1: fluoxetine versus TCAs
Primary outcome: efficacy
1.1 Dichotomous outcome
Twenty‐four studies contributed to this comparison including 2124 participants.
We found no difference in terms of efficacy between fluoxetine and TCAs as a class (OR 0.97, 95% CI 0.77 to 1.22, 24 RCTs, 2124 participants). In head‐to‐head comparisons, only dothiepin/dosulepin was found to be more effective than fluoxetine (OR 2.13, 95% CI 1.08 to 4.20; NNT = 6, 95% CI 3 to 50, 2 RCTs, 144 participants).
1.2 Continuous outcome
Fifty studies contributed to this comparison including 3393 participants.
On this outcome (measured with HDRS or MADRS), we found no differences between fluoxetine and TCAs as a class (SMD 0.03, 95% CI ‐0.07 to 0.14, 50 RCTs, 3393 participants) and between fluoxetine and individual TCAs.
Secondary outcome: acceptability
1.3 Failure to complete due to any cause
Forty‐nine studies contributed to this comparison including 4194 participants.
In terms of participants who dropped out for any cause, fluoxetine was better tolerated than TCAs (OR 0.79, 95% CI 0.65 to 0.96; NNT = 20, 95% CI 13 to 48, 49 RCTs, 4194 participants), in particular amitriptyline (OR 0.62, 95% CI 0.46 to 0.85; NNT = 13, 95% CI 8 to 39, 18 RCTs, 1089 participants).
1.4 Failure to complete due to inefficacy
Thirty‐three studies contributed to this comparison including 2911 participants.
We found no differences in terms of dropouts due to inefficacy (OR 1.29, 95% CI 0.96 to 1.72, 33 RCTs, 2911 participants).
1.5 Failure to complete due to side effects
Forty studies contributed to this comparison including 3647 participants.
The analysis of dropouts due to side effects revealed that amitriptyline (OR 0.41, 95% CI 0.23 to 0.71; NNT = 12, 95% CI 8 to 22, 16 RCTs, 1038 participants), clomipramine (OR 0.30, 95% CI 0.12 to 0.79; NNT = 11 , 95% CI 6 to 46, 2 RCTs, 163 participants), imipramine (OR 0.47, 95% CI 0.26 to 0.86; NNT= 8, 95% CI 6 to 12, 10 RCTs, 1093 participants) and overall TCAs (OR 0.55, 95% CI 0.40 to 0.75; NNT = 14, 95% CI 10 to 20, 40 RCTs, 3647 participants) were less well tolerated than fluoxetine.
Comparison 2: fluoxetine versus heterocyclics
Primary outcome: efficacy
2.1 Dichotomous outcome
Three studies contributed to this comparison including 216 participants.
In terms of dichotomous outcomes, we found no differences between fluoxetine and individual heterocyclics.
2.2 Continuous outcome
Eight studies contributed to this comparison including 561 participants.
In terms of continuous outcomes, we found no differences between fluoxetine and individual heterocyclics.
Secondary: acceptability
2.3 Failure to complete due to any cause
Six studies contributed to this comparison including 444 participants.
In terms of patients who dropped out during the trial for any reason, we found no differences between fluoxetine and individual heterocyclics.
2.4 Failure to complete due to inefficacy
Four studies contributed to this comparison including 262 participants overall.
We found no differences in terms of dropouts due to inefficacy.
2.5 Failure to complete due to side effects
Four studies contributed to this comparison including 262 participants overall.
Similarly, we found no differences in terms of dropouts due to side effects.
Comparison 3: fluoxetine versus other SSRIs
Primary outcome: efficacy
3.1 Dichotomous outcome
Eighteen studies contributed to this comparison including 3238 participants overall.
There was a difference in terms of efficacy in favour of sertraline over fluoxetine (OR 1.37, 95% CI 1.08 to 1.74; NNT = 13, 95% CI 7 to 58, 6 RCTs, 1188 participants).
3.2 Continuous outcome
Twenty‐two studies contributed to this comparison including 4113 participants overall.
In terms of continuous outcomes (measured with HDRS or MADRS), there were inconclusive results.
Secondary outcome: acceptability
3.3 Failure to complete due to any cause
Twenty‐six studies contributed to this comparison including 5033 participants.
In terms of patients who dropped out for any reason, we found a trend in favour of escitalopram over fluoxetine (OR 1.53, 95%, CI 1 to 2.37, 2 RCTs, 578 participants).
3.4 Failure to complete due to inefficacy
Fourteen studies contributed to this comparison including 3371 participants.
No difference was found between fluoxetine and other SSRIs in terms of discontinuation due to inefficacy.
3.5 Failure to complete due to side effects
Twenty‐four studies contributed to this comparison including 4510 participants.
No difference was found between fluoxetine and other SSRIs in terms of discontinuation due to side effects.
Comparison 4: fluoxetine versus SNRIs
Primary outcome: efficacy
4.1 Dichotomous outcome
Fifteen studies contributed to this comparison including 3860 participants.
There was a difference in terms of efficacy in favour of venlafaxine over fluoxetine (OR 1.29, 95%, CI 1.10 to 1.51; NNT =11, 95% CI 8 to 16, 12 RCTs, 3387 participants). No difference was found between fluoxetine and other SNRIs (milnacipran and duloxetine).
4.2 Continuous outcome
Fifteen studies contributed to this comparison including 3310 participants.
There was a difference in terms of efficacy in favour of fluoxetine over milnacipran (measured with HDRS or Montgomery and Asberg Scale for Depression (MADRS)) (SMD ‐0.36, 95% CI ‐0.63 to ‐0.08, 2 RCTs, 213 participants) and a small difference in favour of venlafaxine over fluoxetine (SMD 0.10 95%, CI 0.00 to 0.19,13 RCTs, 3097 participants).
Secondary outcome: Acceptability
4.3 Failure to complete due to any cause
Nineteen studies contributed to this comparison including 3775 participants.
There was no evidence that fluoxetine was associated with a smaller or higher rate of dropout for any reason than the SNRIs.
4.4 Failure to complete due to inefficacy
Eighteen studies contributed to this comparison including 3632 participants.
There was no evidence that fluoxetine was associated with a smaller or higher rate of dropout for inefficacy than the SNRIs.
4.5 Failure to complete due to side effects
Eighteen studies contributed to this comparison including 3732 participants.
The analysis of dropouts due to side effects revealed that fluoxetine had an advantage over venlafaxine (OR 0.72, 95% CI 0.56‐0.94; NNT = 36, 95% CI 20 to 202, 13 RCTs, 2640 participants).
Comparison 5: fluoxetine versus MAOIs or newer ADs
Primary outcome: efficacy
5.1 Dichotomous outcome
Sixteen studies contributed to this comparison including 2567 participants.
There was a difference in terms of efficacy in favour of mirtazapine over fluoxetine (OR 1.46, 95% CI 1.04 to 2.04; NNT = 12, 95% CI 6 to 134, 4 RCTs, 600 participants). No differences were found between fluoxetine and other newer ADs or MAOIs.
5.2 Continuous outcome
Thirteen studies contributed to this comparison including 2029 participants.
We found no differences in terms of efficacy between fluoxetine and MAOIs or newer ADs (measured with HDRS or MADRS).
Secondary outcome: acceptability
5.3 Failure to complete due to any reason
Seventeen studies contributed to this comparison including 2611 participants.
In terms of participants who dropped out during the trial for any reason, fluoxetine performed better in comparison with reboxetine only (OR 0.60, 95%, CI 0.44 to 0.82; NNT = 9, 95% CI 6 to 24, 4 RCTs, 764 participants).
5.4 Failure to complete due to inefficacy
Sixteen studies contributed to this comparison including 2568 participants.
No differences were found between fluoxetine and MAOIs or newer ADs in terms of dropout due to inefficacy.
5.5 Failure to complete due to side effects
Sixteen studies contributed to this comparison including 2157 participants.
No differences were found between fluoxetine and MAOIs or newer ADs in terms of dropout due to side effects. However, we found a trend in favour of fluoxetine over reboxetine (OR 0.41, 95% CI 0.15 to 1.09, 2 RCTs, 211 participants).
Comparison 6: fluoxetine versus other conventional psychotropic drugs
Primary outcome: efficacy
6.1 Dichotomous outcome
Eight studies contributed to this comparison including 1101 participants.
No differences between fluoxetine and any other conventional AD (amineptine, bupropion, pramipezole, tianeptine, trazodone) were found in terms of dichotomous outcome measures.
6.2 Continuous outcome
Thirteen studies contributed to this comparison including 1613 participants.
We found an advantage of fluoxetine over ABT‐200 (SMD ‐1.85, 95% CI ‐2.25 to ‐1.45, 1 RCT, 141 participants) (measured with HDRS or MADRS). No differences in continuous outcome measures were found between fluoxetine and other conventional ADs.
Secondary outcome: acceptability
6.3 Failure to complete due to any cause
Seventeen studies contributed to this comparison including 2419 participants.
In terms of participants who dropped out for any reason, fluoxetine was better tolerated than ABT‐200 (OR 0.18, 95% CI 0.08 to 0.39; NNT = 3, 95% CI 2 to 5, 1 RCT, 144 participants) and pramipexole (OR 0.12, 95% CI 0.03 to 0.42; NNT = 3, 95% CI 2 to 5, 1 RCT, 105 participants).
6.4 Failure to complete due to inefficacy
Fourteen studies contributed to this comparison including 2090 participants.
In terms of dropout due to inefficacy, we found no difference between fluoxetine and other conventional psychotropic drugs. However, due to the large confidence intervals, these results are inconclusive.
6.5 Failure to complete due to side effects
Seventeen studies contributed to this comparison including 2424 participants.
In terms of dropout due to side effects fluoxetine was better tolerated than ABT‐200 (OR 0.08, 95% CI 0.02 to 0.27; NNT = 3, 95% CI 2 to 5, 1 RCT, 144 participants) and pramipexole (OR 0.06, 95% CI 0.01 to 0.50, NNT = 4, 95% CI 2 to 6, 1 RCT, 105 participants).
Comparison 7. fluoxetine versus other non‐conventional AD agents
Primary outcome: efficacy
8.1 Dichotomous outcome
Seven studies contributed to this comparison including 757 participants overall.
No difference between fluoxetine and other non‐conventional agents was found.
8.2 Continuous outcome
Five studies contributed to this outcome including 648 participants overall.
No difference was found on this outcome.
Secondary outcome: acceptability
8.3 Failure to complete due to any cause
Six studies contributed to this comparison including 719 participants overall.
In terms of patients who dropped out for any reason no differences were found between fluoxetine and other non‐conventional ADs.
8.4 Failure to complete due to inefficacy
Two studies contributed to this comparison including 401 participants overall.
No difference was found on this outcome.
8.5 Failure to complete due to inefficacy
Five studies contributed to this comparison including 679 participants overall.
No difference was found on this outcome.
Subgroup analysis
The great majority of included studies had a follow‐up period of between six and 16 weeks.
The subgroup analysis revealed a difference in favour of fluoxetine over nortriptyline, continuous outcome, follow‐up > 16 weeks (SMD ‐0.86, 95% CI ‐1.27 to ‐0.44, 1 RCT, 97 participants), and a difference between fluoxetine and TCAs as a class, follow‐up six to 16 weeks, dichotomous outcome (failure to complete ‐ inefficacy) (OR 1.38, 95% CI 1.02 to 1.87, 28 RCTs, 1053 participants).
For the other comparisons we found no material difference between fluoxetine and other antidepressants both in terms of efficacy and acceptability.
Moreover, with the exception of the TCA group, stratification by each control agent was performed to ascertain whether there were treatment differences between fluoxetine and AD drugs belonging to the same pharmacological class (see results on efficacy and acceptability).
As reported in the methods, sensitivity analyses were not performed in this version of the review. However, we will conduct these analyses in the next update.
Assessment of heterogeneity
For the primary outcomes, I2 indicative of moderate heterogeneity was observed in the comparison between fluoxetine and TCA as a class, (I2 = 55%) and between fluoxetine and imipramine, continuous outcome analyses (I2 = 54%). Substantial heterogeneity was found in the comparison between fluoxetine and the following AD drugs: mianserin (I2 = 78%), paroxetine (I2 = 86%), agomelatine (I2 = 67%) and trazodone (I2 = 62%), desipramine, dothiepin or dosulepin (I2 = 67%), nortryptiline (I2 = 87%), and tianeptine (I2 = 63%). For dichotomous outcomes, moderate heterogeneity was observed in the comparison between fluoxetine and trazodone (I2 = 58%), while substantial heterogeneity was found in the comparison between fluoxetine versus hypericum (I2 = 68%) and versus imipramine (I2 = 68%).
Assessment of publication bias
Visual inspection of funnel plots did not reveal substantial asymmetry in any of the comparisons between fluoxetine and other conventional and unconventional ADs.
Discussion
Summary of main results
This systematic review detected differences between fluoxetine and some comparator ADs. On a dichotomous outcome, fluoxetine was less effective than dothiepin/dosulepin, sertraline, mirtazapine and venlafaxine. On a continuous outcome, fluoxetine was more effective than ABT‐200 and milnacipran, and less effective than sertraline and venlafaxine, although these findings were of borderline statistical significance. However, it is uncertain how these differences translate into clinically meaningful measures. Despite the large number of comparative trials included in this systematic review, the total number of randomised patients was under 25,000. Studies were short, usually eight weeks or less, and the mean size of each trial was around 135 participants, indicating that they were generally underpowered for demonstrating clinically meaningful differences.
In terms of acceptability fluoxetine was better tolerated than TCA, reboxetine, ABT‐200 and pramipexole (dropout due to any reasons). By contrast, escitalopram was better tolerated than fluoxetine, although the result was of borderline statistical significance. Moreover, considering dropout due to side effects, fluoxetine was better tolerated than amitriptyline, clomipramine, imipramine, ABT‐200 and pramipexole. Fluoxetine was also better tolerated than venlafaxine (dropout due to side effects).
Overall completeness and applicability of evidence
Our review currently includes 171 randomised trials with 24,868 participants in total. All studies in the review involved participants with a formal diagnosis of depression on a standardised and validated scale according to DSM‐III or DSM‐IV criteria, and therefore there was considerable homogeneity in the study populations. However, studies were short in duration, for some comparisons the confidence intervals were large, and some analyses were unpowered to demonstrate clinically meaningful differences between treatments. Therefore, interpretation of treatment effects, either where there is a statistically different effect or no statistically different effect, should be made with caution There was also considerable variation in the type of control medication used in the trials. The majority used TCAs, SSRIs and SNRIs, with a minority of trials using other non‐conventional ADs. Most trials provided useful data that were included in the analyses, but in some cases trials provided only efficacy or tolerability data thus limiting the overall completeness of evidence.
Although it has long been argued that placebo controlled trials are required to adequately demonstrate the efficacy of novel AD drugs (Cipriani 2009a), in the present review we focused only on the comparison between fluoxetine and other active treatments. The background logic that guided the development of the present review was based on the need to provide real‐world evidence for patients in need of pharmacological treatment. We therefore made the choice of including only studies that compared fluoxetine with another active treatment, as we reasoned that clinicians need to know how fluoxetine, a reference AD agent, compares with a selection of possible comparator ADs.
Retrieved randomised evidence compared fluoxetine with a selection of possible comparator ADs but only few studies per comparison were found. This inevitably limited the applicability of the evidence pertaining to each pair wise comparison as often confidence intervals were quite wide around treatment estimates.
Although the search was thorough, it is still possible that some unpublished studies have not been identified. It is very difficult to make a fair judgement on this issue as the small number of trials identified per comparison might have hindered the detection of publication bias. However, although we did our very best to retrieve as much data as possible, through asking pharmaceutical companies and study authors to supply all available information, we can conservatively assume that some trial data are still lacking, most of which are likely to be studies with negative findings. We are also aware that other RCTs comparing fluoxetine with other AD drugs may be currently ongoing, and we aim to include these, as well as other unpublished data, in future updates of this review.
Quality of the evidence
The quality of evidence is a crucial issue in translating the results of research into clinical practice. Using high‐quality research evidence is relevant to speedy translation of research in a way that really responds to clinically relevant questions. In the present review only randomised trials were included and the studies were all very similar in design and conduct. However, trial quality is not easy to assess. We note that the majority of included studies failed to report key methodological issues, for example information about randomisation and allocation concealment. Additionally, the reporting of outcome data was often unclear or incomplete and the figures used in the analyses were not immediately understandable. Clearly it is possible that the paucity of information about randomisation and allocation concealment may represent a problem of reporting and not a real defect in study design. However, sometimes discrepancies between published reports and unpublished data posted on drug companies' websites were highlighted, thus confirming that the average quality of clinical trials in the field of ADs is generally low, and that this may represent a potential serious risk of bias (Horder 2011).
Potential biases in the review process
The first limitation is that we made multiple comparisons. By making multiple comparisons we might have committed a type 1 error, that is, identifying and reporting a spurious association. As stated in the review protocol, we did not carry out a Bonferroni correction. As many statistical tests have been used in the review, the findings from this review are better thought of as hypothesis forming rather than hypothesis testing and it would be very reassuring to see the conclusions replicated in future trials and systematic reviews.
Another limitation is that for many of the comparisons, more trials were found presenting data on dropouts than on efficacy. This is because not all gave a 50% reduction in HDRS as their main outcome and therefore a lot of the data from our primary outcome was lost, which could have led to bias. Although this is a well‐known limitation of secondary analyses of data extracted from RCTs, we attempted to control this potential source of bias by extracting continuous efficacy data according to each study’s definition of efficacy. This secondary analysis included many more participants than the primary analysis and the results did not differ substantially from the analysis of the primary outcomes. It is likely, therefore, that the overall comparison was not hampered by the exclusion of these trials.
Another compelling issue is that most of the included studies were sponsored by the drug industry, and data has shown that drug industry studies are more than four times more likely to demonstrate positive effects of the sponsors' drugs in comparison with independent studies (Lexchin 2003). For fluoxetine, it has been shown that the outcome of fluoxetine RCTs varies according to whether this drug was used as a new compound or a reference one, suggesting the presence of bias. This bias may work in favour of fluoxetine in trials where it was the experimental drug and in favour of comparators in trials where fluoxetine was the reference agent (Barbui 2004). A possible explanation for this finding is publication bias. Publication bias might have systematically excluded from publication RCTs failing to show a robust effect of the experimental agents in comparison with reference ones. Alternatively, the common belief that newer medicines are better than old ones, and pharmaceutical marketing pressures to show a positive effect of newer drugs, might have favoured fluoxetine when it was the experimental agent and comparators when fluoxetine was the reference drug.
In this review we decided to focus on treatment response because it is one of the main goals for the treatment of major depressive disorder. The term 'treatment response' describes a state of improvement in the patient’s condition of sufficient quality to result in the treating physician’s impression of at least a moderate degree of global improvement, conventionally defined as a reduction of at least 50% in depressive symptomatology. However, from a clinical point of view, the ultimate goal of the acute treatment phase of major depressive disorder may well be to achieve remission. Full remission from depression correlates with better longer‐term functional recovery, lower risk of relapse and higher level of patients satisfaction than a partial response (without remission). Thus, one important limitation of the present review is that remission was not included as an outcome measure.
Another study limitation is that studies with different duration were lumped together, and this may limit the external validity of study findings. However, the subgroup analysis failed to show material differences, thus suggesting the robustness of the main findings.
From a clinical point of view the analysis of the safety profile of antidepressants remains of crucial importance. In this review, although we included total dropouts as a measure of treatment acceptability. and dropouts due to side effects as a measure of tolerability, individual side effects were not extracted. We acknowledge this limitation and we aim to include data on individual side effects in future updates of this systematic review.
Agreements and disagreements with other studies or reviews
Even though there is a heated debate in the scientific literature (Gartlehner 2010; Gartlehner 2011), there is now robust evidence that there are statistically and clinically significant differences among antidepressants (Cipriani 2009a). Results from this review are consistent with this interpretation and might contribute to the development of an evidence‐based hierarchy of antidepressants to be used by clinicians (both specialists and general practitioners) (Barbui 2011a). Even though fluoxetine was not among the best treatments in terms of efficacy, it remains an important option for physicians when an AD is to be prescribed for moderate to severe major depression.
Authors' conclusions
Implications for practice.
The main finding of the present study is that there are differences in terms of efficacy and tolerability between fluoxetine and certain ADs, but the clinical meaning of these differences is uncertain and no definitive implications for clinical practice can be drawn. The better efficacy profile of sertraline and venlafaxine (and possibly other ADs) over fluoxetine may be clinically meaningful, as suggested by other reviews. In addition to efficacy data, treatment decisions should also be based on other considerations, for example, the fluoxetine long half‐life, the fact that fluoxetine inhibits CYP‐P450 isoenzymes more than some others (and this factor has to be considered particularly for people also taking other medications), the drug toxicity, the patient acceptability and the costs.
Implications for research.
Results described in this systematic review come from a set of randomised studies that are in many cases financially supported by pharmaceutical industries. Industry‐sponsored trials tend to follow a standard design which involves short‐term, double‐blind, parallel‐group studies of patients with acute episodes or exacerbations of chronic illness. Moreover, it is known that economic support by drug manufacturers can strongly influence the progress of research and its results. Consequently, there is a risk that these studies do not provide adequate information to clinicians in real‐world settings. Trials comparing two or more active treatments need to be much larger and of better quality than the studies that we identified for this review. More clinically meaningful outcome measures in trials of antidepressants, such as the ability to work or admission to hospital, are needed. Considering the methodological limitation of standard systematic reviews that rely only on evidence from direct comparisons, and given the wide spectrum of available comparisons for the treatment of major depression, the use of the methodology of multiple treatments meta‐analysis (MTM) may provide a more informative and clinically useful summary of the results that can be used to guide treatment decisions. Moreover, an Individual Patient Data (IPD) meta‐analysis would provide a more specific approach to those patients or illness characteristics related to clinically meaningful outcomes.
What's new
| Date | Event | Description |
|---|---|---|
| 16 July 2013 | New search has been performed | Studies published between 2005 and 2012 added. |
| 16 July 2013 | New citation required but conclusions have not changed | Major update of methods and new studies, conclusions not changed. |
History
Protocol first published: Issue 2, 2003 Review first published: Issue 4, 2005
| Date | Event | Description |
|---|---|---|
| 1 November 2008 | Amended | Converted to new review format. |
| 23 August 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We would like to thank the CCDAN Editorial Team for their support, information and advice. We also would like to thank authors that provided additional data to be used in the present report (Professor Yusuf Moosa, Professor Homayoun Amini).
CRG funding acknowledgement
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Depression, Anxiety and Neurosis Group.
Disclaimer
The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health.
Data and analyses
Comparison 1. Fluoxetine versus TCAs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 24 | 2124 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.77, 1.22] |
| 1.1 Fluoxetine vs Amitriptyline | 11 | 777 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.68, 1.28] |
| 1.2 Fluoxetine vs Clomipramine | 1 | 94 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.27, 1.45] |
| 1.3 Fluoxetine vs Desipramine | 2 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [0.56, 5.15] |
| 1.4 Fluoxetine vs Dothiepin/dosulepin | 2 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 2.13 [1.08, 4.20] |
| 1.5 Fluoxetine vs Doxepine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.54] |
| 1.6 Fluoxetine vs Imipramine | 5 | 761 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.41, 1.35] |
| 1.7 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.55, 1.78] |
| 1.8 Fluoxetine vs Trimipramine | 1 | 41 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [0.56, 7.45] |
| 2 End‐point score on rating scale | 50 | 3393 | Std. Mean Difference (IV, Random, 95% CI) | 0.03 [‐0.07, 0.14] |
| 2.1 Fluoxetine vs Amiptriptyline | 19 | 1023 | Std. Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.09, 0.29] |
| 2.2 Fluoxetine vs Clomipramine | 5 | 372 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.31, 0.10] |
| 2.3 Fluoxetine vs Desipramine | 4 | 147 | Std. Mean Difference (IV, Random, 95% CI) | 0.27 [‐0.32, 0.86] |
| 2.4 Fluoxetine vs Dothiepin/dosulepin | 4 | 266 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.27, 0.59] |
| 2.5 Fluoxetine vs Imipramine | 12 | 1063 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.21, 0.19] |
| 2.6 Fluoxetine vs Lofepramine | 1 | 183 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.16, 0.42] |
| 2.7 Fluoxetine vs Nomifensine | 1 | 28 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐1.12, 0.38] |
| 2.8 Fluoxetine vs Nortriptyline | 2 | 251 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐1.20, 0.24] |
| 2.9 Fluoxetine vs Trimipramine | 2 | 60 | Std. Mean Difference (IV, Random, 95% CI) | 0.41 [‐0.10, 0.92] |
| 3 Failure to complete ‐ Total | 49 | 4194 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.65, 0.96] |
| 3.1 Fluoxetine vs Amitriptyline | 18 | 1089 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.46, 0.85] |
| 3.2 Fluoxetine vs Clomipramine | 2 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.38, 1.14] |
| 3.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.16, 1.24] |
| 3.4 Fluoxetine vs Dothiepin/dosulepin | 5 | 478 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.92, 2.69] |
| 3.5 Fluoxetine vs Doxepine | 4 | 323 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.49, 1.32] |
| 3.6 Fluoxetine vs Imipramine | 12 | 1225 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.51, 1.21] |
| 3.7 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.24, 1.04] |
| 3.8 Fluoxetine vs Nomifensine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 6.33 [0.67, 60.16] |
| 3.9 Fluoxetine vs Nortriptyline | 3 | 448 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.36, 1.47] |
| 3.10 Fluoxetine vs Trimipramine | 1 | 41 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.41, 9.78] |
| 4 Failure to complete ‐ Inefficacy | 33 | 2911 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.96, 1.72] |
| 4.1 Fluoxetine vs Amitriptyline | 13 | 835 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.44, 1.88] |
| 4.2 Fluoxetine vs Clomipramine | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 7.37 [0.37, 145.75] |
| 4.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.20, 5.35] |
| 4.4 Fluoxetine vs Dothiepin/dosulepin | 3 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.49, 3.66] |
| 4.5 Fluoxetine vs Doxepine | 3 | 283 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.60, 4.92] |
| 4.6 Fluoxetine vs Imipramine | 10 | 1093 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.97, 2.05] |
| 4.7 Fluoxetine vs Nortriptyline | 1 | 205 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.07, 2.03] |
| 5 Failure to complete ‐ Side Effects | 40 | 3647 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.40, 0.75] |
| 5.1 Fluoxetine vs Amitriptyline | 16 | 1038 | Odds Ratio (M‐H, Random, 95% CI) | 0.41 [0.23, 0.71] |
| 5.2 Fluoxetine vs Clomipramine | 2 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 0.30 [0.12, 0.79] |
| 5.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.04, 1.68] |
| 5.4 Fluoxetine vs Dothiepin/dosulepin | 5 | 478 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [0.59, 7.16] |
| 5.5 Fluoxetine vs Doxepine | 3 | 283 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.44, 1.53] |
| 5.6 Fluoxetine vs Imipramine | 10 | 1093 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.26, 0.86] |
| 5.7 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.16 [0.02, 1.38] |
| 5.8 Fluoxetine vs Nortriptyline | 1 | 205 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.42, 1.77] |
1.1. Analysis.
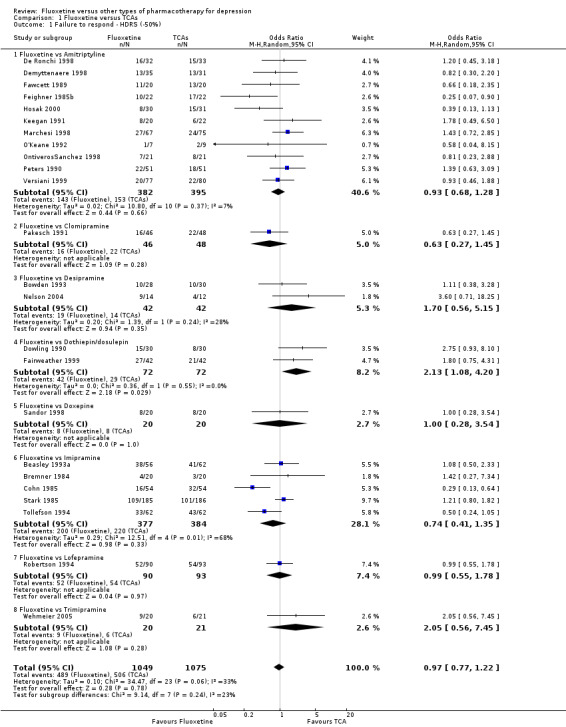
Comparison 1 Fluoxetine versus TCAs, Outcome 1 Failure to respond ‐ HDRS (‐50%).
1.2. Analysis.
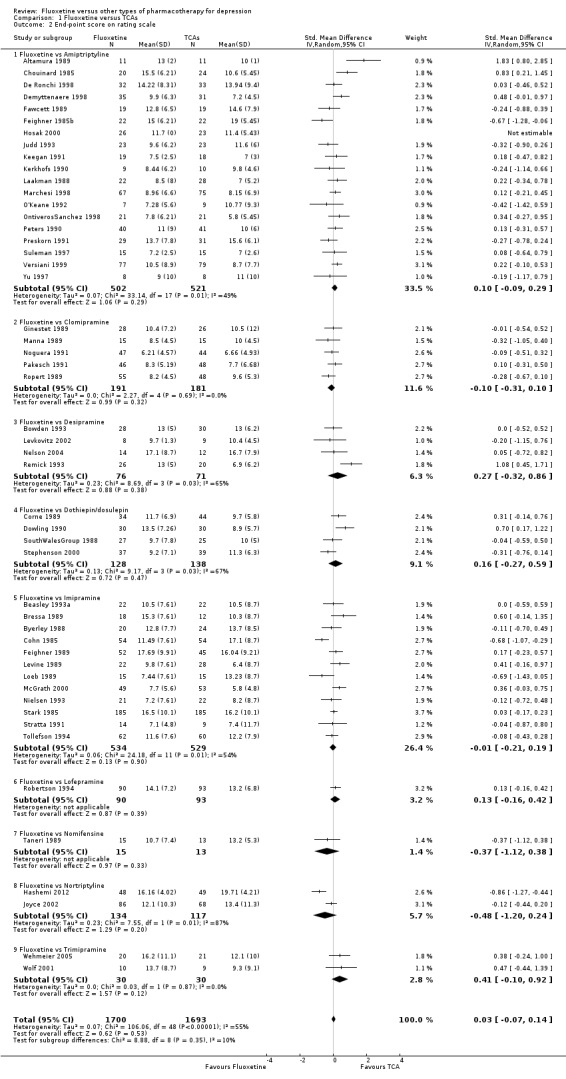
Comparison 1 Fluoxetine versus TCAs, Outcome 2 End‐point score on rating scale.
1.3. Analysis.
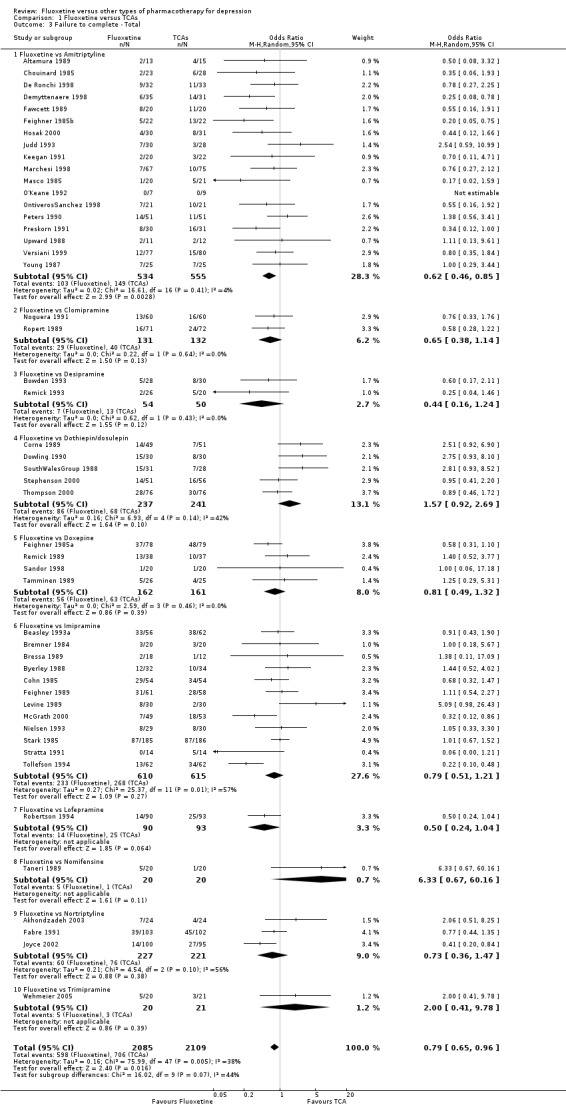
Comparison 1 Fluoxetine versus TCAs, Outcome 3 Failure to complete ‐ Total.
1.4. Analysis.
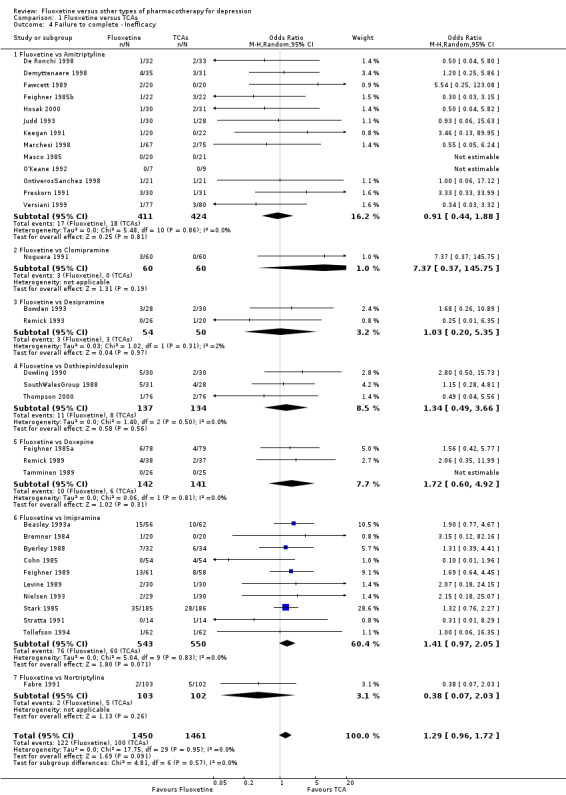
Comparison 1 Fluoxetine versus TCAs, Outcome 4 Failure to complete ‐ Inefficacy.
1.5. Analysis.
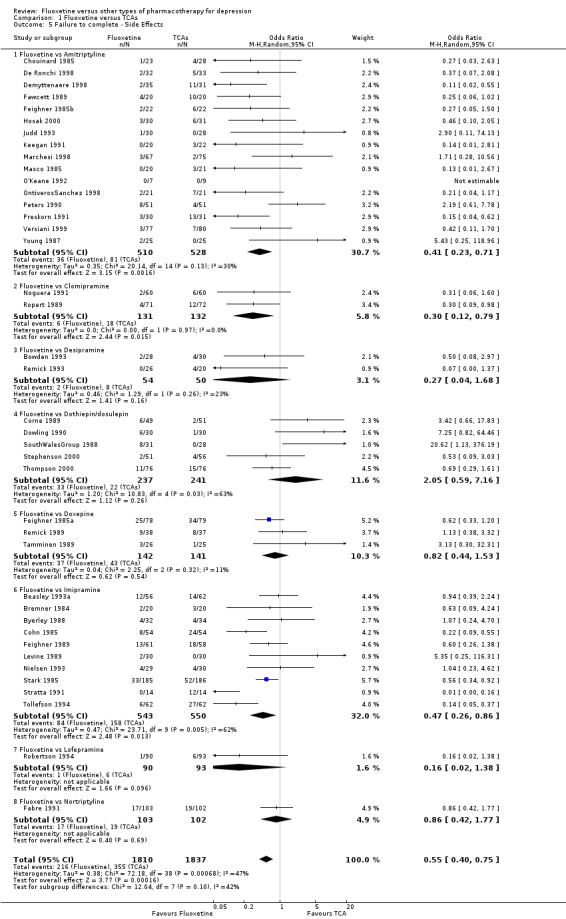
Comparison 1 Fluoxetine versus TCAs, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 2. Fluoxetine versus heterocyclics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Maprotiline | 2 | 163 | Odds Ratio (M‐H, Random, 95% CI) | 1.95 [0.91, 4.18] |
| 1.2 Fluoxetine vs Mianserin | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.27, 2.38] |
| 2 End‐point score on rating scale | 8 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Maprotiline | 5 | 433 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.15, 0.23] |
| 2.2 Fluoxetine vs Mianserin | 3 | 128 | Std. Mean Difference (IV, Random, 95% CI) | 0.43 [‐0.38, 1.23] |
| 3 Failure to complete ‐ Total | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Maprotiline | 4 | 351 | Odds Ratio (M‐H, Random, 95% CI) | 1.75 [0.90, 3.41] |
| 3.2 Fluoxetine vs Mianserin | 2 | 93 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.18, 2.25] |
| 4 Failure to complete ‐ Inefficacy | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs Maprotiline | 3 | 209 | Odds Ratio (M‐H, Random, 95% CI) | 2.54 [0.33, 19.19] |
| 4.2 Fluoxetine vs Mianserin | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 2.27 [0.38, 13.63] |
| 5 Failure to complete ‐ Side Effects | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs Maprotiline | 3 | 209 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.15, 1.93] |
| 5.2 Fluoxetine vs Mianserin | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.23, 4.70] |
2.1. Analysis.
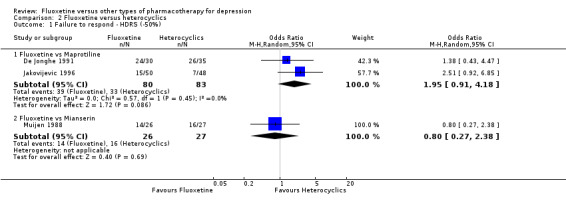
Comparison 2 Fluoxetine versus heterocyclics, Outcome 1 Failure to respond ‐ HDRS (‐50%).
2.2. Analysis.
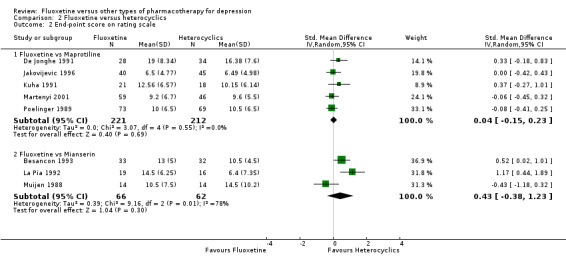
Comparison 2 Fluoxetine versus heterocyclics, Outcome 2 End‐point score on rating scale.
2.3. Analysis.
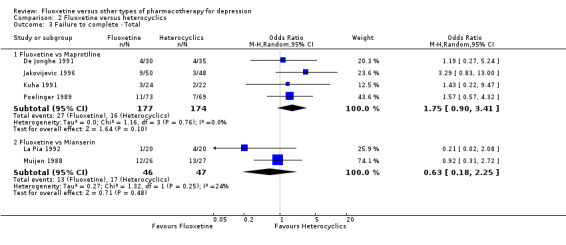
Comparison 2 Fluoxetine versus heterocyclics, Outcome 3 Failure to complete ‐ Total.
2.4. Analysis.
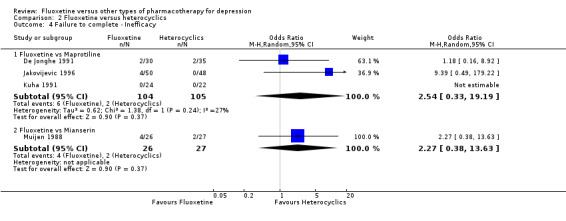
Comparison 2 Fluoxetine versus heterocyclics, Outcome 4 Failure to complete ‐ Inefficacy.
2.5. Analysis.
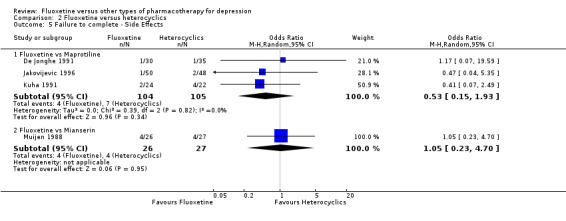
Comparison 2 Fluoxetine versus heterocyclics, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 3. Fluoxetine versus other SSRIs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.20, 1.79] |
| 1.2 Fluoxetine vs Escitalopram | 1 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.56, 1.85] |
| 1.3 Fluoxetine vs Fluvoxamine | 1 | 177 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.52, 1.74] |
| 1.4 Fluoxetine vs Paroxetine | 9 | 1574 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.93, 1.65] |
| 1.5 Fluoxetine vs Sertraline | 6 | 1188 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [1.08, 1.74] |
| 2 End‐point score on rating scales | 21 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Citalopram | 3 | 661 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.10, 0.21] |
| 2.2 Fluoxetine vs Escitalopram | 1 | 231 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.19, 0.33] |
| 2.3 Fluoxetine vs Paroxetine | 11 | 2061 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.26, 0.24] |
| 2.4 Fluoxetine vs Sertraline | 7 | 1160 | Std. Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.03, 0.20] |
| 3 Failure to complete ‐ Total | 25 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Citalopram | 3 | 732 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.60, 1.27] |
| 3.2 Fluoxetine vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [1.00, 2.37] |
| 3.3 Fluoxetine vs Fluvoxamine | 2 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.36, 1.37] |
| 3.4 Fluoxetine vs Paroxetine | 10 | 1848 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.81, 1.20] |
| 3.5 Fluoxetine vs Sertraline | 9 | 1591 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.93, 1.49] |
| 4 Failure to complete ‐ Inefficacy | 13 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs Citalopram | 3 | 732 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.48, 1.56] |
| 4.2 Fluoxetine vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.46, 6.53] |
| 4.3 Fluoxetine vs Paroxetine | 4 | 1005 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.41, 1.39] |
| 4.4 Fluoxetine vs Sertraline | 5 | 1056 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.68, 1.77] |
| 5 Failure to complete ‐ Side Effects | 23 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs Citalopram | 3 | 732 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.34, 1.20] |
| 5.2 Fluoxetine vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.64, 2.12] |
| 5.3 Fluoxetine vs Fluvoxamine | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.14, 7.71] |
| 5.4 Fluoxetine vs Paroxetine | 9 | 1509 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.62, 1.16] |
| 5.5 Fluoxetine vs Sertraline | 9 | 1591 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.92, 1.70] |
3.1. Analysis.
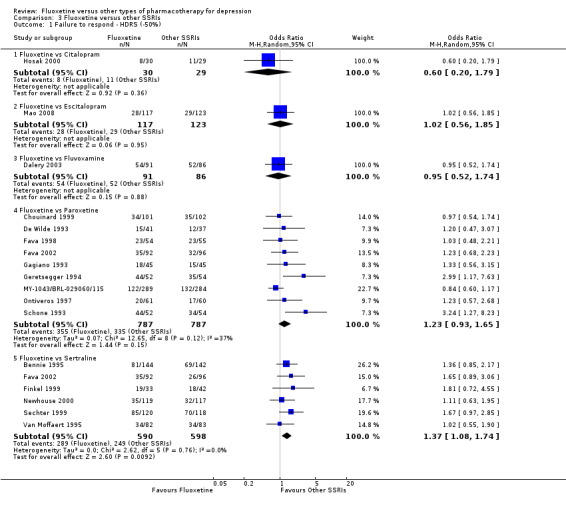
Comparison 3 Fluoxetine versus other SSRIs, Outcome 1 Failure to respond ‐ HDRS (‐50%).
3.2. Analysis.
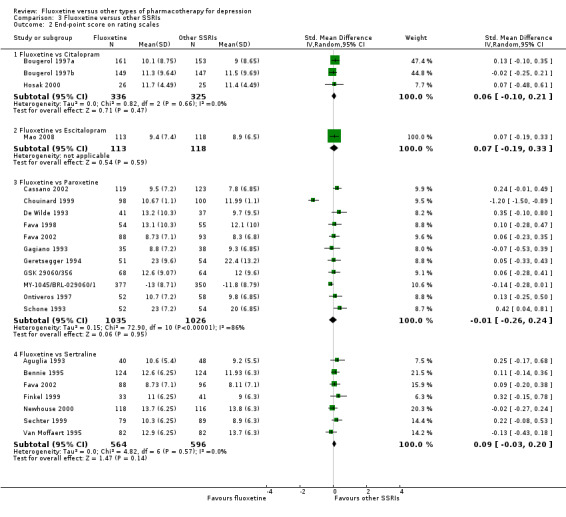
Comparison 3 Fluoxetine versus other SSRIs, Outcome 2 End‐point score on rating scales.
3.3. Analysis.
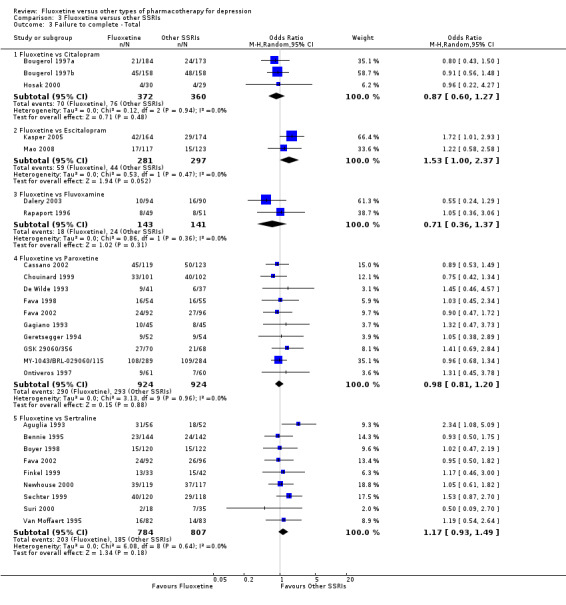
Comparison 3 Fluoxetine versus other SSRIs, Outcome 3 Failure to complete ‐ Total.
3.4. Analysis.
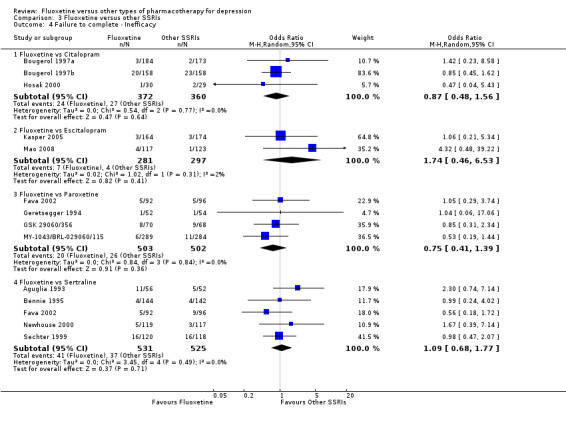
Comparison 3 Fluoxetine versus other SSRIs, Outcome 4 Failure to complete ‐ Inefficacy.
3.5. Analysis.
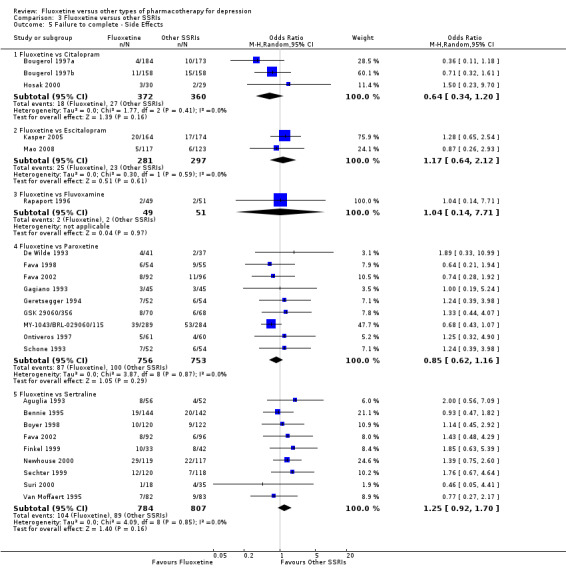
Comparison 3 Fluoxetine versus other SSRIs, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 4. Fluoxetine versus SNRIs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Duloxetine | 1 | 103 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.61, 3.25] |
| 1.2 Fluoxetine vs Milnacipran | 2 | 370 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.78, 1.84] |
| 1.3 Fluoxetine vs Venlafaxine | 12 | 3387 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [1.10, 1.51] |
| 2 End‐point score on rating scale | 15 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Milnacipran | 2 | 213 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.63, ‐0.08] |
| 2.2 Fluoxetine vs Venlafaxine | 13 | 3097 | Std. Mean Difference (IV, Random, 95% CI) | 0.10 [0.00, 0.19] |
| 3 Failure to complete ‐ Total | 19 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Duloxetine | 2 | 532 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.53, 1.52] |
| 3.2 Fluoxetine vs Milnacipran | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.42] |
| 3.3 Fluoxetine vs Venlafaxine | 14 | 2683 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.74, 1.06] |
| 4 Failure to complete ‐ Inefficacy | 18 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs Duloxetine | 2 | 432 | Odds Ratio (M‐H, Random, 95% CI) | 3.33 [0.92, 12.11] |
| 4.2 Fluoxetine vs Milnacipran | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.68, 2.30] |
| 4.3 Fluoxetine vs Venlafaxine | 13 | 2640 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.91, 1.89] |
| 5 Failure to complete ‐ Side Effects | 18 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs Duloxetine | 2 | 532 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.07, 1.23] |
| 5.2 Fluoxetine vs Milnacipran | 3 | 560 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.81, 2.76] |
| 5.3 Fluoxetine vs Venlafaxine | 13 | 2640 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.56, 0.94] |
4.1. Analysis.
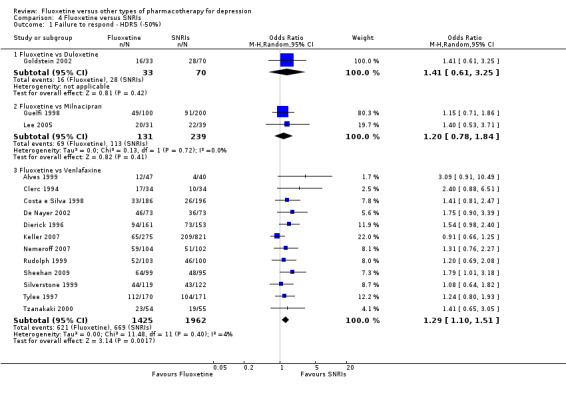
Comparison 4 Fluoxetine versus SNRIs, Outcome 1 Failure to respond ‐ HDRS (‐50%).
4.2. Analysis.
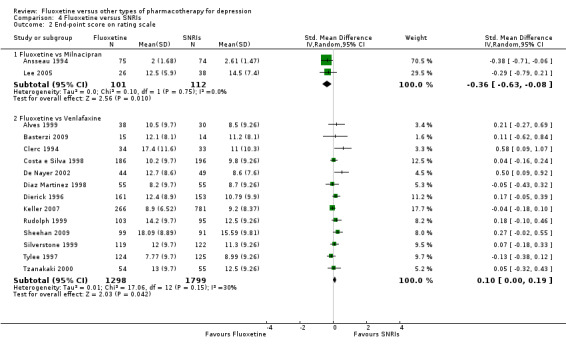
Comparison 4 Fluoxetine versus SNRIs, Outcome 2 End‐point score on rating scale.
4.3. Analysis.
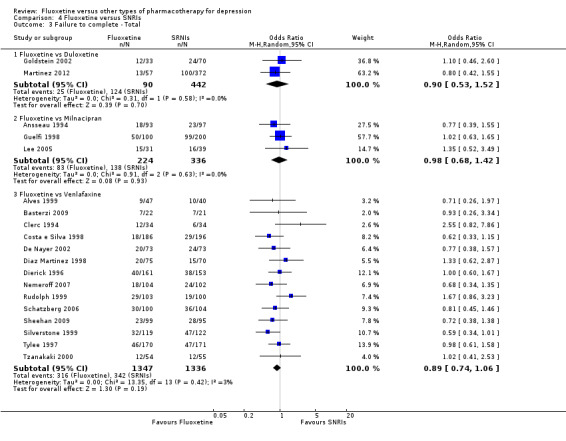
Comparison 4 Fluoxetine versus SNRIs, Outcome 3 Failure to complete ‐ Total.
4.4. Analysis.
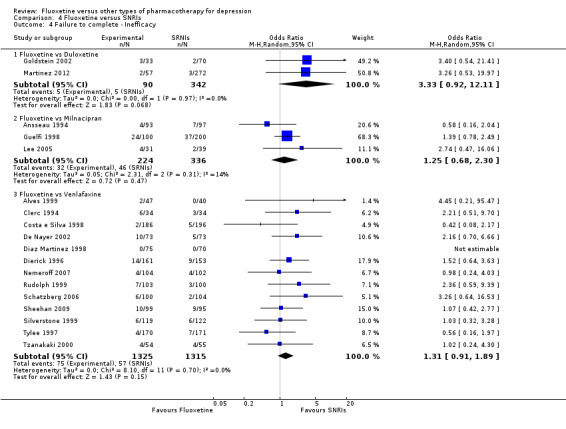
Comparison 4 Fluoxetine versus SNRIs, Outcome 4 Failure to complete ‐ Inefficacy.
4.5. Analysis.
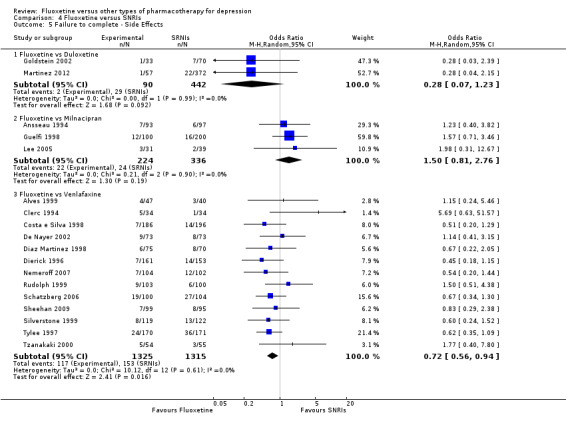
Comparison 4 Fluoxetine versus SNRIs, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 5. Fluoxetine versus MAOIs or newer ADs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 16 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Agomelatine | 1 | 515 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.99, 2.09] |
| 1.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [1.04, 2.04] |
| 1.3 Fluoxetine vs Moclobemide | 7 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.92, 1.75] |
| 1.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.27, 7.34] |
| 1.5 Fluoxetine vs Reboxetine | 3 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.55, 1.10] |
| 2 End‐point score on rating scales | 13 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Agomelatine | 3 | 1213 | Std. Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.18, 0.23] |
| 2.2 Fluoxetine vs Mirtazapine | 1 | 31 | Std. Mean Difference (IV, Random, 95% CI) | 0.57 [‐0.15, 1.29] |
| 2.3 Fluoxetine vs Moclobemide | 6 | 540 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.04, 0.30] |
| 2.4 Fluoxetine vs Phenelzine | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.67, 0.57] |
| 2.5 Fluoxetine vs Reboxetine | 2 | 205 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.31, 0.40] |
| 3 Failure to complete ‐ Total | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.59, 2.49] |
| 3.2 Fluoxetine vs Mirtazapine | 3 | 301 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.51, 1.68] |
| 3.3 Fluoxetine vs Moclobemide | 7 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.70, 1.47] |
| 3.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.01, 4.01] |
| 3.5 Fluoxetine vs Reboxetine | 4 | 764 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.44, 0.83] |
| 4 Failure to complete ‐ Inefficacy | 16 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.41, 2.88] |
| 4.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.71, 2.96] |
| 4.3 Fluoxetine vs Moclobemide | 6 | 679 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.32, 1.56] |
| 4.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.5 Fluoxetine vs Reboxetine | 3 | 464 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.47, 1.77] |
| 5 Failure to complete ‐ Side Effects | 16 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.73, 3.08] |
| 5.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.54, 1.66] |
| 5.3 Fluoxetine vs Moclobemide | 7 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.54, 2.01] |
| 5.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.26] |
| 5.5 Fluoxetine vs Reboxetine | 2 | 211 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.10, 1.61] |
5.1. Analysis.
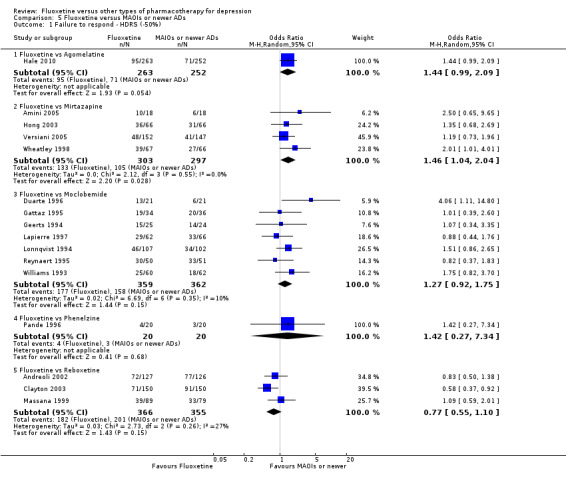
Comparison 5 Fluoxetine versus MAOIs or newer ADs, Outcome 1 Failure to respond ‐ HDRS (‐50%).
