

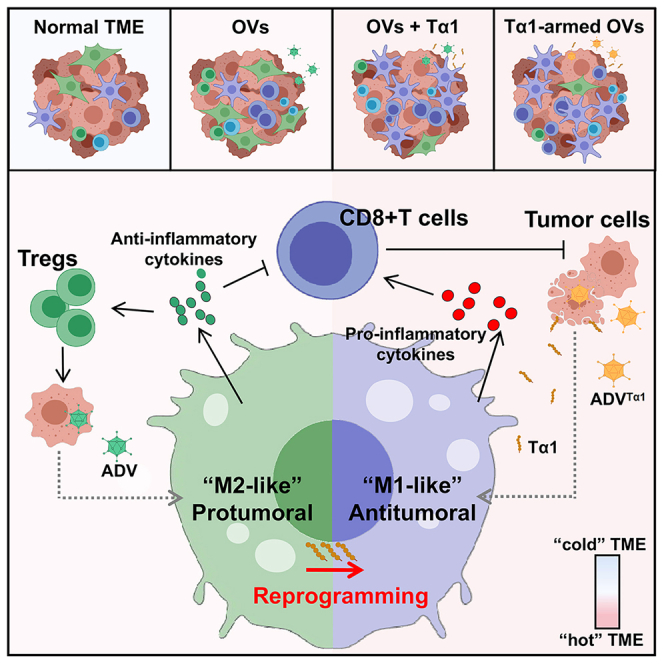
Summary
Although oncolytic adenoviruses are widely studied for their direct oncolytic activity and immunomodulatory role in cancer immunotherapy, the immunosuppressive feedback loop induced by oncolytic adenoviruses remains to be studied. Here, we demonstrate that type V adenovirus (ADV) induces the polarization of tumor-associated macrophages (TAMs) to the M2 phenotype and increases the infiltration of regulatory T cells (Tregs) in the tumor microenvironment (TME). By selectively compensating for these deficiencies, thymosin alpha 1 (Tα1) reprograms “M2-like” TAMs toward an antitumoral phenotype, thereby reprogramming the TME into a state more beneficial for antitumor immunity. Moreover, ADVTα1 is constructed by harnessing the merits of all the components for the aforementioned combinatorial therapy. Both exogenously supplied and adenovirus-produced Tα1 orchestrate TAM reprogramming and enhance the antitumor efficacy of ADV via CD8+ T cells, showing promising prospects for clinical translation. Our findings provide inspiration for improving oncolytic adenovirus combination therapy and designing oncolytic engineered adenoviruses.
Keywords: thymosin α1, oncolytic virus, oncolytic adenovirus, M2 polarization, M1 polarization, tumor associated macrophages, TAM, regulatory T cells, Tregs, tumor immunotherapy, tumor microenvironment, TAM reprogramming
Graphical abstract
Highlights
-
•
ADV-infected tumor cells induce M2 polarization of TAMs, requiring direct cell contact
-
•
Thymosin alpha 1 (Tα1) improves the antitumor efficacy of oncolytic adenovirus
-
•
Tα1 orchestrates TAMs to drive antitumor CD8+ T cell immunity during OV treatment
-
•
Potential clinical translation of ADVTα1 in improving outcomes for patients with cancer
Liu et al. report that the type V adenovirus (ADV) induces M2 polarization of tumor-associated macrophages in a direct cell contact-dependent manner. Tα1 reprograms ADV-induced “M2-like” macrophages toward an antitumoral phenotype, thereby reprogramming the TME into a state more beneficial for antitumor immunity.
