

Summary
Deep mass spectrometry-based proteomic profiling of rare cell populations has been constrained by sample input requirements. Here, we present a protocol for droplet-based one-pot preparation for proteomic samples (DROPPS), an accessible low-input platform that generates high-fidelity proteomic profiles of 100–2,500 cells. We describe steps for depositing cellular material, cell lysis, and digesting proteins in the same microliter-droplet well. We anticipate DROPPS will accelerate biology-driven proteomic research for a multitude of rare cell populations.
For complete details on the use and execution of this protocol, please refer to Waas et al.1
Subject areas: cell biology, flow cytometry, proteomics
Graphical abstract
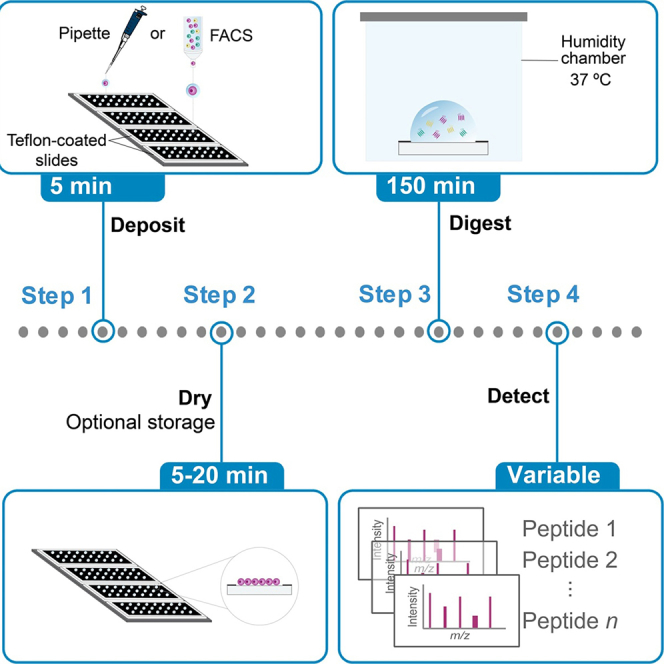
Highlights
-
•
DROPPS is an accessible low-input proteomic method for 100–2,500 cells
-
•
Cells can be deposited with standard pipette or cell sorter
-
•
LC-MS/MS-compatible reagents are used for cell lysis and protein digestion
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Deep mass spectrometry-based proteomic profiling of rare cell populations has been constrained by sample input requirements. Here, we present a protocol for droplet-based one-pot preparation for proteomic samples (DROPPS), an accessible low-input platform that generates high-fidelity proteomic profiles of 100–2,500 cells. We describe steps for depositing cellular material, cell lysis, and digesting proteins in the same microliter-droplet well. We anticipate DROPPS will accelerate biology-driven proteomic research for a multitude of rare cell populations.
Before you begin
In-depth proteomic interrogation of cells, tissues, and fluids is often performed with bottom-up mass spectrometry (MS) workflows, where proteins are extracted and enzymatically digested to obtain peptides.2,3,4 Despite advances in instrument sensitivity – which enable deep proteomic profiling from < 1 μg – the analysis of rare cell populations or other sample-limited systems is impeded by the loss of material incurred by sample preparation. Accordingly, proteomic methods often call for 10,000s–1,000,000s of cells (roughly, 10–100’s μg) as starting material.5 Recent single-cell or low-input proteomic methods typically leverage low-volumes and minimal manual manipulation to reduce losses during sample preparation.6,7,8 Despite these technical advances, many low-input methods only support specific study design (e.g., isobaric labeling with booster channel) or require specialized and costly equipment (e.g., microfabrication capabilities, microfluidic cell isolation systems).
To address this limitation, we have developed an accessible low-input proteomic method for studying rare populations of cells, Droplet-based one-pot preparation for proteomic samples (DROPPS), that is simple to implement and easily integrates with fluorescence activated cell sorting (FACS). DROPPS utilizes commercially available and economical materials to enable reproducible and biologically-informative proteomic analysis of small numbers of cells (100s - 1,000s of cells, or 20 ng–1 μg) from in vitro and in vivo systems. Overall, we demonstrate that DROPPS is an accessible proteomics workflow to enhance and accelerate biological discovery for rare cell populations.