5.2. Analysis.
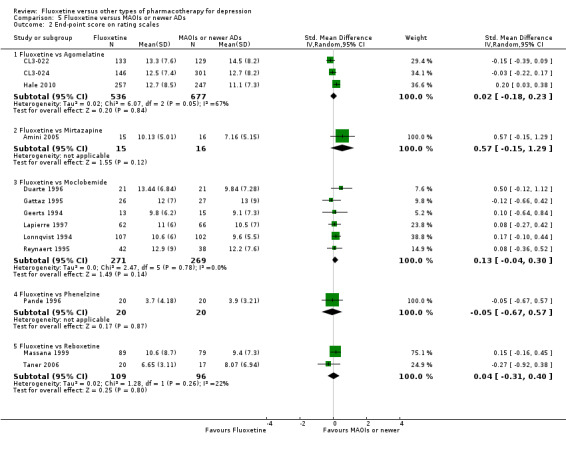
Comparison 5 Fluoxetine versus MAOIs or newer ADs, Outcome 2 End‐point score on rating scales.
5.3. Analysis.
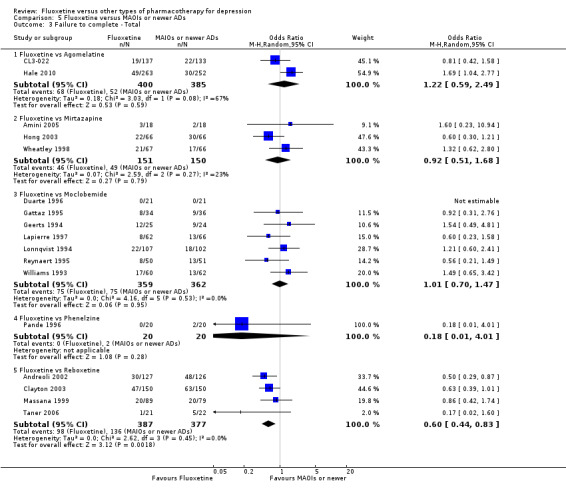
Comparison 5 Fluoxetine versus MAOIs or newer ADs, Outcome 3 Failure to complete ‐ Total.
5.4. Analysis.
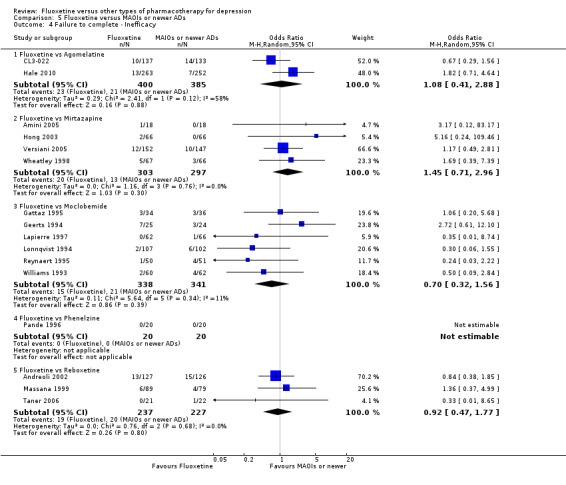
Comparison 5 Fluoxetine versus MAOIs or newer ADs, Outcome 4 Failure to complete ‐ Inefficacy.
5.5. Analysis.
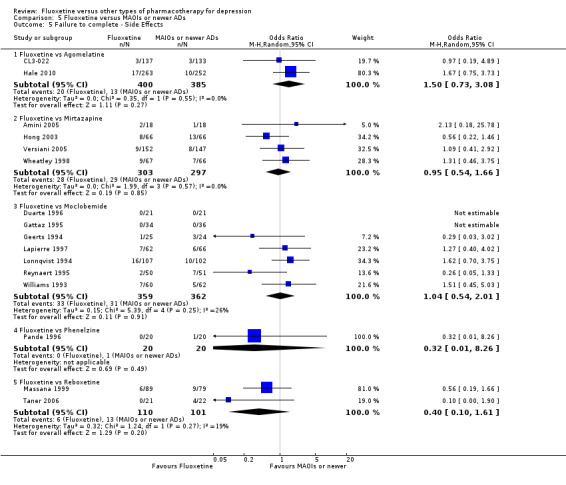
Comparison 5 Fluoxetine versus MAOIs or newer ADs, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 6. Fluoxetine versus other conventional psychotropic drugs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Amineptine | 1 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.13, 1.04] |
| 1.2 Fluoxetine vs Bupropion | 2 | 436 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.48, 1.43] |
| 1.3 Fluoxetine vs Pramipexole | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.24, 1.26] |
| 1.4 Fluoxetine vs Tianeptine | 1 | 387 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.75, 1.67] |
| 1.5 Fluoxetine vs Trazodone | 3 | 110 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.13, 1.86] |
| 2 End‐point score on rating scales | 13 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs ABT‐200 | 1 | 141 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.85 [‐2.25, ‐1.45] |
| 2.2 Fluoxetine vs Amisulpride | 1 | 268 | Std. Mean Difference (IV, Random, 95% CI) | 0.17 [‐0.07, 0.41] |
| 2.3 Fluoxetine vs Nefazodone | 4 | 271 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.30, 0.18] |
| 2.4 Fluoxetine vs Tianeptine | 3 | 730 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.40, 0.10] |
| 2.5 Fluoxetine vs Trazodone | 4 | 203 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.25 [‐0.76, 0.26] |
| 3 Failure to complete ‐ Total | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs ABT‐200 | 1 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.08, 0.39] |
| 3.2 Fluoxetine vs Amineptine | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.17, 2.21] |
| 3.3 Fluoxetine vs Amisulpride | 1 | 281 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.81, 2.38] |
| 3.4 Fluoxetine vs Bupropion | 2 | 436 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.67, 1.48] |
| 3.5 Fluoxetine vs Nefazodone | 3 | 161 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.22, 1.31] |
| 3.6 Fluoxetine vs Pramipexole | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.12 [0.03, 0.42] |
| 3.7 Fluoxetine vs Tianeptine | 3 | 830 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.69, 1.33] |
| 3.8 Fluoxetine vs Trazodone | 4 | 230 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.23, 1.13] |
| 4 Failure to complete ‐ Inefficacy | 14 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs ABT‐200 | 1 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 0.24 [0.03, 2.20] |
| 4.2 Fluoxetine vs Amineptine | 1 | 63 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.19, 5.57] |
| 4.3 Fluoxetine vs Amisulpride | 1 | 281 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.43, 3.10] |
| 4.4 Fluoxetine vs Bupropion | 2 | 436 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.33, 4.10] |
| 4.5 Fluoxetine vs Nefazodone | 3 | 161 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.05, 10.71] |
| 4.6 Fluoxetine vs Pramipexole | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.05, 4.51] |
| 4.7 Fluoxetine vs Tianeptine | 3 | 830 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.27, 2.53] |
| 4.8 Fluoxetine vs Trazodone | 2 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 0.23 [0.04, 1.51] |
| 5 Failure to complete ‐ Side Effects | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs ABT‐200 | 1 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 0.08 [0.02, 0.27] |
| 5.2 Fluoxetine vs Amineptine | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 0.52 [0.03, 7.82] |
| 5.3 Fluoxetine vs Amisulpride | 1 | 281 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.33, 1.82] |
| 5.4 Fluoxetine vs Bupropion | 2 | 436 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.45, 2.25] |
| 5.5 Fluoxetine vs Nefazodone | 4 | 286 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.32, 1.81] |
| 5.6 Fluoxetine vs Pramipexole | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.06 [0.01, 0.50] |
| 5.7 Fluoxetine vs Tianeptine | 3 | 830 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.71, 1.80] |
| 5.8 Fluoxetine vs Trazodone | 3 | 110 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.20, 2.19] |
6.1. Analysis.
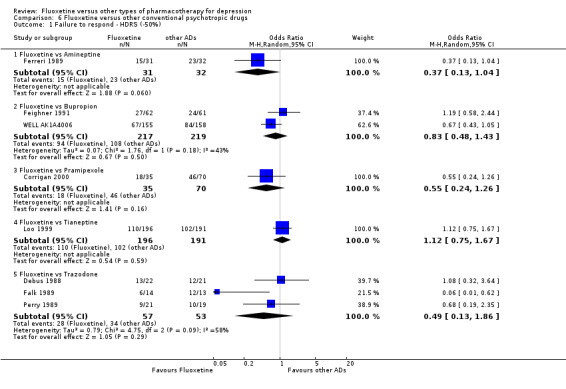
Comparison 6 Fluoxetine versus other conventional psychotropic drugs, Outcome 1 Failure to respond ‐ HDRS (‐50%).
6.2. Analysis.
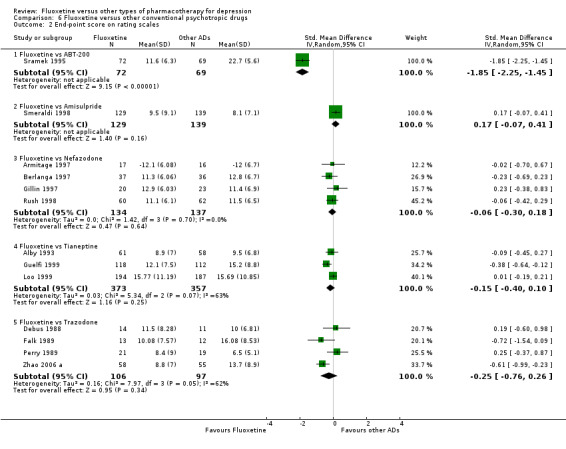
Comparison 6 Fluoxetine versus other conventional psychotropic drugs, Outcome 2 End‐point score on rating scales.
6.3. Analysis.
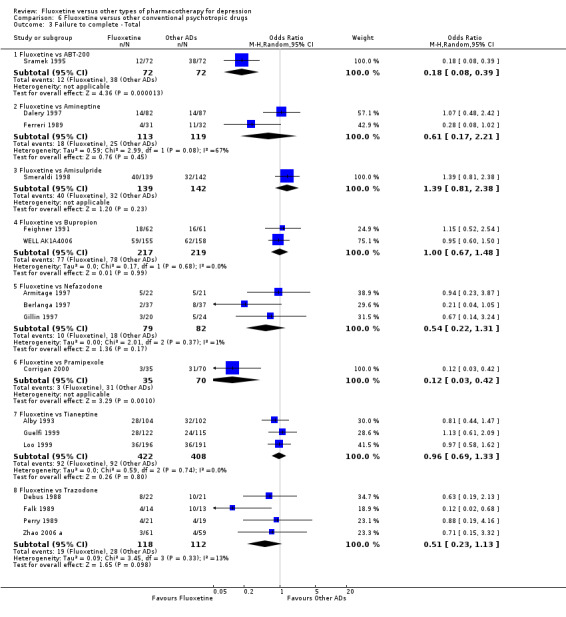
Comparison 6 Fluoxetine versus other conventional psychotropic drugs, Outcome 3 Failure to complete ‐ Total.
6.4. Analysis.
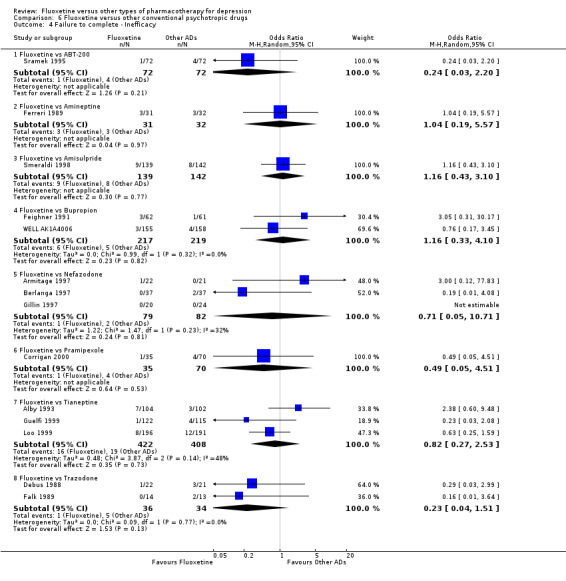
Comparison 6 Fluoxetine versus other conventional psychotropic drugs, Outcome 4 Failure to complete ‐ Inefficacy.
6.5. Analysis.
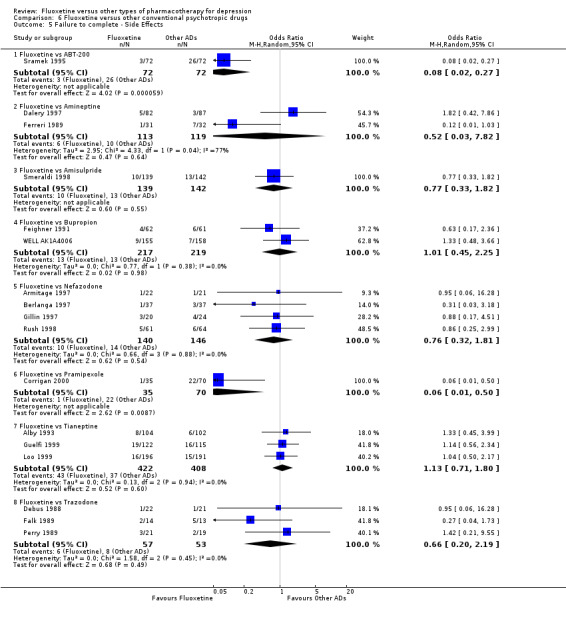
Comparison 6 Fluoxetine versus other conventional psychotropic drugs, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 7. Fluoxetine versus other non‐conventional AD agents.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Failure to respond ‐ HDRS (‐50%) | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Crocus Sativus | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.11, 2.60] |
| 1.2 Fluoxetine vs Hypericum | 6 | 717 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.73] |
| 2 End‐point score on rating scales | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Hypericum | 5 | 648 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.02, 0.29] |
| 3 Failure to complete ‐ Total | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Crocus Sativus | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 17.18] |
| 3.2 Fluoxetine vs Hypericum | 5 | 679 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.58, 1.85] |
| 4 Failure to complete ‐ Inefficacy | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Fluoxetine vs Hypericum | 2 | 401 | Odds Ratio (M‐H, Random, 95% CI) | 4.70 [0.22, 99.39] |
| 5 Failure to complete ‐ Side Effects | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Fluoxetine vs Hypericum | 5 | 679 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.56, 2.64] |
7.1. Analysis.
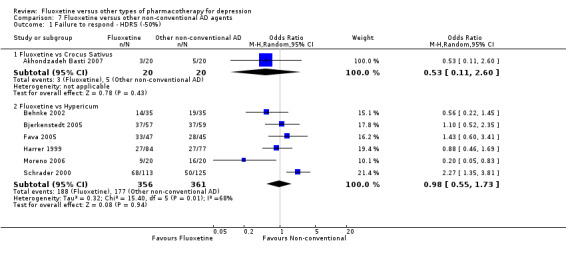
Comparison 7 Fluoxetine versus other non‐conventional AD agents, Outcome 1 Failure to respond ‐ HDRS (‐50%).
7.2. Analysis.
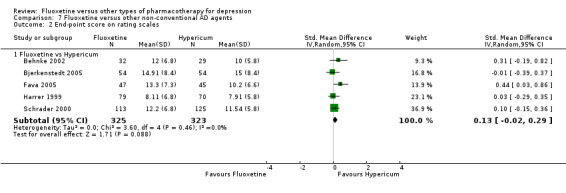
Comparison 7 Fluoxetine versus other non‐conventional AD agents, Outcome 2 End‐point score on rating scales.
7.3. Analysis.
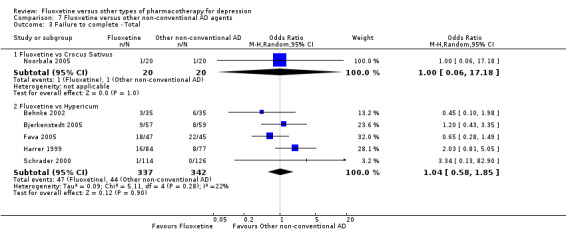
Comparison 7 Fluoxetine versus other non‐conventional AD agents, Outcome 3 Failure to complete ‐ Total.
7.4. Analysis.
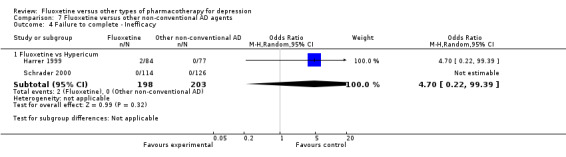
Comparison 7 Fluoxetine versus other non‐conventional AD agents, Outcome 4 Failure to complete ‐ Inefficacy.
7.5. Analysis.
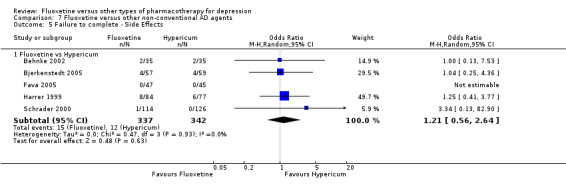
Comparison 7 Fluoxetine versus other non‐conventional AD agents, Outcome 5 Failure to complete ‐ Side Effects.
Comparison 8. Subgroup analysis for fluoxetine versus TCAs: failure to respond.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 5 | 341 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.35, 1.27] |
| 1.1 Fluoxetine vs Amitriptyline | 3 | 207 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.19, 1.65] |
| 1.2 Fluoxetine vs Clomipramine | 1 | 94 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.27, 1.45] |
| 1.3 Fluoxetine vs Imipramine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.27, 7.34] |
| 2 follow‐up 6‐16 weeks | 18 | 1742 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.80, 1.31] |
| 2.1 Fluoxetine vs Amitriptyline | 8 | 570 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.75, 1.50] |
| 2.2 Fluoxetine vs Desipramine | 2 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [0.56, 5.15] |
| 2.3 Fluoxetine vs Dothiepin/dosulepin | 2 | 144 | Odds Ratio (M‐H, Random, 95% CI) | 2.13 [1.08, 4.20] |
| 2.4 Fluoxetine vs Doxepine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.54] |
| 2.5 Fluoxetine vs Imipramine | 4 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.36, 1.34] |
| 2.6 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.55, 1.78] |
8.1. Analysis.
Comparison 8 Subgroup analysis for fluoxetine versus TCAs: failure to respond, Outcome 1 follow‐up <6 weeks.
8.2. Analysis.
Comparison 8 Subgroup analysis for fluoxetine versus TCAs: failure to respond, Outcome 2 follow‐up 6‐16 weeks.
Comparison 9. Subgroup analysis for fluoxetine versus TCAs: endpoint score.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 12 | 541 | Std. Mean Difference (IV, Random, 95% CI) | 0.18 [‐0.15, 0.50] |
| 1.1 Fluoxetine vs Amiptriptyline | 6 | 290 | Std. Mean Difference (IV, Random, 95% CI) | 0.39 [‐0.25, 1.02] |
| 1.2 Fluoxetine vs Clomipramine | 2 | 124 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.00 [‐0.36, 0.35] |
| 1.3 Fluoxetine vs Desipramine | 1 | 26 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.72, 0.82] |
| 1.4 Fluoxetine vs Imipramine | 2 | 60 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐1.31, 1.22] |
| 1.5 Fluoxetine vs Trimipramine | 1 | 41 | Std. Mean Difference (IV, Random, 95% CI) | 0.38 [‐0.24, 1.00] |
| 2 follow‐up 6‐16 weeks | 36 | 2727 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.06, 0.14] |
| 2.1 Fluoxetine vs Amiptriptyline | 13 | 733 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.07, 0.22] |
| 2.2 Fluoxetine vs Clomipramine | 3 | 248 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.40, 0.10] |
| 2.3 Fluoxetine vs Desipramine | 3 | 121 | Std. Mean Difference (IV, Random, 95% CI) | 0.33 [‐0.47, 1.12] |
| 2.4 Fluoxetine vs Dothiepin/dosulepin | 4 | 266 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.27, 0.59] |
| 2.5 Fluoxetine vs Imipramine | 10 | 1003 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.21, 0.19] |
| 2.6 Fluoxetine vs Lofepramine | 1 | 183 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.16, 0.42] |
| 2.7 Fluoxetine vs Nortriptyline | 1 | 154 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.44, 0.20] |
| 2.8 Fluoxetine vs Trimipramine | 1 | 19 | Std. Mean Difference (IV, Random, 95% CI) | 0.47 [‐0.44, 1.39] |
| 3 follow‐up >16 weeks | 1 | 97 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.86 [‐1.27, ‐0.44] |
| 3.1 Fluoxetine vs Nortriptyline | 1 | 97 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.86 [‐1.27, ‐0.44] |
9.1. Analysis.
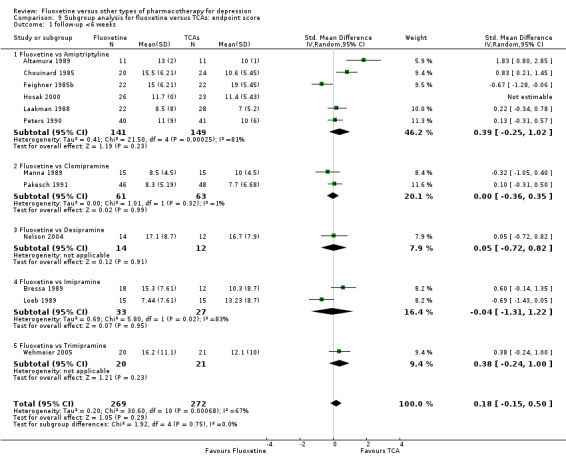
Comparison 9 Subgroup analysis for fluoxetine versus TCAs: endpoint score, Outcome 1 follow‐up <6 weeks.
9.2. Analysis.
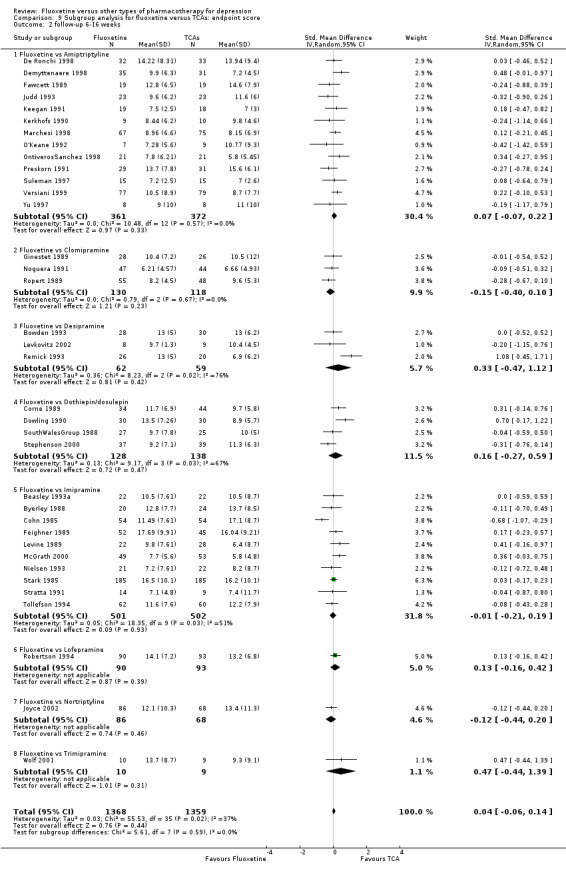
Comparison 9 Subgroup analysis for fluoxetine versus TCAs: endpoint score, Outcome 2 follow‐up 6‐16 weeks.
9.3. Analysis.
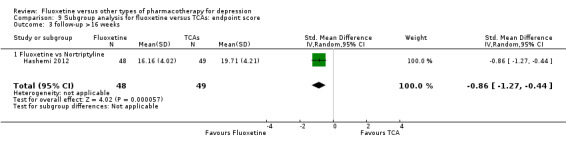
Comparison 9 Subgroup analysis for fluoxetine versus TCAs: endpoint score, Outcome 3 follow‐up >16 weeks.
Comparison 10. Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ total.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 11 | 663 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.47, 1.05] |
| 1.1 Fluoxetine vs Amitriptyline | 6 | 309 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.29, 1.13] |
| 1.2 Fluoxetine vs Doxepine | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.29, 5.31] |
| 1.3 Fluoxetine vs Imipramine | 3 | 98 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.10, 3.13] |
| 1.4 Fluoxetine vs Nortriptyline | 1 | 205 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.44, 1.35] |
| 2 follow‐up 6‐16 weeks | 36 | 3450 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.63, 0.98] |
| 2.1 Fluoxetine vs Amitriptyline | 12 | 780 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.44, 0.90] |
| 2.2 Fluoxetine vs Clomipramine | 2 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.38, 1.14] |
| 2.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.16, 1.24] |
| 2.4 Fluoxetine vs Dothiepin/dosulepin | 5 | 478 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.92, 2.69] |
| 2.5 Fluoxetine vs Doxepine | 3 | 272 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.44, 1.40] |
| 2.6 Fluoxetine vs Imipramine | 9 | 1127 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.51, 1.27] |
| 2.7 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.24, 1.04] |
| 2.8 Fluoxetine vs Nortriptyline | 2 | 243 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.17, 3.93] |
10.1. Analysis.
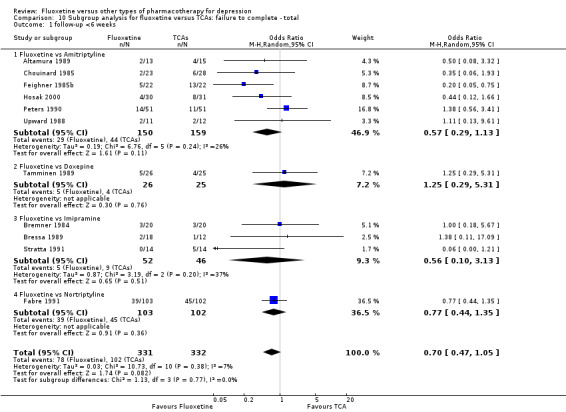
Comparison 10 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ total, Outcome 1 follow‐up <6 weeks.
10.2. Analysis.
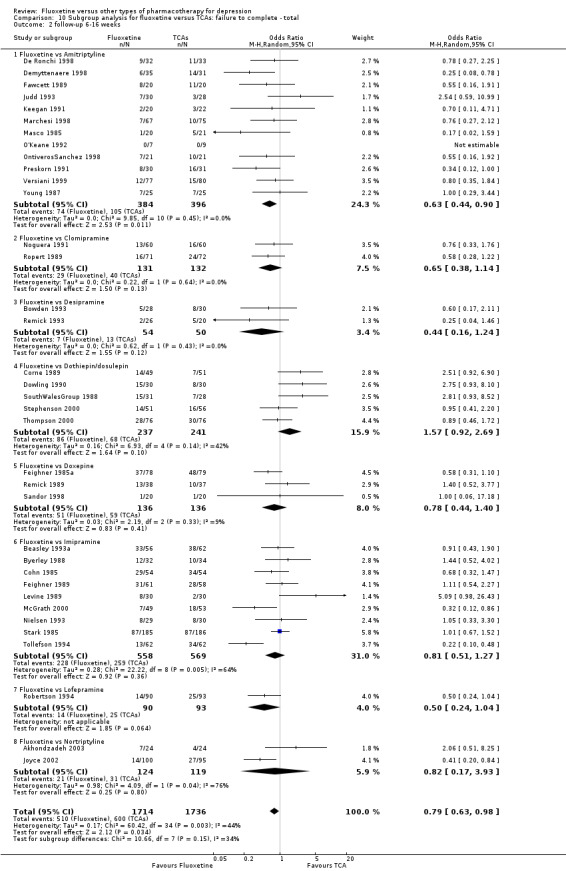
Comparison 10 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ total, Outcome 2 follow‐up 6‐16 weeks.
Comparison 11. Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ inefficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 5 | 401 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.16, 1.50] |
| 1.1 Fluoxetine vs Amitriptyline | 2 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.07, 2.09] |
| 1.2 Fluoxetine vs Doxepine | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.3 Fluoxetine vs Imipramine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 3.15 [0.12, 82.16] |
| 1.4 Fluoxetine vs Nortriptyiline | 1 | 205 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.07, 2.03] |
| 2 follow‐up 6‐16 weeks | 28 | 2510 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [1.02, 1.87] |
| 2.1 Fluoxetine vs Amitriptyline | 11 | 730 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.50, 2.47] |
| 2.2 Fluoxetine vs Clomipramine | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 7.37 [0.37, 145.75] |
| 2.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.20, 5.35] |
| 2.4 Fluoxetine vs Dothiepin/dosulepin | 3 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.49, 3.66] |
| 2.5 Fluoxetine vs Doxepine | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.60, 4.92] |
| 2.6 Fluoxetine vs Imipramine | 9 | 1053 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.96, 2.04] |
11.1. Analysis.
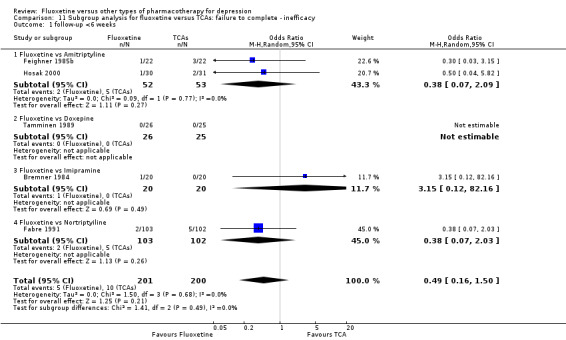
Comparison 11 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ inefficacy, Outcome 1 follow‐up <6 weeks.
11.2. Analysis.
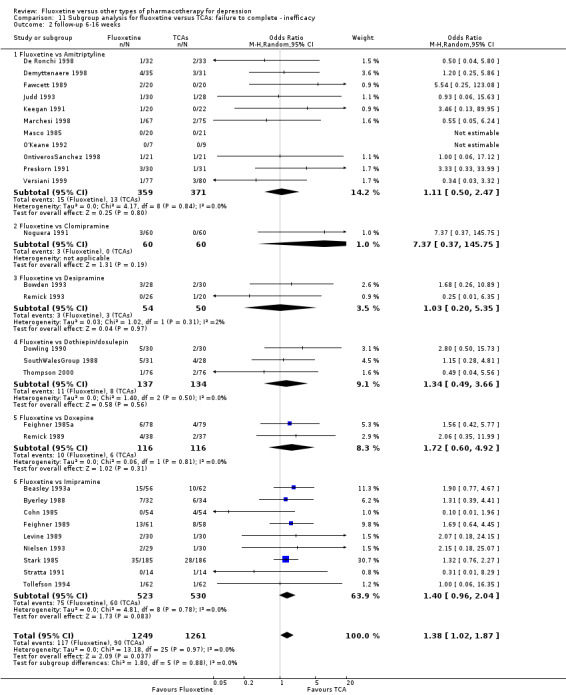
Comparison 11 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ inefficacy, Outcome 2 follow‐up 6‐16 weeks.
Comparison 12. Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ side effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 7 | 554 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.46, 1.43] |
| 1.1 Fluoxetine vs Amitriptyline | 4 | 258 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.21, 1.82] |
| 1.2 Fluoxetine vs Doxepine | 1 | 51 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [0.30, 32.31] |
| 1.3 Fluoxetine vs Imipramine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.09, 4.24] |
| 1.4 Fluoxetine vs Nortriptyline | 1 | 205 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.42, 1.77] |
| 2 follow‐up 6‐16 weeks | 33 | 3093 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.36, 0.72] |
| 2.1 Fluoxetine vs Amitriptyline | 12 | 780 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.18, 0.61] |
| 2.2 Fluoxetine vs Clomipramine | 2 | 263 | Odds Ratio (M‐H, Random, 95% CI) | 0.30 [0.12, 0.79] |
| 2.3 Fluoxetine vs Desipramine | 2 | 104 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.04, 1.68] |
| 2.4 Fluoxetine vs Dothiepin/dosulepin | 5 | 478 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [0.59, 7.16] |
| 2.5 Fluoxetine vs Doxepine | 2 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.42, 1.28] |
| 2.6 Fluoxetine vs Imipramine | 9 | 1053 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.25, 0.87] |
| 2.7 Fluoxetine vs Lofepramine | 1 | 183 | Odds Ratio (M‐H, Random, 95% CI) | 0.16 [0.02, 1.38] |
12.1. Analysis.
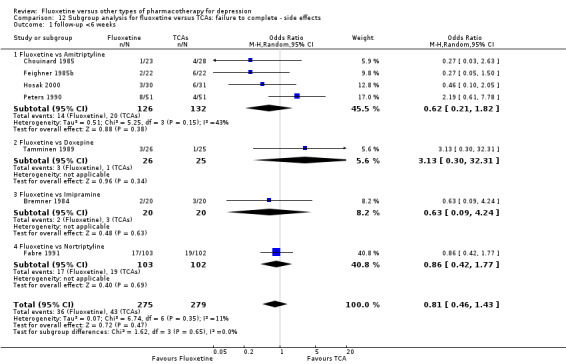
Comparison 12 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ side effects, Outcome 1 follow‐up <6 weeks.
12.2. Analysis.
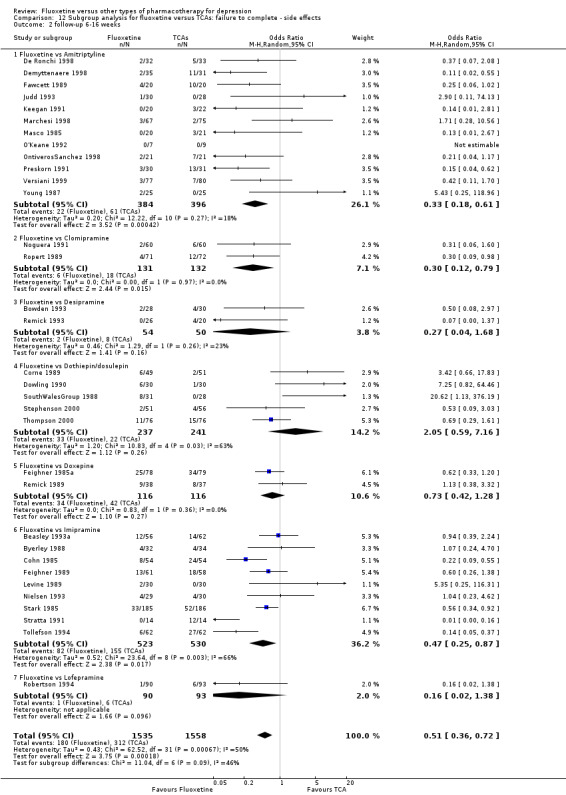
Comparison 12 Subgroup analysis for fluoxetine versus TCAs: failure to complete ‐ side effects, Outcome 2 follow‐up 6‐16 weeks.
Comparison 13. Subgroup analysis for fluoxetine versus heterocyclics: endpoint score.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Maprotiline | 2 | 181 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.34, 0.46] |
| 2 follow‐up 6‐16 weeks | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Maprotiline | 3 | 252 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.20, 0.30] |
| 2.2 Fluoxetine vs Mianserin | 3 | 128 | Std. Mean Difference (IV, Random, 95% CI) | 0.43 [‐0.38, 1.23] |
13.1. Analysis.
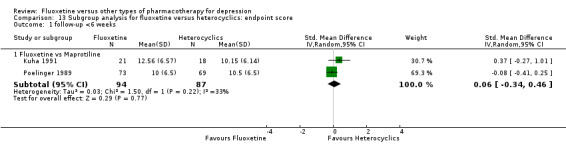
Comparison 13 Subgroup analysis for fluoxetine versus heterocyclics: endpoint score, Outcome 1 follow‐up <6 weeks.
13.2. Analysis.
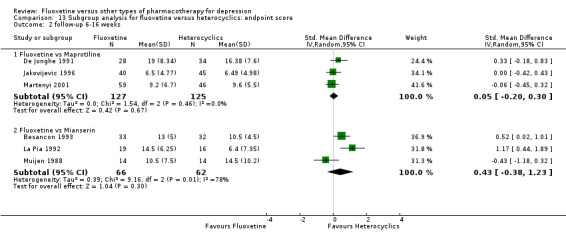
Comparison 13 Subgroup analysis for fluoxetine versus heterocyclics: endpoint score, Outcome 2 follow‐up 6‐16 weeks.
Comparison 14. Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ total.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Maprotiline | 2 | 188 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.63, 3.75] |
| 2 follow‐up 6‐16 weeks | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Maprotiline | 2 | 163 | Odds Ratio (M‐H, Random, 95% CI) | 2.06 [0.75, 5.63] |
| 2.2 Fluoxetine vs Mianserin | 2 | 93 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.18, 2.25] |
14.1. Analysis.
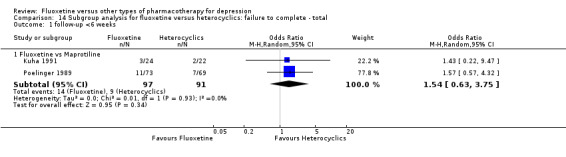
Comparison 14 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ total, Outcome 1 follow‐up <6 weeks.
14.2. Analysis.
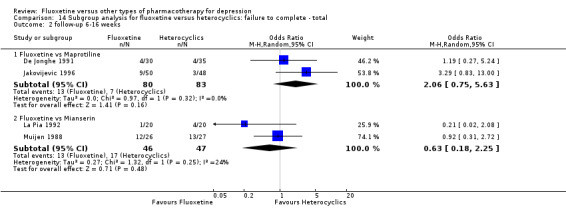
Comparison 14 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ total, Outcome 2 follow‐up 6‐16 weeks.
Comparison 15. Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ inefficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Maprotiline | 1 | 46 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 follow‐up 6‐16 weeks | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Maprotiline | 2 | 163 | Odds Ratio (M‐H, Random, 95% CI) | 2.54 [0.33, 19.19] |
| 2.2 Fluoxetine vs Mianserin | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 2.27 [0.38, 13.63] |
15.1. Analysis.
Comparison 15 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ inefficacy, Outcome 1 follow‐up <6 weeks.
15.2. Analysis.
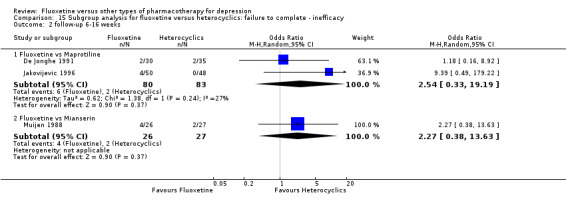
Comparison 15 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ inefficacy, Outcome 2 follow‐up 6‐16 weeks.
Comparison 16. Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ side effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Maprotiline | 1 | 46 | Odds Ratio (M‐H, Random, 95% CI) | 0.41 [0.07, 2.49] |
| 2 follow‐up 6‐16 weeks | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Maprotiline | 2 | 163 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.11, 4.38] |
| 2.2 Fluoxetine vs Mianserin | 1 | 53 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.23, 4.70] |
16.1. Analysis.
Comparison 16 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ side effects, Outcome 1 follow‐up <6 weeks.
16.2. Analysis.
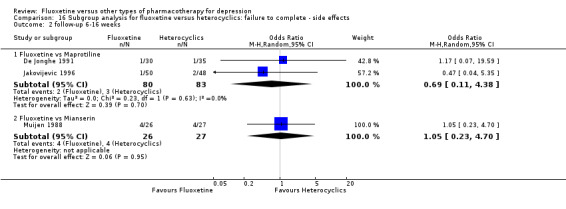
Comparison 16 Subgroup analysis for fluoxetine versus heterocyclics: failure to complete ‐ side effects, Outcome 2 follow‐up 6‐16 weeks.
Comparison 17. Subgroup analysis for fluoxetine versus other SSRIs: failure to respond.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.20, 1.79] |
| 2 follow‐up 6‐16 weeks | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Fluvoxamine | 1 | 177 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.52, 1.74] |
| 2.2 Fluoxetine vs Paroxetine | 9 | 1574 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.93, 1.65] |
| 2.3 Fluoxetine vs Sertraline | 5 | 950 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [1.00, 1.71] |
| 2.4 Fluoxetine vs Escitalopram | 1 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.56, 1.85] |
| 3 follow‐up >16 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Sertraline | 1 | 238 | Odds Ratio (M‐H, Random, 95% CI) | 1.67 [0.97, 2.85] |
17.1. Analysis.
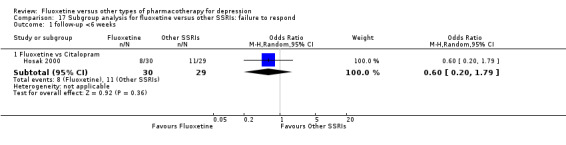
Comparison 17 Subgroup analysis for fluoxetine versus other SSRIs: failure to respond, Outcome 1 follow‐up <6 weeks.
17.2. Analysis.
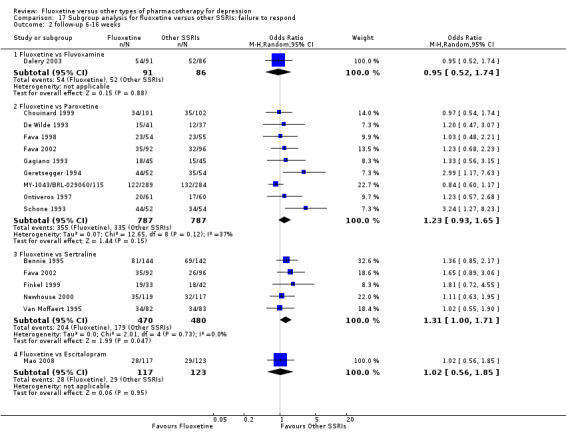
Comparison 17 Subgroup analysis for fluoxetine versus other SSRIs: failure to respond, Outcome 2 follow‐up 6‐16 weeks.
17.3. Analysis.
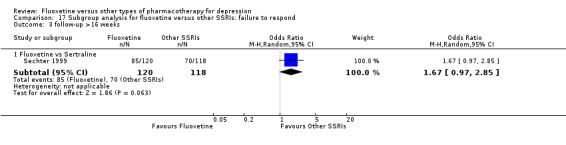
Comparison 17 Subgroup analysis for fluoxetine versus other SSRIs: failure to respond, Outcome 3 follow‐up >16 weeks.
Comparison 18. Subgroup analysis for fluoxetine versus other SSRIs: endpoint score.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 51 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.48, 0.61] |
| 2 follow‐up >16 weeks | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Paroxetine | 1 | 242 | Std. Mean Difference (IV, Random, 95% CI) | 0.24 [‐0.01, 0.49] |
| 2.2 Fluoxetine vs Sertraline | 1 | 168 | Std. Mean Difference (IV, Random, 95% CI) | 0.22 [‐0.08, 0.53] |
| 3 follow‐up 6‐16 weeks | 18 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Citalopram | 2 | 610 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.10, 0.21] |
| 3.2 Fluoxetine vs Paroxetine | 10 | 1819 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.31, 0.24] |
| 3.3 Fluoxetine vs Sertraline | 6 | 992 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.06, 0.19] |
| 3.4 Fluoxetine vs Escitalopram | 1 | 231 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.19, 0.33] |
18.1. Analysis.
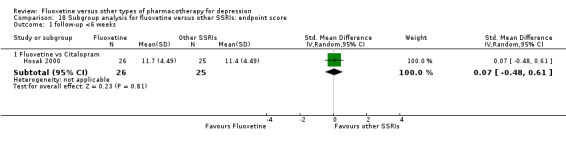
Comparison 18 Subgroup analysis for fluoxetine versus other SSRIs: endpoint score, Outcome 1 follow‐up <6 weeks.
18.2. Analysis.
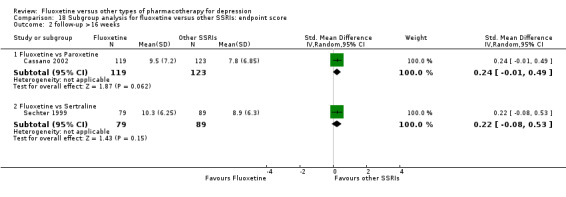
Comparison 18 Subgroup analysis for fluoxetine versus other SSRIs: endpoint score, Outcome 2 follow‐up >16 weeks.
18.3. Analysis.
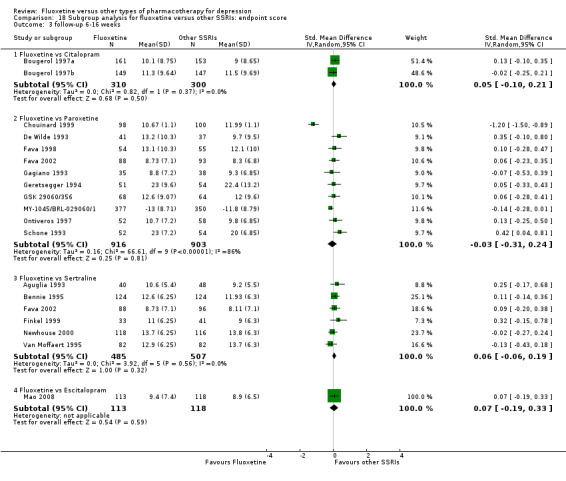
Comparison 18 Subgroup analysis for fluoxetine versus other SSRIs: endpoint score, Outcome 3 follow‐up 6‐16 weeks.
Comparison 19. Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ total.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.22, 4.27] |
| 2 follow‐up 6‐16 weeks | 21 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Citalopram | 2 | 673 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.59, 1.27] |
| 2.2 Fluoxetine vs Fluvoxamine | 2 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.36, 1.37] |
| 2.3 Fluoxetine vs Paroxetine | 9 | 1606 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.81, 1.25] |
| 2.4 Fluoxetine vs Sertraline | 7 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.85, 1.48] |
| 2.5 Fluoxetine vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [1.00, 2.37] |
| 3 follow‐up >16 weeks | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Paroxetine | 1 | 242 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.53, 1.49] |
| 3.2 Fluoxetine vs Sertraline | 2 | 480 | Odds Ratio (M‐H, Random, 95% CI) | 1.33 [0.84, 2.09] |
19.1. Analysis.
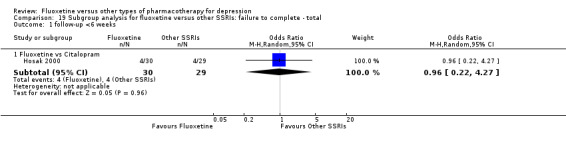
Comparison 19 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ total, Outcome 1 follow‐up <6 weeks.
19.2. Analysis.
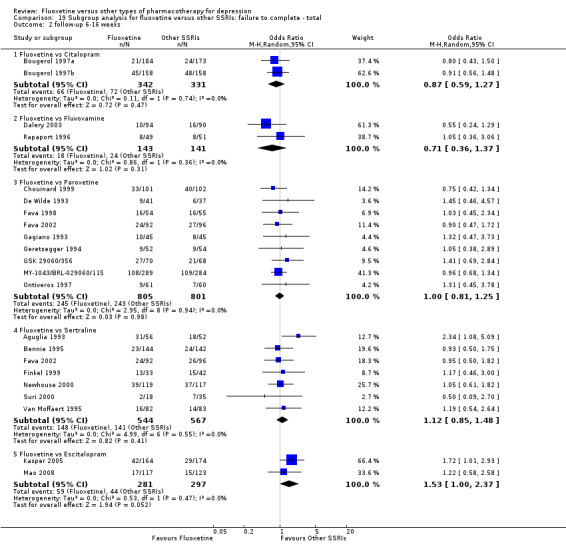
Comparison 19 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ total, Outcome 2 follow‐up 6‐16 weeks.
19.3. Analysis.
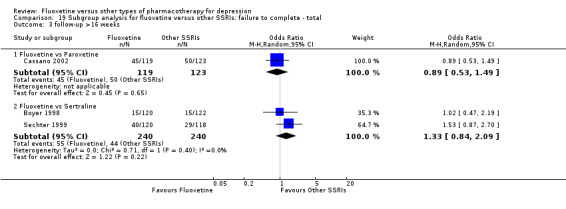
Comparison 19 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ total, Outcome 3 follow‐up >16 weeks.
Comparison 20. Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ inefficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.04, 5.43] |
| 2 follow‐up >16 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Sertraline | 1 | 238 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.47, 2.07] |
| 3 follow‐up 6‐16 weeks | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Citalopram | 2 | 673 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.49, 1.65] |
| 3.2 Fluoxetine vs Paroxetine | 4 | 1005 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.41, 1.39] |
| 3.3 Fluoxetine vs Sertraline | 4 | 818 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.61, 2.29] |
| 3.4 Fluoxetine vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.46, 6.53] |
20.1. Analysis.
Comparison 20 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ inefficacy, Outcome 1 follow‐up <6 weeks.
20.2. Analysis.
Comparison 20 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ inefficacy, Outcome 2 follow‐up >16 weeks.
20.3. Analysis.
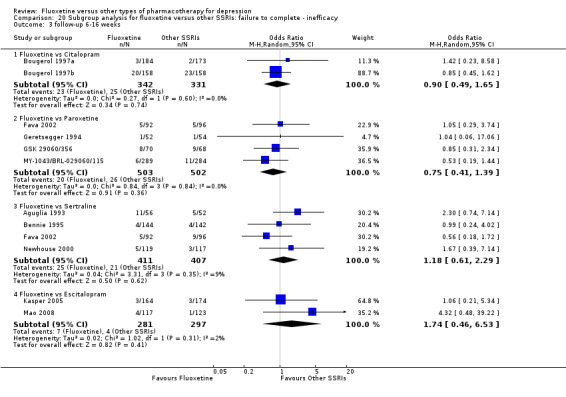
Comparison 20 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ inefficacy, Outcome 3 follow‐up 6‐16 weeks.
Comparison 21. Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ side effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Citalopram | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 1.5 [0.23, 9.70] |
| 2 follow‐up 6‐16 weeks | 20 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Citalopram | 2 | 673 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.29, 1.12] |
| 2.2 Fluoxetine vs Fluvoxamine | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.14, 7.71] |
| 2.3 Fluoxetine vs Paroxetine | 9 | 1509 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.62, 1.16] |
| 2.4 Fluoxetine vs Sertraline | 7 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.85, 1.71] |
| 2.5 Fluoxetina vs Escitalopram | 2 | 578 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.64, 2.12] |
| 3 follow‐up >16 weeks | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Fluoxetine vs Sertraline | 2 | 480 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.72, 2.76] |
21.1. Analysis.
Comparison 21 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ side effects, Outcome 1 follow‐up <6 weeks.
21.2. Analysis.
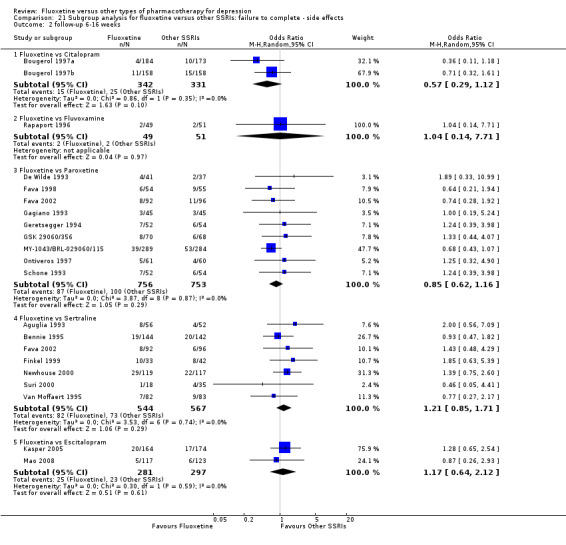
Comparison 21 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ side effects, Outcome 2 follow‐up 6‐16 weeks.
21.3. Analysis.
Comparison 21 Subgroup analysis for fluoxetine versus other SSRIs: failure to complete ‐ side effects, Outcome 3 follow‐up >16 weeks.
Comparison 22. Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to respond.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Moclobemide | 1 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.39, 2.60] |
| 2 follow‐up 6‐16 weeks | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Agomelatine | 1 | 515 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.99, 2.09] |
| 2.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [1.04, 2.04] |
| 2.3 Fluoxetine vs Moclobemide | 6 | 651 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.90, 1.89] |
| 2.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.27, 7.34] |
| 2.5 Fluoxetine vs Reboxetine | 3 | 721 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.55, 1.10] |
22.1. Analysis.
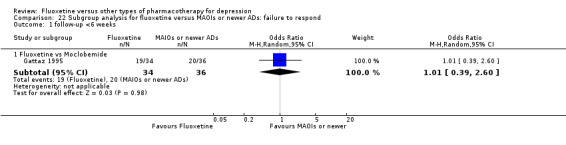
Comparison 22 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to respond, Outcome 1 follow‐up <6 weeks.
22.2. Analysis.
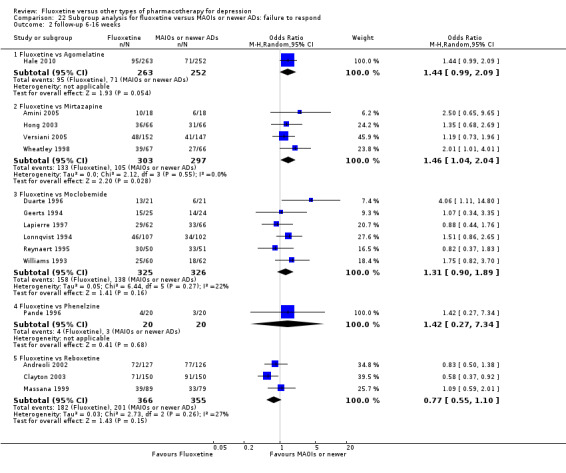
Comparison 22 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to respond, Outcome 2 follow‐up 6‐16 weeks.
Comparison 23. Subgroup analysis for fluoxetine versus MAOIs or newer ADs: endpoint score.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Moclobemide | 1 | 53 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.66, 0.42] |
| 2 follow‐up 6‐16 weeks | 12 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Agomelatine | 3 | 1213 | Std. Mean Difference (IV, Random, 95% CI) | 0.02 [‐0.18, 0.23] |
| 2.2 Fluoxetine vs Mirtazapine | 1 | 31 | Std. Mean Difference (IV, Random, 95% CI) | 0.57 [‐0.15, 1.29] |
| 2.3 Fluoxetine vs Moclobemide | 5 | 487 | Std. Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.02, 0.33] |
| 2.4 Fluoxetine vs Phenelzine | 1 | 40 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.67, 0.57] |
| 2.5 Fluoxetine vs Reboxetine | 2 | 205 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.31, 0.40] |
23.1. Analysis.
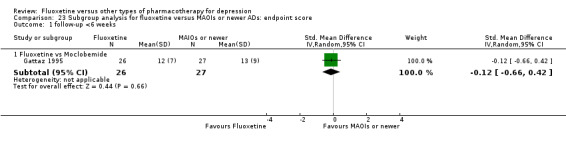
Comparison 23 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: endpoint score, Outcome 1 follow‐up <6 weeks.
23.2. Analysis.
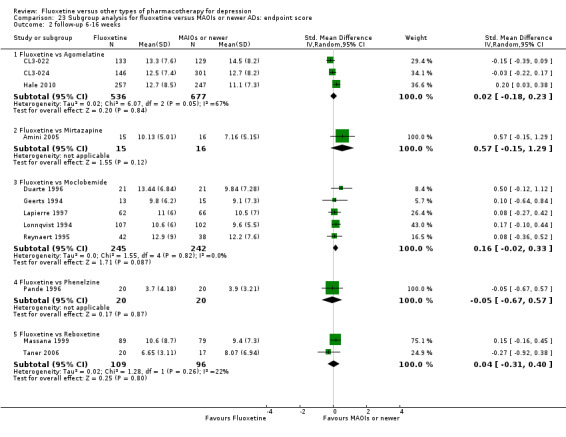
Comparison 23 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: endpoint score, Outcome 2 follow‐up 6‐16 weeks.