Introduction
Cancer is a major disease affecting human health and has become the second leading cause of death worldwide.1 Over the past few decades, many cancer immunotherapies, including monoclonal antibodies, adoptive cell therapies, checkpoint inhibitors, and oncolytic viruses (OVs), have emerged as the most promising therapies for cancer treatment because of their ability to provide a long-lasting and effective clinical response for patients with cancer.2,3
OVs are a new class of immunotherapy agents that can promote tumor regression by preferentially replicating in tumor cells, inducing immunogenic cell death, and stimulating host antitumor immunity.4,5,6 Adenovirus is one of the most studied and promising OVs, and numerous clinical trials on adenovirus are currently being conducted.7 Unfortunately, oncolytic adenoviruses usually produce limited antitumor efficacy due to some potential limitations of the adenovirus itself, including antiviral immune responses, off-target infection, and the induction of an immunosuppressive feedback loop.8,9,10 Many types of engineered biomaterials have been developed as systematic delivery tools to protect adenoviruses from immune clearance in the blood and to enhance tumor homing.11,12,13 However, the induction of an immunosuppressive feedback loop by adenovirus, as well as the therapeutic strategies to compensate for these deficiencies, remains poorly studied. In the present study, we observed that treatment with type V adenovirus (ADV) (a selective replicating type V adenovirus whose biological function has been demonstrated in previous studies10) induced macrophages to polarize toward an “M2-like” pro-tumoral phenotype and resulted in an expansion of regulatory T cells (Tregs).
For enhanced antitumor efficacy of oncolytic adenoviruses, current treatments mainly involve combination with at least one other treatment or anticancer drug, such as immune checkpoint inhibitors, cytokines, cell therapy, and cyclophosphamide, or modification of the adenovirus using genetic engineering.14,15,16,17 Compared with some direct “two-stones-kill-one-bird” treatment strategies, exploring the therapeutic disadvantages of oncolytic adenoviruses during treatment and then compensating for these deficiencies through combination therapy or genetic modification may be a better prospective therapeutic approach.
Thymosin alpha 1 (Tα1), a thymus-derived immunomodulatory peptide, is widely believed to regulate the immune activity of innate immune cells, such as polymorphonuclear leukocytes, dendritic cells (DCs), and macrophages.18,19 Notably, a recent study has shown that Tα1 improves the curative effect of chemotherapy by reversing efferocytosis-induced M2 polarization of macrophages via activation of the TLR7/SHIP1 axis.20
Based on our findings of the transformation of tumor-associated macrophages (TAMs) to the M2 phenotype, increased immune infiltration of Tregs within the tumor microenvironment (TME) after ADV treatment, and the ability of Tα1 to modulate immune cells in previous studies,18,19 we hypothesize that Tα1 could enhance antitumor efficacy by reversing the ADV-induced expansion of immunosuppressive cells, and we confirm this hypothesis in a variety of solid tumor models and in vitro cell experiments. Furthermore, we engineer a recombinant oncolytic adenovirus (ADVTα1) expressing Tα1, which has superior immunomodulatory effects and antitumor efficacy compared to combination therapy, demonstrating good potential for clinical application. Collectively, our study identifies the advantages of oncolytic adenovirus combination immunotherapy and the design of oncolytic engineered adenoviruses.
Results
ADV inhibits tumor growth and increases immune infiltration with the expansion of Tregs and “M2-like” macrophages in the TME
Using a breast cancer model to test the antitumor potential of ADV (Figure 1A), we found that intra-tumoral injection of ADV efficiently restrained tumor growth in BALB/c wild-type (WT) mice relative to that of the PBS group (Figure 1B), and no significant toxicity was observed during the treatment, as shown by mouse body weight (Figure 1C). In addition, we found that oncolytic adenovirus therapy effectively prolonged survival (Figure 1D).
Figure 1.

ADV treatment showed antitumor activity and effectively increased tumor immune infiltration
(A) Experimental schematic of mice from Figures 1B–1M: 4T1 tumor-bearing wild-type (WT) mice were administered ADV or vehicle control (PBS) starting on day 10 when the tumor volume reached approximately 50–100 mm3. Tumor-infiltrating leukocytes (TILs) from the tumor and spleen were assessed by flow cytometry (day 19, day 24, day 35; n = 6 biological replicates); s.c., subcutaneous; i.t., intra-tumoral.
(B–D) 4T1-tumor-bearing WT mice were administered ADV or vehicle control (PBS) starting on day 10 when the tumor reached approximately 50–100 mm3 in volume (n = 10 biological replicates). (B) Tumor growth and individual tumor growth. (C) Body weight. (D) Survival.