The protocol below describes the specific steps for using MCF10A cells. However, we have also used this protocol in HCC1187, MDA-MB-157, Hs-578-T, and epithelial cells isolated from murine mammary glands.
CRITICAL: Protein contaminants can come from many sources, including from contact with skin. Gloves are essential for every step of this protocol. Thus, it is critical that all glassware which encounters MS reagents be thoroughly rinsed with MS-grade acetonitrile, water and the respective buffer before addition of final buffers. Maintaining clean equipment, reagents, and bench space dedicated to proteomic sample preparation is recommended for best results.
Material acquisition (slides and holder)
Timing: variable (up to 2 months, depends on vendor and shipping)
-
1.
Place order for Teflon-coated microscope slides from Tekdon (see key resources table for specifications).
Note: The included template (File S1) has 2 mm diameter wells – as described in Waas et al.1 – however, other well sizes may be more suitable for different applications.
-
2.Make a holder for microscope slides using the provided model (Files S2 and S3).
-
a.Using printer-appropriate software, place the top and bottom components of the plate holder into the print space.
-
b.Convert the stl file to G-code using printer and material-appropriate settings.Note: For the holder used in Waas et al., PrusaSlicer was used to generate G-code with a layer height of 0.15 mm and 15% infill for Prusament polylactic acid (PLA). Printing was performed on an Original i3 Prusa MK3S. Different printers and materials may require different settings.
-
c.Print top and bottom components using the G-code.
-
d.Glue top and bottom component of holder together.
-
i.Allow components to reach room temperature after printing.
-
ii.Find a flat surface to work on.
-
iii.Apply glue to both components.
-
iv.Place components together taking care to align the edges.
-
v.Place a heavy, flat object (e.g., box or book) on top of slide holder to press the components together.Note: Please contact the authors if 3D-printing is inaccessible or prohibitively expensive in your area. If it is cheaper to ship, they will send a slide holder.Note: Designing a model which can be printed as a single component is possible for more advanced 3D printing operators.
-
i.
-
a.
Wash slides
-
3.Wash slide using 100% ethanol.
-
a.Hold the slide by the edges, pour ethanol from a 50 mL conical tube over the Teflon-coated side of microscope slide.Alternative: You can use a wash bottle to dispense the ethanol.
-
b.Allow excess ethanol to drain off the slide.
-
c.Rotate the slide and pour ethanol over the other (uncoated) side of the slide.
-
d.Allow excess ethanol to drain off the slide.Note: This step is to remove any particulates that are present on the slide. Repeat Step 3 if particulates are still present. You can use a clean pipette tip to dislodge any particulate that doesn’t wash off.
-
a.
-
4.Dry the slides.
-
a.Dab the excess ethanol off the slide by touching the edge of the slide to a Kimwipe.CRITICAL: Kimwipes are particulate free, which is why they are used for this step. Avoid use of standard paper towels.
-
b.Stand up the slides, resting the non-coated side on a tube rack or pipette box on top of a fresh Kimwipe and allow ethanol to evaporate.Optional: If further sterilization of slides is desired or required, slides can be autoclaved.
 Pause point: Cleaned slides can be stored in a sealed, clean container at room temperature indefinitely.
Pause point: Cleaned slides can be stored in a sealed, clean container at room temperature indefinitely.
-
a.
Cell collection, staining, and sorting
-
5.
Subculture MCF10A cells to be analyzed in 25 cm2 culture flask according to laboratory or vendor specifications. These criteria are cell line dependent.
-
6.Collect the cells from a 25 cm2 culture flask that is 70%–80% confluent.Note: The number of cells here is in excess of what is needed for cell sorting purposes, but the suggestion to use an entire flask ensures that sufficient material is present for cell counting and to minimize the effect of loss during washing. If it can be avoided, it is advised to avoid washing, staining, and cell sorting with < 1 million cells.
-
a.Remove the culture medium.Note: For Waas et al.,1 MCF10A cells were grown in DMEM:F12 media supplemented with 5% horse serum, 20 ng/mL epidermal growth factor (EGF), 10 μg/mL insulin, 500 ng/mL hydrocortisone, 100 ng/mL cholera toxin, and penicillin-streptomycin (100 U/mL penicillin, 100 μg/mL streptomycin).