Comparison 24. Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ total.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Moclobemide | 1 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.31, 2.76] |
| 2 follow‐up 6‐16 weeks | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetina vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.59, 2.49] |
| 2.2 Fluoxetine vs Mirtazapine | 3 | 301 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.51, 1.68] |
| 2.3 Fluoxetine vs Moclobemide | 6 | 651 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.68, 1.53] |
| 2.4 Fluoxetine vs Reboxetine | 4 | 764 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.44, 0.83] |
24.1. Analysis.
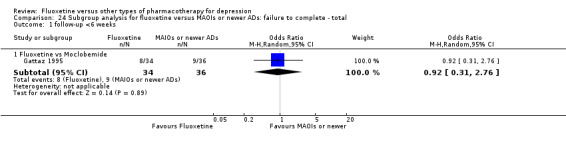
Comparison 24 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ total, Outcome 1 follow‐up <6 weeks.
24.2. Analysis.
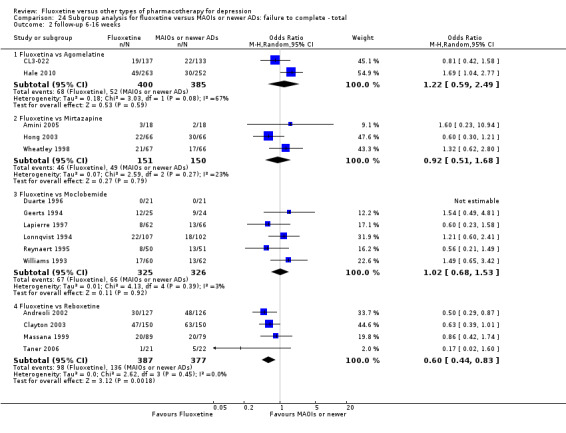
Comparison 24 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ total, Outcome 2 follow‐up 6‐16 weeks.
Comparison 25. Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ inefficacy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Moclobemide | 1 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.20, 5.68] |
| 2 follow‐up 6‐16 weeks | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.41, 2.88] |
| 2.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.71, 2.96] |
| 2.3 Fluoxetine vs Moclobemide | 5 | 609 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.23, 1.65] |
| 2.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.5 Fluoxetine vs Reboxetine | 3 | 464 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.47, 1.77] |
25.1. Analysis.
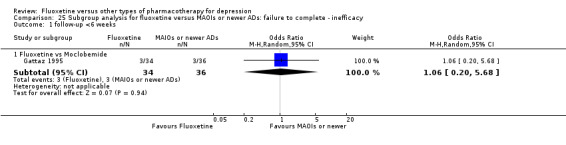
Comparison 25 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ inefficacy, Outcome 1 follow‐up <6 weeks.
25.2. Analysis.
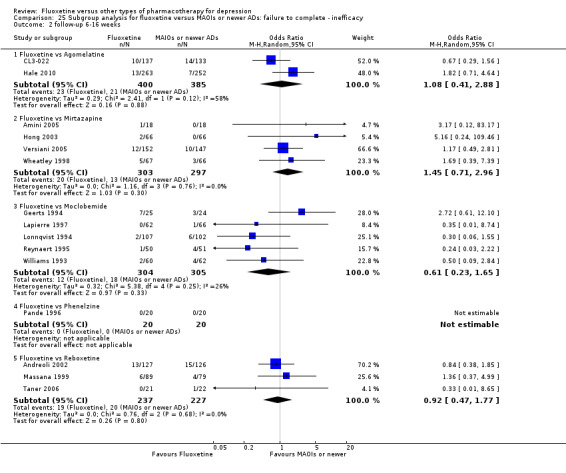
Comparison 25 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ inefficacy, Outcome 2 follow‐up 6‐16 weeks.
Comparison 26. Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ side effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 follow‐up <6 weeks | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Fluoxetine vs Moclobemide | 1 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 follow‐up 6‐16 weeks | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Fluoxetine vs Agomelatine | 2 | 785 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.73, 3.08] |
| 2.2 Fluoxetine vs Mirtazapine | 4 | 600 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.54, 1.66] |
| 2.3 Fluoxetine vs Moclobemide | 6 | 651 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.54, 2.01] |
| 2.4 Fluoxetine vs Phenelzine | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.26] |
| 2.5 Fluoxetine vs Reboxetine | 2 | 211 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.10, 1.61] |
26.1. Analysis.
Comparison 26 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ side effects, Outcome 1 follow‐up <6 weeks.
26.2. Analysis.
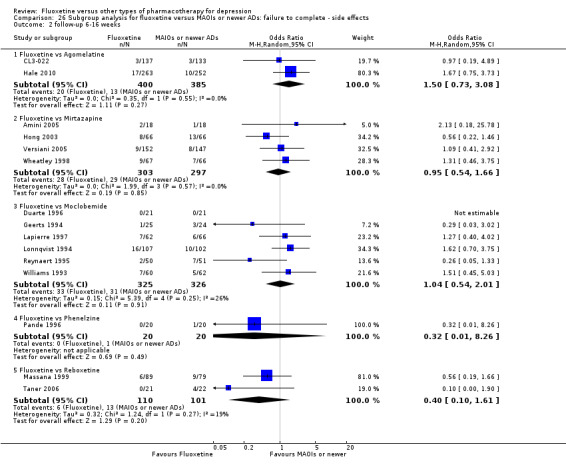
Comparison 26 Subgroup analysis for fluoxetine versus MAOIs or newer ADs: failure to complete ‐ side effects, Outcome 2 follow‐up 6‐16 weeks.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Aguglia 1993.
| Methods | Eight‐week double‐blind, multicentre study | |
| Participants | Outpatients suffering from a major depressive episode according to DSM‐III‐R, with a baseline score on Hamilton Rating Scale for Depression‐17 Item (HDRS‐17) of at least 18, recruited from nine separated psychiatric clinics. Age: 18 years or more Exclusion criteria: depression secondary to other conditions, concomitant illness of renal, cardiac or hepatic origin; hypersensitivity to other antidepressants, likelihood of poor compliance, risk of suicide, peptic ulcer history, an improvement of greater than 25% in the HDRS score during a pre‐treatment placebo washout period. | |
| Interventions | Fluoxetine: 56 participants Sertraline: 52 participants Fluoxetine dose range: 20‐60 mg/day Sertraline dose range: 50‐150 mg/day Benzodiazepines were allowed for hypnotic use and as maintenance treatment for pre‐existing anxiety | |
| Outcomes | HDRS‐17 and Hamilton Rating Scale for Anxiety (HAM‐A), Montgomery and Asberg Scale for Depression (MADRS), Zung Self‐Rating Scale for Anxiety, Leeds Sleep Evaluation Questionnaire, Clinical Global Impression Scale, including severity (CGI‐S) and improvement (CGI‐I) | |
| Notes | Seventy‐five per cent of the patients were women. Higher percentage of patients with a family history of psychiatric illness in the fluoxetine group. Higher percentage of patients with severe depression in the fluoxetine group (30.4%) than in the sertraline group (13.7%). Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information about randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double‐blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Incoherence between denominators |
| Selective reporting (reporting bias) | High risk | No follow‐up scores reported |
| Other bias | Unclear risk | Funding: unclear information |
Akhondzadeh 2003.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients meeting DSM‐IV diagnostic criteria for major depression, with a minimum baseline score of 20 on the Hamilton Rating Scale for Depression‐17 Item (HDRS‐17). Age range: 19‐54 years Exclusion criteria: any other psychiatric primary disease, current or past history of bipolar disorder, use of anxiolytic or MAOI or tryptophan, organic mental disorder, epilepsy, suicidal tendencies, any severe general disease, pregnancy, lactation. | |
| Interventions | Fluoxetine: 24 participants Nortriptyline: 24 participants Fluoxetine dose: 60 mg/day Nortriptyline dose: 150 mg/day | |
| Outcomes | Primary outcome: HDRS‐17 | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned". No further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "were put in capsules to provide similar appearance with fluoxetine". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if raters were independent |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for discontinuation reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Mean endpoint scores and standard deviation reported only in figure. Side effects reported |
| Other bias | Unclear risk | Insufficient information |
Akhondzadeh Basti 2007.
| Methods | Eight‐week double‐blind, randomised trial | |
| Participants | Outpatients meeting DSM‐IV diagnostic criteria for major depression, with a minimum baseline score of 18 and not superior to 25 on the Hamilton Rating Scale for Depression‐17 Item (HDRS‐17). Age range: 19‐55 years Exclusion criteria: patients posed a significant risk of suicide at any time during participation, persons who scored greater than 2 on the suicide item of the HDRS, any clinical significant deterioration during the trial, pregnancy and women not using medically accepted means of birth control. | |
| Interventions | Fluoxetine: 20 participants Petal of Crocus sativus: 20 participants Fluoxetine dose: 10 mg/day Petal of Crocus sativus dose: 15 mg/day | |
| Outcomes | Primary outcome: HDRS‐17, remission was defined as an endpoint score of 7 or less | |
| Notes | Funding: independent study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomised to receive capsule of petal of Crocus or fluoxetine in a 1:1 ratio using computer code", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "capsules (...) did not have any different taste compared to fluoxetine", "the person who administered the medication, raters and patients were blind to assignment", not clear if blindness was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "the person who administered the medication, raters and patients were blind to assignment", not clear if blindness was successful |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of responders reported with denominator. Reasons for dropout not reported |
| Selective reporting (reporting bias) | Unclear risk | Mean endpoint scores and standard deviation reported only in figures. Side effects reported |
| Other bias | Low risk | Funding: independent study |
Alby 1993.
| Methods | Twelve‐week double‐blind study | |
| Participants | Outpatients suffering from a major depressive episode, recurrent depression or disthymia according to DSM‐III‐R, with a score of at least 25 on the HARD Humeur, Agoisse, Ralentissement, Danger (HARD), Ferreri anxiety rating diagram (FARD), Hopkins Symptom check‐list (HSCL). Age range: 25‐65 years Exclusion criteria: not reported | |
| Interventions | Fluoxetine: 104 participants Tianeptine: 102 participants Fluoxetine dose: 20 mg/day Tianeptine dose: 37.5 mg/day Benzodiazepines allowed for severe anxiety or sleep disorders | |
| Outcomes | HARD and on the FARD scales | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropout rate reported with reasons. Scores at endpoint are reported with denominator |
| Selective reporting (reporting bias) | Unclear risk | Full side effects reported. Other outcomes not clearly stated |
| Other bias | High risk | Last author's affiliation was IRIS and this company produces tianeptine |
Altamura 1989.
| Methods | Five‐week double‐blind randomised study | |
| Participants | Inpatients fulfilling DSM‐III criteria for major depressive episode and scoring at least 18 on Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 65 years Exclusion criteria: not reported | |
| Interventions | Fluoxetine: 13 participants Amitriptyline: 15 participants Fluoxetine dose: 20 mg/day Amitriptyline dose: 75 mg/day | |
| Outcomes | HDRS‐17 score | |
| Notes | Elderly patients only Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised". No other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of dropouts reported, but reasons not specified Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported only in figure. Only most common side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Alves 1999.
| Methods | Twelve‐week double‐blind randomised multicentre study | |
| Participants | Outpatients meeting DSM‐IV diagnostic criteria for major depression, with a minimum baseline score of 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), recruited from three clinical sites. Age range: 18‐65 years Exclusion criteria: known sensitivity to venlafaxine or fluoxetine, a history of any clinically significant cardiac, hepatic or renal disease or abnormalities on a screening physical examination, ECG or laboratory tests, with any mental or neurologic disorder and breast‐feeding women; used of any investigational drug, antipsychotic drug, electroconvulsive therapy or sumatriptan within 30 days of baseline, fluoxetine within 21 days and MAOIs within 14 days. | |
| Interventions | Fluoxetine: 47 participants Venlafaxine: 40 participants Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐150 mg/day | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) | |
| Notes | Patients in the fluoxetine group had more chronic histories of depression at baseline. Predominance of females in the whole study. Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were allocated to treatments groups using a balanced randomisation from randomly permuted blocks" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "the investigators were provided individual sealed envelopes" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Efficacy analysis were performed on ITT basis using a last observation carried forward method (LOCF)", but scores reported without denominator. Reasons and number of dropouts reported |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported (at last 7% incidence). Mean endpoint scores and standard deviation reported |
| Other bias | High risk | First author's affiliation was Wyeth‐Lederle, Portugal, and this company produces venlafaxine |
Amini 2005.
| Methods | Six‐week double‐blind randomised trial | |
| Participants | In and outpatients meeting DSM‐IV diagnostic criteria for major depression, with a minimum baseline score of 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐60 years Exclusion criteria: history of recent suicidal attempt (1 month or less before the study) or actual ideation; pregnancy or lactation; history or current diagnosis (DSM‐IV) of dysthymic disorder, bipolar disorder, any psychotic disorder, eating disorders, personality disorders and mental retardation; current diagnosis (DSM‐IV) of anxiety disorders (except for specific phobia), mental disorder due to general medical condition, substance or alcohol abuse or dependency (except for nicotine and caffeine) and postpartum depression; current diagnosis of any serious systemic medical illnesses; treatment with any antidepressant drugs within 1 week, or with MAOIs within 5 weeks, or electroconvulsive therapy within 3 months ago; BMI more than 30 and white blood count more than 4000/mm or neutrophil more than 1500/mm. | |
| Interventions | Fluoxetine: 18 participants
Mirtazapine: 18 participants
Fluoxetine dose: 20 mg/day Mirtazapine dose: 30 mg/day |
|
| Outcomes | Mean decrease in HDRS‐17 score from baseline | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double blind", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double blind", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Double blind", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "A observed cases analysis over the 6 weeks was the primary efficacy analysis (...) an intention to treat analysis with LOCF was also performed...all discussion of result is based on observed cases (OC) analysis". Number of randomised, and number of lost during the follow‐up reported. Number of responders reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Side‐effects are reported. Vital signs and body weight not reported Mean endpoint scores (HDRS) reported in figures and without standard deviations |
| Other bias | Unclear risk | Funding: unclear |
Andreoli 2002.
| Methods | Eight‐week double‐blind, randomised multicentre study | |
| Participants | In and outpatients meeting DSM‐III‐R diagnostic criteria for major depression, with a minimum baseline score of 22 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), recruited from 33 clinical sites. Age range: 18‐65 years Exclusion criteria: history of unresponsiveness to antidepressant treatment, association with endocrine disorders, substance abuse, drug hypersensitivity, chronic respiratory insufficiency, or gastro‐intestinal, hepatic or renal disease, ECT within 6 months of baseline, high risk of suicide, pregnancy or absence of adequate contraception measures. | |
| Interventions | Fluoxetine: 127 participants Reboxetine: 126 participants Placebo: 128 participants Fluoxetine dose range: 20‐40 mg/day Reboxetine dose range: 8‐10 mg/day Chloral hydrate (0.5‐1 g) was allowed as hypnotic | |
| Outcomes | Primary outcome: absolute change in the HDRS‐21 total score Secondary outcomes: Clinical Global Impression Scale (CGI) Severity and Improvement, Montgomery and Asberg Scale for Depression (MADRS), Quality of Sleep Questionnaire | |
| Notes | Response: decrease of at least 50% in the HDRS total score Remission: total score less than 10 Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | "Double blind", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double blind", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Double blind", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of randomised and number of lost during follow‐up reported, but the reasons for dropout were not clear. Only most common side effects were reported Quote: "Statistical analysis was carried out on the intent to treat population" |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects were reported Mean endpoint scores and standard deviation reported |
| Other bias | Unclear risk | Funding: unclear |
Ansseau 1994.
| Methods | Six‐week double‐blind, randomised multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 25 on Montgomery and Asberg Scale for Depression (MADRS) and of at least 4 on Clinical Global Impression Scale (CGI). Age range: 19‐68 years Exclusion criteria: serious or uncontrolled medical illness, major anxiety, agitation, suicide risk, resistance during the current episode to at least two antidepressants, substance abuse or dependence, concomitant therapy with lithium, MAOIs, long‐acting neuroleptic. | |
| Interventions | Fluoxetine: 93 participants Milnacipram: 97 participants Fluoxetine dose: 20 mg/day Milnacipram dose: 100 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression‐24 item (HDRS‐24), MADRS, CGI | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned". No further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for drop‐out reported. Rating scale scores reported without denominator |
| Selective reporting (reporting bias) | High risk | Endpoint scores reported only in figures. Side effects reported |
| Other bias | High risk | Last author's affiliation was Laboratories Pierre Fabre Medicament, and this company produces milnacipran |
Armitage 1997.
| Methods | Eight‐week double‐blind, randomised trial | |
| Participants | Patients meeting DSM‐III‐R diagnostic criteria for non psychotic, moderate to severe major depression disorder and a minimum score of 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). All patients were required to have subjective sleep disturbance (score of 1 or 2 on at least one of the sleep items of the HDRS‐17). Age range: 18‐55 years Exclusion criteria: patients with current general medical condition, history of head trauma or current DSM‐III‐R axis I disorder in the categories of organic mental syndromes or disorders, bipolar disorder, schizophrenia, delusional disorder, or psychotic disorder NOS. Shift workers, or those with any evidence of independence sleep disorder (such as narcolepsy, bruxism, sleep apnoea) were ineligible. Patients with a history of psychoactive substance abuse, to be pregnant, lactating, or sexual active without an adequate method of contraception, previous participation in a nefazodone trial, significant suicide risk, or known hypersensitivity to trazodone, etoperidone, fluoxetine or metachlorophenylpiperazine were other exclusion criteria. |
|
| Interventions | Fluoxetine: 22 participants Nefazodone: 12 participants Fluoxetine dose range: 20‐40 mg/day Nefazodone dose range: 400‐500 mg/day | |
| Outcomes | HDRS‐17 total score and sleep parameters | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double dummy capsule‐dosing scheme" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Mean baseline and endpoint scores reported without standard deviations. Number and reasons for dropout specified |
| Selective reporting (reporting bias) | Unclear risk | Side effects not reported |
| Other bias | High risk | This study was supported in part by Bristol‐Myers Squibbs. This company produces nefazodone |
Bakish 1997.
| Methods | Twelve‐week double‐blind, randomised study | |
| Participants | Patients meeting DSM‐III‐R diagnostic criteria for major affective disorder, unipolar and had met the diagnostic criteria for major depressive disorder episode for at least one month before entering the study, with a minimum baseline score of 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17), with a score of two or more on item one. Age range: 23‐54 years Exclusion criteria: concurrent DSM‐III‐R Axis I diagnosis or unstable medical condition, suicidal patients or having receiving fluoxetine within 36 days preceding enrolment. |
|
| Interventions | Fluoxetine: 9 participants Paroxetine: 12 participants Fluoxetine dose range: 20‐80 mg/day Paroxetine dose range: 20‐50 mg/day | |
| Outcomes | Response: decrease of at least 50% in the HDRS‐17 total score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Baseline and endpoint scores for each arm not reported. Number and reasons for dropout not specified |
| Selective reporting (reporting bias) | High risk | Side effects not reported. Mean baseline and endpoint score reported only in the whole sample |
| Other bias | High risk | Quote: "this study was supported by grants from SmithKline Beecham", and this company produces paroxetine |
Basterzi 2009.
| Methods | Six‐week double‐blind randomised trial | |
| Participants | Patients diagnosed with major depression (MD) or MD‐recurrent according to DSM‐IV diagnostic criteria. Exclusion criteria: any additional axis I or axis II DSM‐IV diagnosis, current pregnancy, acute or chronic infections within the past month, autoimmune, allergic, neoplastic, or endocrine disease and other acute physical disorders, including surgery or infarction of the hearth or brain within the past 6 months, patients exposed to any drug including antidepressants, non‐steroidal anti‐inflammatory drugs and oral contraceptives in the past 4 weeks. | |
| Interventions | Fluoxetine: 21 participants
Venlafaxine: 22 participants Placebo: 21 participants Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐150 mg/day |
|
| Outcomes | Response was defined as a 50% reduction in the index total Hamilton Rating Scale for Depression (HDRS) score | |
| Notes | Funding: by academy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "consequently randomised", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if raters were independent and unclear if blinding was successful |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reason for dropout not clearly reported. Scores reported without denominator |
| Selective reporting (reporting bias) | High risk | Adverse effects not reported. Number of responders only reported for the whole sample (not for each study arm) |
| Other bias | Low risk | Funding: by academy |
Beasley 1993a.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐70 years Exclusion criteria: psychosis, organic mental disorder, substance abuse active within 1 year | |
| Interventions | Fluoxetine: 56 participants Imipramine: 62 participants Fluoxetine dose range: 40‐80 mg/day Imipramine dose range: 150‐300 mg/day Chloral hydrate (max 1g) and flurazepam (max 30 mg) were allowed as hypnotic | |
| Outcomes | HDRS‐21, Raskin, Covi, Clinical Global Impression Severity and Improvement Scales (CGI) | |
| Notes | Response: decrease of at least 50% in the total score Remission: total score less than 7 One patient on fluoxetine committed suicide Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "efficacy data were analysed in accordance with ITT principle", but scores reported without denominator. Study completion rates and reasons for study discontinuations reported |
| Selective reporting (reporting bias) | Unclear risk | Only side‐effect over 5% reported. Vital signs reported |
| Other bias | High risk | Authors' affiliation was Psychopharmacology Division, Lilly Reasearch Laboratories, Eli Lilly and Company, Indianapolis. This company produces fluoxetine |
Behnke 2002.
| Methods | Six‐week double‐blind, randomised multicentre study | |
| Participants | Patients with ICD‐10 depression, with a score between 16 and 24 points on Hamilton Rating Scale for Depression (HDRS‐17). Age range: 18‐73 years Exclusion criteria: participation in a clinical study less than 4 weeks, pregnancy and lactation, insufficient contraception, suicide risk, dementia, or other severe intellectual impairment, chronic alcohol or drug abuse or dependence, severe cardiac, liver, kidney or respiratory insufficiency, neoplasia, Parkinson's or Alzheimer's disease, hypersensitivity to an ingredient of the Hypericum perforatum, febrile illness, anaemia, thyroid or parathyroid disease, pituitary insufficiency. | |
| Interventions | Fluoxetine: 35 participants Hypericum: 35 participants Fluoxetine dose: 40 mg/day Hypericum dose: 300 mg/day | |
| Outcomes | HDRS‐17, von Zerssen Depression Scale (VZD), Clinical Global Impression (CGI) Scale | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of dropouts reported, but reasons not specified. Endpoint scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Side effects not reported. Mean endpoint scores and standard deviation reported |
| Other bias | High risk | Last author's affiliation was PhytoPharm Consulting, Istitute for Phytopharmaceuticals, Berlin; probably this company produces Hypericum perforatum |
Bennie 1995.
| Methods | Six‐week double‐blind, randomised multicentre study | |
| Participants | Outpatients with a diagnosis of major depression or bipolar disorder, depressed, according to DSM‐III‐R, scoring at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17), and with a higher on the Raskin Depression Scale than on the Covi Anxiety Scale. Age: over 18 years Exclusion criteria: pregnant or lactating women, women of childbearing potential not practicing a reliable method of contraception, patients with previous treatment with sertraline or fluoxetine, treated with MAOI within two weeks or other antidepressants medication within one week of double‐blind therapy, treated with reserpine or methyl‐dopa, likely to require additional treatments with psychoactive medication, ECT or intensive psychotherapy during the study; failure to respond to previous antidepressant therapy at clinically appropriate dosages, use of ECT to treat a previous episode of depression, a history of severe allergies or multiple adverse events associated with pharmacotherapy, the presence of significant medical disease; psychiatric history including another Axis I disorder and significant suicide risk. | |
| Interventions | Fluoxetine: 144 participants Sertraline: 142 participants Fluoxetine dose range: 20‐40 mg/day Sertraline dose range: 50‐100 mg/day Chloral hydrate (max 1 g) and temazepam (max 20 mg) were allowed as hypnotic | |
| Outcomes | Primary outcome: HDRS‐17, Clinical Global Impression Severity and Improvement Scales (CGI S‐I) Secondary outcomes: Hamilton Rating Scale for Anxiety, the Raskin Depression Scale and Covi Anxiety Scale, self‐rated Leeds Sleep Questionnaire | |
| Notes | Patients with concomitant medical conditions were allowed to participate in the study provided that the conditions were clearly not associated with the illness of the study and that any required medications were not psychoactive agents. One patient attempted suicide in the fluoxetine group. Funding: by industry |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned, in equal proportion", no other information |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number and reasons for dropout reported. Scores reported with denominator |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations. Only adverse events occurring at least in 3% of the sample reported |
| Other bias | High risk | Quote: "supported by a research grant from international Pharmaceuticals group, Pfizer, Inc, New York". This company produces sertraline |
Berlanga 1997.
| Methods | Eight‐week double‐blind, randomised two‐centre study | |
| Participants | Outpatients with a diagnosis of moderate to severe major depressive episode without psychotic features or bipolar disorder of the depressed type according to DSM‐III‐R, with a total score of least 18 points on Hamilton Rating Scale for Depression‐17 item (HDRS‐17) at baseline. Age: over 18 years Exclusion criteria: concomitant organic mental disorder, psychoactive substance abuse disorder, schizophrenia or other psychotic disorder or any medical condition that contraindicated treatment with antidepressants; pregnancy or lactating; women of childbearing potential not practicing a reliable method of contraception. | |
| Interventions | Fluoxetine: 37 participants Nefazodone: 37 participants Fluoxetine dose range: 20‐40 mg/day Nefazodone: 400‐500 mg/day Concomitant psychotropic medication was prohibited, but occasionally use of benzodiazepines for severe anxiety or insomnia was allowed | |
| Outcomes | HDRS‐17, Hamilton Rating Scale for Anxiety, Clinical Global Impression, Patient Global Assessment | |
| Notes | One patient attempted suicide in the fluoxetine group. Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised". No other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "this was a double‐blind trial", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐dummy", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "this was a double‐blind trial", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of dropouts reported. Scores at rating scales were reported with denominator |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations. Only adverse effect occurred in at least 10% of the sample were reported |
| Other bias | High risk | Quote: "supported by Bristol‐Myers Squibb Pharmaceutical Research Institute, Wallingford, CT". This company produces nefazodone |
Besancon 1993.
| Methods | Eight‐week double‐blind, randomised study | |
| Participants | Outpatients with a diagnosis of depressive episode less than 2 months duration, according to DSM‐III criteria, with a minimum score of 25 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐65 years Exclusion criteria: absence of resistance to mianserin or fluoxetine, absence of associated psychotropic treatment, with the exception of prazepam (40 mg/day). |
|
| Interventions | Fluoxetine: 33 participants Mianserin: 32 participants Fluoxetine dose range: 20‐40 mg/day Mianserin dose range: 60‐90 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Hamilton Rating Scale for Anxiety (HAM‐A), MADRS | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients were allocated at random", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of dropouts reported. Mean endpoint scores not clearly reported. No further information |
| Selective reporting (reporting bias) | Unclear risk | SIde effects not clearly reported. No further information |
| Other bias | High risk | Last author's affiliation was Organon, and this company produces mianserin |
Bhurgri 2011.
| Methods | Twelve‐week randomised study | |
| Participants | Patients with a diagnosis of major depressive episode, single episode or recurrent, according to DSM‐IV criteria, with a minimum score of 18 on the Hamilton Rating Scale for Depression‐24 item (HDRS‐24). Age range: 30‐50 years Exclusion criteria: DSM‐IV diagnosis of acute or chronic mental disorder, a Mini Mental State Examination score less than 23, concomitant use of any psychotropic drugs (except intermittent use of chloral Hydate or diazepam specifically for sleep), presence of another axis I psychiatric disorder, or any unstable medical condition that might interfere with safety or the interpretation of results. |
|
| Interventions | Fluoxetine: 96 participants Nortriptyline: 96 participants Fluoxetine dose range: 20‐80 mg/day Nortriptyline dose range: 25‐100 mg/day | |
| Outcomes | HDRS‐24 and Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned". No other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "a double dummy‐procedure was used to preserve the blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double dummy‐procedure was used to preserve the blind". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a double dummy‐procedure was used to preserve the blind". No further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Mean endpoint score not reported. Number and reasons for drop‐out not reported |
| Selective reporting (reporting bias) | High risk | Mean score reported at sixth week and without number of patients. Side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Bjerkenstedt 2005.
| Methods | Six‐week double‐blind randomised trial | |
| Participants | Outpatients meeting the DSM‐IV criteria for an acute, recurrent episode of Major Depressive Disorder (MDD) with mild or moderate intensity, aged 18‐70 years, a minimum total score of 21 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), history of at least two episodes of non‐psychotic MDD, capacity and willingness to give informed consent and to comply with study procedures. Exclusion criteria: a diagnosis of psychotic mental disorder, other disorders requiring concomitant psychoactive medication; MAOI treatment within 14 days prior to entry; history of treatment resistant MDD (at least two different antidepressants over 6 weeks at sufficient doses) from at least two previous depressive episodes, risk of suicide; history of seizure disorder, alcohol or substance abuse; other serious unstable acute or chronic medical illness; severely impaired hepatic or renal function, pregnancy, breast feeding, or use of inadequate contraceptives in fertile women; known intolerance or hypersensibility to study medications, substantial placebo response at the end of placebo run‐in phase; treatment with any investigation drug during 3 months prior to inclusion; participation in another clinical trial within 30 days before the start of the study. | |
| Interventions | Fluoxetine: 57 participants Hypericum: 59 participants Placebo: 58 participants Fluoxetine dose: 20 mg/day Hypericum dose: 900 mg/day |
|
| Outcomes | Primary outcome: change in HDRS‐21 total score Secondary endpoint: change in total Montgomery and Asberg Scale for Depression (MADRS) score and Clinical Global Impression (CGI) score Response was defined as a 50% reduction in the HDRS‐21 total score |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "a double dummy technique with matching placebos for each active treatment was applied. Thus, both placebos were identical in shape, weight, colour, smell and taste to their corresponding verum formulations", no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "all investigators and personnel, actively involved in the trial, were blinded to group assignment until the database was closed", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons for and number of dropouts reported. Number of responders in the text is different from number reported in the table. The so called "ITT population" in this study was different from the randomised sample |
| Selective reporting (reporting bias) | Unclear risk | The duration of the study was not clearly reported. Adverse experiences reported |
| Other bias | High risk | Quote: "the study was funded by a grant from Lichtwer Pharma GmbH, Berlin, Germany", and this company produces hypericum extract LI 160 |
Bougerol 1997a.
| Methods | Eight‐week double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for a major depressive disorder or bipolar disorder. The severity of depression should be 25 or more on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐65 years Exclusion criteria: pregnancy, lactation, failure to use a safe contraceptive method, alcohol or drug abuse within the last year, patients with severe somatic, neurological or psychiatric disease, treatment with MAOI within 2 weeks prior to entry the trial, hypersensitivity to study drugs, suicide risk. | |
| Interventions | Fluoxetine: 158 participants Citalopram: 158 participants Fluoxetine dose: 20 mg/day Citalopram dose range: 20‐40 mg/day Concomitant psychotropic medication was prohibited, but use of benzodiazepines for insomnia was allowed | |
| Outcomes | Primary outcome: MADRS score Secondary outcomes: Hamilton Rating Scale for Depression ‐17 item (HDRS‐17), Clinical Global Impression (CGI) scores | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double dummy" no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "all efficacy analyses were made on the basis of the efficacy group, whereas the tolerability analyses were made on the basis of the ITT population" |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only when recorded in at least 5 patients |
| Other bias | High risk | Quote: "sponsored by Lundbeck, Copenaghen. This company also delivered the citalopram tablets and bought (in bulk) the fluoxetine capsules (active and placebo) from Eli LIlly, England, and packed them" |
Bougerol 1997b.
| Methods | Eight‐week double‐blind, multicentre study | |
| Participants | Outpatients (primary care) fulfilling DSM‐III‐R criteria for a major depressive disorder. The severity of depression should be 22 or more on the Montgomery and Asberg Scale for Depression (MADRS) score. Age range: 18‐70 years Exclusion criteria: pregnancy, lactation, failure to use a safe contraceptive method, alcohol or drug abuse within the last year, patients with severe somatic, neurological or psychiatric disease, treatment with MAOI within 2 weeks prior to entry the trial, hypersensitivity to study drugs, suicide risk. | |
| Interventions | Fluoxetine: 184 participants Citalopram: 173 participants Fluoxetine dose: 20 mg/day Citalopram dose: 20 mg/day Concomitant psychotropic medication was prohibited, but use of benzodiazepines for insomnia | |
| Outcomes | Primary outcome: MADRS score Secondary outcomes: Hamilton Rating Scale for Depression‐17 item (HDRS‐17), Clinical Global Impression (CGI) scores | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "all efficacy analyses were made on the basis of the efficacy group, whereas the tolerability analyses were made on the basis of the ITT population" |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only when recorder in at least 5 patients |
| Other bias | High risk | Quote: "sponsored by Lundbeck, Copenaghen. This company also delivered the citalopram tablets and bought (in bulk) the fluoxetine capsules (active and placebo) from Eli LIlly, England, and packed them" |
Bowden 1993.
| Methods | Six‐week double‐blind, randomised, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a total score of at least 20 on Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐60 years Exclusion criteria: use of heterocyclics antidepressant drugs within 7 days or MAOI within 14 days of starting active treatment; patients with other significant medical disorders. | |
| Interventions | Fluoxetine: 28 participants Desipramine: 30 participants Fluoxetine dose range: 20‐60 mg/day Desipramine dose range: 150‐250 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Patient self‐rated Global Improvement (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all medication were prepared in identical capsules and administered by use of the double dummy technique", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale reported without denominator. Number and reasons for dropout reported |
| Selective reporting (reporting bias) | Unclear risk | No standard deviations reported. No endpoint scores. Vital signs measures not reported |
| Other bias | High risk | Quote: "this research was supported in part by a grant from Eli Lilly and Company", this company produces fluoxetine |
Boyer 1998.
| Methods | Twenty‐six‐week double‐blind, randomised, multicentre study | |
| Participants | Outpatients (primary care) fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 20 at Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐65 years Exclusion criteria: pregnancy, lactation, failure to use a safe contraceptive method; concurrent major psychiatric disorders, such as anxiety disorder, dementia, somatoform disorders, agoraphobia, social phobia, any history of schizophrenia, psychosis or personality disorder; severe concurrent medical illness; alcohol or drug dependence; serious adverse reactions related to medicines; previous treatment with antidepressant for less than 3 week; major suicide risk. | |
| Interventions | Fluoxetine: 120 participants Sertraline: 122 participants Fluoxetine dose range: 20‐60 mg/day Sertraline dose range: 50‐150 mg/day | |
| Outcomes | MADRS and Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the MADRS total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Criteria and number of drop‐out reported. Rating scale reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Scores at rating scales were reported without standard deviations. Side effects reported |
| Other bias | High risk | Last author's affiliation was Pfizer France, Orsay. This company produces sertraline |
Bremner 1984.
| Methods | Five‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling Research Diagnostic Criteria (RDC) criteria for major depressive disorder, with a score of at least 20 on Hamilton Rating Scale for Depression (HDRS), of 8 on Raskin Depression Scale (RDS). Age range: 23‐69 years Exclusion criteria: suicide risk, history of schizophrenia or other psychotic state likely to be aggravated by imipramine, organic brain disease, history of seizures; glaucoma, chronic urinary retention or serious cardiovascular disease; history of multiple adverse reaction to drugs, drug or alcohol abuse, pregnancy. | |
| Interventions | Fluoxetine: 20 participants Imipramine: 20 participants Fluoxetine dose range: 60‐80 mg/day Imipramine dose range: 125‐300 mg/day | |
| Outcomes | HDRS, RDS, Covi Anxiety scale (CAS), Clinical Global Impressions (CGI) | |
| Notes | Patients over 65 years in the imipramine group only Funding: by academy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the study drugs and placebo were supplied as identical capsules". No other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if raters were independent and unclear if blinding was successful |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Criteria and number of dropouts reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Scores at rating scales were reported without standard deviations |
| Other bias | Low risk | Funding: by academy |
Bressa 1989.
| Methods | Five‐week, double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression, with a score of at least 20 on Hamilton Rating Scale for Depression (HDRS). Age: not stated Exclusion criteria: suicidal ideas, psychosis, seizure disorders, serious cardiac, renal or hepatic disease, alcoholism or drug abuse, use of antidepressant drug with the preceding 14 days, concurrent medication potentially interacting. |
|
| Interventions | Fluoxetine: 18 participants Imipramine: 12 participants Fluoxetine dose range: 20‐60 mg/day imipramine dose range: 75‐175 mg/day |
|
| Outcomes | HDRS, Clinical Global Impression (CGI) scores | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised schedule". No other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear if raters were independent and unclear if blinding was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of drop‐out reported, but unclear reasons for dropout. Rating scale scores reported without denominators. Vital signs and side effects not reported |
| Selective reporting (reporting bias) | Unclear risk | No secondary endpoint scores. No standard deviations reported for HDRS score |
| Other bias | Unclear risk | Funding: unclear |
Byerley 1988.
| Methods | Six‐week double‐blind, randomised, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression (duration of at least 1 month) with a score of at least 20 on Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: not stated Exclusion criteria: psychotic symptoms bipolar illness, schizophrenia, active drug or alcohol abuse, significant medical illness | |
| Interventions | Fluoxetine: 32 participants Imipramine: 34 participants Placebo: 29 participants Fluoxetine dose range: 40‐80 mg/day Imipramine dose range: 150‐300 mg/day Intermittent administration of flurazepam for insomnia (15‐30 mg) | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) scores | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomisation was carried out by using a table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "capsules looked identical", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reason for dropout during the study were reported. Withdrawals were not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | No detailed endpoint scores at CGI. Side effects reported only with percentage, without denominator |
| Other bias | High risk | The study was supported, in part, by Eli Lilly. This company produces fluoxetine |
Cassano 2002.
| Methods | Fifty‐two week double‐blind, randomised, multicentre study | |
| Participants | Outpatients fulfilling ICD‐10 criteria for major depression, with a Mini Mental State Examination (MMSE) score of at least 22 and a Hamilton Rating Scale for Depression (HDRS‐21) score of at least 18. Age: over 65 years Exclusion criteria: concurrent major medical disorders, dementia, any history of schizophrenia, psychosis; alcohol or drug dependence; major suicide risk; use of long‐acting neuroleptic drugs within 6 months or oral neuroleptics within 2 weeks before the study entry; ECT; daily use of benzodiazepines within 8 weeks or SSRI within 4 weeks, MAOI within 3 weeks, TCA within 1 week before the study entry. | |
| Interventions | Fluoxetine: 119 participants Paroxetine: 123 participants Fluoxetine dose range: 20‐60 mg/day Paroxetine dose range: 20‐40 mg/day | |
| Outcomes | HDRS‐21, Clinical Anxiety Scale, Buschke Selective Reminding Test (BSRT), Blessed Information and Memory Test (BIMT), Clifton Assessment Scale (CLAS), Cancellation Task Test (CTT), Wechsler Paried Word Test (WPW), Mini Mental Sate Evaluation (MMSE) and Clinical Global Impression (CGI) | |
| Notes | Depression response: total score less than 10 on the HDRS‐21 Anxiety response: total score less than 8 on the Covi Anxiety scale (CAS) Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear if raters were independent and unclear if blinding was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominator |
| Selective reporting (reporting bias) | High risk | Standard deviations not reported (HDRS). No endpoint scores (CGI, CAS) |
| Other bias | High risk | The study was supported by SmithKline. This company produces paroxetine |
Chouinard 1985.
| Methods | Five‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling Research Diagnostic Criteria (RDC) criteria for major depressive disorder, with a score of at least 21 on Hamilton Rating Scale for Depression (HDRS‐17) and of at least 8 on the Raskin Depression Scale (RAS). Age range: 21‐70 years Exclusion criteria: physical illness, schizophrenia, schizoaffective illness, chronic or acute organic brain syndrome, mental deficiency, alcoholism, epilepsy, drug addiction. |
|
| Interventions | Fluoxetine: 23 participants Amitriptyline: 28 participants Fluoxetine dose range: 40‐80 mg/day Amitriptyline dose range: 100‐300 mg/day Benzodiazepines were allowed for agitation and insomnia | |
| Outcomes | Primary outcome: HDRS‐17, Clinical Global Impression (CGI), Efficacy Index‐Side Effects rating (EISE) Secondary outcomes: Hamilton Rating Scale for Anxiety (HAM‐A) and Zung Depression Scale (SDS) |
|
| Notes | One patient attempted suicide in the fluoxetine group Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned. Assigment was stratified to ensure balanced distribution of male and female patients to each of the two study drug regimen". No other information provided |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind conditions in identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without standard deviations. Reasons for withdrawal reported, but withdrawals not included in analysis |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only with an incidence over 10% |
| Other bias | Unclear risk | Funding: unclear |
Chouinard 1999.
| Methods | Twelve‐week double‐blind, randomised, multicentre study | |
| Participants | Patients fulfilling DSM‐III criteria for major depressive disorder, with a score of at least 20 on Hamilton Rating Scale for Depression (HDRS‐21). Age: not stated Exclusion criteria: significant concurrent illness including renal, hepatic, cardiovascular or neurological disease, non‐stabilised diabetes, other current Axis I psychiatric diagnosis; organic brain syndrome, past or present abuse of alcohol or drugs; pregnancy or lactating; ECT; continuous lithium therapy in preceding 2 months, use of important psychotropic drug, current therapy with an anticoagulant or type 1 antiarrhythmic. |
|
| Interventions | Fluoxetine: 101 participants Paroxetine: 102 participants Fluoxetine dose range: 20‐80 mg/day Paroxetine dose range: 20‐50 mg/day Chloral hydrate was allowed just during the first two weeks of the study | |
| Outcomes | Primary outcomes: HDRS‐21, Clinical Global Impression Secondary outcomes: HDRS‐21 anxiety and somatization scores | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 total score and/or a total score less than 10 Two participants dropped out (1 in the fluoxetine and 1 in the paroxetine group) due to attempted suicide Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the efficacy analysis included all randomised patients who underwent at least one on‐therapy efficacy evaluation". Number of patients responding to treatment reported with denominator. Reason for withdrawal reported |
| Selective reporting (reporting bias) | Low risk | Effect side reported only with an incidence over 10% |
| Other bias | High risk | Funding by SmithKline, and this company produces paroxetine |
CL3‐022.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for single or recurrent episode of Major Depressive Disorder (MDD), with or without melancholic features, without atypical features, without psychotic features and a score of at least 22 on Hamilton Rating Scale for Depression (HDRS). Morover decrease in HDRS total score should not be more than 20% between start of run‐in and inclusion visit and a severity of illness of at least 4 on CGI. Age range: 18‐59 years Exclusion criteria: not specified |
|
| Interventions | Fluoxetine: 137 participants Agomelatine: 133 participants Fluoxetine dose: 20 mg/day Agomelatine dose: 25 mg/day | |
| Outcomes | Primary outcome: HDRS score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information |
| Other bias | High risk | Funding: by industry |
CL3‐024.
| Methods | Six‐week double‐blind, randomised, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for single or recurrent episode of Major Depressive Disorder (MDD), with or without melancholic features, without atypical features, without psychotic features and a score of at least 22 on Hamilton Rating Scale for Depression (HDRS). Age range: 18‐59 years Exclusion criteria: not specified |
|
| Interventions | Fluoxetine: 148 participants
Agomelatine 25mg: 150 participants Agomelatine 50mg: 151 participants Fluoxetine dose: 20 mg/day Agomelatine dose range: 25‐50 mg/day |
|
| Outcomes | Primary outcome: HDRS score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information |
| Other bias | High risk | Funding: by industry |
Clayton 2003.
| Methods | Eight‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 22 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: other psychiatric diagnoses, have received Tricyclics (TCAs) within the previous 14 days and fluoxetine within 14 days or 28 days when resistant to treatment, uncontrolled medical or metabolic illness, use of illicit drugs, history of DSM‐IV substance abuse in the previous 12 months. | |
| Interventions | Fluoxetine: 150 participants Reboxetine: 150 participants Fluoxetine dose: 20‐40 mg/day Reboxetine dose: 8‐10 mg/day | |
| Outcomes | Primary outcome: HDRS score Response: decrease of at least 50% in the HDRS score |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of withdrawals reported, but reasons not clearly described. Mean scores reported only at the baseline and not at the endpoint |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported partially. Baseline mean scores reported without standard deviations |
| Other bias | High risk | Funding by Pharmacia Corporation |
Clerc 1994.
| Methods | Six‐week double‐blind, randomised, multicentre study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with melancholia, with a score of at least 25 on the Montgomery and Asberg Scale for Depression (MADRS). Age: over 18 years Exclusion criteria: medical illness, psychotherapy or ECT during the study duration | |
| Interventions | Fluoxetine: 34 participants Venlafaxine: 34 participants Fluoxetine dose: 40 mg/day Venlafaxine dose: 200 mg/day | |
| Outcomes | Primary outcome: Hamilton Rating Scale for Depression (HDRS‐21), MADRS, Clinical Global Impression Scale (CGI) | |
| Notes | Response: decrease of at least 50% in the HDRS or in the MADRS total scores, or a CGI score of 1 or 2 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind medication, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind medication, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind medication, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "analysis are on the ITT patients and use the last observation carried forward (LOCF) method" |
| Selective reporting (reporting bias) | Unclear risk | Treatment‐emergent study events reported only if it was reported by three or more patients |
| Other bias | High risk | Quote: "this work was supported by a grant from Wyeth‐Ayerst Research, Paris, France", this company produces venlafaxine |
Cohn 1985.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depressive illness, with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS). Age range: 20‐64 years Exclusion criteria: concomitant physical condition or history of conditions that could interfere with therapy | |
| Interventions | Fluoxetine: 54 participants Imipramine: 54 participants Placebo: 57 participants Fluoxetine dose range: 20‐80 mg/day Imipramine dose range: 75‐300 mg/day | |
| Outcomes | HDRS, Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Clinical Global Impression (CGI) Severity and Improvement | |
| Notes | One patient attempted suicide in the fluoxetine group Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind study. Placebo and the study drugs were supplied as identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without standard deviations. Reasons for dropout not clear. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only over 10% |
| Other bias | Unclear risk | Funding: unclear |
Corne 1989.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients (general practice) fulfilling Research Diagnostic Criteria (RDC) criteria for primary unipolar major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS‐17). Age range: 18‐70 years Exclusion criteria: physical illness, use of other antidepressant medication, pregnancy, potential childbearing, lactation | |
| Interventions | Fluoxetine: 49 participants Dothiepin: 51 participants Fluoxetine dose range: 20‐60 mg/day Dothiepine dose range: 50‐100 mg/day | |
| Outcomes | HDRS‐17 score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomly assigned, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double dummy technique was employed", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominator. Effect side reported. Number of patients randomised and number lost during follow‐up not clear |
| Selective reporting (reporting bias) | High risk | Means and standard deviations reported only in figures |
| Other bias | High risk | Second author had affiliation in Eli Lilly, this company produces fluoxetine |
Corrigan 2000.
| Methods | Eight‐week double‐blind, randomised study | |
| Participants | Patients fulfilling DSM‐III‐R criteria for major depression (single or recurrent episode, with or without melancholia and without psychotic features). Age range: 18‐65 years Exclusion criteria: clinically relevant disease, clinically significant changes on the ECG, lifetime history of hypomania/mania, psychotic disorder, dementia, borderline or antisocial personality disorders, history of a serious suicidal attempting the past 12 months, pregnancy or lactation, non‐responders to at least two trials of antidepressant treatment in the past, use of fluoxetine in the past 6 months or use of another investigational drug within one month prior to the baseline visit. | |
| Interventions | Fluoxetine: 35 participants Pramipexole 1 mg: 35 participants Pramipexole 5 mg: 33 participants Placebo: 35 participants Fluoxetine dose: 20 mg/day | |
| Outcomes | Primary outcomes: Hamilton Rating Scale for Depression (HDRS‐17), Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity Secondary outcomes: Beck Depression Inventory, CGI Improvement | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomised clinical trial”, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "results are reported for the observed‐case analysis, for which no missing data were replaced". Number randomised, and number lost during follow‐up reported |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations. Adverse events were reported with a frequency of at least 10% |
| Other bias | High risk | Authors' affiliation was in Pharmacia&Upjohn Inc, and this company produces pramipexole |
Costa e Silva 1998.
| Methods | Eight‐week double‐blind, randomised, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and depressive symptoms for at least 1 month before study entry. Age range: 18‐60 years Exclusion criteria: pregnancy, absence of methods of contraception, known sensitivity to fluoxetine or venlafaxine, history of significant cardiac, renal or hepatic disease, clinically significant abnormalities on a screening examination, ECG, laboratory tests, acute suicide tendency, seizures, history or presence of any psychotic disorder not associated with depression, drug or alcohol dependence within the past year, psychotherapy, use of fluoxetine, antipsychotic drugs, ECT, MAOI within the past 14 days, any other antidepressant, anxiolitics, sedative‐hypnotic drugs (but zopiclone) within 7 days before baseline. | |
| Interventions | Fluoxetine: 186 participants Venlafaxine: 196 participants Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐125 mg/day | |
| Outcomes | Primary outcomes: HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity and Improvement | |
| Notes | Response: decrease of at least 50% in the HDRS or in the MADRS; or a CGI‐I score of 1 or 2 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if raters were independent and unclear if blinding was successful |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviations. Side effects reported only if they occurred at least in 5% of the patients |
| Other bias | High risk | Quote: "supported by a grant from Wyeth‐AYerst International". This company produces venlafaxine |
Dalery 1997.
| Methods | Twelve‐week double‐blind, randomised, multicentre study | |
| Participants | Patients fulfilling DSM‐III‐R criteria for major depression (single or recurrent), with a score of at least 20 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐70 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 82 participants Amineptine: 87 participants Fluoxetine dose: 20 mg/day Amineptine dose: 200 mg/day Anxiolitics and non‐barbiturate hypnotics were allowed | |
| Outcomes | MADRS, Clinical Global Impression (CGI), Mood Anxiety Retardation and Danger (MARD) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "random allocation". No further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons of attrition not clear. Ratings scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Data at follow‐up not reported. Adverse effects not reported |
| Other bias | Unclear risk | Funding: unclear |
Dalery 2003.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐70 years Exclusion criteria: acute suicidal ideation, dementia, history of epilepsy, alcoholism in the previous 6 months, other psychoactive substance, pregnancy, lactation, absence of contraception, hepatic, renal, pulmonary, endocrine, cardiac disease, previous failure with SSRI therapy, concomitant use of lithium, warfarin, carbamazepine, theophyline, insulin, hypoglycaemic agents, MAOI or ECT in the previous 2 weeks. | |
| Interventions | Fluoxetine: 94 participants Fluvoxamine: 90 participants Fluoxetine dose: 20 mg/day Fluvoxamine dose: 100 mg/day | |
| Outcomes | Primary outcome: area under the curve of the change in HDRS‐17 total score from baseline Secondary outcomes: numbers of HDRS‐17 responders, Clinical Global Impression (CGI) Severity and Improvement, Clinical Anxiety Scale (CAS), Irritability Depression and Anxiety Scale (IDAS) total score and sub‐scores, Beck Scale for Suicide Ideation (SSI), Sleep Evaluation and the HDRS‐17 total and subtotal scores | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons and number of dropouts were not clear. Rating scale scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations |
| Other bias | High risk | Quote: "the study was supported by a grant from Solvay Pharmaceuticals". This company produces maprotiline |
De Jonghe 1991.
| Methods | Six‐week double‐blind, randomised, two‐site study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depressive disorder without psychotic features, with a score of at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐70 years Exclusion criteria: high suicide risk, other psychiatric diagnosis, somatic disease which could contraindicate treatment with fluoxetine or maprotiline, history of hypersensitivity, severe allergies, multiple severe reactions to drugs, lactation, pregnancy or pregnancy wish, MAOI use within 2 weeks before starting the trial. | |
| Interventions | Fluoxetine: 30 participants Maprotiline: 35 participants Fluoxetine dose range: 40‐80 mg/day Maprotiline dose range: 50‐150 mg/day Only oxazepam was allowed as hypnotic or anxiolytic, if absolutely required | |
| Outcomes | HDRS‐17, Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Clinical Global Impression (CGI) Severity and Improvement | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominator. Number and reasons of dropouts reported |
| Selective reporting (reporting bias) | Unclear risk | Vital signs not reported |
| Other bias | High risk | Quote: "the study was supported by Eli Lilly Nederlands". This company produces fluoxetine |
De Nayer 2002.
| Methods | Twelve‐week double‐blind, randomised, multicentre study | |
| Participants | Outpatients with a score between 18 and 25 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and minimum baseline of 8 on the Covi Anxiety Scale (CAS), and considered by the investigator to be moderately depressed. Age range: 18‐70 years Exclusion criteria: pregnancy, childbearing potential, absence of contraceptive method, psychiatric disease or personality disorder, known clinically significant laboratory abnormalities, use of antipsychotic drug or ECT within 30 days of baseline, use of fluoxetine within 21 and MAOI within 14 days before baseline; patients who previously failed to respond to venlafaxine or fluoxetine, high suicide risk. | |
| Interventions | Fluoxetine: 73 participants Venlafaxine: 73 participants Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐150 mg/day Lormetazepam was allowed (2 mg) as hypnotic | |
| Outcomes | Primary outcomes: HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity Secondary outcome: CAS | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 or in the MADRS total score Remission: total score less than 8 on the HDRS‐21 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear if blinding was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominator. Number and reason for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviation |
| Other bias | High risk | Funding by Wyeth, and this company produces venlafaxine |
De Ronchi 1998.
| Methods | Ten‐week double‐blind, randomised, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 16 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 60 years Exclusion criteria: mental organic disorder, Mini Mental State Examination (MMSE) less than 24, high suicide risk, history of alcohol or drug abuse, severe physical illness, epilepsy, schizophrenia. | |
| Interventions | Fluoxetine: 32 participants Amitriptyline: 33 participants Fluoxetine dose: 20 mg/day Amitriptyline dose range: 50‐100 mg/day Patients taking lorazepam 5 mg/day for at least 6 months before enrolment were allowed to continue; triazolam was allowed (0.25 mg/day) during the first 2 weeks for insomnia | |
| Outcomes | HDRS‐17, Montgomery and Asberg Scale for Depression (MADRS), Covi Anxiety Scale (CAS), Clinical Global Impression (CGI) Severity and Improvement | |
| Notes | Response: decrease of at least 50% in the HDRS‐17 total score or a total score less than 10 Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised trial", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "all ratings were conducted under double blind condition", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominators. Number and reasons for discontinuation not clear |
| Selective reporting (reporting bias) | Unclear risk | Incidence of adverse effects not clear |
| Other bias | Unclear risk | Funding: unclear |
De Wilde 1993.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Patients fulfilling DSM‐III criteria for major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐65 years Exclusion criteria: pregnancy, lactation, severe concomitant disease, schizophrenia, abuse of alcohol or drugs, severe risk of suicide, ECT in the previous 3 months, MAOI or oral neuroleptics in the previous 14 days, depot neuroleptics in the previous 4 weeks, patients receiving lithium. | |
| Interventions | Fluoxetine: 41 participants Paroxetine: 37 participants Fluoxetine dose range: 20‐60 mg/day Paroxetine dose range: 20‐40 mg/day Temazepam or other short‐acting benzodiazepines were permitted as hypnotic | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Hopkins Symptoms Check List (HSLC), Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "the Last Observation Carried Forward (LOCF) data set was used". Scores in follow‐up were reported without denominator. Reasons for withdrawal not clear |
| Selective reporting (reporting bias) | High risk | No follow‐up scores (MADRS, HDRS, HSLC) |
| Other bias | High risk | Funding by Smithkline, and this company produces paroxetine |
Debus 1988.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: over 18 years Exclusion criteria: pregnancy, lactation, absence of contraception, history of glaucoma, suicidal risk, history serious medical conditions, seizures, history of severe allergies, multiple adverse medication reactions or known allergy, other DSM‐III diagnosis including substance abuse, bipolar disorder, schizophrenia, schizoaffective disorder, paranoid disorder, organic mental disorder, other psychotropic medications, with the exception of some hypnotics, use of fluoxetine or MAOI within the past 4 weeks. | |
| Interventions | Fluoxetine: 22 participants Trazodone: 21 participants Fluoxetine dose range: 20‐60 mg/day Trazodone dose range: 50‐400 mg/day | |
| Outcomes | HDRS‐21, Inventory for Depressive Symptomatology ‐ Clinician Version (IDS‐C) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported with denominator, but withdrawal not included in analysis. Side effects reported |
| Selective reporting (reporting bias) | Unclear risk | No endpoint scores (IDS‐C). Scores without standard deviation (HDRS) |
| Other bias | High risk | Quote: "supported in part by Ely Lilly". This company produces fluoxetine |
Demyttenaere 1998.