(E–L) Percentages (top) and total cells normalized to g (gram) tumor tissue (bottom) of TILs. (E) CD3+CD4+ T cells, (F) CD3+CD8+ T cells, (G) CD11b+F4/80+ macrophages, (H) CD3−CD49b+ NK cells, (I) CD11c+CD86+ DCs, (J) CD25+Foxp3+ Tregs, (K) “M1-like” macrophages, (L) “M2-like” macrophages within the TME of mice (n = 6 biological replicates).
(M) Percentages (top) and total cells normalized to g tumor tissue (bottom) of CD4+ T cells (left) and CD8+ T cells (right) within the spleen of mice (n = 6 biological replicates). The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The therapeutic effect of ADV in vivo is due not only to its direct oncolytic function but also to increased immune infiltration in the TME.21,22 Therefore, we next assessed the systemic immunoregulatory consequences of ADV administration throughout tumor development via profiling the immune response at an early, intermediate, and late time points. With the flow cytometry gating strategy to analyze immune cells in the 4T1 model and H22 model (Figures S2A–S2D), we observed that ADV treatment effectively transformed the tumor from “cold” to “hot,” which was characterized by increases in the percentages and absolute number of CD3+CD4+ T cells, CD3+CD8+ T cells, and CD11b+F4/80+ macrophages at all time points assessed (Figures 1E–1G and S1B–S1D), However, the ADV group and the PBS group had comparable populations of natural killer (NK) cells and DCs at all time points assessed (Figures 1H, 1I, S1E, and S1F). Notably, ADV treatment led to an expansion of CD25+Foxp3+ Tregs in the 4T1 model and H22 model at all time points assessed (Figures 1J and S1G). Similarly, ADV treatment reprogrammed “M1-like” macrophages into “M2-like” macrophages within the TME (Figures 1K, 1L, S1H, and S1I). Furthermore, the percentages and number of CD4+ T cells and CD8+ T cells changed within the spleen only at the early point after ADV treatment, and there were no differences at intermediate and late time points in the 4T1 model (Figure 1M). Interestingly, the percentages and number of CD4+ T cells and CD8+ T cells within the spleen did not change at all time points assessed in the H22 model (Figures S1J and S1K). Based on these results, we concluded that ADV selectively mediated immune responses in the TME, and that changes in CD4+ T cells and CD8+ T cells in the spleen at the early point after ADV treatment were due to immune responses triggered by adenovirus infection.
Collectively, ADV treatment has antitumor activity and effectively increases tumor immune infiltration while causing an expansion of some immunosuppressive cells, such as Tregs and “M2-like” macrophages.
Tα1 effectively enhances the antitumor effect of ADV in a variety of solid tumor models
Tregs and pro-tumoral “M2-like” TAMs are involved in tumor growth, invasion, and metastasis.23,24,25 Tα1, a 28-amino acid peptide isolated from thymic tissue, has been shown to be a Toll-like receptor (TLR)-9 and TLR-2 agonist. Importantly, Tα1 could change macrophage phenotypes and regulate the viability of T cells.26,27 To further explore a more potent antitumor immunotherapy for potential clinical application, we attempted to use Tα1 in combination with ADV therapy to improve antitumor efficacy by compensating for the therapeutic shortcomings of ADV (Figure 2A). We found that ADV combined with Tα1 significantly delayed tumor growth and prolonged survival in a variety of solid tumor models, and no drug toxicity occurred during treatment in any treatment group (Figures 2B–2J and 2M).
Figure 2.

Strong antitumor effect of ADV combined with Tα1 in multiple tumor models
(A) Schematic representation of experimental design and treatment timeline. WT mice were injected subcutaneously with tumor cells. When the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups and treated with an intra-tumoral injection of 0.1 mL of PBS or ADV (2 × 108 PFU) every 2 days for a total of three times. Tα1 (0.25 mg/kg) was injected subcutaneously into the peritumoral site once daily starting with the first dose of ADV; s.c., subcutaneous; i.t., intra-tumoral.
(B–D) 4T1 tumor-bearing mice were treated with different administrations, starting on day 10 after tumor injection (n = 6–9 biological replicates). (B) Tumor growth. (C) Survival. (D) Body weight.