-
b.Rinse the cells with 4 mL of PBS, no calcium, no magnesium (PBS−/−).
-
c.Add 2 mL of TrypLE Express Enzyme and incubate at 37°C for 3–5 min.
-
d.Collect the cells by gentle trituration and add to a tube containing 8 mL of culture medium.Note: If additional subculturing of cells is desired, it would be acceptable to split the cells between subculturing and cell staining and sorting. It is advised to proceed to washing, staining, and sorting with ≥ 1 million cells.
-
e.Centrifuge the cell suspension at 200 × g for 5 min.
-
f.Remove the supernatant.
-
g.Resuspend the cell pellet in 2 mL of PBS−/−.
-
h.Repeat the steps 6e–6g.
-
i.Place the cell suspension on ice.Note: Even if the cells are grown in suspension, they may benefit from dissociation to minimize doublets or larger aggregates.Note: Some cell sorters or facilities may require cell suspension to be within a certain range of concentrations. If so, count the cells by hemocytometer and adjust accordingly.Note: Some cell sorters or facilities may require cell suspension to be filtered to remove aggregates of cells. If so, filter cells through an appropriately sized filter.CRITICAL: Avoid the use of serum or albumin in the PBS−/−. The presence of high abundance proteins will compromise the depth and quality of the obtained mass spectrometry results.
-
a.
-
7.Stain cells for viability.
-
a.Add 4′,6-diamidino-2-phenylindole (DAPI) to the cell suspension at a final concentration of 0.5 μg/mL.
-
b.Incubate for 10 min on ice.
-
a.
Note: If the cells are not brought to cell sorter immediately, it is advisable, though not required, to remove cells from the DAPI working solution and resuspend in PBS until shortly before sorting. Incubation for too long can possibly lead to over-staining and higher background fluorescence.
-
8.Sort the cells into wells of Teflon-coated slides.Note: For the protocol in Waas et al.,1 a BD FACSAria Fusion Flow Cytometer was used. Similar instruments with the capacity to sort into plates should be compatible.
-
a.Bring an additional set of 4 slides to the cell sorter to align the cell sorter with the slides on the slide holder.Note: The slides used for alignment/positioning can be re-used between experiments.
-
b.Using sheath buffer, align the positioning of the slide holder with the stream from the cell sorter using 50 events.Note: Larger droplets, associated with increased event count, will sometimes migrate to the well due to the hydrophobicity of the Teflon even if alignment is not optimal.
-
c.Employing doublet- and dead cell-exclusion, sort 500 cells into each well.Note: The maximum number of cells which can be sorted into a 2 mm diameter well is ∼3,000 but ultimately depends on sort rate and sort mode (e.g., purity or recovery). Consult with the operator of your cell sorter to discuss these details in the context of your experimental system. For Waas et al.1 flow rates of 1–5 (arbitrary units, BD Fusion) were used and sorting was performed using “2-way recovery” mode.Note: If a larger number of cells is desired, alternative well sizes can be employed. In preliminary tests of the DROPPS slides, we tested 4 mm diameter wells which were able to have 10,000 cells sorted into them.
-
d.Place the slide holder with slides in biosafety cabinet until the droplets of fluid in each well evaporate.Note: There may not be complete evaporation due to the hygroscopicity of the salts in the sheath buffer depending on the humidity of the surrounding environment. It is not essential for the wells to be completely dry, but at minimum there should not be a visible droplet which rises above the surface of the slide.Alternatives: Cells in suspension at the desired concentration can be deposited onto slides using a pipette (maximum volume of 10 μL).Pause point: Slides with dried cells can be stored in microscope slide box in a −80°C freezer up to one year. It is recommended to keep the slide box in a freezer bag to minimize frost formation on the box or slides.
-
a.
Humidity chamber preparation
-
9.
Layer the inside of a large container with paper towels. Fill to a depth of 1 cm with water. This is the outer humidity container.
Note: This container needs to be larger than the inner humidity container and be able to fit within your 37°C incubator. We used a biohazardous sample transporter but similar sized, water-resistant containers with sealed lids are acceptable.
-
10.
Layer the inside of a small container with a paper towel. Fill to a depth of 1 cm with water. This is the inner humidity container.