| Methods | Nine‐week double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 15 on the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 18‐60 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 35 participants Amitriptyline: 31 participants Fluoxetine dose: 20 mg/day Amitriptyline dose: 150 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind design", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind design", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind design", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominator. Reasons for dropouts not clear |
| Selective reporting (reporting bias) | Unclear risk | No endpoint scores (CGI). Adverse events not reported |
| Other bias | High risk | Quote: "we are indebted to Eli Lilly Belgium for financial support for the present study", and this company produces fluoxetine |
Demyttenaere 2004.
| Methods | Twenty‐two‐week double‐blind randomised study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depression disorder. Age range: 22‐63 years Exclusion criteria: other DSM‐IV Axis I disorders | |
| Interventions | Fluoxetine: 42 participants Paroxetine: 43 participants Fluoxetine dose: 20 mg/day Paroxetine dose: 20 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS‐17) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind trial", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind trial", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind trial", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Mean scores at rating scales only referred to the whole sample (not to each arm). Number and reasons for dropouts not clear |
| Selective reporting (reporting bias) | Unclear risk | The type of adverse events was not reported |
| Other bias | High risk | Quote: "Eli Lilly Benelux provided logistic and material support for this study", and this company produces fluoxetine |
Diaz Martinez 1998.
| Methods | Eight‐week randomised, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐55 years Exclusion criteria: lactation, childbearing potential, previous treatment with venlafaxine or fluoxetine, history of clinically significant medical disease, abnormalities on ECG or laboratory tests, acute suicidal tendencies, history of seizure disorder, organic mental disorder, bipolar disorder, history of any psychotic disorder not associated with depression, current use of investigational drugs, antipsychotic drugs, ECT within the previous 30 days or MAOI or paroxetine within the previous 14 days, use of antidepressant or hypnotic drugs, but zopiclone (7.5 mg), history of drug or alcohol abuse. | |
| Interventions | Fluoxetine: 75 participants Venlafaxine: 70 participants Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐150 mg/day Only zopiclone was allowed for insomnia | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression, Symptom Checklist 61 Item (SCL‐61) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Open‐label" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominator. Number and reasons for withdrawal reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviations. Adverse events reported over 5% |
| Other bias | High risk | Quote: "this study was supported by Wyeth‐Ayerst International, Saint David's, Pennsylvania": Tihs company produces venlafaxine |
Dierick 1996.
| Methods | Eight‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 18‐83 years Exclusion criteria: history of clinically significant disease, abnormalities on ECG or laboratory tests, acute suicidal tendencies, history of seizure disorder, organic mental disorder, bipolar disorder or personality disorder, history of any psychotic disorder not associated with depression, venlafaxine or fluoxetine hypersensitivity or use within 2 months of baseline, current use of investigational drugs, antipsychotic drugs, ECT or MAOI within the previous 14 days, use of antidepressant drug within 7 days, use of any anxiolytic that could not be withdrawn at baseline, drug or alcohol abuse within 2 years of the start of the study. | |
| Interventions | Fluoxetine: 161 participants Venlafaxine: 153 participants Fluoxetine dose: 20 mg/day Venlafaxine dose range: 75‐150 mg/day | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) scales | |
| Notes | Response: decrease of at least 50% in the HDRS or MADRS total score, or a score of 1 or 2 on the CGI Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Scores reported with denominator. Number and reasons for withdrawals reported |
| Selective reporting (reporting bias) | Unclear risk | No CGI endpoint scores reported. Only most common (over 5%) side effects reported |
| Other bias | High risk | Quote: "this study was supported by Wyeth‐Ayerst Research" and this company produces venlafaxine |
Dowling 1990.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression (unipolar), with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS‐17). Age range: 18‐75 years Exclusion criteria: significant physical illness, lactation, pregnancy, history of schizophrenia or drug or alcohol abuse, current use of antidepressant. | |
| Interventions | Fluoxetine: 30 participants Dothiepin: 30 participants Fluoxetine dose range: 20‐40 mg/day Dothiepine dose range: 100‐200 mg/day Benzodiazepines were allowed for sedation at the discretion of the doctor | |
| Outcomes | HDRS, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity and Improvement, Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blind", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all patients took identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropouts were reported. Scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects were reported. Mean scores were reported without standard deviations. No endpoint scores (MADRS, CGI) |
| Other bias | High risk | In the aknowledgements authors thank Eli Lilly Company. This company produces fluoxetine and probably the study was supported by this industry |
Duarte 1996.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for double depression (dysthymia and major depression), with a score of at least 16 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: suicidal tendencies, delusional depression, severe organic disease, alcoholism, drug abuse, ongoing ECT or structured psychotherapy. |
|
| Interventions | Fluoxetine: 21 participants Moclobemide: 21 participants Fluoxetine dose: 20 mg/day Moclobemide dose: 300 mg/day Use of single benzodiazepines was allowed at discretion of the doctor | |
| Outcomes | Primary outcomes: percentage of responders defined as decrease of at least 50% in the HDRS Secondary outcomes: endpoint score at HDRS, percentage of end of treatment Clinical Global Impression (CGI) very good and good responses | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double blinding was achieved by appropriate drug packaging and formulation", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blinding was achieved by appropriate drug packaging and formulation", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blinding was achieved by appropriate drug packaging and formulation", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominators. Mean scores and standard deviations reported only in figure and they were not clear |
| Selective reporting (reporting bias) | Unclear risk | No vital signs and side effects reported. Quote: "no clinically significant changes in vitals signs were recorded" |
| Other bias | High risk | The last author's affiliation was Hoffmann–La Roche Ltd, and this company produces moclobemide |
Fabre 1991.
| Methods | Five‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression (single episode or recurrent). Age range: 18‐65 years Exclusion criteria: concurrent diagnosis of bipolar disorder or schizophrenia, hyperactivity or agitation, presence of hyper thyroidism or a clinically unstable medical condition, history of narrow angle glaucoma, urinary retention, seizures or substance abuse, MAOI use within 14 days of baseline, pregnancy, lactation, potential childbearing, history of allergy to the study drugs. | |
| Interventions | Fluoxetine: 103 participants Nortriptyline: 102 participants Fluoxetine dose range: 20‐40 mg/day Nortriptyline dose range: 50‐100 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Zung Depression Scale, Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "patients received two bottles of identical capsules". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if blinding was successful |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for withdrawal reported. No endpoint scores (CGI, Zung Depression Scale) reported |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations. Only most common adverse events were reported |
| Other bias | Unclear risk | Funding: unclear |
Fairweather 1999.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients (general practice) fulfilling DSM‐III‐R criteria for major depression. Age range: 18‐70 years Exclusion criteria: concurrent illness, concomitant use of psychotropic medication, long‐term treatment with benzodiazepines |
|
| Interventions | Fluoxetine: 42 participants Dothiepin: 42 participants Fluoxetine dose: 20 mg/day Dothiepine dose range: 75‐150 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Leeds Sleep Evaluation Questionnaire (LSEQ) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "drugs and placebos were packaged in identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominators. Number and reasons for dropouts not clear |
| Selective reporting (reporting bias) | Unclear risk | Only most common adverse events were reported |
| Other bias | High risk | Quote: "the research grant provided by Lilly Industries", and Eli LIlly produces fluoxetine |
Falk 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for unipolar major depression (single or recurrent), with the present episode lasting 4 weeks or more and with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: over 62 years Exclusion criteria: serious medical illness, unstable cardiac arrhythmia, seizure disorders, history of allergy to either drug, severe psychosis, suicidal symptoms or DSM‐II diagnosis of schizophrenia, bipolar disorder, organic mental disorder, substance abuse disorder within the past year or paranoid disorders, use of either drugs within 1 month preceding study entry, MAOI in the prior 14 days or other antidepressants at the time of entry. |
|
| Interventions | Fluoxetine: 14 participants Trazodone: 13 participants Fluoxetine dose range: 20‐60 mg/day Trazodone dose range: 50‐400 mg/day Only use of benzodiazepines and chloral hydrate for sleep were allowed | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Treatment Emergent Symptom Scale (TESS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all capsules were identical", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported without denominators. Reasons and number of dropouts described |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported. Scores reported for each follow‐up with standard deviations |
| Other bias | High risk | Quote: "this research was supported by a grant from Eli Lilly and Company", and this company produces fluoxetine |
Fava 1998.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for moderate to moderately severe major depression without a history of mania or hypomania, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), of at least 8 on the Raskin Depression Scale (and grater than Covi Anxiety Scale (CAS) score). Mean age: 41.3 years Exclusion criteria: schizophrenia, adjustment disorder, bipolar disorder, panic disorder, social phobia, obsessive compulsive disorder, psychotic depression, atypical depression, serious concomitant medical illness, significant abnormal laboratory values, history of seizure disorder, high suicidal risk, recent history of alcohol or drug abuse, use other psychotropic drug within 14 days of baseline, ECT within 3 months of baseline, any investigational drug within 30 days of baseline, previous treatment with paroxetine, pregnancy, childbearing potential without contraceptive. | |
| Interventions | Fluoxetine: 54 participants Paroxetine: 55 participants Placebo: 19 participants Fluoxetine dose range: 20‐80 mg/day Paroxetine dose range: 20‐50 mg/day | |
| Outcomes | HDRS‐21, Covy Anxiety Scale (CAS), Raskin Depression Scale | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "we chose to conduct all analyses with an ITT approach". Number and reasons for dropout reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. Primary and secondary endpoint reported with standard deviations |
| Other bias | High risk | Quote: "this study was supported by SmithKline Beecham Pharmaceuticals" and this industry produces paroxetine |
Fava 2002.
| Methods | Ten‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depression or atypical major depression, with a baseline score of at least 16 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 18 years Exclusion criteria: pregnancy, lactation, suicide risk, serious medical illness, seizure disorders, presence of any of the following diagnosis: organic mental disorder, substance use disorder, schizophrenia, delusional disorder, psychotic disorders not elsewhere classified, bipolar disorder, antisocial personality disorder, mood congruent or mood incongruent features, history of multiple adverse drug reactions, concomitant use of any antidepressants, anxiolytic or other psychotropic medication within 7 days prior study entry, with the exception of chloral hydrate, hyper‐ or hypothyroidism, use of MAOI within 2 weeks of active therapy, lack of response to the treatment of a current major depressive episode by any SSRI. | |
| Interventions | Fluoxetine: 92 participants Sertraline: 96 participants Paroxetine: 96 participants Fluoxetine dose range: 20‐60 mg/day Sertraline dose range: 50‐200 mg/day Paroxetine dose range: 20‐60 mg/day | |
| Outcomes | Primary outcome: total score on the HDRS‐17 Secondary outcomes: improvement on the Clinical Global Impression (CGI) Severity scale and HDRS sleep disturbance, cognitive disturbance (COG) factors |
|
| Notes | Response: decrease of at least 50% in the HDRS‐17 total Remission: total score of maximum 7 on the HDRS‐17 at the endpoint Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported without denominators. Mean scores and standard deviations reported at each follow‐up |
| Selective reporting (reporting bias) | Unclear risk | Only adverse events reported by at least 10% of the patients described |
| Other bias | High risk | Quote: "supported by a grant from Ely Lilly ", and this company produces fluoxetine |
Fava 2005.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for current major depression episode of at lest 2 weeks duration, Hamilton Rating Scale for Depression (HDRS) score at least 16. Age range: 18‐65 years Inclusion criteria: negative pregnancy test within 5 days before study start in women of childbearing potential, use of adequate contraception. Exclusion criteria: pregnancy, lactation, or non use of medically accepted contraception, current serious suicide or homicidal risk, serious or instable medical illness, history of seizure disorders, presence of any of the following diagnosis: organic mental disorder, substance use disorder, including alcohol, active within the last 6 months, schizophrenia, delusional disorder, psychotic disorders not elsewhere classified, bipolar disorder, antisocial personality disorder; history of multiple adverse drug reactions or allergy to the study drugs; mood congruent or mood incongruent psychotic features, concomitant use of other psychotropic drugs within 14 days before baseline, other investigational psychotropic drug within 40 days, fluoxetine within 40 days or any other investigational drug within 1 month, hypothyroidism; failure to respond during the course of current MDE to at least 2 adequate antidepressant trials; any other condition which, in the investigator judgement, may pose significant risk to the patient's health or may decrease the chances of obtaining reliable data to achieve the objectives of the study; mental condition rendering the patients unable to understand nature, scope and risk of the study; history or suspicion of unreality, poor cooperation, or non compliance with medical treatment. |
|
| Interventions | Fluoxetine: 47 participants
St John's wort: 45 participants
Fluoxetine dose: 20 mg/day St John's wort dose: 900 mg/day |
|
| Outcomes | Primary outcome: total score on the HDRS Secondary outcome: improvement on the Clinical Global Impression (CGI) scale and Beck Depression Inventory (BDI) score |
|
| Notes | Response: decrease of at least 50% in the HDRS total Remission: total score of maximum 8 on the HDRS at the endpoint Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number and reasons for dropout reported. Primary and secondary endpoint scores reported |
| Selective reporting (reporting bias) | Low risk | Only most frequent (at least 10%) adverse events reported. Scores reported with standard deviations |
| Other bias | High risk | Quote: "the study was supported by a grant of Lichtwer Pharma AG (Berlin, Germany)", and this company produces St. John's wort |
Fawcett 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for unipolar major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Mean age: 39.9 in the fluoxetine group, 44.5 in the amitriptyline one Exclusion criteria: significant medical illness, concomitant medication with any potential psychiatric side effect, psychotic features, any other DSM‐III Axis I diagnosis other than unipolar major depression. | |
| Interventions | Fluoxetine: 20 participants Amitriptyline: 20 participants Fluoxetine dose range: 20‐60 mg/day Amitriptyline dose range: 50‐200 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression for Severity and Improvement (CGI S‐I), Patient Global Impression (PGI) | |
| Notes | Improvement: a decrease of at least 50% on the total HDRS‐21 score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "(patients) were randomly assigned to fluoxetine treatment", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Mean endpoint scores reported without denominators. Adverse events not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Mean endpoint scores and standard deviations reported (HDRS, CGI) |
| Other bias | High risk | Supported by Eli Lilly, and this company produces fluoxetine |
Feighner 1985a.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for unipolar major depression (single or recurrent episode), with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS) and Raskin Depression Scale (RDS) score of at least 8 and equal or greater to the Covi Anxiety Scale (CAS) score. Age: over 64 years Exclusion criteria: history of, or current conditions that might put them at risk or that precluded evaluation of the results | |
| Interventions | Fluoxetine: 78 participants Doxepine: 79 participants Fluoxetine dose range: 20‐80 mg/day Doxepine dose range: 50‐250 mg/day | |
| Outcomes | HDRS, Clinical Global Impression (CGI) Severity, RDS, CAS scores | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the study drugs and placebo were supplied in identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominators. Reasons and number of early termination not clear |
| Selective reporting (reporting bias) | High risk | Adverse effects were described only when reported by more than 10% of the sample. Mean scores reported without standard deviations |
| Other bias | Unclear risk | Funding: unclear |
Feighner 1985b.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling Research Diagnostic Criteria criteria for unipolar major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS) and Raskin Depression Scale (RDS) score of at least 8. Age range: 19‐69 years Exclusion criteria: serious illness or condition that contraindicated the use of amitriptyline or that could make patients unsuitable for study. | |
| Interventions | Fluoxetine: 22 participants Amitriptyline: 22 participants Fluoxetine dose range: 20‐80 mg/day Amitriptyline dose range: 75‐300 mg/day Only chloral hydrate (max 1 g) was allowed for sleep and one benzodiazepine for agitation | |
| Outcomes | HDRS, RAS, Covi Anxiety Scale (CAS), Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "to ensure the double blind, study drugs were divided into daytime and bedtime doses", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Mean endpoint score and standard deviation at CGI not reported. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Mean endpoint scores (HDRS, RDS, CAS) reported without standard deviations. Side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Feighner 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for unipolar major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS) and Raskin Depression Scale (RDS) score of at least 8 and equal or greater to the Covi Anxiety score (CAS). Age range: 18‐70 years Exclusion criteria: pregnancy, non‐contraception, serious suicide risk, organic brain syndrome, schizophrenia, seizures, drug or alcohol abuse within the past year, contraindication to imipramine. | |
| Interventions | Fluoxetine: 61 participants Imipramine: 58 participants Placebo: 59 participants Fluoxetine dose range: not stated Imipramine dose range: not stated | |
| Outcomes | HDRS, RDS, CAS, Clinical Global Impression (CGI), Patient Global Improvement Scale (PGI) | |
| Notes | Improvement: a moderately or markedly improved on the CGI or a decrease of at least 50% on the total HDRS score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Categorical endpoint not reported |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons of early termination were reported. Side effects reported by more than 10% of the sample described |
| Other bias | High risk | Quote: "(study) performed at the Feighner Research Institute". This institute usually receives founds by pharmacological industries |
Feighner 1991.
| Methods | Six‐week randomised, double‐blind two‐centre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for non‐psychotic major depressive episode, lasting between 4 weeks to 2 years, single or recurrent, which was not secondary to another pre‐existing psychiatric or medical condition, with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS‐21). Age: over 18 years Exclusion criteria: seizures, current diagnosis or history of hepatic or renal dysfunction, anorexia or bulimia, other unstable medical disorder, pregnancy, lactation, childbearing potential, alcohol or substance abuse within the past year, use of psychoactive drug within 1 week of baseline, previous treatment with bupropion or fluoxetine, high suicidal risk. | |
| Interventions | Fluoxetine: 62 participants Bupropion: 61 participants Fluoxetine dose range: 20‐80 mg/day Bupropion dose range: 225‐450 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Severity and Improvement, Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double dummy technique was used to maintain the double blind", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Responders denominator was different from the number of randomised. Reasons for discontinuation were unclear |
| Selective reporting (reporting bias) | High risk | Scores reported without standard deviations. Most frequent (reported at least 5%) adverse events reported |
| Other bias | High risk | Quote: "supported by a grant from Burroghs Wellcome, Co.", and this industry produces bupropion |
Ferreri 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression, with a score between 18 and 25 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) . Age range: 18‐65 years Exclusion criteria: organic brain disease, seizures, other serious illness, hyperthyroidism, allergy, drug or alcohol abuse, use of MAOI within 2 week, serious suicidal risk, pregnancy and lactation. | |
| Interventions | Fluoxetine: 31 participants Amineptine: 32 participants Fluoxetine dose: 20 mg/day Amineptine dose: 200 mg/day | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "(treatments) were administered in identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported without denominators. Number and reasons for early termination reported |
| Selective reporting (reporting bias) | Unclear risk | End‐point scores reported without standard deviations. Side effects not clearly reported |
| Other bias | Unclear risk | Funding: unclear |
Finkel 1999.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS‐24) . Age: over 70 years Exclusion criteria: any significant medical problem, criteria for any other Axis I psychiatric or neurological disorder, any cognitive impairment, suicidal risk, drug abuse or dependence, any medical contraindication to study medications, history of failure to respond to either ECT or adequate trials with two or more antidepressants. | |
| Interventions | Fluoxetine: 33 participants Sertraline: 42 participants Fluoxetine dose range: 20‐40 mg/day Sertraline dose range: 50‐100 mg/day | |
| Outcomes | HDRS‐24, Hamilton Rating Scale for Anxiety (HAM‐A), Clinical Global Impression (CGI) Severity and Improvement, Profile Of Mood States (POMS), Quality of Life Enjoyment and satisfaction Questionnaire (Q‐LES‐Q). | |
| Notes | Response: decrease of at least 50% in the HDRS‐24 total Remission: total score of maximum 7 on the HDRS‐24 at the week 10 and 12 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Responders at endpoint reported without denominators. Number and reasons for withdrawal were reported |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Primary endpoint scores reported in figures and without standard deviations |
| Other bias | Unclear risk | Funding: unclear |
Gagiano 1993.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 18 on the Hamilton Rating Scale for Depression‐ 21 item (HDRS‐21). Age: 18‐65 years Exclusion criteria: pregnancy, lactation, hepatic, renal, neurological, gastrointestinal, or severe cardiovascular disease, schizophrenia, organic brain syndrome, unstable diabetes, recent treatment with MAOI, neuroleptics, lithium therapy, ECTin the previous 3 months, alcohol or drug abuse, severe risk of suicide. | |
| Interventions | Fluoxetine: 45 participants Paroxetine: 45 participants Fluoxetine dose range: 20‐60 mg/day Paroxetine dose range: 20‐40 mg/day | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " double dummy technique", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for withdrawal were not clear. Denominator of responders was different from number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Scores were reported without standard deviations. Only adverse events occurred over 10% were reported |
| Other bias | Unclear risk | Funding: unclear |
Gattaz 1995.
| Methods | Four‐week randomised, double‐blind, two‐centre study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐65 years Exclusion criteria: serious allergies, drug and alcohol abuse, resistance to a previous treatment with an antidepressant prescribed at an effective dosage during at least 3 weeks, and therapy with MAOI in the last 14 days, or with fluoxetine in the last 5 weeks. | |
| Interventions | Fluoxetine: 34 participants Moclobemide: 36 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 300‐600 mg/day Chloral hydrate and low dose of diazepam as hypnotic or/and anxiolytic were allowed | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the HDRS‐17 total Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double dummy, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominators. Response rate was based on patients completed the trials and not on randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons for dropouts reported. Endpoint scores at CGI not reported. Side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Geerts 1994.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depression without psychotic features, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐70 years Exclusion criteria: suicidal intent, any other psychiatric illness, severe organic disease, alcoholism and drug abuse, use of MAOI in the preceding 2 week, use of an antidepressant drug in the previous 4 days, or any investigational drug in the preceding 4 weeks, patients who ever received fluoxetine or moclobemide. | |
| Interventions | Fluoxetine: 25 participants Moclobemide: 24 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 300‐600 mg/day Only lithium and bromazepam were allowed | |
| Outcomes | Final score of less than 10 or a decrease of at least 50% from baseline on the HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote:"patients received capsules of identical appearance and taste", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | End‐point scores reported without denominators. Analysis of HDRS scores were based on completers, instead of on randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons of withdrawals reported. Adverse effects reported |
| Other bias | High risk | Last author's affiliation was Roche Industry and this company produces moclobemide |
Geretsegger 1994.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS). Age range: 61‐85 years Exclusion criteria: improvement of more than 20% on the HDRS during the placebo run‐in period (3‐7 days), severe renal, hepatic or gastrointestinal disease, cardiovascular disease, glaucoma, neurological disease, senile dementia, schizophrenia, organic brain syndrome, prostatic hypertrophy or diabetes, or were considered at serious risk of suicide, lithium therapy, electroconvulsive therapy during the previous 3 months, monoamine oxidase inhibitors or oral neuroleptics in the previous 2 weeks, depot neuroleptics in the previous 4 weeks and know alcohol abuse. | |
| Interventions | Fluoxetine: 52 participants Paroxetine: 54 participants Fluoxetine dose range: 20‐60 mg/day Paroxetine dose range: 20‐40 mg/day | |
| Outcomes | Endpoint score on HDRS‐17, on Montgomery–Åsberg Depression Rating Scale (MADRS) and on Clinical Global Impression (CGI) | |
| Notes | Funding: industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of drop‐out was reported, but reasons for withdrawn were reported only partially |
| Selective reporting (reporting bias) | Unclear risk | Mean end point score not reported. Side effects reported partially |
| Other bias | High risk | Funding: by industry |
Ghaeli 2004.
| Methods | Eight‐week randomised, double‐blind study | |
| Participants | Patients fulfilling DSM‐IV criteria for major depression disorder. Age range: 18‐65 years Exclusion criteria: diabetes mellitus, history of myocardial infarction and other heart disease, pregnancy, electroconvulsive therapy within 6 months before the study. | |
| Interventions | Fluoxetine: 19 participants
Imiprimamine: 24 participants
Fluoxetine dose range: 20‐40 mg/day
Imipramine dose range: 75‐200 mg/day Benzodiazepines allowed when needed for anxiety, agitation or sleep |
|
| Outcomes | Priamry outcome: fasting blood glucose (FBG) levels | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Initial number of randomised patients was different from the sum of the number of the patients in the different groups. Result on primary outcome (FBG) reported |
| Selective reporting (reporting bias) | High risk | Number and reasons for withdrawals not clearly reported. Adverse effects not reported |
| Other bias | Unclear risk | Funding: unclear |
Gillin 1997.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for non‐psychotic, moderate to severe major depressive disorder, with a score of at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 21‐55 years Exclusion criteria: patients engaged in shift work and with a primary sleep disorder independent of affective disturbance, current general medical condition, history of psychoactive substance use disorder within 12 months prior to study entry, current DSM‐III Axis I disorder (organic mental syndrome, bipolar disorder‐depressive, and schizophrenia, delusional disorder, psychotic disorder NOS), pregnancy, lactation, not use of contraception. | |
| Interventions | Fluoxetine: 20 participants Nefazodone: 24 participants Fluoxetine dose range: 20‐60 mg/day Nefazodone dose range: 200‐500 mg/day | |
| Outcomes | HDRS‐17, Inventory of Depressive Symptomatology (IDS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no information about randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐dummy dosing scheme", no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Endpoint scores reported without denominators. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Only the most frequently occurring adverse events (at least in 10% of patients) reported |
| Other bias | High risk | Quote: "this study was supported by Bristol‐Myers‐Squibb Pharmaceutical Research Institute", that produces nefazodone |
Ginestet 1989.
| Methods | Eight‐week randomised, double‐blind study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depression with melancholia, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐70 years Exclusion criteria: known hypersensitivity to clomipramine, narrow angle glaucoma, risk of chronic urinary retention, no improvement or lack of efficacy with previous treatment with clomipramine at least 200 mg/day during 6 weeks, organic brain disease, history of seizures, serious illness including cardiovascular, hepatic, renal, respiratory, hematologic disease, hyperthyroidism, history of severe allergy or multiple adverse drug reaction, recent history of drug or alcohol abuse, concurrent administration of other psychotropic drug except some benzodiazepines, use of MAOIs, pregnancy, lactation. | |
| Interventions | Fluoxetine: 28 participants Clomipramine: 26 participants Fluoxetine dose range: 20‐80 mg/day Clomipramine dose range: 50‐200 mg/day Only oxazepam (50‐300 mg/day) as hypnotic or anxiolytic was allowed | |
| Outcomes | HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Covi Anxiety Scale (CAS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | End‐point scores and standard deviations reported. Dropouts not included in the analysis |
| Selective reporting (reporting bias) | High risk | Number but not reasons for dropouts reported. Side effects occurred on at least 3 occasion reported |
| Other bias | Unclear risk | Funding: unclear |
Goldstein 2002.
| Methods | Eight‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for non‐psychotic major depressive disorder, with a score of at least 15 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and at least 4 on the Clinical Global Impression‐Severity of Illness (CGI). Age range: 18‐65 years Exclusion criteria: any primary DSM‐IV Axis I diagnosis other than major depressive disorder or any anxiety disorder as a primary diagnosis within the past year with the exception of specific phobias, history of substance abuse or dependence within the past year or a positive urine drug screen at study entry, failure of 2 or more adequate courses of antidepressant therapy during the present episode. | |
| Interventions | Fluoxetine: 33 participants Duloxetine: 70 participants Placebo: 70 participants Fluoxetine dose: 20 mg/day Duloxetine dose range: 40‐120 mg/day | |
| Outcomes | Primary outcome: HDRS‐17 Secondary outcomes: Montgomery and Asberg Scale for Depression (MADRS), CGI, Patient Global Impresion (PGI), Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: " this study used double‐blind placebo lead in such that investigators and patients did not know when randomisation occurred and when active study drug was first administered", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " this study used double‐blind placebo lead in such that investigators and patients did not know when randomisation occurred and when active study drug was first administered", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: " this study used double‐blind placebo lead in such that investigators and patients did not know when randomisation occurred and when active study drug was first administered", further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominators. Number and reasons for dropout not clearly reported |
| Selective reporting (reporting bias) | High risk | Mean scores reported without standard deviations. Only common treatment emergent adverse events reported |
| Other bias | High risk | First author's affiliation was Lilly Corporate Center and this company produces fluoxetine |
GSK 29060/356.
| Methods | Eight‐week, multicentre, randomised, double‐blind study | |
| Participants | Patients suffering from a major depressive episode according to DSM‐III‐R, with a baseline score on Hamilton Rating Scale for Depression‐17 Item (HDRS‐17) of at least 18, and an HDRS item 10 score of 1 or more. Age: 18 years or more Exclusion criteria: severe co‐existing disease not stabilised with medication, neurological disorders, DSM‐III diagnosis of schizophrenia, bipolar disorder or psychotic depression, or who met criteria for substance dependence and abuse within the past 6 months, ECT or fluoxetine within 3 months preceding baseline, any investigational drug within 30 days from baseline, MAOIs within 2 weeks preceding baseline, lithium treatment in the past 8 weeks, currently receiving Type 1C antiarrhythmics or oral anticoagulants, patients posing a suicidal risk, pregnant or lactating, hypersensitive to fluoxetine or patients with clinically significant abnormal laboratory values at retest oral screening. | |
| Interventions | Fluoxetine: 70 participants Paroxetine: 68 participants Fluoxetine dose: 20 mg/day Paroxetine dose: 20 mg/day | |
| Outcomes | Primary outcomes: HDRS and Hamilton Rating Scale for Anxiety (HAM‐A) Secondary outcomes: Clinical Global Impression Scale (CGI), including severity and improvement |
|
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information about randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "double‐blind". No further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind". No further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All ITT analyses used last observation carried forward (LOCF) data set. Number and reasons for dropout reported. Mean endpoint scores (HDRS) reported with standard deviations |
| Selective reporting (reporting bias) | Unclear risk | Most frequent side effects reported. Number of responders not reported |
| Other bias | Unclear risk | Funding: unclear information |
Guelfi 1998.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depression for less than 3 months, with a score of at least 22 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐70 years Exclusion criteria: serious or uncontrolled medical illness, no remission between episodes, depression with psychotic features, dysthymia, personality disorder, lack of response to antidepressants, ECT or neuroleptics, major risk of suicide, schizophrenia and dependence of psychoactive substances (DSM‐III‐R) during the previous six months, use of MAOI in the previous 2 weeks, fluoxetine in the previous 4 weeks, long‐acting neuroleptics or ECT in the previous 3 months, pregnancy, lactation, not use of contraception. | |
| Interventions | Fluoxetine: 100 participants Milnacipram 100 mg group: 100 participants Milnacipram 200 mg group: 100 participants Fluoxetine dose: 20 mg/day Only oxazepam (max 50 mg/day) or chloral hydrate (max 2 g/day) as hypnotic or anxiolytic were allowed | |
| Outcomes | Primary outcome: change in the total score on the HDRS‐17 Secondary outcomes: change in the total score Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the MADRS and HDRS‐17 total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominators. Number and reasons for withdrawal were reported |
| Selective reporting (reporting bias) | Unclear risk | Scores reported without standard deviations. Endpoint scores at MADRS not reported |
| Other bias | High risk | Quote: "this study was sponsored by Pierre Fabbre Medicament, Boulogne, France" and this company produces milnacipram |
Guelfi 1999.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients (general practice) fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 25 on the Montgomery and Asberg Scale for Depression (MADRS) and a Mini Mental State Examination (MMSE) of at least 24. Age: over 65 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 122 participants Tianeptine: 115 participants Fluoxetine dose: 20 mg/day Tianeptine dose range: 20‐37.5 mg/day | |
| Outcomes | Primary outcome: change in the total score on the MADRS Secondary outcomes: total number of responders at endpoint, total number of remissions at endpoint, mean variation on the Geriatric Depression Scale (GDS) |
|
| Notes | Response: decrease of at least 50% in the MADRS total score Remission: total score less than 10 on the MADRS Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "deux groupes de traitment ont été constitués par tirage au sorte", randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Mean endpoint scores reported with denominators. Dropouts reported with reasons |
| Selective reporting (reporting bias) | Low risk | Side effects reported |
| Other bias | High risk | Author's affiliation was Eli Lilly and this company produces fluoxetine |
Hale 2010.
| Methods | Eight‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients, aged 18 to 65 years and presented with MDD of severe intensity according to DSM‐IV‐TR criteria, with a score of at least 25 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and a Clinical Global Impression (CGI) score of at least 4. Exclusion criteria: seasonal pattern, psychotic features, post‐partum onset, suicidal intent and/or known suicidal tendencies for the current episode, bipolar disorder, anxiety symptoms (such as panic attacks, obsessive compulsive disorder, PTSD) drug abuse or dependency, resistant to fluoxetine for current episode, treatment with electroconvulsive therapy or formal psychotherapy within 3 months, or light therapy started within the earlier two weeks, not responders to the administration of an appropriate dose of two different early antidepressant treatments for at least four weeks each, patients with neurologic disorders or severe uncontrolled organic disorders. |
|
| Interventions | Fluoxetine: 263 participants Agomelatine: 252 participants Fluoxetine dose range: 20‐40 mg/day Agomelatine dose range: 25‐50 mg/day |
|
| Outcomes | Primary outcomes: responders to treatment were defined by a decrease of at least 50% in total score from baseline Secondary outcomes: patients with a score of 1 or 2 at CGI scale were considered responders |
|
| Notes | Response: decrease of at least 50% in the HDRS total score Remission: total score less than 6 on the HDRS Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "both investigator and patients were blind (...) during the entire duration of the study, all patients two capsules orally in the morning and one in the evening, irrespective the treatment and daily dosage allocated...the appearance and taste of the study treatment were the same from the inclusion to the end of the study for all patients. The packaging and labelling were identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation clearly reported. Mean endpoint scores reported with standard deviation, but without denominator |
| Selective reporting (reporting bias) | Low risk | Adverse events reported for more than 2% of the subjects. Secondary outcome scores (CGI) reported |
| Other bias | High risk | Quote: "this study was supported by Servier. Authors have received honoraria, research grants or both, from Servier". This company produces agomelatine |
Harrer 1999.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients (general practice) fulfilling ICD‐10 criteria for mild depressive episode, with a Mini Mental State Examination (MMSE) of at least 25. Age range: 60‐80 years old Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 79 participants Hypericum: 70 participants Fluoxetine dose: 20 mg/day Hypericum dose: 800 mg/day | |
| Outcomes | Primary outcomes: change in the total score on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) | |
| Notes | Response: decrease of at least 50% in the HDRS‐17 total score or a total score of less than 10 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...randomised in blocks of four for a total of 32 patients at each centre, it was ensured that equal numbers of patients from each sample and with each degree of severity were randomly allocated to each trial centre" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "patients were asked to take coated tablets twice daily...", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominator. Number and reasons for withdrawal reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviation. Adverse drug reactions reported |
| Other bias | High risk | The last author's affiliation was Dr Loges co.gmbh and this company produces St John's wort extract LoHyp‐57 |
Hashemi 2012.
| Methods | Twenty four‐week randomised, double‐blind study | |
| Participants | Patients with a diagnosis of Major Depressive Disorder (MDD), who responded to the drugs in 8 weeks. Age range: 15‐60 years Exclusion criteria: patients received any antidepressant drug previously, had criteria for grief, adjustment disorder, MDD with psychotic features, concomitant axis II or III disorder, bipolar disorder or schizophrenia, pregnancy or breastfeeding. | |
| Interventions | Fluoxetine: 49 participants Nortriptyline: 48 participants Fluoxetine dose range: 20‐60 mg/day Nortriptyline dose range: 50‐150 mg/day | |
| Outcomes | Primary outcome: change in the total score of Beck Depression Inventory (BDI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "both patients and the evaluating team were unaware of treatment allocation", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "both patients and the evaluating team were unaware of treatment allocation", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "both patients and the evaluating team were unaware of treatment allocation", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported with standard deviations, but without denominators |
| Selective reporting (reporting bias) | Unclear risk | Adverse drug reactions reported. Number of withdrawal reported only in the total sample and without specify reasons for discontinuation |
| Other bias | Unclear risk | Funding: unclear |
Hong 2003.
| Methods | Six‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive episode (lasting between 1 week and 1 year), with a score of at least 15 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐75 years Exclusion criteria: pregnancy, lactation, actual suicide risk, history of current diagnosis of bipolar disorder, schizophrenia, psychotic symptoms, organic mental disorder, current diagnosis on DSM‐IV of anxiety or eating disorder, epilepsy, alcohol or substance abuse in the previous 6 months, serious medical diseases. | |
| Interventions | Fluoxetine: 66 participants Mirtazapine: 66 participants Fluoxetine dose range: 20‐40 mg/day Mirtazapine dose range: 30‐45 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominator. Mean scores reported without standard deviations |
| Selective reporting (reporting bias) | Unclear risk | Only main reasons for premature discontinuation reported. Adverse events reported for more than 5% of the subjects |
| Other bias | Unclear risk | Funding: unclear |
Hosak 2000.
| Methods | Four‐week, randomised and open study | |
| Participants | Hospitalized patients. Diagnoses for inclusion (according to the ICD‐10) were: bipolar affective disorder, most recent episode depressed (8 participants); major depressive episode, single (44 participants), major depressive episode, recurrent (38 participants). Average age: 44.5 years (SD: 14.3) |
|
| Interventions | Citalopram: 29 participants Amitriptyline: 31 participants Fluoxetine: 30 participants Citalopram dose range: 20‐60 mg/day Amitriptyline dose range: 150‐300 mg/day Fluoxetine: 20‐60 mg/day | |
| Outcomes | Mean change on Hamilton Depression Rating Scale‐21 item (HDRS‐21) | |
| Notes | Study report published only in Czech Funding: unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the subjects were randomised to the study antidepressant using computer randomisation program (Excel) at the beginning of the initial hospitalisation at the Dpt. of Psychiatry in Hradec Kralovc" |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information provided |
| Selective reporting (reporting bias) | High risk | No information provided |
| Other bias | Unclear risk | Funding: unclear |
Jakovijevic 1996.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for major depressive episode without psychotic features, with a score between 18 and 26 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 40‐65 years Exclusion criteria: past history of hypersensitivity, to fluoxetine or maprotiline, history or presence of gastrointestinal, liver or kidney disease, pregnancy, lactation, history of seizures or serious brain damage, current evidence of clinically important cardiovascular or hematopoietic disease, urinary retention or glaucoma with closed angle, abnormal findings in physical examination, laboratory tests and ECG at admission, evidence of substance use disorder within the past 6 months or currently, use of MAOIs within 2 weeks before the study. | |
| Interventions | Fluoxetine: 50 participants Maprotiline: 48 participants Fluoxetine dose range: 20‐40 mg/day Maprotiline dose range: 75‐150 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) scores | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominator of the responders was different from the number of randomised patients. Number and reasons for withdrawal were not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Endpoint score at CGI and vital signs not reported. Side effects not clearly described |
| Other bias | High risk | Funding: unclear, probably funded by a pharmaceutical company that produces maprotiline |
Joyce 2002.
| Methods | Six‐week randomised, open study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder. Mean age: 31.6 years Exclusion criteria: current moderate to severe alcohol or drug dependence, history of mania (hypomanic patients were included), schizophrenia or severe antisocial personality disorder, major physical illness, use of drugs within 2 weeks of study entry (with the exception of oral contraceptive or occasional hypnotic drugs for sleep). | |
| Interventions | Fluoxetine: 100 participants Nortriptyline: 95 participants Fluoxetine dose range: 10‐80 mg/day Nortriptyline dose range: 50‐175 mg/day | |
| Outcomes | Primary outcomes: improvement greater than 60% from baseline on the Montgomery and Asberg Scale for Depression (MADRS) (response) and 2 months sustained improvement (recovery) Secondary outcomes: Hamilton Rating Scale for Depression (HDRS), Symptom Checklist‐90 (SCL‐90) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not double blind |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not double blind |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not double blind |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominators. Number of drop‐out reported, but reasons for withdrawal not clearly described |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores at SCL‐90 and Simpson‐Angus Scale (SAS) not reported. Side effects not reported |
| Other bias | High risk | Quote: "this project also received a grant from Lottery Health and an unrestricted grant from Eli Lilly (New Zealand)". Eli Lilly produces fluoxetine |
Judd 1993.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder (1 month minimum duration of episode), with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 21‐63 years Exclusion criteria: organic mental disorder, substance use disorder, schizophrenia or schizoaffective disorder, paranoid or other psychotic disorder, bipolar disorder, significant physical illness, history of seizures, drug allergy, glaucoma or urinary retention, use of other psychotropic medication (including lithium), pregnancy, lactation. | |
| Interventions | Fluoxetine: 30 participants Amitriptyline: 28 participants Fluoxetine dose: 20 mg/day Amitriptyline dose range: 50‐200 mg/day Only temazepam or chloral hydrate were allowed | |
| Outcomes | HDRS‐17 score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "medication was given in matching capsules". Unclear if raters were independent and unclear if blinding was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominators. Number and reasons for withdrawal reported |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Endpoint scores reported with maximum, minimum, mean and standard deviation |
| Other bias | High risk | Quote: "the authors are grateful to Eli Lilly, Australia for financial support for this study". Eli Lilly company produces fluoxetine |
Kasper 2005.
| Methods | Eight‐week randomised, double‐blind, multicentre study | |
| Participants | Patients from general practice and specialist settings fulfilling DSM‐IV‐R criteria for major depressive disorder, with a score of at least 22, and maximum 40 on the Montgomery and Asberg Scale for Depression (MADRS), and at least 22 on the Mini Mental State Exams (MMSE) at the screening visit. Age: over 65 years Exclusion criteria: subjects met the DSM‐IV criteria for mania or any bipolar disorder, schizophrenia, or any psychotic disorder, obsessive‐compulsive disorder, eating disorders, or mental retardation or any pervasive developmental or cognitive disorder; had a MADRS score over 5 on item 10 (suicidal thoughts); receiving treatment with antipsychotic, antidepressant, hypnotics, anxiolytics (except oxazepam: 30 mg/day maximum; temazepam: 10mg/day maximum; zolpidem: 5mg/day maximum), antiepileptics, barbiturates, chloral hydrate, antiparkinsonian drugs, diuretics, lithium, sodium valproate or carbamazepine; were receiving electroconvulsive treatment, receiving treatment with behavior therapy or psychotherapy, had received treatment with any investigational drug within 30 days before entry; had a history of schizophrenia, psychotic disorder or drug abuse; had a history of severe drug allergy or hypersensitivity (including to citalopram); or had a lack of response to more than one antidepressant treatment (including citalopram) during the present depressive episode. | |
| Interventions | Fluoxetine: 164 participants Escitalopram: 174 participants Fluoxetine dose: 20 mg/day Escitalopram dose: 10 mg/day |
|
| Outcomes | Changes from baseline in MADRS total score at final assessment Response: at least 50% reduction on the MADRS total score from baseline Secondary outcome: changes in Clinical Global Impression (CGI) score |
|
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominators and only in percentage. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported only in figure and without standard deviations. Side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Keegan 1991.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R or DIS criteria for unipolar major depression, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐70 years Exclusion criteria: any serious psychiatric disorder other than depression, such as schizophrenia, bipolar disorder, panic or obsessive disorder, alcohol or drug abuse within the past six months, serious medical disorders, use of psychoactive drugs that could affect mood. | |
| Interventions | Fluoxetine: 20 participants Amitriptyline : 22 participants Fluoxetine dose range: 20‐80 mg/day Amitriptyline dose range: 100‐250 mg/day Only small amounts of benzodiazepines or chloral hydrate for sleep and anxiety were allowed | |
| Outcomes | Diagnostic Interview Schedule, HDRS‐21, Beck Depression Inventory (BDI), Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "study drugs were packaged as identical capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Rating scale scores reported without denominators. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported with standard deviations. Only 12 most common side effects reported |
| Other bias | High risk | Quote: "the study was supported by Eli Lilly, Canada Inc.". Eli Lilly Company produces fluoxetine |
Keller 2007.
| Methods | Ten‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for Major Depressive Episode (MDD), had experienced depressive symptoms for at least 1 month prior to the start of the study, and had a recurrent depression (at least 3 episode of MMD, with at least 2 episode in the past 5 years and a interval of at least 2 months between the end of the previous episode and the beginning of the current episode) and a total score on Hamilton Rating Scale for Depression‐17 item (HDRS‐17) of at least 20 at screening and 18 at the randomisation. Age: 18 years or older Exclusion criteria: patients who failed a trial of fluoxetine, venlafaxine, or venlafaxine ER during the current episode or were treatment resistant, hypersensitivity to venlafaxine or fluoxetine, serious medical disease, cancer, seizure disorder, bipolar disorder, eating disorder, other axis I disorder, substance dependence/abuse within 6 months, axis II disorder, any psychotic disorder, post‐partum depression; pregnancy, breastfeeding or not using a medically acceptable method of birth control; use of the following drugs: any investigational drug, antipsychotic drug, fluoxetine, monoamine oxidase inhibitor within 30 days or other antidepressant within 14 days; ECT within 3 months, any anxiolytic, sedative‐hypnotic drug, sumatriptan or any other psychotropic drug or substance within 7 days, or any non‐psychopharmacologic drug with psychotropic effects within 7 days of randomisation, unless a stable dose of the drug had been maintained for at least 1 month. | |
| Interventions | Fluoxetine: 275 participants Venlafaxine : 821 participants Fluoxetine dose range: 20‐60 mg/day Venlafaxine dose range: 75‐300 mg/day |
|
| Outcomes | Primary outcome: HDRS‐17 Secondary outcomes: Hamilton Rating Scale for Anxiety (HAM‐A), Inventory of Depressive Symptomatology (IDS), Clinical Global Impression (CGI) Response: at least 50% reduction from baseline HDRS‐17 total score |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout not clearly reported. Denominatosr of the responders was different from the number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. Mean endpoint score (HDRS) reported with standard deviations |
| Other bias | High risk | Quote: "this clinical trial and analysis were sponsored by Wyeth Research, Collegeville", and this company produces venlafaxine |
Kerkhofs 1990.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Inpatients fulfilling Research Diagnostic Criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐64 years Exclusion criteria: concurrent medical disorder | |
| Interventions | Fluoxetine: 16 participants Amitriptyline: 18 participants Fluoxetine dose range: 40‐60 mg/day Amitriptyline dose range: 100‐150 mg/day Only oxazepam (max 100 mg/day) was allowed | |
| Outcomes | HDRS, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) Severity and Improvement, Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly allocated, according to predetermined schedule", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominator reported for responders was different from the number of randomised patients. Reasons for withdrawal were not reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse events were reported. Mean scores were reported with standard deviations |
| Other bias | High risk | Last author's affiliation was Eli Lilly Benelux and this company produces fluoxetine |
Kuha 1991.
| Methods | Five‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling Research Diagnostic Criteria for unipolar major depressive episode, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and 8 on the Raskin Depression Scale (RDS). Age range: 18‐65 years Exclusion criteria: serious non‐stabilised somatic illness, drug or alcohol abuse, evidence of dementia, depressive schizophrenic, serious suicide risk, concurrent administration of other psychotropic drug (with the exclusion of benzodiazepines or chloral hydrate for insomnia or anxiety). | |
| Interventions | Fluoxetine: 24 participants Maprotiline: 22 participants Fluoxetine dose range: 20‐60 mg/day Maprotiline dose range: 50‐150 mg/day | |
| Outcomes | HDRS, RDS, Covi Anxiety Scale (CAS), Patient Global Impression (PGI), Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for withdrawal reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviations. Only most predominant (4%) adverse events reported |
| Other bias | Unclear risk | Funding: unclear |
Kwon 1996.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for unipolar major depression, drug free for a minimum of 2 weeks. Mean age: 44,31 years (SD=9,31) Exclusion criteria: previously received ECT; neurological disorders and major illness. |
|
| Interventions | Twenty participants were randomly assigned to a 6‐weeks of treatment with fluoxetine or amitriptyline No other information about the interventions |
|
| Outcomes | Hamilton Rating Scale for Depression (HDRS) was assessed at baseline and at the end of the 6th week Response: a reduction of at least 50% of the HDRS score |
|
| Notes | Funding: by academy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "randomly assigned by an another psychiatrist who was blind to the rating of HDRS", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Randomly assigned by an another psychiatrist who was blind to the rating of HDRS", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only the total number of randomised patients was reported, the number of participants in each group was not reported. Number and reason for dropout not reported |
| Selective reporting (reporting bias) | High risk | Side effects not reported. Baseline and endpoint score reported for the whole group |
| Other bias | Low risk | Quote: "this work was supported by grant N°. 02‐94‐158 from the Seoul National University Hospital research found" |
La Pia 1992.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and 20 on the Mini Mental State Examination (MMSE). Age range: 60‐80 years Exclusion criteria: history of serious allergies or alcohol and drug abuse in the last year, diagnosis of schizophrenia, dementia, glaucoma, prostatic hypertrophia, recent stroke, serious internal disease, and/or surgical conditions that could interfere with study drugs. | |
| Interventions | Fluoxetine: 20 participants Mianserin: 20 participants Fluoxetine dose: 20 mg/day Mianserin dose: 40 mg/day | |
| Outcomes | HDRS‐21, Geriatric Depression Scale (GDS), Geriatric Rating Scale (GRS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "the statistical evaluation was conducted on the patients who completed the trial". The analysis was not conducted on ITT basis |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons of adverse events reported. Endpoint scores reported without standard deviations |
| Other bias | Unclear risk | Funding: unclear |
Laakman 1988.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | Outpatients with depressive syndrome with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS) and 8 on the Raskin Depression Scale (RDS). Age range: 19‐74 years Exclusion criteria: severe organic illness, evidence of psychosis, psychopathic disorder, addictive illness, suicide tendencies, a period of less than 4 weeks since the last treatment with amitriptyline or neuroleptics. | |
| Interventions | Fluoxetine: 63 participants Amitriptyline : 65 participants Fluoxetine dose range: 20‐60 mg/day Amitriptyline dose range: 50‐150 mg/day Chloral derivative was allowed (eventually changed in flurazepam or nitrazepam only if its effects was inadequate) | |
| Outcomes | HDRS, Clinical Global Impression (CGI), RDS , Covi Anxiety Scale (CAS), Patient Global Impression (PGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "four identical capsules were given" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Rating scale scores reported without denominators. Reasons for withdrawals not clearly described |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported without standard deviations. Side effects partially reported |
| Other bias | Unclear risk | Funding: unclear |
Lapierre 1997.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 18 on the first 17 items of the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐64 years Exclusion criteria: marked suicide risk, major depressive episode associated with moo‐incongruent psychotic features, bipolar disorder, acute confusional state, epileptic or seizure disorder, mental retardation, history of unstable diabetes or clinically significant physical disease, known sensitivity to moclobemide, MAOI, fluoxetine or other SSRIs, history of alcohol or substance abuse within the last 6 months, treatment with MAOI within the past 2 weeks, fluoxetine within the past 5 weeks, try‐ or heterocyclics antidepressants or lithium or daytime benzodiazepines within the past week, ECT within the past 3 months, concomitant use of medication known to affect the action of moclobemide or fluoxetine, use of any investigational drug within the past 3 months, pregnancy, lactation, absence of contraception. | |
| Interventions | Fluoxetine: 62 participants Moclobemide: 66 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 200‐600 mg/day | |
| Outcomes | Primary outcome: Hamilton Rating Scale for Depression (HDRS‐21) Secondary outcomes: Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the MADRS total score and a total score of less than 10 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominator reported for responders was different from the number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons for premature termination reported. Only most frequently reported adverse events were described |
| Other bias | High risk | Quote: "this study was supported by Hoffmann‐La Roche", and this company produces moclobemide |
Lee 2005.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS) total score and over 21 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐64 years Exclusion criteria: previous treatment with either study drugs, hypersensitivity reaction to study drugs, non‐responder to more than two antidepressant drugs, history of severe renal or liver disease, diagnosis of non‐depressive axis I disorders according to DSM‐IV criteria; participation in another study in the past 3 months, pregnancy, high suicidal risk, history of psychotic episodes or patients requiring electroconvulsive therapy. | |
| Interventions | Fluoxetine: 62 participants Moclobemide: 66 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 200‐600 mg/day | |
| Outcomes | Primary outcomes: HDRS score and MADRS score Response: a decrease of 50% or more of the initial score on the HDRS or MADRS, or a Clinical Global Impression (CGI) rating of "remarkably improved" or "moderately improved" |
|
| Notes | Remission: a score of 7 or less on HDRS or a score of 8 or less on MADRS Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported. ITT number was different from the number of randomised |
| Selective reporting (reporting bias) | Unclear risk | Mean baseline and end‐point scores reported with standard deviations. Adverse events reported |
| Other bias | High risk | Quote: "funding for this study was provided by Bukwang Pharm, Seoul, Korea", and this company produces milnacipran |
Levine 1989.
| Methods | Six‐week randomised, double‐blind, two‐centre study | |
| Participants | In‐ and outpatients fulfilling Research Diagnostic Criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS). Mean age: 46.1 (fluoxetine) and 45.4 (imipramine) years Exclusion criteria: significant physical illness, history of drug abuse, schizophrenia, duration of illness more than 1 year. | |
| Interventions | Fluoxetine: 30 participants Imipramine: 30 participants Fluoxetine dose range: 40‐60 mg/day Imipramine dose range: 75‐150 mg/day Only temazepam was allowed for night sedation | |
| Outcomes | HDRS, Montgomery and Asberg Scale for Depression (MADRS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly allocated", no other information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominator reported for responders was different from the number of randomised number. Number and reasons for withdrawals not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviations. Side effects not clearly reported |
| Other bias | Unclear risk | Funding: unclear |
Levkovitz 2002.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for non‐psychotic depressive episode (no longer than 5 months), with a score of at least 21 on the Hamilton Rating Scale for Depression (HDRS‐17) and no more than 2 previous antidepressive drugs given for the current episode and no medication for 3‐5 days before first assessment. Age range: 25‐50 years Exclusion criteria: psychotic state, significant past head injury, severe neurological disease of physical illness, history of drug addiction or alcoholism, ECT in the last year, suicide risk, or suicide attempt in the last year. | |
| Interventions | Fluoxetine: 8 participants Desipramine: 9 participants Fluoxetine dose: 20 mg/day Desipramine dose range: 125‐200 mg/day | |
| Outcomes | HDRS‐17 and Clinical Global Impression | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no information about randomisation procedure |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Quote: "Both the psychiatrist and the patient knew the name of the medication, but this information was withheld from examiner", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Both the psychiatrist and the patient knew the name of the medication, but this information was withheld from examiner", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Both the psychiatrist and the patient knew the name of the medication, but this information was withheld from examination", not clear if blinding of the examinator was successful |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for withdrawals was not clearly reported. Rating scale scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported with standard errors. Side effects not reported |
| Other bias | Unclear risk | Funding: unclear |
Loeb 1989.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | Patients fulfilling DSM‐III criteria for major depressive episode, with a score of at least 18 on the first 17 items of the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: pregnancy, serious vascular disease, hyperthyroidism, glaucoma, urinary retention, hepatic, respiratory or renal marked failure, hematological disease, organic brain disease, seizures, alcohol and/or drug abuse. | |
| Interventions | Fluoxetine: 15 participants Imipramine: 15 participants Fluoxetine dose: 20 mg/day Imipramine dose range: 100‐150 mg/day | |
| Outcomes | HDRS, Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for discontinuation not reported. Scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | End point scores reported without standard deviations. Adverse events not reported |
| Other bias | Unclear risk | Funding: unclear |
Lonnqvist 1994.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for predominantly major depressive disorder, with a score of at least 16 on Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 18 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 107 participants Moclobemide: 102 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 300‐450 mg/day Benzodiazepines were permitted only if strongly indicated | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI), Montgomery and Asberg Scale for Depression (MADRS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the study drugs were supplied in identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported. Scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Mean cores reported with standard deviations |
| Other bias | High risk | Last author's affiliation was Roche OY, Espoo, Finland. This company produces moclobemide |
Loo 1999.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling ICD‐10 criteria for depressive episode, recurrent depressive disorder, or bipolar affective disorder (depressive), with a score of at least 25 on the Montgomery and Asberg Scale for Depression (MADRS), requiring an antidepressant treatment. Age range: 18‐65 years Exclusion criteria: severe risk of suicide, acute or chronic psychosis, failure to respond to 2 antidepressants for the current depressive episode, previous history of drug abuse or dependence, severe somatic diseases in evolution, current treatment with barbiturate, buspirone, anti‐epileptic drugs, use of diazepam, lorazepam and alprazolam. | |
| Interventions | Fluoxetine: 196 participants Tianeptine: 191 participants Fluoxetine dose: 20 mg/day Tianeptine dose: 37.5 mg/day | |
| Outcomes | Primary outcome: MADRS global score Secondary outcomes: decrease of at least 50% in MADRS global score (responder patients) and Clinical Global Impression (CGI) scores | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported, but withdrawal was not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Only serious adverse events reported. End point mean scores reported with standard deviations |
| Other bias | Unclear risk | Funding: unclear |
Manna 1989.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 18 on the first 17 items of Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Mean age: 48 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 15 participants Clomipramine: 15 participants Fluoxetine dose: 20 mg/day Clomipramine dose: 75 mg/day | |
| Outcomes | HDRS, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI), Zung Self‐Rating for Depression | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout not reported. Scores reported without denominators |
| Selective reporting (reporting bias) | High risk | Endpoint scores were reported only in figures and they were not clear. Side effects reported only in percentage without denominators |
| Other bias | Unclear risk | Funding: unclear |
Mao 2008.