(E–G) CT26 tumor-bearing mice were treated with different administrations, starting on day 10 after tumor injection (n = 9–10 biological replicates). (E) Tumor growth. (F) Survival. (G) Body weight.
(H–J) H22 tumor-bearing mice were treated with different administrations, starting on day 5 after tumor injection (n = 10 biological replicates).
(H) Tumor growth.
(I) Individual tumor growth.
(J) Body weight.
(K) Experimental schematic of H22 model, the first rechallenge model, and bilateral tumor-bearing model: The cured mice administered immunotherapies were injected subcutaneously with 5 × 106 H22 cells. The cured mice with immunological memory were again rechallenged with 5 × 106 H22 cells and the opposing flank was injected subcutaneously with 5×105 4T1 cells on day 90 after the first rechallenge, naive mice were used as a control.
(L) Tumor growth of H22 model mice in the first rechallenge model (n = 3–6 biological replicates); CR, complete response.
(M) Survival of mice from Figures 2H–2J (n = 10 biological replicates).
(N) Tumor growth of the bilateral tumor-bearing model (n = 4–6 biological replicates); CR/IM, complete response with immunological memory.
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Interestingly, tumor regression was observed in the ADV group and the ADV combined with Tα1 group in the H22 model (Figure 2I). For determination of whether tumor regression mediated by ADV alone and ADV combined with Tα1 therapy has immunological memory, the cured mice underwent the first rechallenge. No tumor burden was observed in the ADV combined with Tα1 group, whereas all naive mice and ADV group mice were susceptible to challenge with H22 cells (Figure 2K). This finding suggests that ADV combined with Tα1 induces long-term immunological memory, whereas ADV may lead to tumor regression by direct oncolytic effects (Figure 2L). Next, mice with immunological memory were subcutaneously inoculated with H22 cells and 4T1 cells to verify whether the antitumor immunity of ADV combined with Tα1 is tumor specific. We found that 4T1 cells rapidly led to the development of solid tumors in all mice, whereas the ADV combined with Tα1 group mice with immunological memory were insensitive to H22 cells (Figure 2N).
Given the aforementioned, these data suggest that Tα1 can significantly enhance the antitumor effect of ADV in a variety of solid tumor models and that ADV combined with Tα1 induces long-term immunological memory and that its antitumor effect is tumor antigen-specific in the H22 model.
Tα1 reverses the phenotype of TAMs and reduces Treg infiltration during ADV treatment
We hypothesized that Tα1 orchestrates immune infiltration in the TME to improve the antitumor effect of ADV. Consistent with this hypothesis, a 4T1 subcutaneous tumor model was constructed, and mice were administered with different immunotherapies (Figure 3A). On day 24 after subcutaneous injection of 4T1 cells into the flanks of mice, we found a significant increase in the percentages and absolute number of tumor-infiltrating CD3+ T cells, CD11b+F4/80+ macrophages, CD3+ CD4+ T cells, and CD3+ CD8+ T cells in the ADV-treated mice compared with the PBS-treated mice, and ADV combined with Tα1 had the strongest effect on increasing immune infiltration, including CD3+ T cells, CD11b+F4/80+ macrophages, CD3+ CD4+ T cells, CD3+ CD8+ T cells, and IFN-γ-producing CD8+ T cells (Figures 3B–3E). In addition, we found that Tα1 alone could increase the populations of infiltrating CD3+ T cells, indicating that Tα1 could modulate immune responses (Figure 3B). Consistent with the previous results, we found that ADV treatment led to an expansion of CD25+Foxp3+ Tregs and downregulation of CD86 and upregulation of CD206 in TAMs from the ADV-treated mice compared with the PBS and Tα1-treated mice (Figures 3F–3H). Surprisingly, the ADV-induced immunosuppressive TME could be reprogrammed by subcutaneous injection of Tα1. ADV combined with Tα1 caused a notable loss of Tregs compared with the ADV treatment (Figure 3F). Similarly, the highest expression of CD86 and the lowest expression of CD206 on TAMs were observed in the ADV combined with Tα1 group compared with all other groups (Figures 3G and 3H). Of note, we found that subcutaneous Tα1 alone did not change the populations of Tregs, CD86+ TAMs, or CD206+ TAMs in the TME compared with that of the PBS group (Figures 3F–3H). To further confirm whether subcutaneous injection of Tα1 could universally reprogram the ADV-induced immunosuppressive TME, we also analyzed tumor immune infiltration in the H22 model after different interventions (Figure S3A). As expected, ADV combined with Tα1 caused the most beneficial immune infiltration for antitumor immunity compared with that of the other groups (Figures S3B–S3F).