Note: This container needs to be larger than the slide holder and the elevated slide platform but smaller than the outer humidity container. We used a glass food storage container for this step but similar sized, water-resistant containers with sealed lids are acceptable.
-
11.Assemble the two-compartment humidity chamber (see Figure 1).
-
a.Place the elevated slide platform inside the inner humidity container on top of wet paper towel.Note: This elevated slide platform needs to be a water-resistant object to hold up the slide holder above the wet paper towel. We used a pipette tip holder. But similar sized objects can also be used.
-
b.Close the inner humidity container and place inside the outer humidity chamber. Place a pre-warmed (at 37°C) paper towel on top of the inner humidity chamber.
-
c.Close the outer humidity container and place inside a 37°C incubator for a minimum of 30 min.
-
a.
Figure 1.

Humidity chamber setup for sample preparation using DROPPS
(A) Three Teflon-coated microscope slides placed inside the 3D-printed holder (which can hold up to four slides). The well-spacing of the slides is 9 mm to be consistent with flow sorting into 96-well format and with standard microchannel pipette.
(B and C) The (B) inside and (C) outside components of the humidity chamber setup referenced in humidity chamber preparation section and Steps 4 and 7.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 4′,6-diamidino-2-phenylindole | Sigma | D9542 |
| Ethanol | Commercial Alcohols | P016EAAN |
| Iodoacetamide | Sigma | I6125 |
| Ammonium bicarbonate | BioShop | AMC107 |
| Tris(2-carboxyethyl) phosphine | Sigma | C4706 |
| n-dodecyl-β-D-maltoside | Sigma | 850520P |
| Invitrosol | Thermo Fisher Scientific | MS10007 |
| Experimental models: Cell lines | ||
| Human: MCF10A | ATCC | CRL-10317 |
| Software and algorithms | ||
| MSFragger (v.3.8) run using FragPipe (v.20.0) | Kong et al.9 and Teo et al.10 | N/A |
| ProteinProphet | Nesvizhskii et al.11 | N/A |
| Percolator | Kall et al.12 | N/A |
| IonQuant v.1.9.8 | Yu et al.13 | N/A |
| Philosopher v.5.0.0 | da Veiga Leprevost et al.14 | N/A |
| Other | ||
| Orbitrap Fusion Tribrid mass spectrometer | Thermo Scientific | N/A |
| EASY-nLC 1000 HPLC system | Thermo Scientific | N/A |
| Acclaim PepMap 100 C18 HPLC column | Thermo Scientific | 164946 |
| EASY-Spray HPLC column | Thermo Scientific | ES903 |
| Teflon-coated microscope slides | Tekdon (https://tekdon.com) | Custom order. Details in File S1 |
| Microscrope slide holder | This study | Template provided as File S2 and S3. Available g-code at NIH 3D: 3DPX-020483 |
| BD FACSAria fusion flow cytometer (or equivalent plate-compatible instrument) | BD Biosciences | N/A |
Materials and equipment
Tris(2-carboxyethyl)phosphine (TCEP, 100 mM, stock in water, aliquot, store in −20°C)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris(2-carboxyethyl) phosphine | 100 mM | 25.01 mg |
| H2O (MS grade) | N/A | 1000 μL |
| Total | N/A | 1000 μL |
Iodoacetamide (IAA, 250 mM, freshly prepare, keep away from light, do not store)
| Reagent | Final concentration | Amount |
|---|---|---|
| Iodoacetamide | 250 mM | 46.225 mg |
| H2O (MS grade) | N/A | 1000 μL |
| Total | N/A | 1000 μL |
n-dodecyl-β-D-maltoside (DDM, 2% w/v, stock in water, aliquot, store in −20°C)
| Reagent | Final concentration | Amount |
|---|---|---|
| n-dodecyl-β-D-maltoside | 2% | 200 mg |
| H2O (MS grade) | N/A | 10 mL |
| Total | N/A | 10 mL |
Ammonium Bicarbonate (AmBic, 1 M, freshly prepare, do not store)
| Reagent | Final concentration | Amount |
|---|---|---|
| Ammonium Bicarbonate | 1 M | 79.06 mg |
| H2O (MS grade) | N/A | 1 mL |
| Total | N/A | 1 mL |
Lysis buffer (freshly prepare, do not store)
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Ammonium Bicarbonate | 100 mM | 10 μL |
| Acetonitrile (MS grade) | 15% | 15 μL |
| Invitrosol (5 ) | 0.5 | 10 μL |
| 2% n-dodecyl-β-D-maltoside | 0.06% | 3 μL |
| 100 mM Tris(2-carboxyethyl) phosphine | 5 mM | 5 μL |
| H2O (MS grade) | N/A | 57 μL |
| Total | N/A | 100 μL |
Digest buffer (freshly prepare, do not store)
| Reagent | Final concentration | Amount |
|---|---|---|
| 250 mM Iodoacetamide | 35 mM | 14 μL |
| Trypsin/Lys-C (MS grade) | variable | variable |
| H2O (MS grade) | N/A | To 100 μL |
| Total | N/A | 100 μL |
Note: 1:10 enzyme: substrate (wt:wt). If the amount of substrate is not known, you can test this by generating a standard curve using a hemocytometer and protein quantitation assay. Alternatively, in cases where material is limited, we have used the estimate of 2,000–5,000 cells is roughly equivalent to 1 μg.