| Methods | Eight‐week randomised, parallel group, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 18 on the first 17 items of Hamilton Rating Scale for Depression (HDRS) and a score of at least 4 on Clinical Global Impression‐Severity (CGI). Age range: 18‐65 years Exclusion criteria: any current primary DSM‐IV axis I diagnosis or any anxiety disorder as a primary diagnosis within the year preceding enrolment, or schizoaffective disorder, a history of substance abuse or dependence within the past year; serious suicidal risk or serious medical illness; currently take St. John's wort or other Chinese herbal medicine for depression were also excluded. |
|
| Interventions | Fluoxetine: 117 participants Escitalopram: 123 participants Fluoxetine dose: 10 mg/day Escitalopram dose: 20 mg/day | |
| Outcomes | Primary outcome: change in HDRS total score Secondary outcome: change in Montgomery and Asberg Scale for Depression (MADRS) total score Response: at least 50% decrease from HDRS baseline score |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " we administered treatments in a double blind fashion using a double‐dummy design", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation clearly reported. Denominator reported for responders was different from the number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported. Secondary endpoint scores (MADRS, CGI) reported |
| Other bias | High risk | Quote: "contract grant sponsor: Xian‐Janssen Pharmaceutical Company (honoraria to authors for conducting this trial)". This company produces escitalopram |
Marchesi 1998.
| Methods | Ten‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 16 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and a summary score of the HDRS items (agitation, psychic anxiety and somatic anxiety) higher than 5 or the score of at least one of the above items higher than 3. Mean age: 44.1 (females) and 42.1 (males) years old Exclusion criteria: serious suicide risk, schizophrenia, epilepsy, organic brain disease, chronic disease such as cardiovascular, renal, hepatic, respiratory, endocrine‐metabolic, urinary disease, glaucoma, use of antidepressants the week before enrolment, use of fluoxetine during the previous month, use of lithium during the previous 6 months. | |
| Interventions | Fluoxetine: 67 participants Amitriptyline : 75 participants Fluoxetine dose: 20 mg/day Amitriptyline dose range: 75‐225 mg/day Bromazepam (max 6 mg) was allowed | |
| Outcomes | Change in HDRS‐17 total score, in agitation/anxiety score and in the response rate | |
| Notes | Response: decrease of at least 50% in the HDRS‐17 total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported. Scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported. Secondary end point scores not reported |
| Other bias | High risk | Two author's affiliation was Eli LIlly Italia, Sesto Fiorentino, Italy. This company produces fluoxetine |
Martenyi 2001.
| Methods | Six‐week randomised, double‐blind, four‐centre study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for non‐psychotic major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐65 years Exclusion criteria: history of any psychoactive disorder, bipolar mood disorder, substance abuse disorder, somatic disorder, glaucoma, urinary retention and/or prostatic disease and known allergy to maprotiline, pregnancy, absence of contraception, use of MAOI within 2 weeks and depot neuroleptics within 4 weeks of study entry, concomitant psychotropic active medication, with the exception of midazolam, max 15 mg, or medazepam, max 5 mg, for insomnia. | |
| Interventions | Fluoxetine: 59 participants Maprotiline : 46 participants Fluoxetine dose: 20 mg/day Maprotiline dose range: 100‐200 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression‐Severity (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "to maintain the blind, all patients took three capsules of study medication every day", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for discontinuation were not reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported with standard deviation. Side effects not reported |
| Other bias | High risk | Quote: "this work was sponsored by Eli Lilly and Company". This company produces fluoxetine |
Martinez 2012.
| Methods | Twelve‐week randomised, open study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for Major Depressive Disorder (MDD), with a score of at least 16 on the Patient Health Questionnaire (PHQ‐9) at baseline and a score of at least 20 on Quick Inventory of Depression Syntomatology Self‐Report scale (QIDS‐SR) at visit 1 and 2. Exclusion criteria: pregnancy, lactation, absence of contraception. Patients taking an investigational drug (bupriopion, sertraline, venlafaxine, paroxetine, fluoxetine or escitalopram) were allowed to participated only if they had not adequately responded to treatment. Patients with a history of bipolar disorder, primary psychotic disorder, cognitive disorder or obsessive‐compulsive disorder, or current (within last 6 months) primary axis I diagnosis of panic disorder, post‐traumatic stress disorder, generalized anxiety disorder, social anxiety disorder, dysthymia, alcohol or eating disorders were excluded. To be at serious risk of suicide, the presence of a serious, unstable medical illness or clinically significant laboratories abnormality, dementia, mental retardation diagnosis, history of substance abuse or dependence within previous 6 months or positive urine drug screen for any substance of abuse, treatment with electroconvulsive therapy, initiation of psychotherapy within 6 weeks before study entry or during study participation were exclusion criteria. | |
| Interventions | Fluoxetine: 57 participants Duloxetine: 372 participants Fluoxetine dose range: 20‐80 mg/day Duloxetine dose range: 30‐120 mg/day | |
| Outcomes | Primary outcome: change in QIDS‐SR total score Secondary measures: Hamilton Rating Scale for Depression (HDRS) |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Quote: "non‐blinded study" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "non‐blinded study" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "non‐blinded study" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation clearly reported. Endpoint mean score reported as a class |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only when occurred in more than 5% of the sample. Mean endpoint scores reported with standard deviation and denominator |
| Other bias | High risk | Quote: "funded by Lilly USA, Indianapolis", and this company produces fluoxetine |
Masco 1985.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive illness, with a score of at least 20 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17), a score of at least 8 on the Raskin Depression Scale (RDS) and greater than the Covi Anxiety Scale (CAS) score. Mean age: 51 years in both groups Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 20 participants Amitriptyline: 21 participants Fluoxetine dose range: 40‐80 mg/day Amitriptyline dose range: 150‐300 mg/day | |
| Outcomes | HDRS‐17, RDS , CAS, Clinical Global Impression (CGI) Improvement and Severity, Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "capsules that looked identical were supplied to all patients in two bottles marked 'morning dose' and 'bedtime dose'", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for early termination reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. End point scores reported without standard deviation |
| Other bias | High risk | Quote: "supported by a grant from Eli Lilly & Company", this company produces fluoxetine |
Massana 1999.
| Methods | Six‐week randomised, double‐blind, multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for depressive episode (lasting between 1 to 8 months), without psychotic features, with a score of at least 22 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: pregnancy, absence of contraception, dysthymia/cyclothymia, substance abuse disorder, high risk of suicide, resistance to antidepressant treatment, history of major depressive disorder associated with endocrine disorder and/or drug hypersensitivity, chronic respiratory insufficiency, a history of seizures or brain injury, a history or current evidence of any other important clinical condition or use of electroconvulsive therapy in the previous 6 months. | |
| Interventions | Fluoxetine: 89 participants
Reboxetine: 79 participants
Fluoxetine dose range: 20‐40 mg/day
Reboxetine dose range: 8‐10 mg/day Chloral hydrate (0.5‐1 mg) for sleep |
|
| Outcomes | Primary outcome: change in the HDRS total score, number of patients showing response (decrease of at least 50% in HDRS total score) and remission (a final score of 10 or less) Seconday outcomes: Clinical Global Impression‐Severity (CGI‐S), Montgomery and Asberg Scale for Depression (MADRS), Patient Global Impression (PGI) |
|
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of withdrawals reported, but reasons for dropout not clearly described. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | SIde effects not clearly reported. Mean endpoint scores reported with standard deviation |
| Other bias | Unclear risk | Funding: unclear |
McGrath 2000.
| Methods | Ten‐week randomised, double‐blind, multicentre study | |
| Participants | Patients fulfilling DSM‐IV criteria for major depressive episode, lasting for at least 1 month and having Columbia criteria for atypical depression. Age range: 18‐65 years Exclusion criteria: significant suicidal risk, pregnancy, lactation, absence of contraception, unstable and serious physical illness, history of seizures, psychosis or organic mental syndrome, substance use disorder within 6 months, history of mania, antisocial personality disorder, history of non‐response to an adequate trial of fluoxetine or imipramine, history of no response to any other SSRIs, hypothyroidism. | |
| Interventions | Fluoxetine: 49 participants Imipramine: 53 participants Placebo: 52 participants Fluoxetine dose range: 20‐60 mg/day Imipramine dose range: 50‐300 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all patients received identical capsules", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of drop out was reported; reasons for dropout were not clearly stated Quote: "in the analyses to follow‐up, we will focus on the ITT group to estimate treatment effects without bias related to attrition", unclear how this analysis was carried out |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported. Only endpoint adjusted scores reported |
| Other bias | High risk | Quote: "supported by Eli Lilly and Company", this company produces fluoxetine |
Moosa 2003.
| Methods | Twelve‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for depressive episode, with a score of at least 17 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: under 50 years Exclusion criteria: history of a serious medical illness, psychotic symptoms, psychoactive substance abuse or dependency, comorbid illness that required pharmacotherapy, or presence of an eating disorder. | |
| Interventions | Fluoxetine: 14 participants Reboxetine: 14 participants Fluoxetine dose: 20 mg/day Reboxetine dose: 150 mg/day | |
| Outcomes | Primary outcome: change in the HDRS‐21 total score | |
| Notes | Funding: by academy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only the total number of randomised patients reported, not the number of the single group. Number and reasons for dropout not reported |
| Selective reporting (reporting bias) | Unclear risk | Side effects not reported. Mean baseline and endpoint score (HDRS) reported |
| Other bias | Low risk | Funding: by academy |
Moreno 2006.
| Methods | Eight‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for mild to moderate, non psychotic major depressive disorder, with a baseline score of at least 10 and a maximum score of 24 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: patients with other types of depression, psychosis, personality disorders, bipolar disorders, suicidal ideation, uncontrolled organic disease, history of alcohol or drug abuse 1 year prior, abnormal laboratory tests or a history of seizures, treated with electroconvulsive therapy or have taken any investigational drug up to 30 days before screening. Patients who used MAO‐inhibitors 2 weeks before the enrolment, benzodiazepines in doses equivalent to diazepam 10mg/day 1 week before the enrolment and patients already treated with fluoxetine. | |
| Interventions | Fluoxetine: 20 participants Hypericum: 20 participants Fluoxetine dose: 20 mg/day Hypericum dose: 900 mg/day | |
| Outcomes | Primary outcome: change in the HDRS total score Seconday outcomes: Clinical Global Impression (CGI) Severity, Montgomery and Asberg Scale for Depression (MADRS), UKU side effects rating scale (UKU) |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons of withdrawals not clearly reported. Endpoint scores and responders reported only in figures |
| Selective reporting (reporting bias) | Unclear risk | SIde effects not reported. Secondary endpoint not reported |
| Other bias | High risk | Quote: "the authors are grateful to Marjan, for the financial support and donation of hypericum perforatum, and to Eli Lilly Brazil, for the donation of fluoxetine", this two pharmaceutical companies produce the two study drugs |
Mowla 2006.
| Methods | Eight‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depression disorder, based on a structured clinical interview. Mean age: 30.8 (SD: 9.9) (fluoxetine) and 35 (SD: 9.4) (nortriptyline) years Exclusion criteria: patients with another DSM‐IV diagnosis, psychotic symptoms, alcohol or other drugs addiction, major physical illness, potentially pregnant women, breast‐feeding or planning to be pregnant in the next two months, patients in whom tricyclics were contraindicated (i.e. narrow‐angle glaucoma, prostatism). | |
| Interventions | Fluoxetine: 36 participants Nortriptyline : 20 participants Fluoxetine dose: 40 mg/day Nortriptyline dose: 150 mg/day |
|
| Outcomes | Primary outcomes: change in Hamilton Rating Scale for Depression (HDRS) and in Clinical Global Impression (CGI) total scores | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the investigator was blind as to medications and he did not personally manage any patients in the study", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of dropouts was reported, but the reasons were not clearly described. Endpoint scores not reported (HDRS, CGI) |
| Selective reporting (reporting bias) | High risk | Side effects not reported. Item in which there was significant improvement reported without mean and standard deviation |
| Other bias | Unclear risk | Funding: unclear |
Muijen 1988.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling Research Diagnostic Criteria for major depressive disorder or bipolar illness, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐65 years Exclusion criteria: serious somatic illness, alcohol or drug abuse, pregnancy, severe depression with indication for hospital admission or ECT, or TCA, neuroleptics in the previous 4 weeks, MAOI in the previous 2 weeks. | |
| Interventions | Fluoxetine: 26 participants Mianserin: 27 participants Placebo: 28 participants Fluoxetine dose range: 20‐80 mg/day Mianserin dose range: 20‐80 mg/day Only temazepam (max 20 mg) nightly for the shortest possible period | |
| Outcomes | HDRS‐17, CGI, Montgomery and Asberg Scale for Depression (MADRS), Patient Global Impression (PGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported. Score reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only severe and moderate side effects reported |
| Other bias | Unclear risk | Funding: unclear |
MY‐1043/BRL‐029060/115.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients with moderate to severe depression (DSM: single episode or recurrent), with a score of at least 18 in the first 17 items of Hamilton Rating Scale for Depression‐21 item (HDRS‐21) both at the screening and baseline visit, the HDRS score could not decrease by more than 25% between screen and baseline visit. The Raskin Depression Scale (RDS) score had to be at least 8 at baseline and must have exceeded the Covi Anxiety Score (CAS). Age: over 18 years Exclusion criteria: patients with a primary psychiatric diagnosis other than depression, or those with a serious concomitant diseases. Patients were also excluded if they had a serious suicidal threat, recent ECT or with substance abuse. | |
| Interventions | Fluoxetine: 289 participants Paroxetine: 284 participants Fluoxetine dose range: 20‐50 mg/day Paroxetine dose range:20‐80 mg/day |
|
| Outcomes | Changes in HDRS total score, RDS and Clinical Global Impression (CGI) Severity of illness and Improvement; CAS, Symptom Checklist‐90 (SCL‐90), Global Assessment of Functioning (GAF) Scale Response: at least 50% reduction on the HDRS‐21 total score from baseline or a score under 10 at HDRS‐21 at the endpoint |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Mean rating scale scores not reported, both at baseline and at endpoint. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Only most frequent side effects reported; baseline and endpoint scores at rating scales were not reported |
| Other bias | High risk | Funding: by Glasko Smith Kline, and this company produces paroxetine |
MY‐1045/BRL‐029060/1.
| Methods | Twelve‐week randomised, double‐blind, multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for Major Depressive Disorder (MDD), with a total score of at least 18 on the first 17 items of the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Total score could not have decreased by more than 25% between the screen and the baseline visits. Age: over 18 years Exclusion criteria: primary psychiatric diagnosis other than MDD, serious suicidal or homicidal risk, substance abuse or dependence, prior ECT (within 3 months of the study), serious concomitant medical conditions, and subjects with a history of hypersensitivity to fluoxetine or who had previously taken paroxetine. | |
| Interventions | Fluoxetine: 351 participants Paroxetine: 357 participants Fluoxetine dose: 20 mg/day Paroxetine dose: 20 mg/day | |
| Outcomes | Primary outcome: HDRS‐21
Secondary outcomes: HDRS‐21 subscale, Raskin Depression rating scale (RDS), Clinical Global Impression (CGI), Symptom Checklist‐90 (SCL‐90), Global Assessment of Functioning (GAF) Scale, Covi Anxiety Scale (CAS) Response: a decrease of 50% from baseline in the HDRS‐21 total score at any time during the 12‐week study |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported. Mean score at endpoint (HDRS) not reported. Responders were reported in percentage, and without denominator |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. Secondary outcome measures reported only at endpoint, and not at the baseline |
| Other bias | High risk | Funding by Glasko Smithkline, and this company produces paroxetine |
Nelson 2004.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Inpatients with unipolar non‐psychotic major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS) after at least one week in the hospital without medication. Age: 21 years and older Exclusion criteria: patients who had more than 30% improvement in the first week remained medication free for two‐weeks and were excluded if the HDRS score drop below 18. Patients with schizophrenia, schizoaffective disorder, bipolar disorder, psychotic depression, active medical illness, substance abuse in the past 6 months, and cluster B personality disorder were excluded. | |
| Interventions | Fluoxetine: 14 participants
Desipramine: 12 participants
Fluoxetine + desipramine: 13 participants
Fluoxetine dose: 20 mg/day
Desipramine mean dose: 293 mg/day (SD: 116.8) Fluoxetine + desipramine dose: 20 mg/day fluoxetine + 98,1 (SD: 45.0) desipramine |
|
| Outcomes | HDRS, Montgomery and Asberg Scale for Depression (MADRS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported without distinguish the study arms. Score reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Side effects not reported. End point score on HDRS and MADRS reported with standard deviation |
| Other bias | Low risk | Quote: "this research was supported in part by National Institute of Mental Health Grants R01‐MH‐47894 and MH‐30020" |
Nemeroff 2007.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive episode, the symptoms is present for at least 1 month, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: over 18 years Exclusion criteria: bipolar or psychotic disorder, history of alcohol or substance abuse within the past year, any clinically significant medical disorders or abnormalities, history of non response to venlafaxine or fluoxetine, patients receiving any study drug within 6 months; electroconvulsive therapy within 3 months, astemizole, cisapride, sumatriptan, terfenadine, any other antidepressant, anxiolytic, sedative‐hypnotic drug within 7 days, pregnancy, lactation. | |
| Interventions | Fluoxetine: 104 participants
Venlafaxine: 102 participants Placebo: 102 participants Fluoxetine dose range: 20‐60 mg/day Venlafaxine dose range: 75‐225 mg/day |
|
| Outcomes | Primary outcome: reduction in total score and item 1 of the HDRS‐21, Montgomery and Asberg Scale for Depression (MADRS), Clinical Global Impression (CGI), Patient Global Impression (PGI) Response: 1) decrease of 50% HDRS‐21 score; 2) decrease of 50% MADRS score; 3) CGI score of 1 or 2; 4) PGI score of 1 or 2 | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons of dropout reported. Denominator reported for responders was different from the number of randomised patients. Mean endpoint scores (HDRS‐21, MADRS, CGI) reported only in figures and without standard deviations |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported, but not clear the number of patients reporting at least one side effects |
| Other bias | High risk | Quote: "this study was funded by a series of grants to the participating research sites from Wyeth Research, Collegeville", and this company produces venlafaxine |
Newhouse 2000.
| Methods | Twelve‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode (single or recurrent), without psychotic features, with a score of at least 18 on the Hamilton Rating Scale for Depression‐ 24 item (HDRS‐24). Age: over 60 years Exclusion criteria: DSM‐III‐R criteria for any other psychiatric disorder, significant cognitive impairment (Mini Mental State Examination less than 24), any medical contraindication to any antidepressant therapy, endocrine, cardiovascular, gastrointestinal, renal disease, failure to respond to ECT in a prior depressive episode or to adequate trials (6 weeks) of 2 or more antidepressants. | |
| Interventions | Fluoxetine: 119 participants Sertraline: 117 participants Fluoxetine dose range: 20‐40 mg/day Sertraline dose range: 50‐100 mg/day Temazepam and chloral hydrate were allowed for sleep | |
| Outcomes | Primary outcome: HDRS‐24 (total and factor scores), Clinical Global Impression (CGI) Severity, Efficacy Secondary outcomes: Montgomery and Asberg Scale for Depression (MADRS), Hamilton Rating Scale for Anxiety (HAM‐A), Profile Of Mood State (POMS), Beck Depression Inventory (BDI), Quality of Life Enjoyment and Satisfaction Questionnaire (Q‐LES‐Q) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double dummy procedure was used to ensured patients and physician blindness to treatment assignment", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominator. Number and reasons for dropout not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. End point scores reported without standard deviation |
| Other bias | High risk | Quote: "supported by a grant from Pfizer", this company produces sertraline |
Nielsen 1993.
| Methods | Eight‐week double‐blind, randomised study | |
| Participants | Outpatients fulfilling DSM‐III and Bech‐Rafaelsen Melancholia Scale criteria for major depressive disorder, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 18‐70 years Exclusion criteria: suicide risk, history of schizophrenia or organic brain dysfunction, history of severe allergies or serious physical illness, recent period of alcohol or alcohol abuse, pregnancy. | |
| Interventions | Fluoxetine: 29 participants Imipramine: 30 participants Fluoxetine dose: 20 mg/day Imipramine dose range: 75‐150 mg/day | |
| Outcomes | HDRS‐21, Bech‐Rafaelsen Melancholia Scale (MES), Clinical Global Impression (CGI), Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Reasons and numbers of dropouts reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most frequent side effects reported. End point scores and standard deviation not clearly reported |
| Other bias | High risk | One of the authors' affiliation was Eli Lilly, Denmark. This company produces fluoxetine |
Noguera 1991.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Patients fulfilling DSM‐III criteria for major depressive disorder, with a score of at least 17 on the first 17 items of the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), a score of at least 8 on the Raskin Depression Scale (RDS), greater than Covi Anxiety Scale (CAS). Age range: 18‐65 years Exclusion criteria: history of manic episode, pregnancy, lactation, absence of contraception, glaucoma, chronic urinary retention, brain or other significant organic illness, schizophrenia, other mental illness or severe suicidal risk, recent history (less than 1 year) of alcohol or drug abuse, concurrent treatment with other psychotropic drug including lithium, use of MAOI less of 2 weeks prior the study entry. | |
| Interventions | Fluoxetine: 60 participants Clomipramine: 60 participants Fluoxetine dose range: 20‐40 mg/day Clomipramine dose: 100 mg/day Chloralzepate (10 mg) for insomnia was allowed | |
| Outcomes | HDRS‐21, CAS, RDS, Patient Global Impression (PGI), Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "were randomly allocated", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " apparently identical capsules in seven doubles enveloped marked 'morning' and 'midday' dose", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Baseline and mean changes in efficacy measures reported with standard deviation |
| Other bias | High risk | One of the authors' affiliation was Eli Lilly Spain, Madrid; this company produces fluoxetine |
Noorbala 2005.
| Methods | Six‐week randomised, double‐blind clinical trial | |
| Participants | Outpatients fulfilling DSM‐IV criteria for depressive episode, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS) and to be free of psychotropic medication for at least 4 weeks before study entry. Age range: 18‐55 years Exclusion criteria: pregnancy, absence of contraception, current cognitive disorder in the last year, bipolar disorder, schizophrenia or schizotypal personality disorder, significant risk o suicide (two or more at the suicide item of the HDRS or to be judged to have significant suicidal ideation in the view of an investigator). | |
| Interventions | Fluoxetine: 20 participants Crocus Sativus: 20 participants Fluoxetine dose: 20 mg/day Crocus sativus dose: 30 mg/day | |
| Outcomes | Primary outcome: change in the HDRS total score | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised in a 1:1 ratio using a computer‐generated code", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "throughout the study the person who administered the medications, rater and patients were blind to assignments", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " throughout the study the person who administered the medications, rater and patients were blind to assignments", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: " throughout the study the person who administered the medications, rater and patients were blind to assignments", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number of withdrawals reported, but reasons for drop‐out not clearly described. Baseline and end‐point scores reported only in figures |
| Selective reporting (reporting bias) | Unclear risk | Side effects not clearly reported |
| Other bias | Unclear risk | Funding: unclear |
Novotny 2002.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for major depressive disorder, (single or recurrent), without psychotic features, with or without melancholia, or bipolar II disorder, current episode depressed, moderate or severe without psychotic features with or without melancholia, with a score of at least 25 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐65 years Exclusion criteria: dysthymia, cyclothymia, double‐depression, psychotic disorder, drug or alcohol abuse or dependence, serious risk of suicide, treatment resistant depression, recurrent ECT, non‐response to previous treatment with fluoxetine or tianeptine, severe hepatic, cardiovascular, neurological, metabolic disease, cancer or allergy, pregnancy, previous treatment with neuroleptics in the previous 2 months, MAOI, fluoxetine lithium, valproates or carbamazepine within 1 month of baseline, other antidepressants, diazepam, lorazepam, alprazolam, bromazepam, barbiturates, buspirone the week before recruitment. | |
| Interventions | Fluoxetine: 91 participants Tianeptine: 87 participants Fluoxetine dose: 20 mg/day Tianeptine dose: 37.5 mg/day Chloralzepate (max 30 mg), oxazepam (max 60 mg) for anxiety and nitrazepam (1 mg) or lorazepam (1 mg) for insomnia. For patients who were usually taking benzodiazepines for at least 1 month before baseline continuation during the trial was allowed | |
| Outcomes | Primary outcome: MADRS total score | |
| Notes | Response: decrease of at least 50% in the MADRS total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for drop out reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores (MADRS, Clinical Global Impression [CGI]) reported only in figures and without standard deviation. Only most frequent adverse events reported |
| Other bias | High risk | Quote: "this work was supported by Servier", and this company produces tianeptine |
O'Keane 1992.
| Methods | Four‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐64 years Exclusion criteria: not specified. Patients were physical healthy, non obese and did not have DSM‐III‐R axis 2 disorders. | |
| Interventions | Fluoxetine: 7 participants Amitryptyline: 9 participants Fluoxetine dose: 20 mg/day Amitryptyline dose: 250 mg/day | |
| Outcomes | Primary outcome: HDRS | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | No information provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | End point scores reported for each patient. All patients complete the study |
| Selective reporting (reporting bias) | Low risk | Side effects not reported |
| Other bias | Unclear risk | Funding: unclear |
Ontiveros 1997.
| Methods | Six‐week randomised, double‐blind two‐centre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐75 years Exclusion criteria: pregnancy, lactation, severe coexisting disease, unstable diabetes, organic brain syndrome, history of alcohol or drug abuse, schizophrenia or psychosis, severe risk of suicide. | |
| Interventions | Fluoxetine: 61 participants Paroxetine: 60 participants Fluoxetine dose: 20 mg/day Paroxetine dose: 20 mg/day | |
| Outcomes | Primary outcome: change from baseline on the HDRS‐21 total score at endpoint Secondary outcomes: change from baseline in the Hamilton sub‐factor scores (anxiety, retardation, sleep disturbance, melancholia, recognition), proportion of patients responding to treatment, change from baseline on the Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 total score Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all active and placebo medication was supplied as identical coloured capsules", no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Only principal reasons for withdrawals reported. Endpoint scores reported with standard deviation |
| Selective reporting (reporting bias) | Unclear risk | Only adverse events reported for more than 5% of the sample reported. Secondary outcome not reported |
| Other bias | High risk | Quote: "paroxetine was supplied by SmithKline Beecham, Mexico" |
OntiverosSanchez 1998.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐65 years Exclusion criteria: pregnancy, lactation, absence of contraception, severe suicide risk, severe medical illness, history of psychosis or of substance abuse in the previous 1 years, hypersensitivity to fluoxetine or amitriptyline, psychotherapy or use of psychotropic drugs (benzodiazepines, too). | |
| Interventions | Fluoxetine: 21 participants Amitriptyline : 21 participants Fluoxetine dose range: 40‐80 mg/day Amitriptyline dose range: 150‐250 mg/day | |
| Outcomes | HDRS‐21, Hamilton Rating Scale for Anxiety (HAM‐A), Clinical Global Impression (CGI), Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Symptom Checklist‐90 (SCL‐90) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported. Scores reported without standard deviation |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Vital signs reported. Scores reported without standard deviation (HDRS, RDS, CAS, SCL‐90) |
| Other bias | High risk | Last author affiliation was Laboratories Eli Lilly y Cia; this company produces fluoxetine |
Pakesch 1991.
| Methods | Four‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling Kielholz/Poeldinger scheme for depression, with a score of at least 11 on the Hamilton Rating Scale for Depression (HDRS‐14). Age range: 19‐79 years Exclusion criteria: organic disease, endogenous depression, organic psychosis, schizophrenia, alcohol or substance abuse, previous treatment with clomipramine, use of neuroleptics. | |
| Interventions | Fluoxetine: 46 participants Clomipramine: 48 participants Fluoxetine dose: 40 mg/day Clomipramine dose: 50 mg/day. Oxazepam (maw 15 mg) or chloral hydrate (max o.25g) were allowed | |
| Outcomes | HDRS‐14, Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only completers were analysed |
| Selective reporting (reporting bias) | Unclear risk | No figures for percentages |
| Other bias | Unclear risk | Funding: unclear |
Pande 1996.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder or dysthymic disorder or depressive disorder NOS and Columbia criteria for atypical depression, with a score of at least 10 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Mean age: 32.8 (fluoxetine) and 34.3 (phenelzine) years Exclusion criteria: pregnancy, serious medical illness, comorbid psychiatric illness, alcohol or drug abuse, participation to a clinical trial in the previous month. | |
| Interventions | Fluoxetine: 20 participants Phenelzine: 20 participants Fluoxetine dose range: 20‐60 mg/day Phenelzine dose range: 45‐90 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI), Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double dummy", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for drop‐out were reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most frequent (at least 20%) treatment emergent adverse events reported. Vital signs described |
| Other bias | High risk | Quote: "supported by a research grant from Eli Lilly & Company, Indianapolis" and this company produces fluoxetine |
Perry 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression (lasting more than 1 month), with a score of at least 20 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 18 years Exclusion criteria: pregnancy, lactation, absence of contraception, serious suicide risk, glaucoma, presence of cardiovascular arrythmias, hypertension, serious medical illness, including hepatic, renal, respiratory, hematologic disease, histiory of seizure, severe allergies or multiple drug reaction, psychotic patients and patients with DSM‐III diagnosis of organic mental disorder, substance abuse disorder within the past year, schizophrenia, paraniod disorder, bipolar disorder, use of MAOI in the past 14 days, lithium or any other psychotropic drug, use of trazodone or fluoxetine within 4 weeks of study entry. | |
| Interventions | Fluoxetine: 21 participants Trazodone: 19 participants Fluoxetine dose range: 20‐60 mg/day Trazodone dose range: 50‐400 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout were reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most frequent (at least 10%) adverse events reported Vital signs described |
| Other bias | Unclear risk | Funding: unclear |
Peters 1990.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling ICD 9 criteria for major unipolar or bipolar depression, with a score of at least 17 on the HDRS, a score of at least 8 on the Raskin Depression Scale (RDS), greater than Covi Anxiety Scale (CAS) score. Age range: 25‐63 years Exclusion criteria: history of psychosis, suicide risk, severe mental diseases, contraindication to amitriptyline, severe organic disease, known drug allergy, use of amitriptyline within 4 weeks of baseline, use of neuroleptics within 2 weeks of study entry. | |
| Interventions | Fluoxetine: 51 participants Amitriptyline: 51 participants Fluoxetine dose: 20 mg/day Amitriptyline dose: 100 mg/day Chloral hydrate or benzodiazepines for insomnia were allowed | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS‐17), Clinical Global Impression (CGI), RDS, CAS | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was unclear which was the last observation carried forward (LOCF) population |
| Selective reporting (reporting bias) | Unclear risk | Three rating scales for depression were listed in methods, but only one was reported |
| Other bias | Unclear risk | Funding: unclear |
Poelinger 1989.
| Methods | Four‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling Kielholz/Poeldinger scheme for depression, with a score of at least 14 on the Hamilton Rating Scale for Depression (HDRS‐14). Age range: 21‐67 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 73 participants Maprotiline: 69 participants Fluoxetine dose: 40 mg/day Maprotiline dose: 75 mg/day Only chloral hydrate and oxazepam were allowed for insomnia | |
| Outcomes | HDRS‐14, Clinical Global Impresison (CGI) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for dropout not clearly reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | End point scores not reported (CGI). Only most common side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Preskorn 1991.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression (lasting more than 1 month), with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS). Age: over 18 years Exclusion criteria: pregnancy, lactation, absence of contraception, contraindication to amitriptyline, medical illness, history of seizures, glaucoma, severe allergies, multiple adverse drug reaction, known allergy to study medication, use of MAOI within 2 weeks, use of other investigational drugs in past 2 weeks, suicidal risk, DSM‐III diagnosis such as substance abuse in the past year, schizophrenia, schizoaffective disorder, bipolar or paranoid disorder. | |
| Interventions | Fluoxetine: 30 participants Amitriptyline: 31 participants Fluoxetine dose range: 20‐60 mg/day Amitriptyline dose range: 50‐200 mg/day Only chloral hydrate was allowed for sleep | |
| Outcomes | HDRS, Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "fluoxetine, amitriptyline and placebo were identical in appearance", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not reported. Endpoint scores reported with mean and standard deviation |
| Other bias | High risk | Quote: "this work was supported in part by Eli Lilly and Company" and this company produces fluoxetine |
Rapaport 1996.
| Methods | Seven‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for current major depressive episode, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and with a minimum score of 2 on the depressive mood item. Age range: 18‐65 years Exclusion criteria: unstable medical condition other Axis 1 diagnosis, acute suicidally, history of substance dependence within 6 months of the baseline, history of seizure disorder. | |
| Interventions | Fluoxetine: 49 participants Fluvoxamine: 51 participants Fluoxetine dose range: 20‐80 mg/day Fluvoxamine dose range: 100‐150 mg/day Only chloral hydrate (max 1 g) was allowed for sleep | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Hamilton Rating Scale for Anxiety (HAM‐A), Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Symptom Checklist‐90 (SCL‐90) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Study medication was provided in identical‐appearing green capsules", no other information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no other information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the ITT‐LOCF patients sample was the patients sample analysed for efficacy with at least one valid efficacy assessment after baseline determination". Rating scale scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviation. Only adverse events reported for more than 20% of the sample. |
| Other bias | High risk | Quote: "this study was supported by grants from Solvay Pharmaceuticals", and this company produces fluvoxamine |
Remick 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III criteria for current major depressive episode, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Measn age: 43 years Exclusion criteria: psychosis, bipolar disorder, concurrent use of any psychoactive medication | |
| Interventions | Fluoxetine: 38 participants Doxepine: 37 participants Fluoxetine dose range: 20‐60 mg/day Doxepine dose range: 100‐200 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " medication was dispensed in opaque gelatine capsules containing either placebo, fluoxetine or doxepine (...) in a dose‐dispensing system administered by a pharmacist", not clear if blindness was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported |
| Other bias | High risk | Quote: "the authors gratefully acknowledge (...) Eli Lilly of Canada for financial support", this company produces fluoxetine |
Remick 1993.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder (lasting 1 month or more), with a score of at least 20 on the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 18‐65 years Exclusion criteria: any abnormalities on laboratory examination, presence of psychosis, bipolar disorder, concurrent use of any psychoactive medication, pregnancy, lactation. | |
| Interventions | Fluoxetine: 26 participants Desipramine: 20 participants Fluoxetine dose range: 20‐60 mg/day Desipramine dose range: 150‐300 mg/day | |
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Patient Global Impression (PGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "medication was dispensed in opaque gelatine capsules containing either placebo, fluoxetine or doxepine...in a dose‐dispensing system administered by a pharmacist", not clear if blindness was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | End point scores not clearly reported. Only most common side effects reported |
| Other bias | High risk | Quote: "the authors wish to aknowledge (...) Eli Lilly for financial support", this company produces fluoxetine |
Reynaert 1995.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 16 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Mean age: 47 years Exclusion criteria: suicide risk, any other psychiatric illness, severe organic disease, alcoholism and drug abuse, use of MAOI in the previous 2 weeks and antidepressants in the previous 4 days or any investigational drugs in the previous 4 weeks, use in the past of fluoxetine or moclobemide. | |
| Interventions | Fluoxetine: 50 participants Moclobemide: 51 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 300‐600 mg/day Lithium and one benzodiazepine were permitted | |
| Outcomes | Primary outcomes: HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the total score or a score of maximum 10 on the HDRS‐17 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout were reported. Denominator reported for responders was different from the number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Secondary outcomes not reported. Side effects and vital signs not clearly reported |
| Other bias | High risk | Last author's affiliation is Roche S. A., Brussels, Belgium and this company produces moclobemide |
Robertson 1994.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder or bipolar disorder (currently depressive), with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐70 years Exclusion criteria: previous use of fluoxetine or lofepramine prior entry to study or during present episode, use of psychoactive drugs (a part from short acting benzodiazepines within 7 days prior entry), use of MAOI within 14 days and depot neuroleptics within 6 months, ECT, serious suicide risk, pregnancy, lactation, absence of contraception, history of glaucoma, cardiovascular disease or urinary retention, significant other medical illness, history of severe allergies or multiple adverse drug reaction, concurrent use of diuretics. | |
| Interventions | Fluoxetine: 90 participants Lofepramine: 93 participants Fluoxetine dose: 20 mg/day Lofepramine dose range: 140‐210 mg/day | |
| Outcomes | HDRS‐17, Montgomery and Asberg Scale for Depression (MADRS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "blindness was maintained by having each subject on lofepramine taking placebo‐fluoxetine in addiction and vice versa", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons and number of dropouts reported. Scores reported with denominator |
| Selective reporting (reporting bias) | Unclear risk | Adverse events experienced by more than five subjects in either group were reported. Scores reported only in figures and without standard deviation |
| Other bias | High risk | One author's affiliation was Lilly Industries Ltd, and this company produces fluoxetine |
Ropert 1989.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for current major depressive disorder, with a score between 18 and 25 on the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 18‐65 years Exclusion criteria: organic brain disease, history of seizures, serious illness, including cardiovascular, hepatic, renal, respiratory, hematologic, hyperthyroidism, history of severe allergy or multiple drug reaction, history (less than 1 year) of drug and alcohol abuse, concurrent administration of psychotropic drugs (except benzodiazepines), MAOI within 2 weeks, serious suicidal risk, pregnancy, lactation. | |
| Interventions | Fluoxetine: 71 participants Clomipramine: 72 participants Fluoxetine dose: 20 mg/day Clomipramine dose: 75 mg/day | |
| Outcomes | HDRS‐21 | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for discontinuation not clearly reported. Analysis not based on the ITT number |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported only in figures and without standard deviation. Only side effects reported by more than 3 patients |
| Other bias | Unclear risk | Funding: unclear |
Rudolph 1999.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for current major depressive disorder, with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: over 18 years Exclusion criteria: recent treatment within 6 months or known hypersensitivity to either study drugs, serious medical conditions, bipolar mood disorder, psychotic disorder not associated with depression, history of drug or alcohol dependence within 1 years of study entry, suicidal patients, pregnancy, lactation. | |
| Interventions | Fluoxetine: 103 participants Venlafaxine: 100 participants Venlafaxine: 100 participants Placebo: 98 participants Fluoxetine dose range: 20‐60 mg/day Venlafaxine dose range: 75‐250 mg/day Chloral hydrate was allowed as hypnotic | |
| Outcomes | Primary outcomes: HDRS‐21 total score and depressed mood items, Montgomery–Åsberg Depression Rating Scale (MADRS) total score, Clinical Global Impression (CGI) Secondary outcome: Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Response: decrease of at least 50% in the total score from baseline on HDRS and MDRS or a CGi score of 1 or 2. Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised in blocks of six using a table of random numbers..." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common adverse events reported. End point scores reported without standard deviation |
| Other bias | High risk | Quote: "Wyeth‐Ayerst Research provided financial support and the study drugs to the investigators". This company produces venlafaxine |
Rush 1998.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III criteria for moderate to severe major depressive disorder, non psychotic, with a score of at least 18 on the first 17 items of the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 19‐55 years Exclusion criteria: engaged in a shift work, independent sleep/wake disorders, significant concurrent general medical conditions, DSM‐III criteria for psychoactive use disorder within 1 year prior to study, other major lifetime Axis I disorders (organic mental syndrome, bipolar, any psychotic, any eating,panic or obsessive‐compulsive disorder), pregnancy, lactation, absence of contraception. | |
| Interventions | Fluoxetine: 61 participants Nefazodone: 64 participants Fluoxetine dose range: 20‐40 mg/day Nefazodone dose range: 200‐500 mg/day | |
| Outcomes | HDRS‐17 total score, Inventory of Depressive Symptomatology (IDS), Clinical Global Impression (CGI) Improvement | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double dummy dosing regimen was employed", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Scores reported with denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported |
| Other bias | High risk | Quote: "this study was sponsored by Bristol‐Myers Squibb Pharmaceutical research Institute", this company produces nefazodone |
Sandor 1998.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depressive disorder, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS‐17). Age range: 18‐75 years Exclusion criteria: serious medical disease, suicidal patients, history of alcohol or substance abuse, treatment resistant depression, bipolar mood disorder, use of antidepressants in the previous 2 weeks and fluoxetine in the previous 6 weeks. | |
| Interventions | Fluoxetine: 20 participants Doxepine: 20 participants Fluoxetine dose range: 20‐60 mg/day Doxepine dose range: 75‐225 mg/day | |
| Outcomes | HDRS‐17 | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no other information about the randomisation procedures |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Quote: "in a double blind manner", no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "in a double blind manner", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "in a double blind manner", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominator reported for responders was different from the number of randomised. Number and reasons for dropout not clearly described |
| Selective reporting (reporting bias) | Unclear risk | End‐point scores not reported. Side effects not reported |
| Other bias | High risk | Quote: "the original study of cardiac effects of fluoxetine and doxepin was supported by Eli Lilly, Canada". Eli Lilly Company produces fluoxetine |
Schatzberg 2006.
| Methods | Eight‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for unipolar depression (single or recurrent, nonpsychotic) with a current episode of at least 4 weeks in duration; had a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age: 65 years Exclusion criteria: bipolar disorder, psychotic disorder not related to depression, current substance abuse or substance dependence within the past year, current suicidal intent, at least 18 at the MMSE score, have received treatment with fluoxetine or venlafaxine in the past six months, electroconvulsive therapy within 3 months serious medical disease, suicidal patients, history of alcohol or substance abuse, any investigational drug or antipsychotic drug within the prior 30 days, use of astemizole, cisapride, sumatriptan, terfenadine, paroxetine, sertraline, or any monoamine oxidase inhibitor within 14 days, use of other antidepressant, anxiolytic, sedative‐hypnotic drug, or any other psychotropic drug or substance within 7 days, patients with a known hypersensitivity to venlafaxine or fluoxetine, renal or hepatic disease, seizure disorder or myocardiac infarction within the prior 6 months, and patients with a severe, acute or unstable medical illness. | |
| Interventions | Fluoxetine: 100 participants Venlafaxine: 104 participants Placebo: 96 participants Fluoxetine dose range: 20‐60 mg/day Venlafaxine dose range: 75‐225 mg/day |
|
| Outcomes | Decrease in HDRS‐21 score, Montgomery–Åsberg Depression Rating Scale (MADRS) score and Clinical Global Impression (CGI) score. Response: decrease of at least 50% HDRS score from baseline to endpoint |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomisation was by number in six‐patients unit with equal numbers of each treatment", no other information about the randomisation procedures |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "medication for each patient was packaged individually and code‐labelled with the study number and a unique patient randomisation number. Units were distributed to study sites according to the lowest available randomisation number", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "medication for each patient was packaged individually and code‐labelled with the study number and a unique patient randomisation number. Units were distributed to study sites according to the lowest available randomisation number", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout described. Number of responders is reported only in figures and in percentage. Endpoint scores not reported |
| Selective reporting (reporting bias) | Unclear risk | Baseline scores reported without standard deviation. Side effects reported |
| Other bias | High risk | Quote: "funding for this study provided by Wyeth Research", and this company produces venlafaxine |
Schone 1993.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 18 on the first 17 items of the Hamilton Rating Scale for Depression (HDRS‐21). Age range: 65‐85 years old Exclusion criteria: severe physical illness, senile dementia, schizophrenia, organic brain syndrome, alcohol abuse, ECT during the previous 3 months, MAOI in the previous 2 weeks, depot neuroleptics in the previous 4 weeks, oral neuroleptics in the previous 2 weeks. | |
| Interventions | Fluoxetine: 52 participants Paroxetine: 54 participants Fluoxetine dose range: 20‐60 mg/day Paroxetine dose range: 20‐40 mg/day Temazepam (15‐30 mg) was allowed for sleep | |
| Outcomes | HDRS‐21, Montgomery–Åsberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), Mini Mental State Examination (MMSE), Sandoz Clinical Assessment‐Geriatric (SCAG) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Scores reported without denominator. Number and reasons for dropout not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Only those adverse events occurred in at least 5% of participants were reported. Endpoint score at HDRS‐21 was reported only in figures and without standard deviation |
| Other bias | High risk | Quote: "this research was supported by SmithKline Beecham Pharmaceuticals" and this company produces paroxetine |
Schrader 2000.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling ICD 10 criteria for mild to moderate depression, with a score between 16 and 24 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Mean age: 46.5 years Exclusion criteria: history of alcohol and substance abuse, dementia, history of seizures, glaucoma, pituitary deficiency, suicidal ideation, thyroid or parathyroid pathology, Parkinson's disease, pregnancy, any serious concomitant medical conditions, MAOI in the previous 2 weeks, SSRI in the previous 5 weeks. | |
| Interventions | Fluoxetine: 114 participants Hypericum: 126 participants Fluoxetine dose: 20 mg/day Hypericum dose: 500 mg/day | |
| Outcomes | Primary outcome: change from baseline to endpoint on the HDRS‐21 Secondary outcomes: change in depression and anxiety/somatization subscores of the HDRS‐21, Clinical Global Impression (CGI) items 1‐3, responder rates | |
| Notes | Response: decrease of at least 50% in the total score or a score of maximum 10 on the HDRS‐21 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " treatment blindness was assured by 'double dummies', whereby hypericum active and placebo tablets were dispensed together with capsules containing fluoxetine or placebo" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout clearly reported. Primary outcome scores (HDRS‐21) and secondary reported |
| Selective reporting (reporting bias) | Unclear risk | Only those adverse events occurred in at least 2% of participants were reported. Endpoint scores reported with standard deviation |
| Other bias | Unclear risk | Funding: unclear |
Sechter 1999.
| Methods | Twenty‐four‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 20 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: 18‐65 years Exclusion criteria: pregnancy, absence of contraception, use of anticoagulants, serotoninergic drugs, MAOI or lithium, antihypertensive, epilepsy, organic brain disease, malignancy, severe disease or surgical intervention in the pervious 4 weeks, dermatological, haematological, endocrine, respiratory, cardiovascular, renal, hepatic, neurologic diseases, severe allergies or known fluoxetine allergy, previous treatment with sertraline, failure to respond to three or more previous antidepressant treatment, history of alcohol or drug dependence, psychosis, personality disorders, significant suicide risk. | |
| Interventions | Fluoxetine: 120 participants Sertraline: 118 participants Fluoxetine dose range: 20‐60 mg/day Sertraline dose range: 50‐150 mg/day | |
| Outcomes | Change from baseline to endpoint on the HDRS‐17 and Clinacal Global Impression (CGI), Covi Anxiety Scale (CAS), Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS‐17 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | End point scores reported without denominators. Number and reasons for discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Mean scores reported without standard deviations. Treatment‐related adverse events were reported |
| Other bias | High risk | Second author's affiliation was Pfizer, and this company produces sertraline |
Sheehan 2009.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Inpatients fulfilling DSM‐IV criteria for the melancholic subtype of MDD of at least 1 month duration, with a score of at least 24 on the first 21 items of the Hamilton Rating Scale for Depression (HDRS‐21). (In patients at the moment of randomisation, then patients could complete the protocol as outpatients if, in the opinion of the investigator, the response to treatment was sufficient to allow discharge from hospital). Age range: adults, over 18 years old Exclusion criteria: severe or poorly controlled medical illnesses, known hypersensibility to either study drug, treatment with either study drug within 3 months, or myocardial infarction within 6 months before the start of the study, patients with clinically significant abnormalities on the physical examination, electrocardiogram, laboratory tests, or urine test, pregnancy, lactation, absence of contraception. Patients with active suicidal ideation, history of seizures, the presence or history of an organic mental disorder, mania or hypomania or psychotic disorder; electroconvulsive therapy within 3 months, any investigational or antipsychotic drug within 30 days, or astemizole, cisapride, sumatriptan, terfenadine, or any monoamine oxidase inhibitor within 14 days; patients could not have taken any other antidepressant, anxiolytic, sedative‐hypnotic, or other psychotropic drug within 2 days before the start of double blind treatment; any non psychopharmacological drug with psychotropic effects in the last 2 days, and history of alcohol or drug dependence or abuse within 1 year before double‐blind treatment. | |
| Interventions | Fluoxetine: 99 participants Venlafaxine: 95 participants Fluoxetine dose range: 60‐80 mg/day Venlafaxine dose range: 225‐375 mg/day |
|
| Outcomes | Hamilton Rating Scale for Depression (HDRS‐21) total score, Clinical Global Impression (CGI) Improvement , Montgomery–Åsberg Depression Rating Scale (MADRS) total score Response: decrease of at least 50% in the total score on the HDRS and MADRS, or a score of 1‐2 on the CGI‐I |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were enrolled by investigators and assigned to blinded treatment by a computerized randomisation process generated by the sponsor. Medication was randomised in blocks of six patients, lots containing equal numbers of each treatment." |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number and reasons for withdrawal reported. Scores reported with denominators. Mean scores (HDRS, MADRS, CGI) and standard deviations reported clearly at baseline and at the endpoint |
| Selective reporting (reporting bias) | Unclear risk | Only most common (10%) side effects reported |
| Other bias | High risk | Quote: "this study was funded by Wyeth Pharmaceuticals and administered by Quitiles", and this company produces venlafaxine |
Silverstone 1999.
| Methods | Twelve‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 20 on the first 17 items on the Hamilton Depression (HDRS‐21) and a score of at least 8 on the Covi Anxiety Scale (CAS) and symptoms of depression for at least 1 month before study entry. Age: over 18 Exclusion criteria: pregnancy, lactation, absence of contraception, history of clinically significant medical disease, clinically significant abnormalities on a physical examination, ECG or laboratory tests, suicide risk, history of seizure disorder, organic mental disorder, bipolar disorder, history of mania or any psychotic disorder not associated with depression, use of any investigational drug, ECT within 30 days, fluoxetine within 28 days, MAOI or paroxetine within 14 days, any other antidepressant, antipsychotic, anxiolytic, sedative‐hypnotic drug or psychotropic or substance within 7 days of the start of the study, history of drug abuse within 6 months. | |
| Interventions | Fluoxetine: 119 participants Venlafaxine: 122 participants Placebo: 118 participants Fluoxetine dose range: 20‐60 mg/day Venlafaxine dose range: 75‐225 mg/day Chloral hydrate (max 1 g) or zopiclone (max 7.5 mg) for sleep | |
| Outcomes | Primary outcomes: HDRS‐21, Hamilton Rating Scale for Anxiety (HAM‐A) and Clinical Global Impression (CGI) Improvement Secondary outcomes: CAS, HDRS mood items, Hospital Anxiety and Depression Scale (HADS), Clinical Global Impression (CGI) Severity, HDRS and HAM‐A response rate | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS and HAM‐A, or a score of 1 on the CGI‐I Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for withdrawal reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects reported. Endpoint scores reported without standard deviation |
| Other bias | High risk | Quote: "This study was supported by Wyeth‐Ayerst Research, Philadelphia". This company produces venlafaxine |
Smeraldi 1998.
| Methods | Twelve‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for dysthymia or a single episode of major depression partial remission, with a score between 14 and 26 on the Montgomery–Åsberg Depression Rating Scale (MADRS). Age range: 18‐70 years Exclusion criteria: experience of inefficacy or intolerance to the study drug, suicidal risk, abuse or dependence on psychoactive substances, use of antidepressants or psychoactive drug in the previous 2 weeks, discontinuation of continuous or occasional use of benzodiazepines in the previous 2 weeks, need for psychoactive agents other than the study drug, severe debilitation, clinically relevant concomitant disease, cancer, pheochromocytoma, Parkinson's syndrome, pregnancy, absence of contraception, previous evidence of poor compliance, participation in a clinical trial in the previous 6 months. | |
| Interventions | Fluoxetine: 139 participants
Amisulpride: 142 participants Fluoxetine dose: 20 mg/day Amisulpride dose: 50 mg/day |
|
| Outcomes | Primary outcome: a reduction of at least 50% on the MADRS total score Secondary outcomes: change at endpoint on MADRS, Hamilton Rating Scale for Anxiety (HAM‐A), Sheean Disability Scale (SDS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "a total of 281 patients were included. The intention to treat analysis consisted of 268 patients". Scores at rating scales were reported without denominators. Number and reasons for dropout were reported |
| Selective reporting (reporting bias) | Unclear risk | Treatment emergent adverse events reported. Endpoint scores not reported |
| Other bias | High risk | Synthelabo Clinical Research (Limito di Pioltello, Milano) participated at the study, and this company produces amisulpride |
SouthWalesGroup 1988.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 16‐70 years Exclusion criteria: pregnancy, absence of contraception, ECT, use of adequate doses of tricyclics in the previous 4 weeks, use of MAOI in the previous 10 days, history of sensitivity to drugs. | |
| Interventions | Fluoxetine: 31 participants Dothiepin: 28 participants Fluoxetine dose range: 60‐80 mg/day Dothiepine dose range: 150‐225 mg/day Temazepam for night sedation was allowed | |
| Outcomes | Global Assessment of Severity (GAS), Clinical Global Impression (CGI), HDRS‐17, Beck and Rafaelsen Mania Scale (MAS), Montgomery–Åsberg Depression Rating Scale (MADRS) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "placebo tablets resembled the opposite active medication", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | End point scores (MADRS, CGI, MAS) not reported Quote: "there was no significant differences between numbers remaining in the trial in the two groups at any time point". No ITT analysis |
| Selective reporting (reporting bias) | Unclear risk | Number and reasons for attrition reported. Only main side effects reported. Vital signs not reported |
| Other bias | High risk | Quote: "we would like to thank (...) Eli Lilly", and this company produces fluoxetine |
Sramek 1995.