Figure 3.

Tα1 can further increase immune infiltration while reversing the phenotype of TAMs and reducing Treg infiltration
(A) Schematic representation of experimental design and treatment timeline. WT mice were injected subcutaneously with 5 × 105 4T1 cells. When the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups and treated with an intra-tumoral injection of 0.1 mL of PBS or ADV (2 × 108 PFU) every 2 days for a total of three times. Tα1 (0.25 mg/kg) was injected subcutaneously in the peritumoral site once daily starting with the first dose of ADV. TILs from the tumor were assessed by flow cytometry (day 24; n = 7 biological replicates); s.c., subcutaneous; i.t., intra-tumoral.
(B) Percentages (left) and total cells normalized to g (gram) tumor tissue (right) of CD3+ T cells, TAMs, NK cells, and DCs within the TME of mice (n = 6 biological replicates).
(C–H) Representative plots (left), percentages (top), and total cells normalized to g tumor tissue (bottom) of TILs. (C) CD3+CD4+ T cells, (D) CD25+Foxp3+ Tregs, (E) CD3+CD8+ T cells, (F) IFN-γ+CD8+ T cells, (G) “M1-like” macrophages, and (H) “M2-like” macrophages within the TME of mice (n = 6 biological replicates).
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The ADV we used as a pathogen-associated molecular pattern can activate innate immunity.28,29 Meanwhile, Tα1 has been reported to activate myeloid cells through the TLR signaling pathway,18 and ADV combined with Tα1 increased the percentage and absolute number of DCs in the 4T1 model (Figure 3B). Therefore, we utilized CD11c-dtr transgenic mice (CD11c promoter directs the expression of a diphtheria toxin receptor, and administration of diphtheria toxin allows for the depletion of DC populations.) to address the real contribution of DCs (Figure S3G). Indeed, a single dose of intraperitoneal diphtheria toxin allows for the depletion of DC populations (Figure S3H). We then monitored the effect of combination therapy on tumor progression and found that DCs did not play a major role in the antitumor effects of ADV combined with Tα1 therapy (Figures S3I–S3K).
In summary, these data clearly demonstrate that Tα1 reprograms the TME toward a more beneficial state for antitumor immunity during combination therapy.
The antitumor effect of ADV combined with Tα1 is mediated by macrophages and CD8+ T cells
Given the changes in macrophage phenotype and the recruitment of CD8+ T cells induced by ADV combined with Tα1, to confirm their critical role during treatment, we depleted macrophages or CD8+ T cells in 4T1-bearing mice and H22-bearing mice by anti-CSF1R or anti-CD8a antibodies, respectively (Figure 4A). Flow cytometry showed that macrophages and CD8+ T cells were rarely found in the tumor tissue from mice after antibody injection compared with that of the control mice (Figures S4A and S4C). By observing tumor growth and survival in mice, we found that depletion of macrophages and CD8+ T cells impaired the efficacy of ADV combined with Tα1 (Figures 4B–4E).
Figure 4.

Macrophages and CD8+ T cells mediate the antitumor activity of combination therapy
(A) Experimental schematic of mice from Figures 4B–4E and S4A–S4D: tumor-bearing mice were administered different immunotherapies and treated with intraperitoneal injections of anti-CSF1R or anti-CD8a; s.c., subcutaneous; i.t., intra-tumoral; i.p., intra-peritoneal.
(B and C) 4T1 model. (B) Tumor growth and (C) survival of 4T1 tumor-bearing mice (n = 10 biological replicates).
(D and E) H22 model. (B) Tumor growth and (C) survival of H22 tumor-bearing mice (n = 10 biological replicates).