Alternatives: There are a variety of reagents which can be used for reduction and alkylation which have different reported benefits and risks.15,16
Step-by-step method details
Cell lysis, protein solubilization and reduction
In this step, lysis buffer containing organic solvent, surfactants, and a reducing agent are added atop of the dried cellular material. This completes the cell lysis which was initiated by evaporation and freezing. The components in the solution are intended to solubilize, denature, and reduce proteins which will enable more complete digestion.
-
1.
Allow the slides to come to room temperature.
Note: It is advised to allow the slides to come to room temperature in a biosafety cabinet or chemical fume hood as the air flow can reduce condensation and deposition of airborne particulate.
-
2.
Place the slides into the slide holder.
-
3.
Add 2.5 μL of lysis buffer to each well.
-
4.
Place the slides in the pre-heated (at 37°C) humidity chamber and place the humidity chamber in the incubator at 37°C.
-
5.
Incubate the slides for 30 min to allow time for lysis and reduction of protein disulfide bonds.
Enzymatic digestion of proteins
In this step, proteins are alkylated and enzymatically digested. The alkylation is intended to block disulfide bonds from reforming – this increases accessibility to proteolytic enzymes (e.g., Trypsin, Lys-C) and decreases the amount of peptide cross-links. The enzymes cleave proteins into peptides at specific residues.
-
6.
Add 1 μL of digest buffer to each well.
-
7.
Place the slides in the humidity chamber and place the humidity chamber in the incubator at 37°C.
-
8.
Incubate the slides for 2 h to allow time for alkylation and digestion.
Note: We have only tested digest incubations up to 2 h. Longer digest incubations are likely possible but it will be important to monitor samples and ensure that the digest buffer has not evaporated. Top up of digest buffer may be required for longer digest incubations.
Transfer to autosampler plate
In this step, enzymatic digestion is quenched by reducing temperature and acidification. The digested sample is transferred to the plate and the well is washed with additional volume of solution to minimize sample loss.
-
9.
Remove the slides from the incubator.
-
10.
Quickly deposit 5 μL of chilled HPLC grade water with 0.1% formic acid (FA) to each well on the slide (this keeps sample from evaporating).
-
11.
Transfer the sample with a pipette to a vial or plate compatible with the LC autosampler system.
-
12.
Deposit 10 μL of HPLC grade water with 0.1% FA to each well.
-
13.
Transfer the sample to the same vial or plate from step 11.
Mass spectrometry data acquisition
In this step, digested peptides are separated based on their hydrophobicity by reversed-phase liquid chromatography and ionized into the mass spectrometer for detection and quantification.
-
14.
Acquire data on mass spectrometer interfaced with a liquid chromatography system with trap column.