| Methods | Twenty‐week randomised, double‐blind study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive disorder, without melancholia, with a score of at least 21 on the Hamilton Rating Scale for Depression (HDRS‐24) and a score of at least 2 on the item 1 of HDRS‐21 and a score of maximum 18 on the Hamilton Rating Scale for Anxiety (HAM‐A), a score of at least 8 on the Raskin Depression Scale (RDS) and a total Covi Anxiety Scale (CAS) less than RDS. Age range: 18‐65 years Exclusion criteria: any clinically significant hematological, endocrine, cardiological, renal, gastrointestinal, neurological disorder, seizure disorder, significant suicidal risk, other Axis I disorders besides dysthymia, Axis 2 diagnosis of antisocial or borderline disorder, history of substance or alcohol abuse within 6 months, ECT in the previous 6 months, use of MAOI or fluoxetine within 3 weeks, any other antidepressant within the last week, use of benzopines within the last 2 weeks, being in any type of psychotherapy since less than 3 months, or having ended such therapy within 1 month prior the study. | |
| Interventions | Fluoxetine: 72 participants ABT‐200: 72 participants Fluoxetine dose: 20 mg/day ABT‐200 dose: 20 mg/day | |
| Outcomes | HDRS‐21, Montgomery–Åsberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), HAM‐A | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "patients in the ABT‐200 group received one placebo capsule which resembled fluoxetine", not clear if blindness was successful |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported, but included in the analysis only by estimation of outcome |
| Selective reporting (reporting bias) | Unclear risk | End point scores (MADRS, CGI) not reported. Only side effects reported by at least 20% of the sample reported |
| Other bias | High risk | Funding: by industry. (Abbott Laboratories, Abbott Park) |
Stark 1985.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depressive disorder (with a duration of illness of at least 4 weeks), with a score of at least 20 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and a score of at least 8 on the Raskin Depression Scale (RDS). Age range: 18‐70 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 185 participants Imipramine: 186 participants Placebo: 169 participants Fluoxetine dose range: 20‐80 mg/day Imipramine dose range: 75‐300 mg/day | |
| Outcomes | HDRS‐21, RDS, Covi Anxiety Scale (CAS), Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Responders reported without denominators. Number and reasons for dropout reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported only if they occurred at least in 5% of the sample. Endpoint mean scores and standard deviations reported |
| Other bias | High risk | Both authors' affiliation was Lilly Research laboratories, and this company produces fluoxetine |
Stephenson 2000.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Patients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 22 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐70 years Exclusion criteria: concurrent treatment for depressive illness, use of other drugs with psychopharmacological effect, serious risk of suicide, significant cardiac, renal or hepatic disease, pregnancy, lactation, absence of contraception. | |
| Interventions | Fluoxetine: 51 participants Dothiepin: 56 participants Fluoxetine dose: 20 mg/day Dothiepine dose range: 75‐150 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), MADRS, Brief Psychiatric Rating Scale (BPRS), Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double dummy", no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons and number of dropouts not clearly reported. Statistical analysis was not on an ITT basis |
| Selective reporting (reporting bias) | Unclear risk | End‐point mean scores and standard deviations reported. Side effects not clearly reported |
| Other bias | High risk | First author's affiliation was Eli Lilly, and this company produces fluoxetine |
Stratta 1991.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Patients with atypical depression according to Quitkin et al (1988) criteria Mean age: 35 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 14 participants Imipramine: 14 participants Fluoxetine dose: 20 mg/day Imipramine dose range: 75‐125 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Clinical Global Impression (CGI), Covi Anxiety Scale (CAS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Withdrawals were reported, but not included in the analysis. Scores reported without denominators. Side effects not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Endpoint mean scores and standard deviations reported |
| Other bias | Unclear risk | Funding: unclear |
Suleman 1997.
| Methods | Six‐week randomised, single‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 17 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 18‐65 years Exclusion criteria: any physical illness or psychiatric diagnosis beside depressive disorder, drug or alcohol abuse, organic mental disorder, pregnancy or lactation, use of any medication except incidental analgesics and current psychotherapy. | |
| Interventions | Fluoxetine: 15 participants Moclobemide: 15 participants Amitriptyline: 15 participants Fluoxetine dose: 20 mg/day Moclobemide dose: 240 mg/day Amitriptyline dose: 100 mg/day | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, quote "the secret codes were only known to the dispenser", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Single blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Single blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Single blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Endpoint mean scores and standard deviation reported. Scores reported with denominators |
| Selective reporting (reporting bias) | Unclear risk | Effects side not clearly reported |
| Other bias | High risk | Quote: "the drugs used in this study were provided by Mess Eli Lilly Company and Hoffman La Roche Company", and these companies produce study drugs |
Suri 2000.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for unipolar major depressive disorder, with a score of at least 14 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 18‐62 years Exclusion criteria: diagnosis of a mood disorder to a secondary general medical condition, bipolar disorder, substance abuse, history of prior treatment with sertraline or fluoxetine. For patients with a history of substance abuse a period of 30 days of sobriety was required prior to study entry. | |
| Interventions | Fluoxetine: 18 participants Sertraline (50 mg): 17 participants Sertraline (100 mg): 17 participants Fluoxetine dose: 20 mg/day Lorazepam (0.5 mg) was allowed | |
| Outcomes | Primary outcome: a HDRS‐21 score of maximum 7 or a Clinical Global Impression (CGI) score of maximum 2 at endpoint (remission) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Sinlge blind, with a blinder rater, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Sinlge blind, with a blinder rater, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Sinlge blind, with a blinder rater, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported with denominators, but withdrawals not included in the analysis. Reasons and number of discontinuation reported |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported without standard deviations. Side effects not reported |
| Other bias | High risk | Quote: "supported in part from Eli Lilly and Company", this company produces fluoxetine |
Tamminen 1989.
| Methods | Five‐week randomised, double‐blind study | |
| Participants | In‐ and outpatients fulfilling RDC (Research Diagnostic Criteria) for unipolar major depressive disorder with a score of at least 17 on the first 17 items of the Hamilton Rating Scale for Depression (HDRS) and a score of at least 8 and equal to or higher than the Covi Anxiety Scale (CAS) score. Age mean: 40.7 (fluoxetine); 42.7 (doxepin). Exclusion criteria: history of drug abuse, concurrent administration of other psychotropic drugs including lithium. | |
| Interventions | Fluoxetine: 26 participants Doxepine: 25 participants Fluoxetine dose range: 40‐80 mg/day Doxepine dose range: 50‐150 mg/day Chloral hydrate and oxazepam were allowed | |
| Outcomes | HDRS, Clinical Global Impression (CGI), Raskin Depression Scale (RDS), CAS | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the study drugs and placebo were supplied in identical capsules" , no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Endpoint mean scores reported without standard deviations. Only most common side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Taner 2006.
| Methods | Eight‐week single‐blind randomised study | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder lasting at least 1 month and for atypical depression. Exclusion criteria: significant suicide risk, pregnancy, lactation or unwillingness to use effective birth control in women, unstable and serious physical illness, a history of seizures, psychosis or organic mental syndrome, substance abuse disorders within the past 6 months, except for nicotine dependence, history of mania, antisocial personality disorder and use of an antidepressant over the previous month. |
|
| Interventions | Fluoxetine: 21 participants
Reboxetine: 22 participants
Fluoxetine dose range: 40‐80 mg/day Reboxetine dose range: 4‐10 mg/day |
|
| Outcomes | Change in the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) score and in Clinical Global Impression (CGI) score | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Single blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "both psychiatrists, one rated the tests and the other who performed the drug follow‐up, were blinded to each other until the end of the study", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Single blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported, mean endpoint scores (HDRS‐17, CGI, Global Assessment of Functioning [GAF]) reported, but data in the text was different from tables |
| Selective reporting (reporting bias) | High risk | Side effects were described, but the whole number of patients reporting side effects was unclear |
| Other bias | Unclear risk | Funding: unclear |
Taneri 1989.
| Methods | Five‐week randomised double‐blind study | |
| Participants | Outpatients with diagnosis of neurotic or reaction depressive disorder on the ICD, with a score of at least 17 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: suicidality, severe organic disease, diabetes mellitus, glaucoma, hyperthyroidism, pregnancy, hypersensitivity to drug, abnormal liver values, organic psychosis, schizophrenia, psychopathy, addiction to alcohol or drugs, seizures. | |
| Interventions | Fluoxetine: 20 participants Nomifensine: 20 participants Fluoxetine dose: 40 mg/day Nomifensine dose: 150 mg/day Chloral hydrate or benzodiazepines for sleep were allowed | |
| Outcomes | HDRS, Clinical Global Impression (CGI), Symptom Check List of Taneri, Patient Global Impression (PGI), Zung Depression Scale (SDS) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported without denominators. Reasons and number of dropouts reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | End point mean scores and standard deviations reported. Only most common side effects reported |
| Other bias | Unclear risk | Funding: unclear |
Thompson 2000.
| Methods | Twelve‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients (general practice) DSM‐III‐R criteria for major unipolar depression, with a score of at least 12 on the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐70 years Exclusion criteria: suicidal ideation, history of treatment resistant depression, bipolar disorder, organic brain disease, substance use disorder, use of antidepressants within the last 6 months, participation to another study within 3 months, medical contraindication to either drug, pregnancy, lactation, absence of contraception, administration of any other psychotropic medication. | |
| Interventions | Fluoxetine: 76 participants Dothiepin: 76 participants Fluoxetine dose: 20 mg/day Dothiepine dose range: 75‐150 mg/day Concomitant use of benzodiazepines was allowed for insomnia | |
| Outcomes | Primary outcomes (all were dichotomised as above or below 80% of full compliance): pill count, patient completed questionnaire, Medication Event Monitoring System Secondary outcomes: HDRS, Short‐Form Health Survey Questionnaire 36 (SF‐36) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Only main reason for dropout reported. Score reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Adverse events not clearly described. End point scores reported without standard deviations |
| Other bias | High risk | Quote: "this study was carried out by Eli Lilly and Company", and this company produces fluoxetine |
Tignol 1993.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Inpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 24 on the Montgomery and Asberg Scale for Depression (MADRS). Age range: 18‐65 years Exclusion criteria: pregnancy or nursing, severe concomitant physical disease, severe risk of suicide, abuse of alcohol or illicit drugs, schizophrenia or psychosis, organic brain syndrome, history of serious allergic drug reaction, treatment with any investigational compound during the previous 6 months, lithium or ECT in the previous 3 months, depot neuroleptics in th previous month, MAOI or oral neuroleptics in the previous 2 weeks, present use of oral anticoagulant or psychotropic drug (except chloral hydrate: 500 mg for sleep). | |
| Interventions | Fluoxetine: 87 participants Paroxetine: 89 participants Fluoxetine dose: 20 mg/day Paroxetine dose: 20 mg/day | |
| Outcomes | MADRS, Hamilton Rating Scale for Anxiety (HAM‐A), Hospital Anxiety and Depression (14 items), Clinical Global Impression (CGI) Severity | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominators of responders were different from number of randomised patients. Reasons and number of dropouts not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | Only most common adverse events are reported. Endpoint scores reported without standard deviations |
| Other bias | High risk | Quote: "this research was supported by SmithKline Beecham Pharmaceuticals" and this company produces paroxetine |
Tollefson 1994.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive unipolar disorder for at least 1 month, non‐psychotic, and subtype as agitated according Research Diagnostic Criteria, with a score of at least 14 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and a score of 2 or more on at least 2 items of the Agitation Rating Scale (ARS). Age range: 18‐65 years old Exclusion criteria: pregnancy, breast feeding, absence of contraception, serious suicidal risk, contraindication to use study drug, concurrent DSM diagnosis such as organic mental disorder, substance use disorder, schizophrenia and related psychotic disorders, bipolar disorder, severe allergies, drug reactions, use of other psychotropic drugs within 4 weeks. | |
| Interventions | Fluoxetine: 62 participants Imipramine: 62 participants Fluoxetine dose range: 20‐80 mg/day Imipramine dose range: 150‐300 mg/day | |
| Outcomes | Primary outcome: change on Hamilton Rating Scale for Depression (HDRS) from baseline to endpoint Secondary outcomes: percentages of responders, remitters and weekly change from baseline, Clinical Global Impression (CGI), Hamilton Rating Scale for Anxiety (HAM‐A), ARS, HDRS‐17 item 3, HDRS‐17 item 9, Patient Global Impression (PGI) | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS‐17 during at least 4 weeks of treatment Remission: endpoint score of maximum 7 on the HDRS‐17 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reason for withdrawals reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. Mean endpoint scores reported with standard deviations |
| Other bias | High risk | Quote: "study funding was provided through a research grant from Lilly Research Laboratories, a division of Eli Lilly and Company" and this company produces fluoxetine |
Tylee 1997.
| Methods | Twelve‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients (general practice) fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least 19 on the Montgomery–Åsberg Depression Rating Scale (MADRS). Age: over 18 years Exclusion criteria: use of study drugs within 1 month of entry, psychosis, organic mental disorder, bipolar depression, acute suicidal risk, use of psychoactive drug or ECT within 1 month of entry, drug or alcohol dependence, history of clinically significant physical disorder, clinically significant abnormalities (ECG, laboratory test), pregnancy, lactation. | |
| Interventions | Fluoxetine: 170 participants Venlafaxine: 171 participants Fluoxetine dose: 20 mg/day Venlafaxine dose: 75 mg/day | |
| Outcomes | Primary outcome: endpoint score on MADRS and Clinical Global Impression (CGI), and Hamilton Rating Scale for Depression (HDRS) Secondary outcomes: Hospital Anxiety and Depression Scale (HADS) | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS or MADRS and a CGI Improvement of 1 or 2 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "eligible patients were randomised by the permuted blocks method", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "identical capsules" and "In order to maintain blinding, a matched placebo was taken in the evening by patients randomised to received fluoxetine", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for discontinuation reported. Denominator reported for responders was different from the number of randomised patients |
| Selective reporting (reporting bias) | High risk | Adverse events reported only if they occurred in at least 5% of the sample. Endpoint scores reported without standard deviations |
| Other bias | High risk | Quote: "this study was funded by Wyeth Laboratories Ltd." This company produces venlafaxine |
Tzanakaki 2000.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Inpatients fulfilling DSM‐IV criteria for major depression, with melancholia and symptoms lasting at least 1 month before study entry, with a score of at least 25 on the Montgomery–Åsberg Depression Rating Scale (MADRS). Age range: 18‐64 years Exclusion criteria: pregnancy, absence of contraception, known sensitivity to venlafaxine or fluoxetine, history of uncontrolled heart failure within the last 6 months, hepatic or renal disease, clinically significant abnormality (ECG, laboratory tests), acute suicide tendencies, history of seizure disorders, any psychotic disorder not associated with depression, history of alcohol or drug dependence within the past year, use of any investigational drug, antipsychotic drug or ECT within 30 days, fluoxetine within 14 days, MAOI or benzodiazepines within 7 days. | |
| Interventions | Fluoxetine: 54 participants
Venlafaxine: 55 participants
Fluoxetine dose: 60 mg/day
Venlafaxine dose: 225 mg/day Temazepam and oxazepam were allowed for sleep |
|
| Outcomes | Primary outcomes: Hamilton Rating Scale for Depression (HDRS), MADRS, Clinical Global Impression (CGI), Severity and improvement scores at each assessment | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS or MADRS and a CGI improvement of 1 or 2 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for discontinuation reported. Scores reported without denominator |
| Selective reporting (reporting bias) | Unclear risk | Only most common adverse events were reported. Mean endpoint scores reported without standard deviations |
| Other bias | High risk | Quote: "this study was supported by a grant from Wyeth‐Ayerst International". This company produces venlafaxine |
Upward 1988.
| Methods | Four‐week randomised, double‐blind study | |
| Participants | Depressed outpatients Age range: 24‐63 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 11 participants Amitriptyline: 12 participants Fluoxetine dose range: 60‐80 mg/day Amitriptyline dose range: 150‐200 mg/day Temazepam (10‐20 mg) was allowed for sleep | |
| Outcomes | Efficacy data not reported. Only dropout rate | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reason for dropouts reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Endpoint scores reported with standard deviation. Adverse events not reported |
| Other bias | High risk | Quote: "we thank Lilly Industries Ltd for financial support and drug assays", this company produces fluoxetine |
Van Moffaert 1995.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for moderate to severe major depression, with a score of at least 18 on the first 17 items of Hamilton Rating Scale for Depression (HDRS) and a score of at least 3 on the Clinical Global Impression. Age range: 18‐80 years old Exclusion criteria: Montgomery–Åsberg Depression Rating Scale (MADRS) score more than 40, suicidal ideation, history of mania, hypomania or psychosis, comorbid severe psychiatric disorder, organic mood disorder, psychotropic drug dependence, pregnancy, lactation, clinically significant renal, hepatic, cardiovascular, respiratory, cerebrovascular disease, use of concomitant serotonergic drug (including lithium and carbamazepine). | |
| Interventions | Fluoxetine: 82 participants Sertraline: 83 participants Fluoxetine dose range: 20‐40 mg/day Sertraline dose range: 50‐100 mg/day Chloral hydrate and short acting benzodiazepines as hypnotics | |
| Outcomes | HDRS, MADRS, Clinical Global Impression (CGI) | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS or MADRS, or a score less than 10 on the HDRS Funding: by industry |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout reported. Responders denominator is different from number of randomised patients |
| Selective reporting (reporting bias) | Unclear risk | Side effects reported only when occurred at least at 4% of the sample. Endpoint scores reported without standard deviations |
| Other bias | High risk | Quote: "we would like to acknowledge the financial and logistic support of Pfizer Belgium", this company produces fluoxetine |
Versiani 1999.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | Inpatients fulfilling DSM‐IV criteria for major depression, with a score of at least 18 on the first 17 items on the HDRS‐21 and a score of at least 18 on the Hamilton Rating Scale for Anxiety (HAM‐A). Age: over 18 years Exclusion criteria: pregnancy, lactation, absence of contraception, suicidal risk, medical disease, history of allergy to study drugs, previous participation to any antidepressant trial, history of unresponsiveness to fluoxetine or amitriptyline, organic mental disorder, substance abuse, bipolar disorder, melancholic disorder, panic or obsessive compulsive disorder, concomitant medication with psychotropic effect. | |
| Interventions | Fluoxetine: 77 participants Amitriptyline : 80 participants Fluoxetine dose: 20 mg/day Amitriptyline dose range: 50‐250 mg/day | |
| Outcomes | HDRS‐21, HAM‐A, Raskin Depression Scale (RDS), Covi Anxiety Scale (CAS), Clinical Global Impression (CGI), Patient Global Impression (PGI) | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS and a decrease of at least 25% in the total score on the HAM‐A Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reason for dropout reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Only most frequent side effects reported. Mean endpoint scores reported with standard deviation |
| Other bias | High risk | Authors' affiliation was Eli lilly Venezuela, and this company produces fluoxetine |
Versiani 2005.
| Methods | Eight‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐IV criteria for major depressive episode, with a score of at least 25 on the first 17 items of the Hamilton Rating Scale for Depression (HDRS). Age range: 18‐65 years Exclusion criteria: bipolar disorder, depressive disorder NOS, anxiety disorder, schizophrenia, adjustment disorder or psychotic symptoms or borderline personality disorder, eating disorder, post‐partum depression, organic mental illness, epilepsy (treatment with anticonvulsive medication for epilepsy or seizures), alcohol or substance abuse in the past 6 months, a decrease of 25% in the HDRS score from the washout period to the screening measurement, duration of the present episode exceeding 12 months, lack of response to two adequate antidepressant drugs for a duration of at least 6 weeks, actual risk of suicide Montgomery–Åsberg Depression Rating Scale (MADRS) item of 5‐6), unstable medical conditions, participation in another clinical trial within 30 days, MAOI within 3 weeks, fluoxetine for the current episode of depression, ECT within 3 months, depot antipsychotic (2 months) or other psychotropic drugs (1 week), use of benzodiazepines within 2 weeks of treatment start (patients receiving a stable dosage could participate if the dosage was kept the same throughout the trial), pregnancy, lactation, absence of contraception. | |
| Interventions | Fluoxetine: 152 participants Mirtazapine: 147 participants Fluoxetine dose range: 20‐40 mg/day Mirtazapine dose range: 30‐60 mg/day | |
| Outcomes | Primary outcome: change in the HDRS score Secondary outcomes: MADRS score and Clinical Global Impression (CGI) score |
|
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons and number of dropouts not clearly reported. Only baseline scores at rating scales (HDRS, MADRS, CGI) reported |
| Selective reporting (reporting bias) | High risk | Adverse events occurring in more 5% reported. Number of responders reported only in figure and without denominators |
| Other bias | High risk | Quote: "this study was supported by Organon NV, The Netherlands", and this company produces mirtazepine |
Wehmeier 2005.
| Methods | Five‐week randomised, double‐blind, parallel group study | |
| Participants | In and out‐patients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 16 on Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age range: 61‐85 years Exclusion criteria: a reduction of HDRS‐17 total score of more than 25% between the screening visit and the baseline visit, serious suicidal risk, severe organic brain disorder, significant organic illness, a history of seizures, a history of schizophrenia and a recent history (in the last year) of drug or alcohol abuse. | |
| Interventions | Fluoxetine: 20 participants Trimipramine: 21 participants Fluoxetine dose: 20 mg/day Trimipramine dose: 150 mg/day | |
| Outcomes | Primary outcome: HDRS‐17 Secondary outcomes: Montgomery–Åsberg Depression Rating Scale (MADRS), the Adjective Mood Scale (AMS), Clinical Global Impression (CGI), Patient Global Impression (PGI) |
|
| Notes | Response: decrease of at least 50% in the total score on the HDRS or a total score on HDRS lower than 10 Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of dropouts reported, but the reasons were unclear. Last observation carried forward analysis (LOCF) was used to report responders |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoint scores reported with standard deviations. Side effects reported |
| Other bias | High risk | Quote: "this work was supported by Lilly Deutschland, Bad Homburg, Germany", and this company produces fluoxetine |
WELL AK1A4006.
| Methods | Eight‐week randomised, double‐blind, multicentre study | |
| Participants | Out‐patients fulfilling DSM‐IV criteria for recurrent major depression, with a score of at least 20 on Hamilton Rating Scale for Depression‐21 item (HDRS‐21) on day ‐1 and ‐7, were currently experiencing a recurrent major depressive episode and had a normal sexual functioning. Mean age Fluoxetine: 37.1 years (SD: 10.7) Mean age Bupropion: 38.6 years (SD: 12.0) Exclusion criteria: not stated |
|
| Interventions | Fluoxetine: 155 participants Bupropion: 158 participants Fluoxetine dose range: 20‐60 mg/day Bupropion dose range: 150‐400 mg/day | |
| Outcomes | Primary outcomes: change in HDRS‐21; incidence of the orgasm dysfunction | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS‐21 between baseline and endpoint Remission: HDRS‐21 total score dropped to less than 8 between baseline and endpoint Funding: by industry |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Mean score at baseline and endpoint not reported |
| Selective reporting (reporting bias) | Unclear risk | Most frequent side effects reported |
| Other bias | High risk | Funding probably by SmithKline, and this company produces bupropion |
Wheatley 1998.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 21 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and a score of at least 2 on the HDRS‐17 item 1. Age range: 18‐75 years Exclusion criteria: bipolar disorder, depressive disorder NOS, anxiety disorder within the last 2 years, schizophrenia, adjustment disorder, schizotypal or borderline personality disorder, eating disorder within the last 2 years, epilepsy, treatment with anticonvulsive medication for seizures, alcohol or substance abuse in the previous year, post‐partum depression within 1 year after delivery, high risk of suicide, unstable medical conditions, non‐responders to antidepressant treatments, use of MAOI within 2 weeks, previous use of fluoxetine for the current episode of depression, ECT within 3 months, continuous use of benzodiazepines, pregnancy, lactation, absence of contraception. | |
| Interventions | Fluoxetine: 67 participants Mirtazapine: 66 participants Fluoxetine dose range: 20‐40 mg/day Mirtazapine dose range: 15‐60 mg/day Temazepam (20 mg) oxazepam (15 mg) and nitrazepam (5 mg) were allowed | |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI), Quality of Life Enjoyment and Satisfaction Questionnaire (Q‐LES‐Q) | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were allocated to treatment with either mirtazepine or fluoxetine, according to the centrally prepared randomisation list", no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "active medication was prepared as indistinguishable looking tablets and packaging was performed using a double dummy technique", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Scores reported without denominators. Reason and numbers of dropouts reported |
| Selective reporting (reporting bias) | Unclear risk | Adverse events occurred in more than 5% of the sample reported. Endpoint scores not reported |
| Other bias | High risk | Quote: "supported by a clinical research grant from NV Organon, Oss, The Netherlands" and this company produces mirtazapine |
Williams 1993.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III criteria for major depressive episode, with a score of at least 17 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21). Age range: 20‐86 years Exclusion criteria: suicide risk, other psychiatric disorder, alcohol abuse, use of MAOI in the previous 2 weeks, use of other antidepressants in the previous week, pregnancy, lactation, known allergy to trial medication. | |
| Interventions | Fluoxetine: 60 participants Moclobemide: 62 participants Fluoxetine dose range: 20‐40 mg/day Moclobemide dose range: 300‐600 mg/day | |
| Outcomes | Primary outcome: HDRS‐21 Secondary outcome: Clinical Global Impression (CGI) |
|
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported, but not included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Only most common side effects were reported. Mean endpoint scores not reported |
| Other bias | Unclear risk | Funding: unclear |
Winokur 2003.
| Methods | Eight‐week double‐blind randomised study | |
| Participants | Patients fulfilling DSM‐IV criteria for major depressive disorder (based on a semi structured clinical interview), a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21) and a score of at least 4 on the 3 HDRS‐21 sleep item. Age range: 18‐75 years Exclusion criteria: patients with an history of primary sleep disorder, significant medical problems, current alcohol or substance abuse or dependence, psychosis, or suicidal ideation. Psychotropic drugs were discontinued at least 1 week before study initiation and no subject received any prolonged‐acting central nervous system agent during the previous month. |
|
| Interventions | Fluoxetine: 8 participants
Mirtazapine: 8 participants
Fluoxetine dose range: 20‐40 mg/day Mirazapine dose range: 15‐45 mg/day |
|
| Outcomes | Change in the HDRS‐21 score and in Clinical Global Impression (CGI) score | |
| Notes | Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, double dummy, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, double dummy, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, double dummy, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout not clearly reported Only HDRS baseline scores reported |
| Selective reporting (reporting bias) | High risk | Side effects not reported. Number of responders not reported |
| Other bias | High risk | Quote: "funds for this study were provided through on unrestricted educational grant from Organon, Inc" and this industry produces venlafaxine |
Wolf 2001.
| Methods | Five‐week randomised, double‐blind two‐centre study | |
| Participants | In‐ and outpatients fulfilling DSM‐III‐R criteria for major depression, with a score of at least 16 on the Hamilton Rating Scale for Depression‐17 item (HDRS‐17). Age: over 60 years Exclusion criteria: serious suicidal risk, glaucoma, chronic urinary retention, prostatic hypertrophy, significant organic illness, severe organic brain disease, history of seizures, schizophrenia, hypo‐ or hyperthyroidism, history of severe allergy, known allergy to imipramine, history of less than 1 year of alcohol or drug abuse. | |
| Interventions | Fluoxetine: 10 participants Trimipramine: 9 participants Fluoxetine dose: 20 mg/day Trimipramine dose: 150 mg/day | |
| Outcomes | HDRS‐17, Montgomery and Asberg Scale for Depression (MADRS) | |
| Notes | This study focuses on sleep related problems Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "since investigators were blind with regard to treatment", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported. Scores reported without denominators |
| Selective reporting (reporting bias) | Unclear risk | Side effects not clearly reported. Mean endpoint scores and standard deviation reported |
| Other bias | High risk | Quote: "this work was supported by Lilly Deutschland", this company produces fluoxetine |
Young 1987.
| Methods | Six‐week randomised, double‐blind multicentre study | |
| Participants | Outpatients fulfilling RDC criteria for moderate‐severe major depression, with a score of at least 18 on the Hamilton Rating Scale for Depression (HDRS). Age: 20‐65 years Exclusion criteria: schizophrenia, organic features, use of antidepressant drugs or ECT during the 4 weeks before. | |
| Interventions | Fluoxetine: 25 participants Amitriptyline: 25 participants Fluoxetine dose range: 40‐80 mg/day Amitriptyline dose range: 50‐150 mg/day | |
| Outcomes | HDRS, Hamilton Rating Scale for Anxiety (HAM‐A), Beck Depression Inventory Scale (BDI) | |
| Notes | Most patients taking sedatives during study Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "both drugs and placebo were identically formulated", no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: " an independent assessor scored the patients", no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number and reasons for dropout reported, but not included in the analysis |
| Selective reporting (reporting bias) | High risk | Only most common side effects were reported. Endpoint scores (HRSD, HAM‐A) not reported |
| Other bias | Unclear risk | Funding: unclear |
Yu 1997.
| Methods | Six‐week randomised, double‐blind study | |
| Participants | Patients with serious depressive disorder Mean age: 51 years Exclusion criteria: not stated | |
| Interventions | Fluoxetine: 8 participants Amitriptyline: 8 participants Fluoxetine dose: 20 mg/day Amitriptyline dose: 150 mg/day | |
| Outcomes | Hamilton Rating Scale for Depression (HDRS), Hamilton Rating Scale for Anxiety (HAM‐A) | |
| Notes | Funding: unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind, no further information |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided, impossible to evaluate |
| Selective reporting (reporting bias) | Unclear risk | No information provided, impossible to evaluate |
| Other bias | Unclear risk | Funding: unclear |
Zhao 2006 a.
| Methods | Six‐week randomised, open label trial | |
| Participants | Patients fulfilling DSM‐IV criteria for major depression, with a score of at least 18 on Hamilton Rating Scale for Depression‐17 item (HDRS‐17) and a HDRS retardation score of at least 8 (assessed using the score of the first item of depressed mood, the 7th item of interest and activity, the 8th item of retardation and 14 item of decreased sexuality in the HDRS scale. Age: over 18 years Exclusion criteria: past history of any manic or hypomanic episode, any medical record listed as follows: disease in heart, liver, kidney, immune system, neural system, blood system, narrow‐angle glaucoma, past history of allergic reaction to the studied drugs, being medicated with MAOIs in the past 2 weeks, being treated with ECT in the past 6 months, recurrent suicidal ideation or a suicide attempt, lack of therapeutic reaction to fluoxetine or trazodone in past 6 months. | |
| Interventions | Fluoxetine: 61 participants Trimipramine: 59 participants Fluoxetine dose range: 20‐80 mg/day Trimipramine dose range: 100‐300 mg/day | |
| Outcomes | Reduction in HDRS‐17 and reduction in HDRS‐17 retardation factor score; total score of Symptom Checklist‐90‐R (SCL‐90‐R) energy related 5 items; medical outcomes study‐Short Form (SF‐36); 4) cognitive function | |
| Notes | Response: decrease of at least 50% in the total score on the HDRS‐17 from baseline Funding: by industry | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information |
| Allocation concealment (selection bias) | Unclear risk | No information provided |
| Blinding (performance bias and detection bias) All outcomes | High risk | Open label trial, no further information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open label trial, no further information |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open label trial, no further information |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Number and reasons for dropout not clearly reported. Number of the responders not reported |
| Selective reporting (reporting bias) | High risk | Endpoint scores reported with standard deviations. Side effects reported only with percentage |
| Other bias | High risk | Quote: "this study was financially supported by Eli Lilly Asia, Inc", and this company produces fluoxetine |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Baca Baldomero 2005 | Wrong design trial |
| Bitrain 2011 | Wrong design trial |
| Brasseur 1989 | Not RCT |
| Cohn 1989 | Not meeting inclusion criteria |
| Ducher 2008 | Wrong design trial |
| Goodnick 1987 | Wrong design trial: randomisation to different doses of fluoxetine, without any drug comparator |
| Gu 2001 | Not fulfilled inclusion criteria |
| Hunter 2006 | Randomisation to fluoxetine or placebo, no drug comparator |
| Iovieno 2011 | Randomisation to fluoxetine or placebo, no drug comparator |
| Kroenke 2001 | Wrong diagnosis |
| Musgnung 2005 | Wrong design trial: not RCT |
| Nemetz 2005 | Wrong drug comparison: neither antidepressant nor herbal product |
| Peveler 2005 | Wrong design trial |
| Roose 1994 | Wrong design trial: not RCT |
| Schmidt 1999 | Long‐term treatment of depression |
| Serrano‐Blanco 2006 | Wrong inclusion criteria: diagnosis of dysthymia |
| Simon 1996 | Not meeting inclusion criteria |
| Simon 1998 | Not meeting inclusion criteria |
| Simon 1999 | Not meeting inclusion criteria |
| Strik 1998 | Wrong design trial |
Characteristics of studies awaiting assessment [ordered by study ID]
Chen 2006.
| Methods | Six‐week randomised, double‐blind, double dummy clinical trial |
| Participants | Patients with depressive disorder |
| Interventions | 48 participants randomised to fluoxetine or reboxetine Fluoxetine dose: 20 mg/day Reboxetine dose: 8 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
GSK 29060/134.
| Methods | Randomised, double‐blind study |
| Participants | Patients with major depression with associated anxiety |
| Interventions | Fluoxetine and paroxetine |
| Outcomes | Montgomery and Asberg Scale for Depression (MADRS), Hamilton Rating Scale for Anxiety (HAM‐A) |
| Notes | Waiting for translation from Portuguese to English |
Huang 2006a.
| Methods | Eight‐week, randomised study |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Citalopram: 30 participants Fluoxetine: 30 participants |
| Outcomes | Hamilton Depression Rating Scale (HDRS), Clinical Global Impression (CGI) and Treatment Emergent Symptom Scale (TESS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Huang 2006b.
| Methods | Six‐week, randomised study |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Citalopram: 26 participants Fluoxetine: 25 participants Citalopram dose‐range: 20‐60 mg/day Fluoxetine dose‐range: 20‐60 mg/day |
| Outcomes | Hamilton Depression Rating Scale 17‐Item (HDRS‐17) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Jiang 2006.
| Methods | Six‐week randomised, double‐blind, double dummy multicentre trial |
| Participants | Patients with depressive disorder |
| Interventions | Fluoxetine: 145 participants Kaiyuanshen: 144 participants Fluoxetine dose: 20 mg/day Kaiyuanshen dose: 1440 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Li 2005.
| Methods | Six‐week randomised, double‐blind, double dummy multicentre trial |
| Participants | Patients with depressive disorder |
| Interventions | 144 participants randomised to fluoxetine or bupropion 61 patients in Bupropion group and 64 in fluoxetine group completed the study Fluoxetine dose: 20 mg/day Bupropion dose: 300 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Li 2006a.
| Methods | Six‐week randomised, double‐blind, double dummy trial |
| Participants | Patients with depressive disorder, with a score of at least 18 at Hamilton Depression Rating Scale (HDRS) and a score of 14 or greater at the Hamilton Anxiety Scale (HAM‐A) |
| Interventions | 137 participants randomised to fluoxetine or reboxetine 64 patients in Reboxetine group and 68 in fluoxetine group completed the study Fluoxetine dose: 20 mg/day Reboxetine dose: 8 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS), Hamilton Rating Scale for Anxiety (HAM‐A) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Li 2006b.
| Methods | Six‐week randomised, double‐blind, double dummy trial |
| Participants | Patients with depressive disorder |
| Interventions | 228 participants randomised to fluoxetine or reboxetine Fluoxetine dose: unclear Reboxetine dose: unclear |
| Outcomes | Hamilton Depression Rating Scale (HDRS) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Li 2006c.
| Methods | Four‐week, randomised study |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Sixty patients randomised to fluoxetine or reboxetine Fluoxetine dose range: 4‐8 mg Reboxetine dose range: 20‐40 mg |
| Outcomes | Hamilton Depression Rating Scale (HDRS) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Liang 2005.
| Methods | Eight‐week, (likely) randomised study |
| Participants | In‐ and outpatients with depression according to CCMD‐III criteria |
| Interventions | Citalopram: 30 participants Fluoxetine: 30 participants Citalopram dose range: 10‐60 mg/day Fluoxetine: dose range: 10‐40 mg/day |
| Outcomes | Change in Hamilton Depression Rating Scale 24 Item (HDRS‐24) from baseline to endpoint, number of patients who responded to treatment |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Licinio 2004.
| Methods | Eight‐week randomised study |
| Participants | Out‐patients fulfilling DSM‐IV criteria for unipolar major depressive disorder, with a score of at least 18 on the Hamilton Depression Rating Scale‐21 item (HDRS‐21)
Age range: 18‐70 years Exclusion criteria: active suicidal risk or history of life‐threatening suicide attempts |
| Interventions | 272 participants randomised to fluoxetine or desipramine Fluoxetine dose range: 10‐40 mg/day Desipramine dose range: 50‐200 mg/day |
| Outcomes | HDRS‐21. |
| Notes | Funding:unclear. |
Ma 2007.
| Methods | Randomised double‐blind, double dummy multicentre trial |
| Participants | Patients with depressive disorder |
| Interventions | 228 participants randomised to fluoxetine or bupropion |
| Outcomes | Hamilton Depression Rating Scale‐17 item (HDRS‐17) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
NCT00909155.
| Methods | Randomised, double‐blind, controlled study |
| Participants | Patients with depressive disorder |
| Interventions | Participants randomised to fluoxetine or venlafaxine Fluoxetine: 10 participants Venlafaxine: 17 participants |
| Outcomes | Mood and Anxiety Symptoms Questionnaire (MASQ‐AD), Hamilton Depression Rating Scale (HDRS); fMRI response to an emotional regulation task |
| Notes | Funding: by National Grants and industry |
Qin 2006.
| Methods | Eight‐week randomised single‐blind trial |
| Participants | First episode depressive patients |
| Interventions | Eighty participants randomised to fluoxetine or venlafaxine Fluoxetine dose range: 20‐40 mg/day Venlafaxine dose range: 75‐225 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Sackeim 2006.
| Methods | Randomised (unpublished) multi‐site trial |
| Participants | Out‐patients with depression according to DSM‐IV criteria Age range: 18‐60 years |
| Interventions | Fluoxetine: 173 participants Sertraline: 177 participants Fluoxetine range dose: 20‐ 80 mg/day Sertraline range dose: 50‐150mg/day |
| Outcomes | Hamilton Depression Rating Scale‐24 item (HDRS‐24) |
| Notes | Funding: by industry |
Salehi 2009.
| Methods | Randomised clinical trial |
| Participants | Patients with depression according to DSM‐IV criteria |
| Interventions | Fluoxetine: 40 participants Imipramine: 40 participants Fluoxetine dose: 20 mg/day Imipramine dose: 100 mg/day |
| Outcomes | Unclear |
| Notes | Waiting for translation from Arabic to English (only abstract available in English) |
Shen 2005.
| Methods | Randomised double‐blind, multicentre trial |
| Participants | Patients with depressive disorder |
| Interventions | Fluoxetine: 113 participants Reboxetine: 109 participants Fluoxetine dose: 20 mg/day Reboxetine dose: 8 mg/day |
| Outcomes | Hamilton Depression Rating Scale‐17 item (HDRS‐17), Hamilton Rating Scale for Anxiety (HAM‐A), Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Stassen 1999.
| Methods | Six‐week randomised study |
| Participants | Patients fulfilling DSM‐III criteria for major depressive disorder, with a score of at least 15 on the Hamilton Depression Rating Scale‐17 item (HDRS‐17). Age range: 18‐86 years old |
| Interventions | Fluoxetine: 440 participants Moclobemide: 437 participants Fluoxetine dose: 20‐40 mg/day Moclobemide dose: 300‐600 mg/day |
| Outcomes | HDRS‐17, Clinical Global Impression (CGI) |
| Notes | Funding: probably by industry |
Su 2006.
| Methods | Twelve‐week, (likely) randomised study |
| Participants | Patients with first episode major depression |
| Interventions | Fluoxetine: 40 participants Venlafaxine: 40 participants |
| Outcomes | Hamilton Depression rating scale‐17 item (HDRS‐17), Hamilton Rating Scale for Anxiety (HAM‐A), Wechsler Adult Intelligence Scale (WAIS), Wisconsin Card Sorting Test (WCST) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Sun 2005.
| Methods | Six‐week, randomised study |
| Participants | Patients with depression |
| Interventions | Fluoxetine: 51 participants Venlafaxine: 51 participants Fluoxetine dose‐rage: 20‐40 mg/day Venlafaxine dose‐rage: 50‐200 mg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Sun 2006.
| Methods | Eight‐week, (likely) randomised study |
| Participants | Patients with depression |
| Interventions | Sixty participants randomised to fluoxetine or venlafaxine |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Tan 1997.
| Methods | Randomised, double‐blind study |
| Participants | Patients with depressive disorder |
| Interventions | Eighteen participants randomised to fluoxetine or amitriptyline |
| Outcomes | Unclear |
| Notes | Waiting for translation from Chinese to English |
Wang 2006.
| Methods | Six‐week, randomised study |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Sixty patients randomised to fluoxetine or venlafaxine |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Wang 2007a.
| Methods | Unclear |
| Participants | Patients with depressive disorder |
| Interventions | Participants assigned to fluoxetine or paroxetine |
| Outcomes | Quality of life |
| Notes | Waiting for translation from Chinese to English |
Wang 2007b.
| Methods | Six‐week, randomised, double‐blind, double dummy study |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | 48 participants randomised to fluoxetine or bupropion |
| Outcomes | Hamilton Depression rating scale (HDRS), Hamilton Rating Scale for Anxiety (HAM‐A) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Wang 2009.
| Methods | Randomised, double‐blind, double dummy multicentre study |
| Participants | Patients with mild or moderate depression |
| Interventions | High‐dose of Morinda Officinalis Oligose capsule: 119 participants Low‐dose of Morinda Officinalis Oligose capsule: 119 participants Fluoxetine: 118 participants |
| Outcomes | Hamilton Depression Rating Scale‐17 item (HDRS‐17) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Wang 2011.
| Methods | Six‐week randomised study |
| Participants | Out‐patients with major depressive disorder (MDD) |
| Interventions | 117 participants randomised to fluoxetine or venlafaxine |
| Outcomes | Hamilton Depression Rating Scale‐21 item (HDRS‐21) |
| Notes | Abstract from conference |
Xiao 2005.
| Methods | Six‐week, (likely) randomised study |
| Participants | Patients with depression according to CCMD‐II‐R criteria |
| Interventions | Sixty participants randomised to fluoxetine or venlafaxine |
| Outcomes | Hamilton Depression rating scale (HDRS) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Xu 2010.
| Methods | Randomised, open trial |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Sixty patients randomised to fluoxetine or escitalopram Fluoxetine dose range: 20‐40 mg/day Escitalopram dose range: 10‐20bmg/day |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Zhao 2005.
| Methods | Six‐week randomised study |
| Participants | Patients with senile depressive disorder |
| Interventions | 50 participants randomised to fluoxetine or citalopram |
| Outcomes | Hamilton Depression Rating Scale‐17 item (HDRS‐17) and Clinical Global Impression (CGI) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Zhao 2006.
| Methods | Eight‐week randomised, single‐blind trial |
| Participants | Patients with depression according to CCMD‐III criteria |
| Interventions | Citalopram: 30 participants Fluoxetine: 30 participants Citalopram dose range: 20‐60 mg/day Fluoxetine dose range: 20‐60 mg/day |
| Outcomes | Hamilton Depression Rating Scale‐24 item (HDRS‐24) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Zhou 2005.
| Methods | Eight‐week randomised, single‐blind trial |
| Participants | First episode depressive patients aged 60 years or over |
| Interventions | Sixty‐four patients randomised to fluoxetine or venlafaxine |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Zhu 2005.
| Methods | Six‐week randomised trial |
| Participants | Elderly patients with depression |
| Interventions | Sixty patients randomised to fluoxetine or mirtazapine |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Zhu 2006.
| Methods | Six‐week randomised trial |
| Participants | Patients with depression according to CCMD‐III criteria, aged 60 years or over |
| Interventions | Fluoxetine: 23 participants Mirtazapine: 23 participants |
| Outcomes | Hamilton Depression Rating Scale (HDRS) |
| Notes | Waiting for translation from Chinese to English (only abstract available in English) |
Characteristics of ongoing studies [ordered by study ID]
ChiCTR‐TRC‐11001668.
| Trial name or title | ChiCTR‐TRC‐11001668 |
| Methods | Eight‐week, randomised, double‐blind, multicentre study with parallel groups |
| Participants | Outpatients suffering from moderate to severe Major Depressive Disorder, according DSM‐IV‐TR criteria Age range: 18 to 65 years Exclusion criteria: all types of depression other than major depressive disorder and all other psychiatric disorders. Pregnancy, breastfeeding or possibility of becoming pregnant during the study without an effective contraception. |
| Interventions | Agomelatine: 314 participants Fluoxetine: 314 participants Agomelatine dosage range: 25‐50 mg Fluoxetine dosage range: 20‐40 mg |
| Outcomes | Change from baseline to end‐point on Depression‐Hamilton Depression Rating Scale‐17 items (HDRS‐17), Clinical Global (CGI) Improvement, Leeds Sleep Evaluation Questionnaire (LSEQ) and Hamilton Anxiety Rating Scale (HAM‐A) |
| Starting date | August 2006 |
| Contact information | Shu Liang, Prof, +86 10 65610341‐308, shu‐liang@126.com |
| Notes | Funding: by industry |
CTRI/2011/05/001719.
| Trial name or title | CTRI/2011/05/001719 |
| Methods | Eight week, randomised, open‐label study |
| Participants | Patients fulfilling DSM‐IV criteria for Major Depression Disorder, with a score of at least 20 on the Hamilton Depression Rating Scale‐17 item (HDRS‐17), a score of 2 in the first item of the HDRS‐17 at screening and baseline and a score of at least 4 on Clinical Global Impression of Severity (CGI‐S) at the enrolment visit. Exclusion criteria: history of bipolar disorder (I or II), schizophrenia, schizoaffective disorder, eating disorder or obsessive‐compulsive disorder. Patients recording more than 20% reduction on HDRS‐17 score at baseline (at the time of study allocation) as against the same recorded at the time of screening. Patients not responding to the administration of an appropriate dose of two different earlier antidepressant treatments (including fluoxetine) for at least 4 weeks each, for the current and earlier episodes, or not responding to fluoxetine monotherapy for at least 4 weeks. Substance or alcohol abuse in the last 30 days or dependence in the last 6 months,or with a risk of suicidal behavior (scoring 3 on item N°3 of HDRS‐17). Concomitant psychotropic medication, neurologic disorders or serious or uncontrolled diseases, hepatic insufficiency or renal insufficiency, clinically significant abnormalities on physical examination or laboratory test. Patients having participated in any type of clinical study within in the last one month of the screening date, pregnancy, breastfeeding, absence of adequate contraception measures. |
| Interventions | Fluoxetine dose range: 20‐40 mg IN‐ASTR‐001 dose range: 25‐50 mg |
| Outcomes | HDRS‐17 and CGI‐S |
| Starting date | April 2011 |
| Contact information | Kanhei Charan Sahoo, MD, 07966523302, kanheicharan_sahoo@intaspharma.com |
| Notes | Funding: by industry |
EUCTR2007‐002130‐11‐ES.
| Trial name or title | EUCTR2007‐002130‐11‐ES. |
| Methods | Randomised, single‐blind study. |
| Participants | Patients fulfilling DSM‐IV criteria for Major Depression Disorder, with a score of at least 14 on the Hamilton Depression Rating Scale‐17 item (HDRS‐17) and resistant to a SSRI (administered at correct dose for at least 6 weeks). Exclusion criteria: treatment with any antidepressant drugs or psychotherapy, and to be resistant to any investigational drug. |
| Interventions | Fluoxetine dose: 20 mg Venlafaxine range dose: 75‐150 mg Nortriptyline dose: 25 mg |
| Outcomes | HDRS‐17 |
| Starting date | February 2008 |
| Contact information | https://www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2007‐002130‐11 |
| Notes | Funding: by academy |
NCT01204086.
| Trial name or title | NCT01204086. |
| Methods | Six‐week open‐label, randomised trial |
| Participants | Patients fulfilling DSM‐IV criteria for Major Depression Disorder, with a score of at least 16 on the Hamilton Depression Rating Scale (HDRS). Age range: 16‐65 years old. Exclusion criteria: monoamine oxidase inhibitor or antidepressant treatment within two weeks prior to entering the study, diagnosis of substance abuse within the past three months, an organic mental disease, mental retardation or dementia, a serious surgical condition or physical illness, pregnancy, breastfeeding. |
| Interventions | Fluoxetine range dose: 20‐80 mg Venlafaxine range dose 75‐ 225 mg |
| Outcomes | HDRS |
| Starting date | March 2007 |
| Contact information | Po See Chen, MD, +886‐6‐2353535 ext 5213, chenps@mail.ncku.edu.tw |
| Notes | Funding: by academy |
NCT01254305.
| Trial name or title | NCT01254305. |
| Methods | Eight week, randomised, double‐blind study |
| Participants | Patients fulfilling DSM‐IV criteria for major depression disorder, with a minimum duration of 4 weeks. Age range: 18‐65 years old. Exclusion criteria: patients with a suicide risk, history or current diagnosis (DSM‐IV) of any manic or hypomanic episode, schizophrenia or any other psychotic disorder, obsessive‐compulsive disorder, pregnancy, breastfeeding, absence of adequate contraception measures. |
| Interventions | Fluoxetine dose range: 20‐60 mg F2695 SR dose range: 40‐120 mg |
| Outcomes | Clinical Global Impression (CGI) Severity, Patient Global Impression (PGI) Severity |
| Starting date | April 2011 |
| Contact information | Carl Gommoll, MS, Forest Laboratories |
| Notes | Funding: by industry |
Differences between protocol and review
In this update of the review we applied the risk of bias tool to assess the quality of all included studies. However, a formal comparison of intervention effects according to risk of bias was not performed as for most studies the risk of bias was rated as unclear.
A dosage subgroup analysis was not performed as this can be more appropriately examined in a MTM meta‐analysis (see discussion).
Summary of findings tables using the GRADE methodology were added.
Contributions of authors
LRM, CG, MP collected the data; LRM, MP, AC and CB ran the analysis; LRM, CG, MP, DP, TAF, AC and CB drafted and critically revised the manuscript.
Sources of support
Internal sources
Department of Public Health and Community Medicine, Section of Psychiatry, University of Verona, Italy.
Department of Psychiatry, University of Oxford, UK.
External sources
No sources of support supplied
Declarations of interest
LRM, CG, MP, DP, AC, CB: none declared. TAF has received honoraria for speaking at continuing medical education (CME) meetings sponsored by Asahi Kasei, Eli Lilly, GlaxoSmithKline, Mochida, MSD, Otsuka, Pfizer, Shionogi and Tanabe‐Mitsubishi. He is a diplomate of the Academy of Cognitive Therapy. He has received royalties from Igaku‐Shoin, Seiwa‐Shoten and Nihon Bunka Kagakusha. He is on the advisory board for Sekisui Chemicals and Takeda Science Foundation. The Japanese Ministry of Education, Science, and Technology; the Japanese Ministry of Health, Labor and Welfare; and the Japan Foundation for Neuroscience and Mental Health have funded his research projects.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Aguglia 1993 {published data only}
- Aguglia E, Casacchia M, Cassano GB, Faravelli C, Ferrari G, Giordano P, et al. Double‐blind study of the efficacy and safety of sertraline versus fluoxetine in major depression. International Clinical Psychopharmacology 1993;8(3):197‐202. [DOI] [PubMed] [Google Scholar]
Akhondzadeh 2003 {published data only}
- Akhondzadeh S, Faraji H, Sadeghi M, Afkham K, Fakhrzadeh H, Kamalipour A. Double‐blind comparison of fluoxetine and nortriptyline in the treatment of moderate to severe major depression. Journal of Clinical Pharmacy and Therapeutics 2003;28(5):379‐84. [DOI] [PubMed] [Google Scholar]
Akhondzadeh Basti 2007 {published data only}
- Akhondzadeh Basti A, Moshiri E, Noorbala AA, Jamshidi AH, Abbasi SH, Akhondzadeh S. Comparison of petal of Crocus sativus L. and fluoxetine in the treatment of depressed outpatients: a pilot double‐blind randomised trial. Progress in Neuropsychopharmacology & Biological Psychiatry 2007;31(2):439‐42. [DOI] [PubMed] [Google Scholar]
Alby 1993 {published data only}
- Alby JM, Cabane J, Ferreri M, Bougerol T. Efficacy and acceptability of tianeptine in major depressive disorder and in dysthymia (DSM‐IIIR) with somatic complaints: double blind study versus fluoxetine. European Neuropsychopharmacology 1993;33(3):333. [Google Scholar]
- Alby JM, Ferreri M, Cabane J, Bodinat C, Dagens V. Efficacy of tianeptine (Stablon ®) for the treatment of major depression and dysthymia with somatic complaints. A comparative study versus fluoxetine. Annales De Psychiatrie 1993;8(2):136‐41. [Google Scholar]
Altamura 1989 {published data only}
- Altamura AC, Novellis F, Guercetti G, Invernizzi G, Percudani M, Montgomery SA. Fluoxetine compared with amitriptyline in elderly depression: a controlled clinical trial. International Journal of Clinical Pharmacology Research 1989;9(6):391‐6. [PubMed] [Google Scholar]
- Altamura AC, Percudani M, Guercetti G, Invernizzi G. Efficacy and tolerability of fluoxetine in the elderly: a double‐blind study versus amitriptyline. International Clinical Psychopharmacology 1989;Suppl 1:103‐6. [PubMed] [Google Scholar]
Alves 1999 {published data only}
- Alves C, Cachola I, Brandao J. Efficacy and tolerability of venlafaxine and fluoxetine in outpatients with major depression. Primary Care Psychiatry 1999;5(2):57‐63. [Google Scholar]
Amini 2005 {published data only}
- Amini H, Aghayan S, Jalili SA, Akhondzadeh S, Yahyazadeh O, Pakravan‐Nejad M. Comparison of mirtazapine and fluoxetine in the treatment of major depressive disorder: a double‐blind, randomised trial. Journal of Clinical Pharmacy and Therapeutics 2005;30(2):133‐8. [DOI] [PubMed] [Google Scholar]
Andreoli 2002 {published data only}
- Andreoli V, Caillard V, Deo RS, Rybakowski JK, Versiani M. Reboxetine, a new noradrenaline selective antidepressant, is at least as effective as fluoxetine in the treatment of depression. Journal of Clinical Psychopharmacology 2002;22(4):393‐9. [DOI] [PubMed] [Google Scholar]
- Bosc M, Dubini A, Polin V. Development and validation of a social functioning scale, the Social Adaptation Self‐evaluation Scale. European Neuropsychopharmacology 1997;7 Suppl 1:57‐70. [DOI] [PubMed] [Google Scholar]
- Dubini A, Bosc M, Polin V. Do noradrenaline and serotonin differentially affect social motivation and behaviour?. European Neuropsychopharmacology 1997;Suppl 1:S49‐55; discussion S71‐3. [DOI] [PubMed] [Google Scholar]
- Dubini A, Bosc M, Polin V. Noradrenaline‐selective versus serotonin‐selective antidepressant therapy: differential effects on social functioning. Journal of Psychopharmacology 1997;11 Suppl 4:17‐23. [PubMed] [Google Scholar]
- Healy DT. Reboxetine ‐ the first selective noradrenaline reuptake inhibitor. 151st Annual Meeting of the American Psychiatric Association. Toronto, Ontario, Canada. 1998.
- Massana J. Reboxetine versus fluoxetine: benefits in social function. 152nd Annual Meeting of the American Psychiatric Association, Washington DC. 1999.
- Moller H. Reboxetine, the first selective noradrenaline reuptake inhibitor, is more effective at improving social functioning than fluoxetine. 152nd Annual Meeting of the American Psychiatric Association, Washington DC. 1999.
- Versiani M, Montgomery SA, Stahl S, Schwartz G. [The effect of reboxetine on anxiety]. 39th Annual Meeting of the American College of Neuropsychopharmacology, San Juan, Puerto Rico. 2000.
- Versiani MV, Montgomery SA, Stahl SM, Schwartz GE. Effect of the novel selective noradrenaline reuptake inhibitor reboxetine on anxiety. 154th Annual Meeting of the American Psychiatric Association, New Orleans, LA. 2001.