Note: From Waas et al., LC-MS/MS analysis was performed on an Orbitrap Fusion MS coupled to an EASY-nLC 1000 System. Peptides were washed on a pre-column (Acclaim PepMap 100 C18, 3 μm particle size, 75 μm diameter, 20 mm length, Thermo Fisher Scientific) with 60 μL of mobile phase A (0.1% FA in HPLC grade water) at 3 μL/min and separated using a 50 cm EASY-Spray column (2 μm particle size,75 μm diameter, 500 mm length , ThermoFisher) ramping mobile phase B (0.1% FA in HPLC grade acetonitrile) from 0% to 5% in 2 min, 5%–27% in 160 min, 27%–60% in 40 min interfaced online using an EASY-Spray source (ThermoFisher). The Orbitrap Fusion MS was operated in data dependent acquisition mode using a 2.5 s cycle at a full MS resolution of 240,000 with a full scan range of 350–1550 m/z with RF Lens at 60%, full MS automatic gain control at 200%, and maximum inject time at 40 ms. MS/MS scans were recorded in the ion trap with 1.2 Th isolation window, 100 ms maximum injection time, MS/MS automatic gain control at 200%, with a scan range of 200–1400 m/z using Normal scan rate. Ions for MS/MS were selected using monoisotopic peak detection, intensity threshold of 1,000, positive charge states of 2–5, 40 s dynamic exclusion, and then fragmented using higher-energy collisional dissociation with 31% normalized collision energy.
Expected outcomes
The quality and depth of proteomic data resulting from DROPPS will ultimately depend on available equipment, instrumentation, and acquisition parameters. For instrumentation and equipment identical, or equivalent, to those described in this protocol, example of expected results for MCF10A cells and three triple negative breast cancer cell lines (HCC1187, MDA-MB-157, Hs-578-T) are shown (Figure 2). There are many factors to consider for assessing the quality of proteomic results including the depth of coverage and the observed variance. There is a relationship between the number of cells used as input and both proteomic depth and variance. Characterizing this relationship is an important aspect of determining the fitness-for-purpose of a proteomic sample preparation strategy. Another important consideration for proteomic data is batch-to-batch variability, as it can be impractical to acquire all data for all samples of an experimental cohort at one time. Examining these relationships and sources of variance with available equipment and instrumentation is highly suggested as a preliminary step in experimental design.
Figure 2.

Expected results for implementing DROPPS on mammary epithelial cell lines
(A) Cell number titration workflow where 10–2,500 MCF10A cells were deposited per well (n = 3 each) by fluorescent-activated cell sorting and subsequently processed using DROPPS.
(B and C) The (B) peptide counts and (C) protein counts of the individual runs where columns represent the mean values.
(D and E) Distribution of intensity coefficients of variation (CVs) at the (D) peptide and (E) protein level.
(F) Schematic of operator experimental design where 500 MCF10A cells were deposited per well (n = 8 each) by three operators and subsequently processed using DROPPS.
(G and H) The (G) peptide counts and (H) protein counts of the individual samples prepared by each operator.
(I and J) Distribution of (I) peptide intensity or (J) protein intensity CVs.
(K) Workflow for testing inter-batch variation by DROPPS. Sorted cells (500 cells) from three TNBC cell lines were processed separately on different days. Each cell line had seven or eight sample replicates processed each day.
(L and M) The (L) peptide counts and (M) protein counts of the individual samples prepared by each operator.
(N and O) Distribution of (N) peptide intensity or (O) protein intensity CVs within a batch or for combining batches. Boxplots show the median, interquartile ranges, and 95% confidence interval estimate. TNBC: triple negative breast cancer. Altered content from Figures S1 and S2 from Waas et al.1
The results portrayed in Figure 2 depict similar proteomic depth from multiple cell lines. However, each of these lines are human and are similar in origin (mammary epithelium). Species and type of cell are two additional factors which can influence the depth of proteomic results.
Quantification and statistical analysis
In Waas et al., data were analyzed using FragPipe (v.20.0) using MSFragger (v.3.8) to search against a human (Uniprot, 43,392 sequences, accessed 2023-02-08) proteome – canonical plus isoforms. Default settings for LFQ workflow were used using IonQuant v.1.9.8 and Philosopher v.5.0.0 with the following modifications. MaxLFQ min ions was set to 1; MBR RT tolerance was set to 2 min, and MBR top runs was set to 10. The “MaxLFQ intensity” columns are extracted from the “combined_protein.tsv” file output and used for downstream analysis.
Note: We suggest that users match data analysis parameters to empirical results (e.g., observed variance in retention time) and instrument acquisition specifications (e.g., resolving power).
Note: Raw files can be analyzed using various software (e.g. MaxQuant, ProteomeDiscoverer).
Limitations
An important consideration for MS-based proteomics, especially those relying on data-dependent acquisition (DDA), is that a missing value does not mean the protein is absent. Stochastic sampling, modified peptides, ionization suppression, or intensity beneath limit of detection are all technical reasons a protein may be missing a quantitative value in the results. The requirement for cells to be in a single cell suspension for cell sorting means that spatial information can be lost without special experimental design (e.g., micro or microdissection of sample prior to dissociation). Additionally, the dissociation procedure, or even the act of cell sorting may affect the proteome. DROPPS hasn’t been tested on formalin-fixed, paraffin embedded samples or those embedded in optical cutting temperature medium. Reagents in these samples can interfere with LC and MS. If using flow-based cell sorting, there is a limit to the cells which can be sorted into the wells. This has to be determined empirically, as many factors can affect this.
Troubleshooting
Problem 1
Incomplete trypsin digestion (Step 8).
Potential solution
We recommend using a 1:10 (wt:wt) enzyme-to-substrate ratio in a 3.5 μL total volume (2.5 μL lysis buffer + 1 μL of digest buffer) which should efficiently digest up to ∼1.5 μg of protein in a sample. If the protein concentration is not known, an estimate of 200 pg–500 pg/cell can be used for common mammalian cell types. If trypsin digestion is incomplete, using longer incubation times of up to 4 h or addition of a digestion step with more protease (at a 1:50 (wt:wt) enzyme-to-substrate ratio) after 2–4 h may increase the extent of enzymatic cleavage. Ensure that the sample does not contain protease inhibitors that may inhibit trypsin.
Problem 2
Number of protein detections is below the expected range (Various steps).
Potential solution
There are many plausible reasons for low protein detections. One potential explanation can be due to high concentrations of salt in the samples which can interfere with ionization prior to MS analysis. Typical MS proteomics sample preparation workflows involve an offline desalting step yet it can result in sample loss. Consequently, we prefer to use an in-line trap column for desalting as described in the critical note in step 13.
Another reason might be due to the presence of detergents and/or polymer contaminants in the sample. Detergents can hinder ionization and mask the detection of peptides. Though this protocol uses MS-compatible detergents, there may be trace amounts of detergents and/or polymers in samples due to glassware being cleaned with MS incompatible reagents. Thus, it is critical that all glassware which encounters MS reagents be thoroughly rinsed with MS-grade acetonitrile, water and the respective buffer before addition of final buffers.
High abundance of BSA/FBS can also mask the detection of low abundance proteins. It is critical to thoroughly wash cell culture samples with PBS and use FACS buffer that does not contain BSA/FBS.
Finally, there are cell type and cell line differences in protein detection. For example, we typically detect less proteins in mesenchymal/fibroblast-like cells. It may be beneficial to include an epithelial cell processing control to ensure that DROPPS is being performed technically correct and that differences in protein detection are due to cell type biological differences.
Problem 3
Batch-to-batch variation (Various steps).
Potential solution
Inconsistencies in sample preparation can lead to greater batch-to-batch variation than expected. It is critical when designing large studies to use experimental blocking to randomize samples from different conditions across batches. One can also consider the use of spike in standards (e.g., iRT peptides) to evaluate sample-to-sample technical variation.
Problem 4
Evaporation during sample preparation (Steps 4–8).
Potential solution
It is critical to work fast when pipetting minute amounts of solvent on the slides to prevent evaporation at undesired times. Consider using a multi-channel pipette to efficiently add solvent to the slides. Consider performing pipetting steps in a cold room if available.
With our humidity chamber set-up, we do not experience notable sample evaporation during digestion, yet it is a possible concern with new set-ups. As a result, we recommend careful monitoring of samples during digestion in initial DROPPS experiments to determine if there is significant evaporation and if mid-incubation top-up of digest buffer is required. Alternative humidity chamber set-ups or containers may be better for minimizing evaporation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas Kislinger (thomas.kislinger@utoronto.ca).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Matthew Waas (waasmatthew@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze datasets and code.