Ansseau 1994 {published data only}
- Ansseau M, Papart P, Troisfontaines B, Bartholome F, Bataille M, Charles G, et al. Controlled comparison of milnacipran and fluoxetine in major depression. Psychopharmacology 1994;114(1):131‐7. [DOI] [PubMed] [Google Scholar]
Armitage 1997 {published data only}
- Armitage R, Yonkers K, Cole D, Rush AJ. A multicenter, double blind comparison of the effects of nefazodone and fluoxetine on sleep architecture and quality of sleep in depressed outpatients. Journal of Clinical Psychopharmacology 1997;17(3):161‐8. [DOI] [PubMed] [Google Scholar]
- Armitage R, Yonkers K, Rush A, Cole D, Novak K. Comparison of the effects of nefazodone and fluoxetine on sleep architecture and sleep efficiency in depressed patients. 8th ECNP (European College of Neuropsychopharmacology) Congress, Venice, Italy. 1995.
- Rush AJ, Armitage R, Gillin JC, Yonkers KA, Winokur A, Moldofsky H, et al. Comparative effects of nefazodone and fluoxetine on sleep in outpatients with major depressive disorder. Biological Psychiatry 1998;44(1):3‐14. [DOI] [PubMed] [Google Scholar]
Bakish 1997 {published data only}
- Bakish D, Cavazzoni P, Chudzik J, Ravindran A, Hrdina PD. Effects of selective serotonin reuptake inhibitors on platelet serotonin parameters in major depressive disorder. Biological Psychiatry 1997;41(2):184‐90. [DOI] [PubMed] [Google Scholar]
Basterzi 2009 {published data only}
- Başterzi AD, Yazici K, Aslan E, Delialioğlu N, Taşdelen B, Tot Acar S, et al. Effects of fluoxetine and venlafaxine on serum brain derived neurotrophic factor levels in depressed patients. Progress in Neuropsychopharmacology & Biological Psychiatry 2009;33(2):281‐5. [DOI] [PubMed] [Google Scholar]
Beasley 1993a {published data only}
- Beasley CM Jr, Holman SL, Potvin JH. Fluoxetine compared with imipramine in the treatment of inpatient depression. A multicenter trial. Annals of Clinical Psychiatry 1993;5(3):199‐207. [DOI] [PubMed] [Google Scholar]
Behnke 2002 {published data only}
- Behnke K, Jensen GS, Graubaum HJ, Gruenwald J. Hypericum perforatum versus fluoxetine in the treatment of mild to moderate depression. Advances in Therapy 2002;19(1):43‐52. [DOI] [PubMed] [Google Scholar]
Bennie 1995 {published data only}
- Bennie EH, Mullin JM, Martindale JJ. A double‐blind multicenter trial comparing sertraline and fluoxetine in outpatients with major depression. Journal of Clinical Psychiatry 1995;56(6):229‐37. [PubMed] [Google Scholar]
- Flament MF, Lane R. Acute antidepressant response to fluoxetine and sertraline in psychiatric outpatients with psychomotor agitation. International Journal of Psychiatry in Clinical Practice 2001;5(2):103‐9. [DOI] [PubMed] [Google Scholar]
- Flament MF, Lane RM, Zhu R, Ying Z. Predictors of an acute antidepressant response to fluoxetine and sertraline. International Clinical Psychopharmacology 1999;14(5):259‐75. [PubMed] [Google Scholar]
Berlanga 1997 {published data only}
- Berlanga C, Arechavaleta B, Heinze G, Campillo C, Torres M, Caballero A, Apiquian R, Castelli P. A double‐blind comparison of nefazodone and fluoxetine in the treatment of depressed outpatients. Salud Mental 1997;20(3):1‐8. [Google Scholar]
- Berlanga C, Heinze G, Campillo C, Torres M, Caballero A, Apiquián R. Nefazodone and fluoxetine in major depression ‐ a comparison study. Xth World Congress of Psychiatry, Madrid, Spain. 1996.
- Berlanga C, Heinze G, Torres M, Apiquian R, Caballero A. Personality and clinical predictors of recurrence of depression. Psychiatric Services 1999;50(3):376‐80. [DOI] [PubMed] [Google Scholar]
Besancon 1993 {published data only}
- Besancon G, Cousin R, Guitton B, Lavergne F. Double‐blind study of mianserin and fluoxetine in ambulatory therapy of depressed patients. Encephale 1993;19(4):341‐5. [PubMed] [Google Scholar]
- Lavergne F, Berlin I. Symptoms profile during mianserin and fluoxetine treatment in depressed patients. VIth World Congress of Biological Psychiatry, Nice, France. 1997.
- Lavergne F, Berlin I, Payan C, Besancon G. Clinical differences in response to mianserin and fluoxetine in depressive patients. VIIIth European College of Neuropsychopharmacology Congress, Venice, Italy. 1995.
- Lavergne F, Berlin I, Payan C, Besancon G. Clinical differences in response to mianserin and fluoxetine in depressive patients. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
Bhurgri 2011 {published data only}
- Bhurgri GR, Korejo HB, Qureshi MA. Role of fluoxetine and nortriptyline in major depressive disorders. Rawal Medical Journal 2011;36(1):38‐40. [Google Scholar]
Bjerkenstedt 2005 {published data only}
- Bjerkenstedt L, Edman GV, Alken RG, Mannel M. Hypericum extract LI 160 and fluoxetine in mild to moderate depression: a randomised, placebo‐controlled multi‐centre study in outpatients. European Archives of Psychiatry and Clinical Neuroscience 2005;255(1):40‐7. [DOI] [PubMed] [Google Scholar]
Bougerol 1997a {published data only}
- Bougerol T, Scotto JC, Patris M, Strub N, Lemming O, Hopfner Petersen HE. Citalopram and fluoxetine in major depression: Comparison of two clinical trials in a psychiatrist setting and in general practice (first trial). Clinical drug Investigation 1997;14(2):77‐89. [Google Scholar]
- Patris M, Bouchard JM, Bougerol T, Charbonnier JF, Chevalier JF, Clerc G, et al. Citalopram versus fluoxetine: a double‐blind, controlled, multicenter, phase III trial in patients with unipolar major depression treated in general‐practice. International Clinical Psychopharmacology 1996;11(2):129‐36. [PubMed] [Google Scholar]
Bougerol 1997b {published data only}
- Bougerol T, Scotto J‐C, Patris M, Strub N, Lemming O, Hopfner Petersen HE. Citalopram and fluoxetine in major depression: Comparison of two clinical trials in a psychiatrist setting and in general practice (second trial). Clinical drug Investigation 1997;14(2):77‐89. [Google Scholar]
Bowden 1993 {published data only}
- Bowden CL, Schatzberg AF, Rosenbaum A, Contreras SA, Samson JA, Dessain E, et al. Fluoxetine and desipramine in major depressive disorder. Journal of Clinical Psychopharmacology 1993;13(5):305‐11. [PubMed] [Google Scholar]
- Rosenbaum AH, Schatzberg AF, Bowden CL, Samson JA, Schildkraut JJ. MHPG as a predictor of clinical response to fluoxetine. Clinical Neuropharmacology 1992;15:209. [DOI] [PubMed] [Google Scholar]
- Schatzberg AF. Noradrenergic versus serotonergic antidepressants: predictors of treatment response. Journal of Clinical Psychiatry 1998;59 Suppl 14:15‐8. [PubMed] [Google Scholar]
Boyer 1998 {published data only}
- Bisserbe JC, Boyer P, Souetre E, Hotton JM, Troy S. A six month sertraline fluoxetine comparative study in depressed outpatients: outcome and costs. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Boyer P. Dysthymic patients and measurements of change. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Boyer P, Danion JM, Bisserbe JC, Hotton JM, Troy S. Clinical and economic comparison of sertraline and fluoxetine in the treatment of depression A 6‐month double‐blind study in a primary‐care setting in France. PharmacoEconomics 1998;13:157‐69. [DOI] [PubMed] [Google Scholar]
- Danion JM, Boyer P, Troy S, Hotton J. Double blind randomized comparative study of sertraline and fluoxetine in depressive outpatient: Medico‐economic aspect. VIth World Congress of Biological Psychiatry, Nice, France. 1997.
Bremner 1984 {published data only}
- Bremner JD. Fluoxetine in depressed patients: a comparison with imipramine. Journal of Clinical Psychiatry 1984;45(10):414‐9. [PubMed] [Google Scholar]
Bressa 1989 {published data only}
- Bressa GM, Brugnoli R, Pancheri P. A double‐blind study of fluoxetine and imipramine in major depression. International Clinical Psychopharmacology 1989;4 Suppl 1:69‐73. [PubMed] [Google Scholar]
Byerley 1988 {published data only}
- Byerley WF, Reimherr FW, Wood DR, Grosser BI. Fluoxetine, a selective serotonin uptake inhibitor, for the treatment of outpatients with major depression. Journal of Clinical Psychopharmacology 1988;8(2):112‐5. [PubMed] [Google Scholar]
- Reimherr FW, Ward MF, Byerley WF. The introductory placebo washout: a retrospective evaluation. Psychiatry Research 1989;30(2):191‐9. [DOI] [PubMed] [Google Scholar]
Cassano 2002 {published data only}
- Cassano GB, Puca F, Scapicchio PL, Trabucchi M. Paroxetine and fluoxetine effects on mood and cognitive functions in depressed nondemented elderly patients. Journal of Clinical Psychiatry 2002;63(5):396‐402. [PubMed] [Google Scholar]
Chouinard 1985 {published data only}
- Beasley CM Jr, Sayler ME, Potvin JH. Fluoxetine versus amitriptyline in the treatment of major depression: a multicenter trial. International Clinical Psychopharmacology 1993;8(3):143‐9. [DOI] [PubMed] [Google Scholar]
- Chouinard G. A double‐blind controlled clinical trial of fluoxetine and amitriptyline in the treatment of outpatients with major depressive disorder. Journal of Clinical Psychiatry 1985;46:32‐7. [PubMed] [Google Scholar]
Chouinard 1999 {published data only}
- Chouinard G, Saxena B, Belanger MC, Ravindran A, Bakish D, Beauclair L, et al. A Canadian multicenter, double‐blind study of paroxetine and fluoxetine in major depressive disorder. Journal of Affective Disorders 1999;54(1‐2):39‐48. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. A Double‐Blind, Randomized, Multicentre Comparison of Paroxetine and Fluoxetine in the Treatment of Patients With Major Depression With Regard to Antidepressant Efficacy, Tolerance and Anxiolytic Effect. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 1993.
CL3‐022 {unpublished data only}
- European Medicines Agency. CHMP Assessment Report for Thymanax [Procedure No. EMEA/H/C/000916, Doc. Ref. EMEA/97539/2009].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000915/WC500046226.pdf [Accessed 22 October 2012] 2008. [Google Scholar]
- European Medicines Agency. CHMP Assessment Report for Thymanax [Procedure No. EMEA/H/C/000916, Doc. Ref. EMEA/97539/2009].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000916/WC500038315.pdf [Accessed 25 October 2012] 2009. [Google Scholar]
- European Medicines Agency. CHMP Assessment Report for Valdoxan [Procedure No. EMEA/H/C/656, Doc. Ref. EMEA/CHMP/87018/2006].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000656/WC500070527.pdf [Accessed 22 January 2013] 2006. [Google Scholar]
- Hickie IB, Rogers NL. Novel melatonin‐based therapies: potential advances in the treatment of major depression. Lancet 2011;378:621‐31. [DOI] [PubMed] [Google Scholar]
CL3‐024 {unpublished data only}
- European Medicines Agency. CHMP Assessment Report for Thymanax [Procedure No. EMEA/H/C/000916, Doc. Ref. EMEA/97539/2009].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000915/WC500046226.pdf [Accessed 22 October 2012] 2008. [Google Scholar]
- European Medicines Agency. CHMP Assessment Report for Thymanax [Procedure No. EMEA/H/C/000916, Doc. Ref. EMEA/97539/2009].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000916/WC500038315.pdf [Accessed 25 October 2012] 2009.
- European Medicines Agency. CHMP Assessment Report for Valdoxan [Procedure No. EMEA/H/C/656, Doc. Ref. EMEA/CHMP/87018/2006].http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000656/WC500070527.pdf [Accessed 22 January 2013] 2006. [Google Scholar]
- Hickie IB, Rogers NL. Novel melatonin‐based therapies: potential advances in the treatment of major depression. Lancet 2011;378:621‐31. [DOI] [PubMed] [Google Scholar]
Clayton 2003 {published data only}
- Clayton A, Ferguson J, Reisner J, Brown M, Schwartz G, Zajecka J. Reboxetine and sexual side effects. European Neuropsychopharmacology 2002;12 Suppl 3:205. [Google Scholar]
- Clayton AH, Zajecka J, Ferguson JM, Filipiak‐Reisner JK, Brown MT, Schwartz GE. Lack of sexual dysfunction with the selective noradrenaline reuptake inhibitor reboxetine during treatment for major depressive disorder. International Clinical Psychopharmacology 2003;18(3):151‐6. [DOI] [PubMed] [Google Scholar]
- Clayton AH, Zajecka JM, Ferguson JM, Reisner JK, Brown MT, Schwartz GE. Reboxetine and sexual side effects. 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA. 2002.
Clerc 1994 {published data only}
- Clerc GE, Ruimy P, Verdeau Palles J. A double‐blind comparison of venlafaxine and fluoxetine in patients hospitalised for major depression and melancholia. International Clinical Psychopharmacology 1994;9(3):139‐43. [DOI] [PubMed] [Google Scholar]
- Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of unidimensional subscales derived from the Hamilton Depression Rating Scale to antidepressant drug effects. Journal of Psychiatric Research 2002;36(6):437‐48. [DOI] [PubMed] [Google Scholar]
- Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. British Journal of Psychiatry 2001;178:234‐41. [DOI] [PubMed] [Google Scholar]
Cohn 1985 {published data only}
- Beasley CM Jr, Sayler ME, Bosomworth JC, Wernicke JF. High‐dose fluoxetine: efficacy and activating‐sedating effects in agitated and retarded depression. Journal of Clinical Psychopharmacology 1991;11(3):166‐74. [PubMed] [Google Scholar]
- Cohn JB, Wilcox C. A comparison of fluoxetine, imipramine, and placebo in patients with major depressive disorder. Journal of Clinical Psychiatry 1985;46:26‐31. [PubMed] [Google Scholar]
Corne 1989 {published data only}
- Corne SJ, Hall JR. A double‐blind comparative study of fluoxetine and dothiepin in the treatment of depression in general‐practice. International Clinical Psychopharmacology 1989;4(3):245‐54. [DOI] [PubMed] [Google Scholar]
Corrigan 2000 {published data only}
- Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depression and Anxiety 2000;11(2):58‐65. [DOI] [PubMed] [Google Scholar]
Costa e Silva 1998 {published data only}
- Costa e Silva J. Randomized, double‐blind comparison of venlafaxine and fluoxetine in outpatients with major depression. Journal of Clinical Psychiatry 1998;56(7):352‐7. [DOI] [PubMed] [Google Scholar]
- Michelson D. Comparing the efficacy and safety of fluoxetine and venlafaxine in outpatient depression. Journal of Clinical Psychiatry 1999;60(5):338‐9. [DOI] [PubMed] [Google Scholar]
- Paton C. Venlafaxine versus fluoxetine. Journal of Clinical Psychiatry 1999;60(10):708. [DOI] [PubMed] [Google Scholar]
Dalery 1997 {published data only}
- Dalery J, Rochat C, Peyron E, Bernard G. Comparative study of the efficacy and acceptability of amineptine and fluoxetine in patients with major depression. Encephale 1992;18(3):257‐62. [PubMed] [Google Scholar]
- Dalery J, Rochat C, Peyron E, Bernard G. The efficacy and acceptability of amineptine versus fluoxetine in major depression. International Clinical Psychopharmacology 1997;12 Suppl 3:S35‐8. [DOI] [PubMed] [Google Scholar]
Dalery 2003 {published data only}
- Dalery J. Fluvoxamine and fluoxetine in the treatment of depression; similarities and differences. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Dalery J, Honig A. Fluvoxamine versus fluoxetine in major depressive episode: a double‐blind randomised comparison. Human Psychopharmacology 2003;18(5):379‐84. [DOI] [PubMed] [Google Scholar]
Debus 1988 {published data only}
- Beasley CM Jr, Dornseif BE, Pultz JA, Bosomworth JC, Sayler ME. Fluoxetine versus trazodone: efficacy and activating‐sedating effects. Journal of Clinical Psychiatry 1991;52(7):294‐9. [PubMed] [Google Scholar]
- Debus JR, Rush AJ, Himmel C, Tyler D, Polatin P, Weissenburger J. Fluoxetine versus trazodone in the treatment of outpatients with major depression. Journal of Clinical Psychiatry 1988;49(11):422‐6. [PubMed] [Google Scholar]
De Jonghe 1991 {published data only}
- Jonghe F, Ravelli DP, Tuynman Qua H. A randomised, double‐blind study of fluoxetine and maprotiline in the treatment of major depression. Pharmacopsychiatry 1991;24(2):62‐7. [DOI] [PubMed] [Google Scholar]
Demyttenaere 1998 {published data only}
- Demyttenaere K, Mesters P, Boulanger B, Dewe W, Delsemme MH, Gregoire J, et al. Adherence to treatment regimen in depressed patients treated with amitriptyline or fluoxetine. Journal of Affective Disorders 2001;65(3):243‐52. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Ganse E, Gregoire J, Gaens E, Mesters P. Compliance in depressed patients treated with fluoxetine or amitriptyline. International Clinical Psychopharmacology 1998;13(1):11‐7. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Ganse E, Gregoire P, Mesters P. Compliance among patients suffering from major depressive disorder and treated by fluoxetine or amitriptyline. VIth World Congress of Biological Psychiatry, Nice, France. 1997.
Demyttenaere 2004 {published data only}
- Demyttenaere K, Albert A, Mesters P, Dewé W, Bruyckere K, Sangeleer M. What happens with adverse events during 6 months of treatment with selective serotonin reuptake inhibitors?. Journal of Clinical Psychiatry 2005;66(7):859‐63. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Bruffaerts R, Albert A, et al. Development of an antidepressant compliance questionnaire. Acta Psychiatrica Scandinavica 2004;110(3):201‐7. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Mesters P, Dewe W, Boulanger B, Bruyckere K, Sangeleer M, et al. Six month compliance with fluoxetine or paroxetine treatment in depressed outpatients. European Neuropsychopharmacology 2002;12 Suppl 3:208. [Google Scholar]
De Nayer 2002 {published data only}
- Nayer A, Clercq M, Mignon A. Symptom relief obtained with venlafaxine versus fluoxetine in depressed patients with concomitant anxiety. European Neuropsychopharmacology 2000;10 Suppl 3:242. [Google Scholar]
- Nayer A, Geerts S, Ruelens L, Schittecatte M, Bleeker E, Eeckhoutte I, et al. Venlafaxine compared with fluoxetine in outpatients with depression and concomitant anxiety. International Journal of Neuropsychopharmacology 2002;5(2):115‐20. [DOI] [PubMed] [Google Scholar]
- Preud'homme X, Nayer A, Clercq M, Mignon A. Symptom relief obtained with venlafaxine versus fluoxetine in major depressed patients with concomitant anxiety. International Journal of Neuropsychopharmacology 2000;3 Suppl 1:233. [Google Scholar]
De Ronchi 1998 {published data only}
- Ronchi D, Rucci P, Lodi M, Ravaglia G, Forti P, Volterra V. Fluoxetine and amitriptyline in elderly depressed patients: a 10‐week, double‐blind study on course of neurocognitive adverse events and depressive symptoms. Archives of Gerontology and Geriatrics 1998;Suppl 6:125‐1,40. [Google Scholar]
De Wilde 1993 {published data only}
- Wilde J, Spiers R, Mertens C, Bartholome F, Schotte G, Leyman S. A double‐blind, comparative, multicenter study comparing paroxetine with fluoxetine in depressed patients. Acta Psychiatrica Scandinavica 1993;87(2):141‐5. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline. Double‐blind Comparative Multicentre Study Comparing Paroxetine b.d. (twice daily) with Fluoxetine b.d. (twice daily) in Depressed Patients. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com), [29060/064]. clinicaltrials.gov. 1990.
Diaz Martinez 1998 {published data only}
- Diaz Martinez A, Benassinni O, Ontiveros A, Gonzalez S, Salin R, Basquedano G, et al. A randomised, open‐label comparison of venlafaxine and fluoxetine in depressed outpatients. Clinical Therapeutics 1998;20(3):467‐76. [DOI] [PubMed] [Google Scholar]
Dierick 1996 {published data only}
- Dierick M, Ravizza L, Realini R, Martin A. A double‐blind comparison of venlafaxine and fluoxetine for treatment of major depression in outpatients. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 1996;20(1):57‐71. [DOI] [PubMed] [Google Scholar]
- Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of unidimensional subscales derived from the Hamilton Depression Rating Scale to antidepressant drug effect. Journal of Psychiatric Research 2002;36(6):437‐48. [DOI] [PubMed] [Google Scholar]
- Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. British Journal of Psychiatry 2001;178:234‐41. [DOI] [PubMed] [Google Scholar]
Dowling 1990 {published data only}
- Dowling B, Webb MG, Halpin CM, Sangiwa MG. Fluoxetine: a comparative study with dothiepin. Irish Journal of Psychiatry 1990;11(1):3‐7. [Google Scholar]
Duarte 1996 {published data only}
- Duarte A, Mikkelsen H, Delini Stula A. Moclobemide versus fluoxetine for double depression: A randomised double‐blind study. Journal of Psychiatric Research 1996;30(6):453‐8. [DOI] [PubMed] [Google Scholar]
Fabre 1991 {published data only}
- Fabre LF, Scharf MB, Itil TM. Comparative efficacy and safety of nortriptyline and fluoxetine in the treatment of major depression: a clinical study. Journal of Clinical Psychiatry 1991;52(1):62‐7. [PubMed] [Google Scholar]
Fairweather 1999 {published data only}
- Fairweather DB, Stanley N, Hindmarch I. Fluoxetine and dothiepin in depressed patients: a comparison of efficacy and effects on cognition and subjective sleep. Xth European College of Neuropsychopharmacology Congress, Vienna, Austria. 1997.
- Fairweather DB, Stanley N, Yoon JS, Hindmarch I. The effects of fluoxetine and dothiepin on cognitive function in depressed patients in general practice. Human Psychopharmacology 1999;14(5):325‐32. [Google Scholar]
Falk 1989 {published data only}
- Falk WE, Rosenbaum JF, Otto MW, Zusky PM, Weilburg JB, Nixon RA. Fluoxetine versus trazodone in depressed geriatric patients. Journal of Geriatric Psychiatry and Neurology 1989;2(4):208‐14. [DOI] [PubMed] [Google Scholar]
Fava 1998 {published data only}
- Fava M, Amsterdam JD, Deltito JA, Salzman C, Schwaller M, Dunner DL. A double‐blind study of paroxetine, fluoxetine, and placebo in outpatients with major depression. Annals of Clinical Psychiatry 1998;10(4):145‐50. [DOI] [PubMed] [Google Scholar]
Fava 2002 {published data only}
- Fava M, Hoog SL, Judge RA, Kopp JB, Nilsson ME, Gonzales JS. Acute efficacy of fluoxetine versus sertraline and paroxetine in major depressive disorder including effects of baseline insomnia. Journal of Clinical Psychopharmacology 2002;22(2):137‐47. [DOI] [PubMed] [Google Scholar]
- Fava M, Rosenbaum JF, Hoog SL, Krebs W, Dillon JA. Comparison of symptoms after treatment interruption: Evidence of fluoxetine, sertraline and paroxetine. XXIst Collegium Internationale Neuro‐Psychopharmacologicum, Glasgow, Scotland. 1998.
- Fava M, Rosenbaum JF, Hoog SL, Tepner RG, Kopp JB, Sayler M. Fluoxetine versus sertraline and paroxetine in major depression: efficacy and tolerability in patients with high or low baseline insomnia. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Fava M, Rosenbaum JF, Hoog SL, Tepner RG, Kopp JB, Sayler M. Fluoxetine versus sertraline and paroxetine in major depression: tolerability and efficacy in patients with low and high baseline insomnia. IXth Congress of the Association of European Psychiatrists, Copenhagen, Denmark. 1998.
- Hoog SL, Fava M, Rosenbaum JF, Tepner RG, Kopp JB, Sayler M. Fluoxetine vs. sertraline and paroxetine in major depression in patients with high or low baseline insomnia. XXIst Collegium Internationale Neuro‐Psychopharmacologicum, Glasgow, Scotland. 1998.
Fava 2005 {published data only}
- Farabaugh A, Mischoulon D, Fava M, Wu SL, Mascarini A, Tossani E, Alpert J. The relationship between early changes in the HAM‐17 anxiety/somatization factor items and treatment outcome among depressed outpatients. International Clinical Psychopharmacology 2005;20(2):87‐91. [DOI] [PubMed] [Google Scholar]
- Fava M, Alpert J, Nierberg AA, Mischoulon D, Otto MW, Zajecka J, Murck H, Rosenbaum JF. A double‐blind, randomised trial of St. John's Wort, Fluoxetine and placebo in major depressive disorder. Journal of Clinical Psychoparmacology 2005;25(5):441‐7. [DOI] [PubMed] [Google Scholar]
- Fava M, Alpert JE, Nierenberg AA, Mischoulon D, Otto MW, Murck H, et al. A double‐blind, randomized trial of St. John’s Wort, fluoxetine, and placebo in MDD. 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA. 2002.
- Murck H, Fava M, Alpert J, Nierenberg AA, Mischoulom D, Otto MW, Zajecka J, Mannel M, Rosenbaum JF. Hypericum extract in patients with MDD and reversed vegetative signs: re‐analysis from data of a double‐blind, randomised trial of hypericum extract, fluoxetine and placebo. International Journal of Neuropsychopharmacology 2005;8(2):215‐21. [DOI] [PubMed] [Google Scholar]
- Papakostas GI, Crawford CM, Scalia MJ, Fava M. Timing of clinical Improvment and symptom resolution in the treatment of major depressive disorder. Neuropsychobiology 2007;56(2‐3):132‐7. [DOI] [PubMed] [Google Scholar]
- Schulz V. A study on the efficacy of the Hypericum extract LI 160 in 135 depressive patients in the USA [US‐studie zur Wirksamkeit des Hypericum‐Extraktes LI 160 bei 135 Depressiven Patienten]. Zeitschrift fur Phytotherapie 2006;27(3):124‐5. [Google Scholar]
- Tossani E, Mascarini A, Alpert JE, Mischoulon D, Papakostas GI, Ryan JL, et al. Relationship between anxious depression and treatment outcome in outpatients with depression. 157th Annual Meeting of the American Psychiatric Association, New York, NY. 2004.
Fawcett 1989 {published data only}
- Fawcett J, Zajecka JM, Kravitz HM, Edwards J, et al. Fluoxetine versus amitriptyline in adult outpatients with major depression. Current Therapeutic Research 1989;45(5):821‐32. [Google Scholar]
Feighner 1985a {published data only}
- Feighner JP, Cohn JB. Double‐blind comparative trials of fluoxetine and doxepin in geriatric patients with major depressive disorder. Journal of Clinical Psychiatry 1985;46:20‐5. [PubMed] [Google Scholar]
Feighner 1985b {published data only}
- Fairweather DB, Kerr JS, Harrison DA, Moon CA, et al. A double blind comparison of the effects of fluoxetine and amitriptyline on cognitive function in elderly depressed patients. Human Psychopharmacology 1993;8(1):41‐7. [Google Scholar]
- Feighner JP. A comparative trial of fluoxetine and amitriptyline in patients with major depressive disorder. Journal of Clinical Psychiatry 1985;46(9):369‐72. [PubMed] [Google Scholar]
- Kerr JS, Fairweather DB, Hindmarch I. Effects of fluoxetine on psychomotor performance, cognitive function and sleep in depressed patients. International Clinical Psychopharmacology 1993;8(4):341‐3. [DOI] [PubMed] [Google Scholar]
Feighner 1989 {published data only}
- Feighner JP, Boyer WF, Merideth CH, Hendrickson GG. A double‐blind comparison of fluoxetine, imipramine and placebo in outpatients with major depression. International Clinical Psychopharmacology 1989;4(2):127‐34. [DOI] [PubMed] [Google Scholar]
Feighner 1991 {published data only}
- Feighner JP, Gardner EA, Johnston JA, Batey SR, Khayrallah MA, Ascher JA, et al. Double‐blind comparison of bupropion and fluoxetine in depressed outpatients. Journal of Clinical Psychiatry 1991;52(8):329‐35. [PubMed] [Google Scholar]
Ferreri 1989 {published data only}
- Ferreri M. Fluoxetine versus amineptine in the treatment of outpatients with major depressive disorders. International Clinical Psychopharmacology 1989;4 Suppl 1:97‐101. [PubMed] [Google Scholar]
Finkel 1999 {published data only}
- Doraiswamy PM, Krishnan KR, Clary C. Efficacy and safety of sertraline in depressed geriatric patients with vascular disease. 151st Annual Meeting of the American Psychiatric Association, Toronto, Ontario, Canada. 1998.
- Doraiswamy PM, Krishnan KR, Oxman T, Jenkyn LR, Coffey DJ, Burt T, et al. Does antidepressant therapy improve cognition in elderly depressed patients?. Journals of Gerontology Series A‐Biological Sciences & Medical Sciences 2003;58(12):1137‐44. [DOI] [PubMed] [Google Scholar]
- Finkel SI, Richter EM, Clary CM, Batzar E. Comparative efficacy of sertraline vs Fluoxetine in patients age 70 or over with major depression. American Journal of Geriatric Psychiatry 1999;7(3):221‐7. [DOI] [PubMed] [Google Scholar]
- Krishnan KR, Doraiswamy PM, Clary CM. Clinical and treatment response characteristics of late‐life depression associated with vascular disease: A pooled analysis of two multicenter trials with sertraline. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2001;25(2):347‐61. [DOI] [PubMed] [Google Scholar]
- Newhouse P, Finkel S, Richter E. SSRIs in the treatment of depressed out patients aged 70 years and older. VIIIth ECNP (European College of Neuropsychopharmacology) Congress, Venice, Italy. 1995.
- Newhouse P, Ko G, Richter E. Comparison of sertraline and fluoxetine in depressed geriatric outpatients: plasma levels and efficacy. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Newhouse PA, Krishnan KR, Doraiswamy PM, Richter EM, Batzar ED, Clary CM. A double‐blind comparison of sertraline and fluoxetine in depressed elderly outpatients. Journal of Clinical Psychiatry 2000;61(8):559‐68. [DOI] [PubMed] [Google Scholar]
Gagiano 1993 {published data only}
- Gagiano CA. A double blind comparison of paroxetine and fluoxetine in patients with major depression. British Journal of Clinical Research 1993;4:145‐52. [Google Scholar]
- GlaxoSmithKline. A double‐blind comparative multicentre study comparing paroxetine b.d. (twice daily) with fluoxetine (Prozac®) b.d. (twice daily) in depressed patients. [29060/079]. clinicaltrials.gov. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 1991.
Gattaz 1995 {published data only}
- Gattaz WF, Vogel P, Kick H, Delini‐Stula A, Kohler M, Mikkelsen H, et al. Comparative efficacy and tolerability of moclobemide and fluoxetine in major depression. Pharmacopsychiatry 1995;28:179. [Google Scholar]
- Gattaz WF, Vogel P, Kick H, Kohnen R. Moclobemide versus fluoxetine in the treatment of inpatients with major depression. Journal of Clinical Psychopharmacology 1995;15 Suppl 2:35S‐40S. [DOI] [PubMed] [Google Scholar]
- Gattaz WF, Vogel P, Kick H, Kohnen R. Moclobemide versus fluoxetine in the treatment of major depression. Xth World Congress of Psychiatry, Madrid, Spain. 1996. [DOI] [PubMed]
Geerts 1994 {published data only}
- Geerts S, Bruynooghe F, Cuyper H, Demeulemeester F, Haazen L. Moclobemide versus fluoxetine for major depressive episodes. Clinical Neuropharmacology 1994;7 Suppl 1:50‐7. [DOI] [PubMed] [Google Scholar]
Geretsegger 1994 {published data only}
- Geretsegger C, Bohmer F, Ludwig M. Paroxetine in the elderly depressed patient: randomised comparison with fluoxetine of efficacy, cognitive and behavioural effects. International Clinical Psychopharmacology 1994;9(1):25‐9. [PubMed] [Google Scholar]
- GlaxoSmithKline. A Double‐blind Comparative Study Comparing Paroxetine b.d. (twice daily) with Fluoxetine b.d. (twice daily) in Geriatric Patients with Major Depression. [29060/061]. clinicaltrials.gov. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 1991.
- Schone W, Ludwig M. A double‐blind study of paroxetine compared with fluoxetine in geriatric patients with major depression. Journal of Clinical Psychopharmacology 1993;13 Suppl 2:34‐9. [DOI] [PubMed] [Google Scholar]
- Schone W, Ludwig M. Paroxetine in the treatment of depression in geriatric patients. A double‐blind comparative study with fluoxetine. Fortschritte der Neurologie Psychiatrie 1994;62 Suppl 1:16‐8. [PubMed] [Google Scholar]
Ghaeli 2004 {published data only}
- Ghaeli P, Shahsavand E, Mesbahi M, Avarsaji K. Comparing the effects of 8‐week treatment with fluoxetine and imipramine on fasting blood glucose of patients with major depressive disorder. XII World Congress of Psychiatry, Yokohama, Japan. 2002. [DOI] [PubMed]
- Ghaeli P, Shahsavand E, Mesbahi M, Kamkar MZ, Sadeghi M, Dashti‐Khavidaki S. Comparing the effects of 8‐week treatment with fluoxetine and imipramine on fasting blood glucose of patients with major depressive disorder. Journal of Clinical Psychopharmacology 2004;24(4):386‐8. [DOI] [PubMed] [Google Scholar]
Gillin 1997 {published data only}
- Gillin JC, Rapaport M, Erman MK, Winokur A, Albala BJ. A comparison of nefazodone and fluoxetine on mood and on objective, subjective, and clinician‐rated measures of sleep in depressed patients: a double‐blind, 8‐week clinical trial. Journal of Clinical Psychiatry 1997;58(5):185‐92. [DOI] [PubMed] [Google Scholar]
Ginestet 1989 {published data only}
- Ginestet D. Fluoxetine in endogenous depression and melancholia versus clomipramine. International Clinical Psychopharmacology 1989;4 Suppl 1:37‐40. [PubMed] [Google Scholar]
Goldstein 2002 {published data only}
- Demitrack MA, Goldstein DJ, Mallinckrodt C, Lu Y. Efficacy and safety of duloxetine treatment of major depression. 154th Annual Meeting of the American Psychiatric Association, New Orleans; LA. 2001.
- Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack MA. Duloxetine in the treatment of major depressive disorder: a double‐blind clinical trial. Journal of Clinical Psychiatry 2002;63(3):225‐31. [DOI] [PubMed] [Google Scholar]
GSK 29060/356 {unpublished data only}
- GlaxoSmithKline. A double‐blind, multicenter study to compare paroxetine and fluoxetine in the treatment of patients with major depressive disorder with regard to antidepressant efficacy, effects on associated anxiety and tolerability. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 1994.
- GlaxoSmithKline. Extension phase for a double‐blind, multicentre study to compare paroxetine and fluoxetine in the treatment of patients with major depressive disorder with regard to antidepressant efficacy, effects on associated anxiety and tolerability. GSK ‐ Clinical Study Register 1994 (www.gsk‐clinicalstudyregister.com) 1994.
Guelfi 1998 {published data only}
- Guelfi JD, Ansseau M, Corruble E, Samuelian JC, Tonelli I, Tournoux A, et al. A double‐blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatients. International Clinical Psychopharmacology 1998;13(3):121‐8. [DOI] [PubMed] [Google Scholar]
Guelfi 1999 {published data only}
- Guelfi JD. Fluoxetine versus tianeptine in elderly depressed. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Guelfi JD, Bouhassira M, Bonett‐Perrin E, Lancrenon S. Study of the efficacy of fluoxetine versus tianeptine in the treatment of elderly depressed patients, followed in general practice. Encephale 1999;25(3):265‐70. [PubMed] [Google Scholar]
Hale 2010 {published data only}
- Hale A. Efficacy and safety of agomelatine (25 mg/day with potential adjustment to 50 mg) given orally for eight weeks in out‐patients with severe major depressive disorder: a randomised double‐blind, parallel groups, international study versus selective serotonin reuptake inhibitor (SSRI) with a double‐blind extension period of 16 weeks. Controlled‐Trials.com 2008 (http://www.controlled‐trials.com/ISRCTN19313268/servier).
- Hale A, Corral RM, Mencacci C, Ruiz JS, Severo CA, Gentil V. Superior antidepressant efficacy results of agomelatine versus fluoxetine in severe MDD patients: a randomised double‐blind study. International Clinical Psychopharmacology 2010;25(6):305‐14. [DOI] [PubMed] [Google Scholar]
Harrer 1999 {published data only}
- Harrer G, Schmidt U, Kuhn U, Biller A. Comparison of equivalence between the st John's wort extract lohyp‐57 and fluoxetine. Arzneimittelforschung 1999;49(4):289‐96. [DOI] [PubMed] [Google Scholar]
Hashemi 2012 {published data only}
- Hashemi S, Shirazi HG, Mohammadi A, Zadeh‐Bagheri G, Noorian Kh, Malekzadeh M. Nortriptyline versus fluoxetine in the treatment of major depressive disorder: a six‐month, double‐blind clinical trial. Clinical Pharmacology 2012;4:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hong 2003 {published data only}
- Hong CJ, Hu WH, Chen CC, Hsiao CC, Tsai SJ, Ruwe FJL. A double‐blind, randomised, group‐comparative study of the tolerability and efficacy of 6 weeks' treatment with mirtazapine or fluoxetine in depressed Chinese patients. Journal of Clinical Psychiatry 2003;64:921‐6. [DOI] [PubMed] [Google Scholar]
Hosak 2000 {published data only}
- Hosak L, Tuma I. Anxiety in depression. Homeostasis in Health and Disease 1997;38(3):114‐9. [Google Scholar]
- Hosak L, Tuma I. Comparative study of three antidepressants: Preliminary results. Homeostasis in Health and Disease 1996;37(3):138‐9. [Google Scholar]
- Hosak L, Tuma I, Hanus H. A comparative study of three antidepressants with a different mechanism of action in hospitalised patients. Ceska a Slovenska Psychiatrie 1999;95:146‐56. [Google Scholar]
- Hosak L, Tuma I, Hanus H. D‐fenfluramine test in depression: Final results. Ceska a Slovenska Psychiatrie 2000;96(3):126‐30. [Google Scholar]
- Hosák L, Tůma I, Hanus H, Straka L. Costs and outcomes of use of amitriptyline, citalopram and fluoxetine in major depression: exploratory study. Acta Medica (Hradec Kralove) 2000;43(4):133‐7. [PubMed] [Google Scholar]
Jakovijevic 1996 {published data only}
- Jakovljevic M, Vujic D, Aleksandrovsky Y, Szerenberger W, Faltus F, Henigsberg N. Efficacy and tolerability of fluoxetine compared with maprotiline in major depressive episode: Results of Central‐East European Study. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Jakovljevic M, Vujic D, Henigsberg N, Aleksandrovsky Y, Faltus F, Szerenberger W. Central‐East European study of fluoxetine (Portal, Lek) and maprotiline (Ladiomil, Pliva) in major depressive episode. Psychiatria Danubina 1996;8 Suppl 1:1‐5. [Google Scholar]
- Jakovljevic M, VujicD, Henigsberg N. Serotonergic (fluoxetine) versus noradrenergic (maprotiline) antidepressants and suicidality. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
Joyce 2002 {published data only}
- Harley J, Roberts R, Joyce P, Mulder R, Luty S, Frampton C, Kennedy M. Orosomucoid influences the response to antidepressants in major depressive disorder. Journal of Psychopharmacology 2010;24(4):531‐5. [DOI] [PubMed] [Google Scholar]
- Joyce PR, Luty SE, Mulder RT, McKenzie JM, Miller AL, Kennedy MA. Self mutilation, suicide attempts and borderline personality disorder in depressed patients. Australian and New Zealand Journal of Psychiatry, Abstracts of the 40th Congress of the Royal Australian and New Zealand College of Psychiatrists, Sydney, Australia 2005;39(Suppl 1):A107. [Google Scholar]
- Joyce PR, Mulder RT, Luty SE, Sullivan PF, McKenzie JM, Abbott RM, Stevens IF. Patterns and predictors of remission, response and recovery in major depression treated with fluoxetine or nortriptyline. Australian and New Zealand Journal of Psychiatry 2002;36(3):384‐91. [DOI] [PubMed] [Google Scholar]
- Mulder RT, Joyce PR, Frampton CM, Luty SE, Sullivan PF. Six months of treatment for depression: outcome and predictors of the course of illness. The American Journal of Psychiatry 2006;163(1):95‐100. [DOI] [PubMed] [Google Scholar]
- Porter R, Mulder RT, Joyce PR, Luty SE. Tryptophan and tyrosine availability and response to antidepressant treatment in major depression. Journal of Affective Disorders 2005;86(2‐3):129‐34. [DOI] [PubMed] [Google Scholar]
Judd 1993 {published data only}
- Judd FK, Moore K, Norman TR, Burrows GD, Gupta RK, Parker G. A multicenter double blind trial of fluoxetine versus amitriptyline in the treatment of depressive illness. Australian and New Zealand Journal of Psychiatry 1993;27(1):49‐55. [DOI] [PubMed] [Google Scholar]
- Norman TR, Gupta RK, Burrows GD, Parker G, Judd FK. Relationship between antidepressant response and plasma concentrations of fluoxetine and norfluoxetine. International Clinical Psychopharmacology 1993;8(1):25‐9. [DOI] [PubMed] [Google Scholar]
Kasper 2005 {published data only}
- Kasper S, Lemming OM, Swart H. Escitalopram in the long‐term treatment of major depressive disorder in elderly patients. Neuropsychobiology 2006;54(3):152‐9. [DOI] [PubMed] [Google Scholar]
- Kasper S, Swart H, Friis Andersen H. Escitalopram in the treatment of depressed elderly patients. The American Journal of Geriatric Psychiatry 2005;13(10):884‐91. [DOI] [PubMed] [Google Scholar]
Keegan 1991 {published data only}
- Keegan D, Bowen RC, Blackshaw S, Saleh S, Dayal N, Remillard F, et al. A comparison of fluoxetine and amitriptyline in the treatment of major depression. International Clinical Psychopharmacology 1991;6(2):117‐24. [DOI] [PubMed] [Google Scholar]
Keller 2007 {published data only}
- Dunlop BW, Li T, Kornstein SG, Friedman ES, Rothschild AJ, Pedersen R, et al. Concordance between clinician and patient ratings as predictors of response, remission, and recurrence in major depressive disorder [NCT00046020]. Journal of Psychiatric Research 2011;45(1):96‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop BW, Li T, Kornstein SG, Friedman ES, Rothschild AJ, Pedersen R, et al. Correlation between patient and clinician assessments of depression severity in the PREVENT study [NCT00046020]. Psychiatry Research 2010;177(1‐2):177‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MB, Trivedi MH, Thase ME, Shelton RC, Kornstein SG, Nemeroff CB, et al. The Prevention of Recurrent Episodes of Depression with Venlafaxine for Two Years (PREVENT) Study: Outcomes from the 2‐year and combined maintenance phases. Journal of Clinical Psychiatry [Comment in: J Clin Psychiatry. 2008 May;69(5):865‐6; author reply 866; PMID: 18681767] 2007;68(8):1246‐56. [DOI] [PubMed] [Google Scholar]
- Keller MB, Trivedi MH, Thase ME, Shelton RC, Kornstein SG, Nemeroff CB, et al. The Prevention of Recurrent Episodes of Depression with Venlafaxine for Two Years (PREVENT) study: outcomes from the acute and continuation phases. Biological Psychiatry [Erratum appears in Biol Psychiatry. 2008 Apr;63(7):721], [Erratum appears in Biol Psychiatry. 2012 Feb 15;71(4):387] 2007;62(12):1371‐9. [DOI] [PubMed] [Google Scholar]
- Keller MB, Yan B, Ahmed S, Parker‐Zavod E, Pedersen R. Placebo‐controlled trial of venlafaxine ER in prevention of recurrence in patients with recurrent unipolar major depression. Neuropsychopharmacology 2005;30 Suppl 1:175‐6. [Google Scholar]
- Kocsis JH, Thase ME, Trivedi MH, Shelton RC, Kornstein SG, Nemeroff CB, et al. Prevention of recurrent episodes of depression with venlafaxine ER in a 1‐year maintenance phase from the PREVENT Study [NCT00046020]. Journal of Clinical Psychiatry 2007;68(7):1014‐23. [DOI] [PubMed] [Google Scholar]
- Kornstein SG. Beyond remission: Rationale and design of the Prevention of Recurrent Episodes of Depression with Venlafaxine for Two Years (PREVENT) Study [NCT00046020]. CNS Spectrums 2006;11 Suppl 15:28‐34. [DOI] [PubMed] [Google Scholar]
- Kornstein SG. Maintenance therapy to prevent recurrence of depression: summary and implications of the PREVENT study. [NCT00046020]. Expert Review of Neurotherapeutics 2008;8(5):737‐42. [DOI] [PubMed] [Google Scholar]
- Kornstein SG, Kocsis JH, Ahmed S, Thase M, Friedman ES, Dunlop BW, et al. Assessing the efficacy of 2 years of maintenance treatment with venlafaxine extended release 75‐225 mg/day in patients with recurrent major depression: A secondary analysis of data from the PREVENT study [NCT00046020]. International Clinical Psychopharmacology 2008;23(6):357‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapicchio PL. Beyond remission. Prevention of recurrences in long‐term treatment of depression. First data from the PREVENT study [NCT00046020]. Italian Journal of Psychopathology 2008;14(1):80‐7. [Google Scholar]
- Thase ME, Gelenberg A, Kornstein SG, Kocsis JH, Trivedi MH, Ninan P, et al. Comparing venlafaxine extended release and fluoxetine for preventing the recurrence of major depression: Results from the PREVENT study. Journal of Psychiatric Research 2011;45(3):412‐20. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Dunnerb DL, Kornsteinc SG, Thase ME, Zajeckae JM, Rothschild AJ, et al. Psychosocial outcomes in patients with recurrent major depressive disorder during 2 years of maintenance treatment with venlafaxine extended release [NCT00046020]. Journal of Affective Disorders 2010;126(3):420‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kerkhofs 1990 {published data only}
- Kerkhofs M, Rielaert C, Maertelaer V, Linkowski P, Czarka M, Mendlewicz J. Fluoxetine in major depression: efficacy, safety and effects on sleep polygraphic variables. International Clinical Psychopharmacology 1990;5:253‐60. [PubMed] [Google Scholar]
Kuha 1991 {published data only}
- Kuha S, Mehtonen O‐P, Henttonen A, Naarala M. The efficacy of fluoxetine versus maprotiline in depressed patients and by dose. Nordic Journal of Psychiatry ‐ Nordisk Psykiatrisk Tidsskrift 1991;45(2):109‐17. [Google Scholar]
Kwon 1996 {published data only}
- Kwon JS, Youn T, Jung HY. Right hemisphere abnormalities in major depression: quantitative electroencephalographic findings before and after treatment. Journal of Affective Disorders 1996;40(3):169‐73. [DOI] [PubMed] [Google Scholar]
Laakman 1988 {published data only}
- Laakmann G, Blaschke D, Engel R, Schwarz A. Fluoxetine vs amitriptyline in the treatment of depressed out‐patients. British Journal of Psychiatry 1988;153:64‐8. [PubMed] [Google Scholar]
La Pia 1992 {published data only}
- Pia S, Giorgio D, Ciriello R, Sannino A, Simone L, Paoletti C, et al. Evaluation of the efficacy, tolerability, and therapeutic profile of fluoxetine versus mianserin in the treatment of depressive disorders in the elderly. Current Therapeutic Research, Clinical & Experimental 1992;52(6):847‐58. [Google Scholar]
- Pia S, Giorgio D, Ciriello R, Sannino A, et al. Double blind controlled study to evaluate the effectiveness and tolerability of fluoxetine versus mianserin in the treatment of depressive disorders among the elderly and their effects on cognitive‐behavioral parameters. New Trends in Experimental and Clinical Psychiatry 1992;8(4):139‐46. [Google Scholar]
Lapierre 1997 {published data only}
- Lapierre YD, Joffe R, McKenna K, Bland R, Kennedy S, Ingram P, et al. Moclobemide versus fluoxetine in the treatment of major depressive disorder in adults. Journal of Psychiatry and Neuroscience 1997;22(2):118‐26. [PMC free article] [PubMed] [Google Scholar]
Lee 2005 {published data only}
- Lee MS, Ham BJ, Kee BS, Kim JB, Yeon BK, Oh KS, et al. A comparison of the efficacy and safety between milnacipran and fluoxetine in the treatment of patients with depression. International Journal of Neuropsychopharmacology 2002;5 Suppl 1:205. [Google Scholar]
- Lee MS, Ham BJ, Kee BS, Kim JB, Yeon BK, Oh KS, et al. Comparison of efficacy and safety of milnacipran and fluoxetine in Korean patients with major depression. Current Medical Research Opinion 2005;21(9):1369‐75. [DOI] [PubMed] [Google Scholar]
Levine 1989 {published data only}
- Levine S, Deo R, Mahadevan K. A comparative trial of a new antidepressant, fluoxetine. British Journal of Psychiatry 1987;150:653‐5. [DOI] [PubMed] [Google Scholar]
- Levine S, Deo R, Mahadevan K. A comparative trial of a new antidepressant, fluoxetine. International Clinical Psychopharmacology 1989;4 Suppl 1:41‐4. [PubMed] [Google Scholar]
Levkovitz 2002 {published data only}
- Levkovitz Y, Caftori R, Avital A, Richter‐Levin G. The SSRIs drug fluoxetine, but not the noradrenergic tricyclic drug desipramine, improves memory performance during acute major depression. Brain Research Bulletin 2002;58:345‐50. [DOI] [PubMed] [Google Scholar]
Loeb 1989 {published data only}
- Loeb C, Albano C, Gandolfo C. Fluoxetine versus imipramine. International Clinical Psychopharmacology 1989;4 Suppl 1:75‐9. [PubMed] [Google Scholar]
Lonnqvist 1994 {published data only}
- Lonnqvist J, Sihvo S, Syvalahti E, Kiviruusu O. Moclobemide and fluoxetine in atypical depression: a double‐blind trial. Journal of Affective Disorders 1994;32(3):169‐77. [DOI] [PubMed] [Google Scholar]
- Lonnqvist J, Sihvo S, Syvalahti E, Sintonen H, Kiviruusu O, Pitkanen H. Moclobemide and fluoxetine in the prevention of relapses following acute treatment of depression. Acta Psychiatrica Scandinavica 1995;91(3):189‐94. [DOI] [PubMed] [Google Scholar]
- Lonnqvist J, Sintonen H, Syvalahti E, Appelberg B, Koskinen T, Mannikko T, et al. Antidepressant efficacy and quality‐of‐life in depression: a double‐blind study with moclobemide and fluoxetine. Acta Psychiatrica Scandinavica 1994;89(6):363‐9. [DOI] [PubMed] [Google Scholar]
Loo 1999 {published data only}
- Loo H. Double‐blind study comparing tianeptine and fluoxetine in patients with ICD‐10 criteria for depressive disorders with or without somatic syndrome [NR537]. 151st Annual Meeting of the American Psychiatric Association; 1998 May 30 ‐ Jun 4; Toronto, Ontario, Canada. 1998:212.
- Loo H, Saiz‐Ruiz J, Costa e Silva JA, Ansseau M, Herrington R, Vaz‐Serra A, et al. Efficacy and safety of tianeptine in the treatment of depressive disorders in comparison with fluoxetine. Human Psychopharmacology 2001;16 Suppl 1:31‐8. [DOI] [PubMed] [Google Scholar]
- Loo H, Saiz‐Ruiz J, Costa e Silva JA, Ansseau M, Herrington R, Vaz‐Serra A, et al. Efficacy and safety of tianeptine in the treatment of depressive disorders in comparison with fluoxetine. Journal of Affective Disorders 1999;56(2‐3):109‐18. [DOI] [PubMed] [Google Scholar]
Manna 1989 {published data only}
- Manna V, Martucci N, Agnoli A. Double‐blind controlled study on the clinical efficacy and safety of fluoxetine vs clomipramine in the treatment of major depressive disorders. International Clinical Psychopharmacology 1989;4 Suppl 1:81‐8. [PubMed] [Google Scholar]
Mao 2008 {published data only}
- Mao PX, Tang YL, Jiang FJ, Shu L, Gu X, Li M, et al. Escitalopram in major depressive disorder: a multicenter, randomised, double‐blind, fixed‐dose, parallel trial in a Chinese population. Depression and Anxiety 2008;25(1):46‐54. [DOI] [PubMed] [Google Scholar]
Marchesi 1998 {published data only}
- Marchesi C, Ceccherininelli A, Rossi A, Maggini C. Is anxious‐agitated major depression responsive to fluoxetine? A double‐blind comparison with amitriptyline. Pharmacopsychiatry 1998;31(6):216‐21. [DOI] [PubMed] [Google Scholar]
Martenyi 2001 {published data only}
- Martenyi F, Dossenbach M, Mraz K, Metcalfe S. Gender differences in the antidepressive effect: A double‐blind trial of fluoxetine and maprotiline in the treatment of major depression. European Neuropsychopharmacology 2000;10 Suppl 3:221. [DOI] [PubMed] [Google Scholar]
- Martenyi F, Dossenbach M, Mraz K, Metcalfe S. Gender differences in the antidepressive effect? A double blind trial of fluoxetine and maprotiline in the treatment of major depression. International Journal of Neuropsychopharmacology 2000;3 Suppl 1:241. [Google Scholar]
- Martenyi F, Dossenbach M, Mraz K, Metcalfe S. Gender differences in the efficacy of fluoxetine and maprotiline in depressed patients: a double‐blind trial of antidepressants with serotonergic or norepinephrinergic reuptake inhibition profile. European Neuropsychopharmacology 2001;11(3):227‐32. [DOI] [PubMed] [Google Scholar]
Martinez 2012 {published data only}
- Martinez JM, Katon W, Greist JH, Kroenke K, Thase ME, Meyers AL, et al. A pragmatic 12‐week, randomised trial of duloxetine versus generic selective serotonin‐reuptake inhibitors in the treatment of adult outpatients in a moderate‐to‐severe depressive episode. International Clinical Psychopharmacology 2012;27(1):17‐26. [DOI] [PubMed] [Google Scholar]
Masco 1985 {published data only}
- Masco HL, Sheetz MS. Double‐blind comparison of fluoxetine and amitriptyline in the treatment of major depressive illness. Advances in Therapy 1985;2(6):275‐84. [Google Scholar]
Massana 1999 {published data only}
- Massana J. Reboxetine versus fluoxetine: benefits in social function. 152nd Annual Meeting of the American Psychiatric Association, Washington DC. 1999.
- Massana J, Moller HJ, Burrows GD, Montenegro RM. Reboxetine: a double‐blind comparison with fluoxetine in major depressive disorder. International Clinical Psychopharmacology 1999;14(2):73‐80. [DOI] [PubMed] [Google Scholar]
- Moller H. Reboxetine, the first selective noradrenaline reuptake inhibitor, is more effective at improving social functioning than fluoxetine. 152nd Annual Meeting of the American Psychiatric Association, Washington DC. 1999.
McGrath 2000 {published data only}
- Agosti V, McGrath PJ. Comparison of the effects of fluoxetine, imipramine and placebo on personality in atypical depression. Journal of Affective Disorders 2002;71(1‐3):113‐20. [DOI] [PubMed] [Google Scholar]
- McGrath PJ, Stewart JW, Janal MN, Petkova E, Quitkin FM, Klein DF. A placebo‐controlled study of fluoxetine versus imipramine in the acute treatment of atypical depression. American Journal of Psychiatry 2000;157(3):344‐50. [DOI] [PubMed] [Google Scholar]
- McGrath PJ, Stewart JW, Quitkin FM. Fluoxetine treatment of atypical depression. 52nd Annual Meeting of the American Psychiatric Association, Washington DC. 1999.