Acknowledgments
Work in the Kislinger lab was supported by operating grants from the Canadian Institutes of Health Research, the Terry Fox Research Institute PPG, the Canadian Cancer Society, and the Canada Research Chair program. A.K. was supported by the Ontario Graduate Scholarship and Ontario Student Opportunity Trust Fund awards. M. Waas was supported by a CIHR Postdoctoral Fellowship.
Author contributions
M. Waas, M.G., A.K., and C.Z. generated figures. T.K. supervised the study. M. Waas, M.G., and A.K. wrote the manuscript, which all other authors edited and approved.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2024.103397.
Contributor Information
Matthew Waas, Email: waasmatthew@gmail.com.
Thomas Kislinger, Email: thomas.kislinger@utoronto.ca.
Supplemental information
References
- 1.Waas M., Khoo A., Tharmapalan P., McCloskey C.W., Govindarajan M., Zhang B., Khan S., Waterhouse P.D., Khokha R., Kislinger T. Droplet-based proteomics reveals CD36 as a marker for progenitors in mammary basal epithelium. Cell Rep. Methods. 2024;4 doi: 10.1016/j.crmeth.2024.100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nusinow D.P., Szpyt J., Ghandi M., Rose C.M., McDonald E.R., Kalocsay M., Jané-Valbuena J., Gelfand E., Schweppe D.K., Jedrychowski M., et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell. 2020;180:387–402.e16. doi: 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence R.T., Perez E.M., Hernández D., Miller C.P., Haas K.M., Irie H.Y., Lee S.-I., Blau C.A., Villén J. The Proteomic Landscape of Triple-Negative Breast Cancer. Cell Rep. 2015;11:630–644. doi: 10.1016/j.celrep.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D., Eraslan B., Wieland T., Hallström B., Hopf T., Zolg D.P., Zecha J., Asplund A., Li L.H., Meng C., et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019;15 doi: 10.15252/msb.20188503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waas M., Kislinger T. Addressing Cellular Heterogeneity in Cancer through Precision Proteomics. J. Proteome Res. 2020;19:3607–3619. doi: 10.1021/acs.jproteome.0c00338. [DOI] [PubMed] [Google Scholar]
- 6.Leduc A., Huffman R.G., Cantlon J., Khan S., Slavov N. Exploring functional protein covariation across single cells using nPOP. Genome Biol. 2022;23:261. doi: 10.1186/s13059-022-02817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Y., Piehowski P.D., Zhao R., Chen J., Shen Y., Moore R.J., Shukla A.K., Petyuk V.A., Campbell-Thompson M., Mathews C.E., et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018;9:882. doi: 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin K., Zhang T., Lin T.-T., Habowski A.N., Zhao R., Tsai C.-F., Chrisler W.B., Sontag R.L., Orton D.J., Lu Y.-J., et al. Facile One-Pot Nanoproteomics for Label-Free Proteome Profiling of 50–1000 Mammalian Cells. J. Proteome Res. 2021;20:4452–4461. doi: 10.1021/acs.jproteome.1c00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong A.T., Leprevost F.V., Avtonomov D.M., Mellacheruvu D., Nesvizhskii A.I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods. 2017;14:513–520. doi: 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teo G.C., Polasky D.A., Yu F., Nesvizhskii A.I. Fast Deisotoping Algorithm and Its Implementation in the MSFragger Search Engine. J. Proteome Res. 2021;20:498–505. doi: 10.1021/acs.jproteome.0c00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 12.Käll L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 13.Yu F., Haynes S.E., Nesvizhskii A.I. IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.da Veiga Leprevost F., Haynes S.E., Avtonomov D.M., Chang H.Y., Shanmugam A.K., Mellacheruvu D., Kong A.T., Nesvizhskii A.I. Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat. Methods. 2020;17:869–870. doi: 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller T., Winter D. Systematic Evaluation of Protein Reduction and Alkylation Reveals Massive Unspecific Side Effects by Iodine-containing Reagents. Mol. Cell. Proteomics. 2017;16:1173–1187. doi: 10.1074/mcp.M116.064048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suttapitugsakul S., Xiao H., Smeekens J., Wu R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol. Biosyst. 2017;13:2574–2582. doi: 10.1039/c7mb00393e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets and code.