Moosa 2003 {published data only}
- Moosa MY, Panz VR, Jeenah FY, Joffe BI. African women with depression: The effect of imipramine and fluoxetine on body mass index and leptin secretion. Journal of Clinical Psychopharmacology 2003;23(6):549‐52. [DOI] [PubMed] [Google Scholar]
Moreno 2006 {published data only}
- Moreno RA, Teng CT, ALmeida KM, Tavares HJ. Hypericum perforatum versus fluoxetine in the treatment of mild to moderate depression: a randomised double‐blind trial in a Brazilian sample. Revista Brasileira de Psiquiatria 2006;28(1):29‐32. [DOI] [PubMed] [Google Scholar]
Mowla 2006 {published data only}
- Mowla A, Ghanizadeh A, Pani A. A comparison of effects of fluoxetine and nortriptyline on the symptoms of major depressive disorder. Journal of Clinical Psychopharmacology 2006;26(2):209‐11. [DOI] [PubMed] [Google Scholar]
Muijen 1988 {published data only}
- Muijen M, Roy D, Silverstone T, Mehmet A, Christie M. A comparative clinical trial of fluoxetine, mianserin and placebo in depressed outpatients. Acta Psychiatrica Scandinavica 1988;78(3):384‐90. [DOI] [PubMed] [Google Scholar]
MY‐1043/BRL‐029060/115 {published data only}
- GlaxoSmithKline. A multicenter, randomised, double blind, placebo‐controlled comparison of paroxetine and fluoxetine in the treatment of major depressive disorder. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com).
MY‐1045/BRL‐029060/1 {unpublished data only}
- GlaxoSmithKline. A multicenter, randomised, double‐blind, placebo‐controlled comparison of paroxetine and fluoxetine in the treatment of Major Depressive Disorder. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) .
Nelson 2004 {published data only}
- Nelson JC. Synergistic effects of serotonergic and noradrenergic antidepressants. XXIst Collegium Internationale Neuro‐Psychopharmacologicum, Glasgow, Scotland. 1998.
- Nelson JC, Mazure CM, Jatlow PI, Bowers Jr MB, Price LH. Combining norepinephrine and serotonin reuptake inhibition mechanisms for treatment of depression: A double‐blind, randomised study. Biological Psychiatry 2004;55(3):296‐300. [DOI] [PubMed] [Google Scholar]
Nemeroff 2007 {published data only}
- Nemeroff CB, Amchin J. Placebo‐controlled trial of the efficacy and tolerability of venlafaxine and fluoxetine in outpatients with major depression. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Nemeroff CB, Cantillon M. Venlafaxine demonstrates excellent response frequency in trial with fluoxetine and placebo in depressed outpatients. International Journal of Neuropsychopharmacology 2000;3 Suppl 1:191. [Google Scholar]
- Nemeroff CB, Thase ME, EPIC 014 Study Group. A double‐blind, placebo‐controlled comparison of venlafaxine and fluoxetine treatment in depressed outpatients. Journal of Psychiatric Research. 2007;41(3‐4):351‐9. [DOI] [PubMed] [Google Scholar]
Newhouse 2000 {published data only}
- Newhouse P, Finkel S, Richter E. SSRIs in the treatment of depressed out patients aged 70 years and older. VIIIth ECNP (European College of Neuropsychopharmacology) Congress, Venice, Italy. 1995.
- Newhouse P, Ko G, Richter E. Comparison of sertraline and fluoxetine in depressed geriatric outpatients: plasma levels and efficacy. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Newhouse PA, Krishnan KR, Doraiswamy PM, Richter EM, Batzar ED, Clary CM. A double‐blind comparison of sertraline and fluoxetine in depressed elderly outpatients. Journal of Clinical Psychiatry 2000;61(8):559‐68. [DOI] [PubMed] [Google Scholar]
Nielsen 1993 {published data only}
- Nielsen BM, Behnke K, Arup P, Christiansen PE, Geisler A, Ipsen E, et al. A comparison of fluoxetine and imipramine in the treatment of outpatients with major depressive disorder. Acta Psychiatrica Scandinavica 1993;87(4):269‐72. [DOI] [PubMed] [Google Scholar]
Noguera 1991 {published data only}
- Noguera R, Altuna R, Alvarez E, Ayuso JL, Casais L, Udina C. Fluoxetine vs clomipramine in depressed patients: a controlled multicenter trial. Journal of Affective Disorders 1991;22(3):119‐24. [DOI] [PubMed] [Google Scholar]
Noorbala 2005 {published data only}
- Noorbala AA, Akhondzadeh S, Tahmacebi‐Pour N, Jamshidi AH. Hydro‐alcoholic extract of Crocus sativus L. versus fluoxetine in the treatment of mild to moderate depression: A double‐blind, randomised pilot trial. Journal of Ethnopharmacology 2005;97(2):281‐4. [DOI] [PubMed] [Google Scholar]
Novotny 2002 {published data only}
- Novotny V, Faltus F. Tianeptine and fluoxetine in major depression: a 6‐week randomised double‐blind study. Human Psychopharmacology 2002;17(6):299‐303. [DOI] [PubMed] [Google Scholar]
O'Keane 1992 {published data only}
- O'Keane V, McLoughlin D, Dinan TG. D‐fenfluramine‐induced prolactin and cortisol release in major depression: response to treatment. Joournal of Affective Disorders 1992;26(3):143‐50. [DOI] [PubMed] [Google Scholar]
Ontiveros 1997 {published data only}
- GlaxoSmithKline. A Comparative Double‐Blind Study of Paroxetine and Fluoxetine in the Treatment of Depression in Out‐Patients [Trial MY‐0144/BRL 29060/1/CPMS‐135 (PAR 135)]. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 1992.
- Ontiveros A, Garcia‐Barriga C. A double‐blind, comparative study of paroxetine and fluoxetine in out‐patients with depression. British Journal of Clinical Research 1997;8:23‐32. [Google Scholar]
- Ontiveros A, et al. A double blind study with paroxetine vs. fluoxetine in depressive patients. Biological Psychiatry 1995;35:677. [Google Scholar]
OntiverosSanchez 1998 {published data only}
- Ontiveros Sanchez de la Barquera JA, Brandi F, Brunner E. Double‐blind study of fluoxetine vs. amitriptyline in depressive and anxiety symptoms and life quality in adults with major depression [Estudio doble‐ciego sobre fluoxetina vs amitriptilina en los sintomas depresivos y de ansiedad, y calidad de vida de los adultos con depresion mayor]. Salud Mental 1998;21:58‐63. [Google Scholar]
Pakesch 1991 {published data only}
- Pakesch G, Dossenbach M. Efficacy and safety of fluoxetine versus clomipramine in ambulatory patients with a depressive syndrome in a clinical trial with private practitioners [Wirkung und Sicherheit von Fluoxetin versus Clomipramin bei ambulanten Patienten mit einem depressiven Syndrom in einer klinischen Prufung bei niedergelassenen Arzten]. Wiener Klinische Wochenschrift 1991;103(6):176‐82. [PubMed] [Google Scholar]
Pande 1996 {published data only}
- Pande AC, Birkett M, Fechner‐Bates S, Haskett RF, Greden JF. Fluoxetine versus phenelzine in atypical depression. Biological Psychiatry 1996;40(10):1017‐20. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Pande AC, Demitrack MA. Two year follow‐up of atypical depression. Journal of Psychiatric Research 1999;33(1):23‐9. [DOI] [PubMed] [Google Scholar]
Perry 1989 {published data only}
- Fudge JL, Perry PJ, Garvey MJ, Kelly MW. A comparison of the effect of fluoxetine and trazodone on the cognitive functioning of depressed outpatients. Journal of Affective Disorders 1990;18(4):275‐80. [DOI] [PubMed] [Google Scholar]
- Perry PJ, Garvey MJ, Kelly MW, Cook BL, Dunner FJ, Winokur G. A comparative trial of fluoxetine versus trazodone in outpatients with major depression. Journal of Clinical Psychiatry 1989;50(8):290‐4. [PubMed] [Google Scholar]
Peters 1990 {published data only}
- Peters UH, Lenhard P, Metz M. Therapy of depression in the psychiatrist's office ‐ A double‐blind multicenter study [Ambulante Therapie der Depression mit Fluoxetin ‐ eine multizentrische Doppelblindstudie]. Nervenheilkunde 1990;9(1):28‐31. [Google Scholar]
Poelinger 1989 {published data only}
- Poelinger W, Haber H. Fluoxetine 40 mg vs maprotiline 75 mg in the treatment of out‐patients with depressive disorders. International Clinical Psychopharmacology 1989;4(1):47‐50. [PubMed] [Google Scholar]
Preskorn 1991 {published data only}
- Preskorn SH, Silkey B, Beber J, Dorey C. Antidepressant response and plasma concentrations of fluoxetine. Annals of Clinical Psychiatry 1991;3(2):147‐51. [Google Scholar]
Rapaport 1996 {published data only}
- Rapaport M, Coccaro E, Sheline Y, Holland P, Perse T, Fabre L. Comparison of fluvoxamine and fluoxetine in major depression. VIIIth ECNP (European College of Neuropsychopharmacology) Congress, Venice, Italy. 1995.
- Rapaport M, Coccaro E, Sheline Y, Perse T, Holland P, Fabre L, et al. A comparison of fluvoxamine and fluoxetine in the treatment of major depression. Journal of Clinical Psychopharmacology 1996;16(5):373‐8. [DOI] [PubMed] [Google Scholar]
Remick 1989 {published data only}
- Remick RA, Keller FD, Gibson RE, Carter D. A comparison between fluoxetine and doxepin in depressed patients. Current Therapeutic Research 1989;46(5):842‐8. [Google Scholar]
Remick 1993 {published data only}
- Remick RA, Claman J, Reesal R, Gibson RE, et al. Comparison of fluoxetine and desipramine in depressed outpatients. Current Therapeutic Research 1993;53(5):457‐65. [Google Scholar]
Reynaert 1995 {published data only}
- Reynaert C, Janne P, Vause M, Zdanowicz N, Lejeune D. Clinical trials of antidepressants: the hidden face: where locus of control appears to play a key role in depression outcome. Psychopharmacology 1995;119(4):449‐54. [DOI] [PubMed] [Google Scholar]
- Reynaert C, Parent M, Mirel J, Janne P, Haazen L. Moclobemide versus fluoxetine for a major depressive episode. Psychopharmacology 1995;118(2):183‐7. [DOI] [PubMed] [Google Scholar]
Robertson 1994 {published data only}
- Burns RA, Lock T, Edwards DR, Katona CL, Harrison DA, Robertson MM, et al. Predictors of response to amine‐specific antidepressants. Journal of Affective Disorders 1995;35(3):97‐106. [DOI] [PubMed] [Google Scholar]
- Lawrence KM, Katona CL, Abou‐Saleh MT, Robertson MM, Nairac BL, Edwards DR, et al. Platelet 5‐HT uptake sites, labelled with 3H paroxetine, in controls and depressed patients, before and after treatment with fluoxetine or lofepramine. Psychopharmacology 1994;115(1‐2):261‐4. [DOI] [PubMed] [Google Scholar]
- Robertson MM, Abou Saleh MT, Harrison DA, Nairac BL. A double‐blind controlled comparison of fluoxetine and lofepramine in major depressive illness. Journal of Psychopharmacology 1994;8(2):98‐103. [DOI] [PubMed] [Google Scholar]
Ropert 1989 {published data only}
- Ropert R. Fluoxetine versus clomipramine in major depressive disorders. International Clinical Psychopharmacology 1989;4 Suppl 1:89‐95. [PubMed] [Google Scholar]
Rudolph 1999 {published data only}
- Rudolph RL, Feiger AD. A double‐blind, randomised, placebo‐controlled trial of once‐daily venlafaxine extended release (xr) and fluoxetine for the treatment of depression. Journal of Affective Disorders 1999;56(2‐3):171‐81. [DOI] [PubMed] [Google Scholar]
Rush 1998 {published data only}
- Rush AJ, Armitage R, Gillin JC, Yonkers KA, Winokur A, Moldofsky H, et al. Comparative effects of nefazodone and fluoxetine on sleep in outpatients with major depressive disorder. Biological Psychiatry 1998;44(1):3‐14. [DOI] [PubMed] [Google Scholar]
Sandor 1998 {published data only}
- Sandor P, Baker B, Irvine J, Dorian P, McKessok D, Mendlowitz S. Effectiveness of fluoxetine and doxepin in treatment of melancholia in depressed patients. Depression and Anxiety 1998;7(2):69‐72. [PubMed] [Google Scholar]
Schatzberg 2006 {published data only}
- Anonymous. Venlafaxine IR, fluoxetine and placebo effective in reducing depression in elderly. Brown University Geriatric Psychopharmacology Update 2006; Vol. 10, issue 6:3‐4.
- Schatzberg A, Roose S. A double‐blind, placebo‐controlled study of venlafaxine and fluoxetine in geriatric outpatients with major depression. American Journal of Geriatric Psychiatry 2006;14(4):361‐70. [DOI] [PubMed] [Google Scholar]
Schone 1993 {published data only}
- Geretsegger C, Bohmer F, Ludwig M. Paroxetine in the elderly depressed patient: randomized comparison with fluoxetine of efficacy, cognitive and behavioural effects. International Clinical Psychopharmacology 1994;9(1):25‐9. [PubMed] [Google Scholar]
- Schone W, Ludwig M. A double‐blind study of paroxetine compared with fluoxetine in geriatric patients with major depression. Journal of Clinical Psychopharmacology 1993;13 Suppl 2:34‐9. [DOI] [PubMed] [Google Scholar]
- Schone W, Ludwig M. Paroxetine in the treatment of depression in geriatric patientsa double‐blind comparative study with fluoxetine [Paroxetin in der Depressionsbehandlung geriatrischer Patienteneine doppelblinde Vergleichsstudie mit Fluoxetin]. Fortschritte der Neurologie Psychiatrie 1994;62 Suppl 1:16‐8. [PubMed] [Google Scholar]
Schrader 2000 {published data only}
- Friede M, Henneicke von Zepelin HH, Freudenstein J. Differential therapy of mild to moderate depressive episodes (ICD‐10 F 32.0; F 32.1) with St. John's wort. Pharmacopsychiatry 2001;34 Suppl 1:38‐41. [DOI] [PubMed] [Google Scholar]
- Schrader E. Equivalence of St John's wort extract (Ze 117) and fluoxetine: a randomised, controlled study in mild‐moderate depression. International Journal of Psychopharmacology 2000;15(2):61‐8. [DOI] [PubMed] [Google Scholar]
Sechter 1999 {published data only}
- Sechter D, Troy S. Comparison of sertraline and fluoxetine on quality‐of‐life in depressed outpatients. 150th Annual Meeting of the American Psychiatric Association; 1997 May 17‐22; San Diego, CA. 1997.
- Sechter D, Troy S. Double blind randomized comparative study of sertraline and fluoxetine in depressive outpatients. VIth World Congress of Biological Psychiatry, Nice, France. 1997.
- Sechter D, Troy S, Paternetti S, Boyer P. A double‐blind comparison of sertraline and fluoxetine in the treatment of major depressive episode in outpatients. European Psychiatry: the Journal of the Association of European Psychiatrists 1999;14(1):41‐8. [DOI] [PubMed] [Google Scholar]
- Sechter D, Troy S, Rioux P. A double blind comparison of sertraline and fluoxetine in the treatment of major depressive episode in out patients. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
Sheehan 2009 {published data only}
- Sheehan DV, Nemeroff CB, Thase ME, Entsuah R, EPIC 016 Study Group. Placebo controlled inpatient comparison of venlafaxine and fluoxetine for the treatment of major depression with melancholic features. International Clinical Psychopharmacology 2009;24(2):61‐86. [DOI] [PubMed] [Google Scholar]
Silverstone 1999 {published data only}
- Silverstone PH, Ravindran A. Once‐daily venlafaxine extended release (xr) compared with fluoxetine in outpatients with depression and anxiety. Venlafaxine xr 360 study group. Journal of Clinical Psychiatry 1999;60(1):22‐8. [DOI] [PubMed] [Google Scholar]
Smeraldi 1998 {published data only}
- Biondi F, Casadei G. Preliminary results of a randomized double blind study of amisulpride vs fluoxetine in 281 dysthymic patients. XXth Collegium Internationale Neuro‐Psychopharmacologicum, Melbourne, Australia. 1996.
- Biondi F, Casadet G. Low‐dose amisulpride versus fluoxetine in 281 dysthymic patients ‐ a randomized medium‐term (3 months), double‐blind study. 149th Annual Meeting of the American Psychiatric Association, New York, NY. 1996.
- Jori MC, Cesana BM, Casadei G. Onset of action in the pharmacologic treatment of dysthymia. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Smeraldi E. Amisulpride in the treatment of dysthymia: Preliminary results in a comparative double‐blind study with fluoxetine. Nuova Rivista di Neurologia 1995;5 Suppl 2:22‐6. [Google Scholar]
- Smeraldi E. Amisulpride versus fluoxetine in patients with dysthymia or major depression in partial remission: A double‐blind, comparative study. Journal of Affective Disorders 1998;48(1):47‐56. [DOI] [PubMed] [Google Scholar]
- Smeraldi E, Haefele E, Crespi G, Casadei G L, Biondi F, Vigorelli E. Amisulpride versus fluoxetine in dysthymia: Preliminary results of a double‐blind comparative study. European Psychiatry 1996;11 Suppl 3:141‐3. [Google Scholar]
SouthWalesGroup 1988 {published data only}
- South Wales Antidepressant Drug Trial Group. A double‐blind multi‐centre trial of fluoxetine and dothiepin in major depressive illness. International Clinical Psychopharmacology 1988;3(1):75‐81. [DOI] [PubMed] [Google Scholar]
Sramek 1995 {published data only}
- Sramek JJ, Kashkin K, Jasinsky O, Kardatzke D, Kennedy S, Cutler NR. Placebo‐controlled study of ABT‐200 versus fluoxetine in the treatment of major depressive disorder. Depression 1995;3(4):199‐203. [Google Scholar]
Stark 1985 {published data only}
- Stark P, Hardison CD. A review of multicenter controlled studies of fluoxetine vs imipramine and placebo in outpatients with major depressive disorder. Journal of Clinical Psychiatry 1985;46:53‐8. [PubMed] [Google Scholar]
Stephenson 2000 {published data only}
- Stephenson DA, Harris B, Davies RH, Mullin JM, Richardson E, Boardman H, et al. The impact of antidepressants on sleep and anxiety: A comparative study of fluoxetine and dothiepin using the Leeds Sleep Evaluation Questionnaire. Human Psychopharmacology 2000;15(7):529‐34. [DOI] [PubMed] [Google Scholar]
Stratta 1991 {published data only}
- Stratta P, Bolino F, Cupillari M, Casacchia M. A double‐blind parallel study comparing fluoxetine with imipramine in the treatment of atypical depression. International Clinical Psychopharmacology 1991;6(3):193‐6. [DOI] [PubMed] [Google Scholar]
- Stratta P, Cupillari M, Casacchia M. Double‐blind study comparing fluoxetine with imipramine in the treatment of atypical depression. Rivista Sperimentale Di Freniatria E Medicina Legale Delle Alienazioni Mentali 1992;116(1):148‐58. [Google Scholar]
Suleman 1997 {published data only}
- Suleman MI, Sebit MB, Acuda SW, Siziya S. Fluoxetine and moclobemide versus amitriptyline in major depression: a single blind randomised clinical trial in Zimbabwe. Central‐African Journal of Medicine 1997;43(2):38‐40. [Google Scholar]
Suri 2000 {published data only}
- Suri RA, Altshuler LL, Rasgon N, Calcagno J, Frye MA, Gitlin MJ, et al. A single‐blind trial assessing the effectiveness/efficacy of fluoxetine versus sertraline for the treatment of major depression. 153rd Annual Meeting of the American Psychiatric Association, Chicago, IL. 2000.
- Suri RA, Altshuler LL, Rasgon NL, Calcagno JL, Frye MA, Gitlin MJ, et al. Efficacy and response time to sertraline versus fluoxetine in the treatment of unipolar major depressive disorder. Journal of Clinical Psychiatry 2000;61(12):942‐6. [DOI] [PubMed] [Google Scholar]
Tamminen 1989 {published data only}
- Tamminen TT, Lehtinen VV. A double‐blind parallel study to compare fluoxetine with doxepin in the treatment of major depressive disorders. International Clinical Psychopharmacology 1989;4 Suppl 1:51‐6. [PubMed] [Google Scholar]
Taner 2006 {published data only}
- Taner E, Demir EY, Cosar B. Comparison of the effectiveness of reboxetine versus fluoxetine in patients with atypical depression: a single blind, randomised clinical trial. Advances in Therapy 2006;23(6):974‐87. [DOI] [PubMed] [Google Scholar]
- Taner E, Yancar Demir E, Cosar B. Efficacy and tolerability of reboxetine compared to fluoxetine in patients with atypical depression. International Journal of Neuropsychopharmacology 2006;9 Suppl 1:S187. [Google Scholar]
Taneri 1989 {published data only}
- Taneri Z, Kohler R. Fluoxetine versus nomifensine in outpatients with neurotic or reactive depressive disorder. International Clinical Psychopharmacology 1989;4(1):57‐61. [PubMed] [Google Scholar]
Thompson 2000 {published data only}
- McKendrick J, Stephenson DA, Thompson C, Peveler RC. Compliance with antidepressant treatment of depression in primary care in the UK. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Thompson C, Peveler RC, Stephenson D, McKendrick J. Compliance with antidepressant medication in the treatment of major depressive disorder in primary care: a randomised comparison of fluoxetine and a tricyclic antidepressant. American Journal of Psychiatry 2000;157(3):338‐43. [DOI] [PubMed] [Google Scholar]
Tignol 1993 {published data only}
- GlaxoSmithKline. A double‐blind, randomised, multicentre study comparing paroxetine 20mg daily versus fluoxetine 20mg daily in the treatment of adults with major depression with regard to antidepressant efficacy, tolerance and anxiolytic effect. [29060/087]. clinicaltrials.gov. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com). 1991.
- Tignol J. A double‐blind, randomised, fluoxetine‐controlled, multicenter study of paroxetine in the treatment of depression. Journal of Clinical Psychopharmacology 1993;13 Suppl 2:18‐22. [DOI] [PubMed] [Google Scholar]
Tollefson 1994 {published data only}
- Tollefson GD, Greist JH, Jefferson JW, Heiligenstein JH, Sayler ME, Tollefson SL, et al. Is baseline agitation a relative contraindication for a selective serotonin reuptake inhibitor: a comparative trial of fluoxetine versus imipramine. Journal of Clinical Psychopharmacology 1994;14(6):385‐91. [PubMed] [Google Scholar]
- Tollefson GD, Heiligenstein JH, Tollefson SL, Birkett MA, Knight DL, Nemeroff CB. Is there a relationship between baseline and treatment‐associated changes in 3H‐IMI platelet binding and clinical response in major depression?. Neuropsychopharmacology 1996;14(1):47‐53. [DOI] [PubMed] [Google Scholar]
Tylee 1997 {published data only}
- Tylee A, Beaumont G, Bowden MW, Reynolds A. A double‐blind, randomised, 12‐week comparison study of the safety and efficacy of venlafaxine and fluoxetine in moderate to severe major depression in general‐practice. Primary Care Psychiatry 1997;3(1):51‐8. [Google Scholar]
Tzanakaki 2000 {published data only}
- Tzanakaki M, Guazzelli M, Nimatoudis I, Zissis NP. Increased remission rates with venlafaxine compared with fluoxetine in hospitalised patients with major depression and melancholia. International Clinical Psychopharmacology 2000;15(1):29‐34. [DOI] [PubMed] [Google Scholar]
- Tzanakaki M, Ierodiakonou Ch, Dimitriou E, Alevizos V. Randomized, double‐blind comparison of venlafaxine and fluoxetine in hospitalized patients with major depression and melancholia. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
Upward 1988 {published data only}
- Upward JW, Edwards JG, Goldie A, Waller DG. Comparative effects of fluoxetine and amitriptyline on cardiac function. British Journal of Clinical Pharmacology 1988;26(4):399‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Moffaert 1995 {published data only}
- Moffaert M, Bartholome F, Cosyns P, Nayer AR, Mertens C. A controlled comparison of sertraline and fluoxetine in acute and continuation treatment of major depression. Human Psychopharmacology 1995;10(5):393‐405. [Google Scholar]
Versiani 1999 {published data only}
- Versiani M, Ontiveros A, Mazzotti G, Ospina J, Davila J, Mata S, et al. A double‐blind comparison of fluoxetine and amitriptyline in the treatment of major depression with associated anxiety ('anxious depression'). Cross Cultural Psychiatry 1999:249‐58. [DOI] [PubMed] [Google Scholar]
- Versiani M, Ontiveros A, Mazzotti G, Ospina J, Davila J, Mata S, et al. Fluoxetine versus amitriptyline in the treatment of major depression with associated anxiety (anxious depression): a double‐blind comparison. International Clinical Psychopharmacology 1999;14(6):321‐7. [DOI] [PubMed] [Google Scholar]
- Versiani M, Plewes J, Ontiveiros A, Mazzotti G, Ospina J, Davila J, et al. Fluoxetine and amitriptyline in the treatment of major depression with associated anxiety. XXIst Collegium Internationale Neuro‐Psychopharmacologicum, Glasgow, Scotland. 1998.
- Versiani M, Plewes J, Ontiveiros A, Mazzotti G, Ospina J, Davila J, et al. Fluoxetine and ammitriptyline in the treatment of major depression with associated anxiety. XIth European College of Neuropsychopharmacology Congress, Paris, France. 1998.
- Versiani MV, Ontiveiros A, Mazzotti G, Ospina J, Davila J, Mata S, et al. A double‐blind comparison of fluoxetine and amitriptyline in the treatment of major depression with associated anxiety. 151st Annual Meeting of the American Psychiatric Association, Toronto, Ontario, Canada. 1998.
Versiani 2005 {published data only}
- Versiani M, Moreno R, Ramakers‐van Moorsel CJ, Schutte AJ, Comparative Efficacy Antidepressants Study Group. Comparison of the effects of mirtazapine and fluoxetine in severely depressed patients. CNS Drugs 2005;19(2):137‐46. [DOI] [PubMed] [Google Scholar]
Wehmeier 2005 {published data only}
- Wehmeier PM, Kluge M, Maras A, Riemann D, Berger M, Kohnen R, Dittmann RW, Gattaz WF. Fluoxetine versus trimipramine in the treatment of depression in geriatric patients. Pharmacopsychiatry 2005;38(1):13‐6. [DOI] [PubMed] [Google Scholar]
WELL AK1A4006 {unpublished data only}
- GlaxoSmithKline. A multicenter, double blind, placebo‐controlled comparison of the safety and efficacy and effects on sexual functioning of Wellbutrin (bupropion HCI) Sustained Release (SR) and Fluoxetine in outpatients with moderate to severe recurrent Major Depression. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com).
Wheatley 1998 {published data only}
- Kremer CM. Mirtazapine versus fluoxetine ‐ efficacy on symptoms associated with depression. 151st Annual Meeting of the American Psychiatric Association, Toronto, Ontario, Canada. 1998.
- Kremer CM. Mirtazapine vs fluoxetine: Efficacy on symptoms associated with depression. XXIst Collegium Internationale Neuro‐Psychopharmacologicum, Glasgow, Scotland. 1998.
- Wheatley D, Kremer CM. A randomized, double‐blind comparison of mirtazapine and fluoxetine in patients with major depression. 150th Annual Meeting of the American Psychiatric Association, San Diego, CA. 1997.
- Wheatley DP, Moffaert M, Timmerman L, Kremer CM (Mirtazapine‐Fluoxetine Study Group). Mirtazapine: efficacy and tolerability in comparison with fluoxetine in patients with moderate to severe major depressive disorder. Mirtazapine‐Fluoxetine Study Group. Journal of Clinical Psychiatry 1998;59(6):306‐12. [PubMed] [Google Scholar]
Williams 1993 {published data only}
- Williams R, Edwards RA, Newburn GM, Mullen R, Menkes DB, Segkar C. A double‐blind comparison of moclobemide and fluoxetine in the treatment of depressive disorders. International Clinical Psychopharmacology 1993;7(3‐4):155‐8. [DOI] [PubMed] [Google Scholar]
Winokur 2003 {published data only}
- Winokur A, DeMartinis NA 3rd, McNally DP, Gary EM, Cormier JL, Gary KA. Comparative effects of mirtazapine and fluoxetine on sleep physiology measures in patients with major depression and insomnia. Journal of Clinical Psychiatry 2003;64(10):1224‐9. [DOI] [PubMed] [Google Scholar]
- Winokur A, Gary KA, DeMartinis NA, McNally DP, Rodner SA, Cormier JL. Comparative effects of mirtazapine and fluoxetine on sleep continuity measures and daytime sleepiness in patients with major depression and insomnia: An interim report. 39th Annual Meeting of the American College of Neuropsychopharmacology, San Juan; Puerto Rico. 2000.
Wolf 2001 {published data only}
- Gattaz WF, Maras Schmidt AA, Löw Bahro HM, Kohnen Dittmann RW, Wolff Berger RM, Riemann D. Efficacy and safety of fluoxetine versus trimipramine in geriatric depression. Xth World Congress of Psychiatry, Madrid, Spain. 1996.
- Wolf R, Dykierek P, Gattaz WF, Maras A, Kohnen R, Dittmann RW, et al. Differential effects of trimipramine and fluoxetine on sleep in geriatric depression. Pharmacopsychiatry 2001;34(2):60‐5. [DOI] [PubMed] [Google Scholar]
Young 1987 {published data only}
- Young JP, Coleman A, Lader MH. A controlled comparison of fluoxetine and amitriptyline in depressed out‐patients. British Journal of Psychiatry 1987;151:337‐40. [DOI] [PubMed] [Google Scholar]
Yu 1997 {published data only}
- Yu BR, Zhou DP, Zhang F. Fluoxetine and amitriptyline in the treatment of 22 depressive‐disorder cases: a double blind randomised study. Chinese Journal of New Drugs 1997;16(4):259‐60. [Google Scholar]
Zhao 2006 a {published data only}
- Eli Lilly. Depression with lack of motivation; comparison between fluoxetine and trazodone (ID#6706). [Trial B1Y‐GH‐S013; Summary ID#6706] www.lillytrials.com/results/Prozac.pdf.
- Zhao J, Ang Q, Wang J, Sun X, Wei J, Li M, Liu P. A comparative efficacy of fluoxetine and trazodone in depression with remarkable retardation and loss of energy: a randomised open‐label trial. Internal Medicine Journal ‐ Tokyo 2006;13(1):25‐30. [Google Scholar]
References to studies excluded from this review
Baca Baldomero 2005 {published data only}
- Baca Baldomero E, Giner Ubago J, Leal Cercos C, Vallejo Ruiloba J, Garcia Calvo C, Prieto Lopez R. Venlafaxine extended release versus conventional antidepressants in the remission of depressive disorders after previous antidepressant failure: ARGOS study. Depression and Anxiety 2005;22(2):68‐76. [DOI] [PubMed] [Google Scholar]
Bitrain 2011 {published data only}
- Bitran S, Farabaugh AH, Ameral VE, LaRocca RA, Clain AJ, Fava M, Mischoulon D. Do early changes in the HAM‐D‐17 anxiety/somatization factor items affect the treatment outcome among depressed outpatients? Comparison of two controlled trials of St John's wort (Hypericum perforatum) versus a SSRI. International Clinical Psychopharmacology 2011;26(4):206‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brasseur 1989 {published data only}
- Brasseur R. A multicenter open trial of fluoxetine in depressed out patients in Belgium. International Clinical Psychopharmacology 1989;4 Suppl 11:107‐11. [PubMed] [Google Scholar]
Cohn 1989 {published data only}
- Cohn JB, Collins G, Ashbrook E, Wernicke JF. A comparison of fluoxetine imipramine and placebo in patients with bipolar depressive disorder. International Clinical Psychopharmacology 1989;4(4):313‐22. [DOI] [PubMed] [Google Scholar]
Ducher 2008 {published data only}
- Ducher JL, Dalery J. Correlations between Beck's suicidal ideation scale, suicidal risk assessment scale RSD and Hamilton's depression rating scale. Encephale 2008;34(2):132‐8. [DOI] [PubMed] [Google Scholar]
Goodnick 1987 {published data only}
- Goodnick PJ, Fieve RR, Peselow ED, Barouche F, Schlegel A. Double‐blind treatment of major depression with fluoxetine: use of pattern analysis and relation of HAM‐D score to CGI change. Psychopharmacology Bulletin 23;1:162‐3. [PubMed] [Google Scholar]
Gu 2001 {published data only}
- Gu NF, Li HF, Shu L, Zhang HY, et al. Multicenter study of St.John's wort extract in treatment of mild to moderate depression. Chinese Journal of Clinical Pharmacy 2001;10(5):271‐4. [Google Scholar]
Hunter 2006 {published data only}
- Hunter AM, Leuchter AF, Cook LA, Abrams M. Brain functional changes (QEEG cordance) and worsening suicidal ideation and mood symptoms during antidepressant treatment. Acta Psychiatrica Scandinavica 2010;122(6):461‐9. [DOI] [PubMed] [Google Scholar]
- Hunter AM, Leuchter AF, Morgan ML, Cook LA. Changes in brain function (quantitative EEG cordance) during placebo lead‐in and treatment outcomes in clinical trials for major depression. American Journal of Psychiatry. 2006;163(8):1426‐32. [DOI] [PubMed] [Google Scholar]
- Hunter AM, Ravikumar S, Cook LA, Leuchter AF. Brain functional changes during placebo lead‐in and changes in specific symptoms during pharmacotherapy for major depression. Acta Psychiatrica Scandinavica 2009;119(4):266‐73. [DOI] [PubMed] [Google Scholar]
Iovieno 2011 {published data only}
- Iovieno N, Nieuwenhuizen A, Clain A, Baer L, Nierenberg AA. Residual symptoms after remission of major depressive disorder with fluoxetine and risk of relapse. Depression and Anxiety 2011;28(2):137‐44. [DOI] [PubMed] [Google Scholar]
Kroenke 2001 {published data only}
- Aikens JE, Kroenke K, Nease DE Jr, Klinkman MS, Sen A. Trajectories of improvement for six depression‐related outcomes. General Hospital Psychiatry 2008;30(1):26‐31. [DOI] [PubMed] [Google Scholar]
- Kroenke K, West SL, Swindle R, Gilsenan A, Eckert GJ, Dolor R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: A randomised trial. JAMA 2001;286(23):2947‐55. [DOI] [PubMed] [Google Scholar]
Musgnung 2005 {published data only}
- Musgnung J, Trivedi M, Benattia I, Graepel J. Depression remission rates with venlafaxine XR versus SSRIs using treatment algorithms. Psychiatriki 2005;16:217. [Google Scholar]
Nemetz 2005 {published data only}
- Nemets B, Bersudsky Y, Belmaker RH. Controlled double‐blind trial of phenytoin vs. fluoxetine in major depressive disorder. Journal of Clinical Psychiatry 2005;66(5):586‐90. [DOI] [PubMed] [Google Scholar]
Peveler 2005 {published data only}
- Peveler R, Kendrick T, Buxton M, Longworth L, Baldwin D, Moore M, et al. A randomised controlled trial to compare the cost‐effectiveness of tricyclic antidepressants, selective serotonin reuptake inhibitors and lofepramine. Health Technology Assessment 2005;9(16):1‐134. [DOI] [PubMed] [Google Scholar]
Roose 1994 {published data only}
- Roose SP, Glassman AH, Attia E, Woodring S. Comparative efficacy of selective serotonin reuptake inhibitors and tricyclics in the treatment of melancholia. American Journal of Psychiatry 1994;151(12):1735‐9. [DOI] [PubMed] [Google Scholar]
Schmidt 1999 {published data only}
- Schmidt U, Harrer G, Kuhn U, Biller A. Equivalence comparison of the st john's wort extract lohyp‐57 versus fluoxetine HCl. Zeitschrift fur Phytotherapie 1999;20(2):89‐90. [Google Scholar]
Serrano‐Blanco 2006 {published data only}
- Serrano‐Blanco A, Gabarron E, Garcia‐Bayo I, Soler‐Vila M, Caramés E, Peñarrubia‐Maria MT, et al. Effectiveness and cost‐effectiveness of antidepressant treatment in primary health care: a six‐month randomised study comparing fluoxetine to imipramine. Journal of Affective Disorders 2006;91(2‐3):153‐63. [DOI] [PubMed] [Google Scholar]
Simon 1996 {published data only}
- Simon GE, Korff M, Heiligenstein JH, Revicki DA, Grothaus L, Katon W, Wagner EH. Initial antidepressant choice in primary‐care: effectiveness and cost of fluoxetine vs tricyclic antidepressants. JAMA 1996;275(24):1897‐902. [PubMed] [Google Scholar]
Simon 1998 {published data only}
- Simon GE, Heiligenstein JH, Grothaus L, Katon W, Revicki D. Should anxiety and insomnia influence antidepressant selection: a randomised comparison of fluoxetine and imipramine. Journal of Clinical Psychiatry 1998;59(2):49‐55. [DOI] [PubMed] [Google Scholar]
Simon 1999 {published data only}
- Simon GE, Heiligenstein J, Revicki D, VonKorff M, Katon WJ, Ludman E, et al. Long‐term outcomes of initial antidepressant drug choice in a "real world" randomised trial. Archives of Family Medicine 1999;8(4):319‐25. [DOI] [PubMed] [Google Scholar]
Strik 1998 {published data only}
- Strik JJ, Honig A, Lousberg R, Cheriex EC, Praag HM. Cardiac side‐effects of two selective serotonin reuptake inhibitors in middle‐aged and elderly depressed patients. International Clinical Psychopharmacology 1998;13(6):263‐7. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Chen 2006 {published data only}
- Chen JD, Guo XF, Luo Q, Xun GL, Chen YG. A randomised and double‐blinded clinical trial of reboxetine mesylate for treatment of depression. Chinese Journal of New Drugs 2006;15:1678‐9. [Google Scholar]
GSK 29060/134 {published data only}
- GlaxoSmithKline. A randomized, double‐blind, controlled study of paroxetine and fluoxetine in the treatment of patients with major depression with associated anxiety. GSK ‐ Clinical Study Register (www.gsk‐clinicalstudyregister.com) 2005.
- Silva JAC, Ruschel S, Caetano D. Double‐blind, randomised, controlled study of Paroxetine and Fluoxetine in the treatment of patients with major depression and associated anxiety [Estudo Randomizado, Duplo‐cego, Controlado de Paroxetina e Fluoxetina no Tratamento de Pacientes com Depressão Maior Associada à Ansiedade]. Revista da Associacao Medica Brasileira (Sao Paulo) 1996;53(12):92‐6. [Google Scholar]
Huang 2006a {published data only}
- Huang J, Sun Z. A control study of citalopram and fluoxetine in first‐episode senile depression. Journal of Clinical Psychosomatic Diseases 2006;12:175‐6. [Google Scholar]
Huang 2006b {published data only}
- Huang K. Contrast study of citalopram and fluoxetine in treatment of depression. Modern Medicine and Health 2006;20:1456‐7. [Google Scholar]
Jiang 2006 {published data only}
- Jiang R‐H, Du B, Zhang H‐Y. Randomized, double blind, double‐dummy, multicenter, parallel controlled clinical trial of Kaiyuanshen in the treatment of depressive disorder. Chinese Journal of Clinical Pharmacology 2006;22:15‐7. [Google Scholar]
Li 2005 {published data only}
- Li HF, Zhao JP, Kuang WH, Yao PF, Chen JD, Sun XL, et al. Bupropion‐sustained release in treatment of depression (72 patients): a randomised, double‐blind, double‐dummy, multicenter clinical trial. Chinese Journal of New Drugs and Clinical Remedies 2005;24:614‐8. [Google Scholar]
Li 2006a {published data only}
- Li HF, Ma C, Chen YG, Fan JX, Gu NF. Clinical effect of reboxetine in the treatment of anxiety in depressive patients. Chinese Journal of Clinical Pharmacy 2006;15:339‐42. [Google Scholar]
Li 2006b {published data only}
- Li L, Zhang H, Chen J. A control study on the curative effect and reliability of reboxetine with fluoxetine for treatment of depression. Chinese Journal of Behavioral Medical Science 2006;15:721‐2. [Google Scholar]
Li 2006c {published data only}
- Li N, Ji WD, Zhang DH, Li Y. Efficacy and safety of reboxetine versus fluoxetine for the elders with depression. Chinese Journal of New Drugs 2006;15:1682‐4. [Google Scholar]
Liang 2005 {published data only}
- Liang C, Liu L, Zhang X. A comparative study of citalopram and fluoxetine in the treatment of depression. Shandong Archives of Psychiatry 2005;18:82‐3. [Google Scholar]
Licinio 2004 {published data only}
- Licinio J, O'Kirwan F, Irizarry K, Merriman B, Thakur S, Jepson R, et al. Association of a corticotropin‐releasing hormone receptor 1 haplotype and antidepressant treatment response in Mexican‐Americans. Molecular Psychiatry 2004;9:1075‐82. [DOI] [PubMed] [Google Scholar]
- National Institute of General Medical Sciences (NIGMS). Antidepressant Treatment of Mexican‐Americans: UCLA Pharmacogenetics and Pharmacogenomics Research Group. clinicaltrials.gov.
Ma 2007 {published data only}
- Ma X, Gao C, Tan Q. Bupropion SR and fluoxetine in treatment of depression in multicenter clinical trial. Journal of Xi'an Jiaotong University (Medical Sciences) 2007;28:533‐6. [Google Scholar]
NCT00909155 {published data only}
- Kolden G. Brain Imaging Techniques That Predict Antidepressant Responsiveness. www.clinicaltrials.gov/ct2/show/NCT00909155.
- Light SN, Heller AS, Johnstone T, Kolden GG, Peterson MJ, Kalin NH, et al. Reduced right ventrolateral prefrontal cortex activity while inhibiting positive affect is associated with improvement in hedonic capacity after 8 Weeks of antidepressant treatment in major depressive disorder. Biological Psychiatry 2011;70(10):962‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Qin 2006 {published data only}
- Qin S. The efficacy and tolerability of venlafaxine and fluoxetine in treatment of patients with first‐episode depression. Medical Journal of Chinese People Health 2006;18:519‐21. [Google Scholar]
Sackeim 2006 {published data only}
- Sackeim HA, Roose SP, Lavori PW. Determining the duration of antidepressant treatment: application of signal detection methodology and the need for duration adaptive designs (DAD). Biological Psychiatry 2006;59:483‐92. [DOI] [PubMed] [Google Scholar]
Salehi 2009 {published data only}
- Salehi B, Sanjani FG. Comparison of the effect of fluoxetine and imipramine on fasting blood sugar of patients with major depressive disorders, four and eight weeks after treatment [Arabic]. Scientific Journal of Kurdistan University of Medical Sciences 2009;14:45‐51. [Google Scholar]
Shen 2005 {published data only}
- Shen YF, Li HF, Ma C, et al. Comparison of reboxetine with fluoxetine in treatment of depression: a randomised double‐blind multicenter study. Chinese Journal of New Medicines and Clinical Practice 2005;24:619‐23. [Google Scholar]
Stassen 1999 {published data only}
- Stassen HH, Angst J, Delini‐Stula A. Fluoxetine versus moclobemide: Cross‐comparison between the time courses of improvement. Pharmacopsychiatry 1999;32:56‐60. [DOI] [PubMed] [Google Scholar]
Su 2006 {published data only}
- Su H, Jiang K, Lou F, et al. The cognition function comparison study between venlafaxine and fluoxetine hydrochloride in first‐episode major depression treatment. Chinese Journal of Nervous and Mental Diseases 2006;32:32‐8. [Google Scholar]
Sun 2005 {published data only}
- Sun CY, Wang GH, Zhou T, Tian YL. A comparative clinical study for venlafaxine versus fluoxetine in the treatment of depression. Chinese Journal of New Drugs 2005;14:617‐9. [Google Scholar]
Sun 2006 {published data only}
- Sun P, Song L, Sun F. Comparative study of fluoxetine vs venlafaxine in treatment of depressive patients who cured by fluoxetine but had no obvious effect. Medical Journal of Chinese People Health 2006;28:545‐6. [Google Scholar]
Tan 1997 {published data only}
- Tan MG, He Qi, Gao CL, Ren YP, Zhang TL. Fluoxetine and amitriptyline in the treatment of 18 depressive‐disorder cases: a double blind randomised study. Chinese Journal of New Drugs 1997;16:262‐3. [Google Scholar]
Wang 2006 {published data only}
- Wang J, Hu Y, Qi F. Comparison of event‐related potential P300 in the geriatric depression treated with venlafaxine or fluoxetine. Shanghai Archives of Psychiatry 2006;18:94‐7. [Google Scholar]
Wang 2007a {published data only}
- Wang D, Wang X, Li X. Comparison between the effects of fluoxetine and paroxetine on the life quality of the depressive disorder. Nervous Diseases and Mental Health 2007;7:23‐5. [Google Scholar]
Wang 2007b {published data only}
- Wang X, Zhang B, Li J, Sun X‐L. Double‐blind, double‐dummy, randomised controlled trials of bupropion hydrochloride sustained‐release tablets for depression. Chinese Journal of Evidence‐Based Medicine 2007;7:409‐14. [Google Scholar]
Wang 2009 {published data only}
- Wang X‐Q, Zhang H‐Y, Shu L, et al. Efficacy and safety of morinda officinalis oligose capsule in the treatment of mild or moderate depression. Chinese Journal of New Drugs 2009;18:802‐5. [Google Scholar]
Wang 2011 {published data only}
- Wang HC, Chen PS. Genetic polymorphisms of CYP2C19 influence treatment response to antidepresants in a Taiwanese population with major depressive disorder [conference abstract]. Biological Psychiatry [abstracts from the 66th Annual Meeting of the Society of Biological Psychiatry San Francisco, CA United States May 12‐14, 2011] 2011; Vol. 69.
Xiao 2005 {published data only}
- Xiao B, Xie W, Shi Z. A clinical control study of venlafaxine and fluoxetine in the treatment of senile depression. Chinese Journal of Behavioral Medical Science 2005;14:703‐4. [Google Scholar]
Xu 2010 {published data only}
- Xu S‐Q, Cao J, Huang W‐W. Comparison of escitalopram with fluoxetine in old depressive patients. Chinese Journal of New Drugs 2010;19:207‐9. [Google Scholar]
Zhao 2005 {published data only}
- Zhao HD, Wang JL, Xia JM. A comparative study of citalopram and fluoxetine in the treatment of senile depressive disorders. Chinese Mental Health Journal 2005;19:355‐6. [Google Scholar]
Zhao 2006 {published data only}
- Zhao H, Guan T. A control study in the treatment of depressive disorder with citalopram and fluoxetine. Evaluation and Analysis of Drug‐Use in Hospitals of China 2006;6:106‐8. [Google Scholar]
Zhou 2005 {published data only}
- Zhou M, Yao L. The efficacy and tolerability of venlafaxine and fluoxetine in treatment of elderly patients with first‐episode depression. Chinese Journal of Psychiatry 2005;38:157‐60. [Google Scholar]
Zhu 2005 {published data only}
- Zhu J, Jiang X, Zhou D, Zhang F. Comparative study of mirtazapine vs. fluoxetine in treatment of elderly depressive patients. Journal of Clinical Psychological Medicine 2005;15:277‐8. [Google Scholar]
Zhu 2006 {published data only}
- Zhu J. Comparative study of mirtazapine and fluoxetine in the treatment of senile depression. China Journal of Health Psychology 2006;14:546‐7. [Google Scholar]
References to ongoing studies
ChiCTR‐TRC‐11001668 {unpublished data only}
- ChiCTR‐TRC‐11001668. Efficacy and safety of agomelatine with flexible dose (25 mg/day with potential adjustment at 50 mg) given orally for 8 weeks in out‐patients with Major Depressive Disorder. www.chictr.org (accessed 16 July 2012).
CTRI/2011/05/001719 {unpublished data only}
- CTRI/2011/05/001719. An Open‐Label, Randomized, Parallel Group, Prospective, Multicentre Clinical Trial for Evaluation of Efficacy and Safety of IN‐ASTR‐001 in Patients with Major Depressive Disorder. [Clinical Trials Registry ‐ India] www.ctri.nic.in (accessed 16 July 2012).
EUCTR2007‐002130‐11‐ES {unpublished data only}
- EUCTR2007‐002130‐11‐ES. Therapeutic strategies in major depressive disorder resistant to treatment with SSRIs. Pragmatic, parallel group, randomised trial with blinded evaluation [Estrategias terapéuticas en Trastorno Depresivo Mayor resistente a tratamiento con Inhibidores Selectivos de la Recaptación de la Serotonina. Ensayo clínico pragmático, paralelo, aleatorizado con evaluación enmascarada]. [EU Clinical Trials Register] www.clinicaltrialsregister.eu (accessed 16 July 2012).
NCT01204086 {unpublished data only}
- NCT01204086. Pharmacogenomics Studies of Antidepressants. http://www.clinicaltrials.gov/ct2/show/NCT01204086 (accessed 16 July 2012).
NCT01254305 {unpublished data only}
- NCT01254305. Safety and Efficacy of F2695 SR in Adults With Fatigue Associated With Major Depressive Disorder. http://www.clinicaltrials.gov/ct2/show/NCT01204086 (accessed 16 July 2012).
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skew ness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and statistic manual of mental disorders :DSM III. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. DSM‐III‐R: diagnostic and statistical manual of mental disorders. Washington, DC: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). American Journal of Psychiatry 2000;157 Suppl 4:1‐45. [PubMed] [Google Scholar]
APA 2006
- American Psychiatric Association. American Psychiatric Association Practice Guidelines for the Treatment of Psychiatric Disorders: Compendium 2006. Washington, DC: American Psychiatric Association, 2006. [Google Scholar]
Barbui 2004
- Barbui C, Cipriani A, Brambilla P, Hotopf M. "Wish bias" in antidepressant drug trials?. Journal of Clinical Psychopharmacology 2004;24(2):126‐30. [DOI] [PubMed] [Google Scholar]
Barbui 2011a
- Barbui C, Tansella M. Cochrane reviews impact on mental health policy and practice. Epidemiology and Psychiatric Sciences 2011;20(3):211‐4. [DOI] [PubMed] [Google Scholar]
Barbui 2011b
- Barbui C, Cipriani A. Cluster randomised trials. Epidemiology and Psychiatric Sciences 2011;20(4):307‐9. [DOI] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of randomised controlled trials. The CONSORT statement. Journal of American Medical Association JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Cipriani 2005
- Cipriani A, Brambilla P, Furukawa T, Geddes J, Gregis M, Hotopf M, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD004185.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2006a
- Cipriani A, Barbui C, Brambilla P, Furukawa TA, Hotopf M, Geddes JR. Are all antidepressants really the same? The case of fluoxetine: a systematic review. Journal of Clinical Psychiatry 2006;67(6):850‐64. [DOI] [PubMed] [Google Scholar]
Cipriani 2009a
- Cipriani A, Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD006117.pub2] [DOI] [PubMed] [Google Scholar]
Cipriani 2009b
- Cipriani A, Santilli C, Furukawa TA, Signoretti A, Nakagawa A, McGuire H, Churchill R, Barbui C. Escitalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD006532.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2011
- Cipriani A, Barbui C, Salanti G, Rendell J, Brown R, Stockton S, et al. Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple‐treatments meta‐analysis. Lancet 2011;378:1306‐15. [DOI] [PubMed] [Google Scholar]
Cipriani 2012a
- Cipriani A, Koesters M, Furukawa TA, Nosè M, Purgato M, Omori IM, Trespidi C, Barbui C. Duloxetine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2012;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2012b
- Cipriani A, Purgato M, Furukawa TA, Trespidi C, Imperadore G, Signoretti A, Churchill R, Watanabe N, Barbui C. Citalopram versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ciuna 2004
- Ciuna A, Andretta M, Corbari L, Levi D, Mirandola M, Sorio A, Barbui C. Are we going to increase the use of antidepressants up to that of benzodiazepines?. European Journal of Clinical Pharmacology 2004;60(9):629‐34. [DOI] [PubMed] [Google Scholar]
Cohen 1992
- Cohen J. A power primer. Psychological Bulletin 1992;112(1):155‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26:57‐63. [DOI] [PubMed] [Google Scholar]
Freemantle 2000
- Freemantle N, Mason J. The importance of achieving additional drug benefits at a reasonable cost. Pharmacoeconomics 2000;17(4):319‐24. [DOI] [PubMed] [Google Scholar]
Furukawa 2002
- Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the 'number needed to treat'? An empirical study of summary effect measures in meta‐analyses. International Journal of Epidemiology 2002;31(1):72‐6. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Gartlehner 2010
- Gartlehner G, Chapman A, Strobelberger M, Thaler K. Differences in efficacy and safety of pharmaceutical treatments between men and women: An umbrella review. PLoS One 2010;5:e11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gartlehner 2011
- Gartlehner G, Hansen RA, Morgan LC, Thaler K, Lux L, Noord M, et al. Comparative benefits and harms of second‐generation antidepressants for treating major depressive disorder: an updated meta‐analysis. Annals of Internal Medicine 2011;155(11):772‐85. [DOI] [PubMed] [Google Scholar]
Geddes 2000
- Geddes JR, Freemantle N, Mason J, Eccles MP, Boynton J. SSRIs versus other antidepressants for depressive disorder. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001851] [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org, 2011. [Google Scholar]
Horder 2011
- Horder J, Matthews P, Waldmann R. Placebo, Prozac and PLoS: significant lessons for psychopharmacology. Journal of Psychopharmacology 2011;25(10):1277‐88. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326(7400):1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marshall 2009
- Marshall KP, Georgievskava Z, Georgievsky I. Social reactions to Valium and Prozac: a cultural lag perspective of drug diffusion and adoption. Research in Social and Administrative Pharmacy 2009;5(2):94‐107. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Nagakawa 2009
- Nakagawa A, Watanabe N, Omori IM, Barbui C, Cipriani A, McGuire H, et al. Milnacipran versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD006529.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2010
- National Institute for Clinical Excellence. Depression: management of depression in primary and secondary care ‐ NICE guidance. National Institute for Clinical Excellence, 2010. [Google Scholar]
Odgaard‐Jensen 2011
- Odgaard‐Jensen J, Vist GE, Timmer A, Kunz R, Akl EA, Schünemann H, et al. Randomisation to protect against selection bias in healthcare trials. Cochrane Database of Systematic Reviews 2001, Issue 4. Art. No.: MR000012. [DOI: 10.1002/14651858.MR000012.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Omori 2010
- Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, et al. Fluvoxamine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD006114.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Spitzer 1972
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives General Psychiatry 1978;35(6):773‐82. [DOI] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta‐analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119‐29. [DOI] [PubMed] [Google Scholar]
Trespidi 2011
- Trespidi C, Barbui C, Cipriani A. Why it is important to include unpublished data in systematic reviews. Epidemiology and Psychiatric Sciences 2011;20(2):133‐5. [DOI] [PubMed] [Google Scholar]
Watanabe 2011
- Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, Churchill R, Furukawa TA. Mirtazapine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2011;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1977
- World Health Organization. Manual of the international statistical classification of diseases, injuries, and causes of death. Geneva: World Health Organization, 1977. [Google Scholar]
WHO 1992
- World Health Organization. The Tenth Revision of the International Classification of Diseases and Related Health Problems (ICD‐10). Geneva: World Health Organization, 1992. [Google Scholar]
WHO 2006
- World Health Organization. WHO Collaborative Centre for Drug Statistics Methodology. http://www.whocc.no/atcddd/.
Wong 2005
- Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nature Reviews Drug Discovery 2005;4(9):764‐74. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cochrane 2005
- Cipriani A, Brambilla P, Furukawa T, Geddes J, Gregis M, Hotopf M, Malvini L, Barbui C. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD004185.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]