(F–I) The nude mice model. (F) Experimental schematic of mice from Figures 4G–4I: 4T1-bearing immunodeficient nude mice were administered different immunotherapies starting on day 10. Tumor-associated macrophages (TAMs) were assessed by flow cytometry (n = 6 biological replicates). (G) Tumor growth. (H) The proportion of TAMs. (I) Representative plots (left) and percentages (right) of “M1-like” macrophages (top) and “M2-like” macrophages (bottom) within the TME of mice.
(J) Experimental schematic of mice from Figures 4K–4N and S4J–S4L: 4T1-bearing mice were administered different interventions and treated with intraperitoneal injections of anti-CSF1R or anti-CD8a. Tumor-infiltrating CD8+ T cells were assessed by flow cytometry (n = 6 biological replicates); s.c., subcutaneous; i.t., intra-tumoral; i.p., intra-peritoneal.
(K and L) Representative plots (K), percentages, and total cells (L) normalized to g (gram) tumor tissue of CD3+ CD8+ T cells within the TME of mice (n = 6 biological replicates).
(M and N) Representative plots (M) and percentages (N) of CD25+ CD8+ T cells, CD69+ CD8+ T cells, and IFN-γ+ CD8+ T cells within the TME of mice (n = 6 biological replicates).
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Macrophages can not only control the progression of tumors through phagocytosis but also secrete some proinflammatory cytokines to favor CD8+ T cell-mediated antitumor responses. CD8+ T cells can also affect the phenotype of TAMs by IFN-γ.30,31,32 To explore the relationship between macrophages and CD8+ T cells during ADV combined with Tα1 treatment, we subcutaneously injected 4T1 cells into immunodeficient nude mice (Figure 4F). In this setting, neither subcutaneous injection of Tα1 alone nor intra-tumoral injection of ADV alone inhibited tumor growth, and even the therapeutic effect of Tα1 alone was abolished completely (Figure 4G). In addition, there was no significant difference in tumor growth between the groups with ADV combined with Tα1 and ADV alone (Figure 4G), indicating the critical role of T cells in our model. To further explore whether the polarization of macrophages can be regulated in the absence of T cells, we profiled TAM populations and phenotypes in 4T1 breast cancer tumors on day 14 after different treatments. The percentage of 4T1 tumor-infiltrating TAMs was similar among all groups (Figure 4H). Strikingly, we found that ADV alone still induced TAMs in the pro-tumoral “M2-like” phenotype, characterized by downregulation of CD86 and upregulation of CD206 in TAMs, compared with PBS (Figure 4I). Concordantly, CD86 and CD206 expression in TAMs regulated by ADV could be reversed by Tα1 (Figure 4I).
We next investigated whether ADV combined with Tα1 regulated CD8+ T cell by macrophages and whether this regulatory capacity is impaired in mice treated with anti-CSF1R (Figure 4J). In contrast to ADV combined with Tα1, ADV combined with Tα1 mice treated with anti-CSF1R were insufficient to recruit and activate CD8+ T cells, characterized by reducing tumor-infiltrating CD3+ CD8+ T cells, CD25+ CD8+ T cells, CD69+ CD8+ T cells, and IFN-γ+ CD8+ T cells (Figures 4K–4N). Interestingly, there was no difference in PD1+ CD8+ T cells across all treatment groups, yet the percentage of TIM3+ CD8+ T cells in the tumor was reduced after ADV combined with Tα1 (Figures S4F and S4G). In summary, these findings confirm that the tumor-suppressing efficacy of ADV combined with Tα1 requires macrophages and CD8+ T cells and suggest that ADV combined with Tα1 mediates a better antitumor immune response than ADV alone by reprogramming TAMs and then activating CD8+ T cells.
Tα1 reverses the M2 polarization of macrophages induced by ADV in vitro
To investigate the effects of ADV treatment on the polarization of macrophages, we used 4T1 cells, ADV, ADV-infected 4T1 cells (4T1ADV cells), conditioned medium obtained from 4T1 cell cultures (4T1-CM), or 4T1ADV-CM to stimulate the RAW264.7 macrophage-like cell line in vitro. Subsequently, changes in the phenotypic markers of macrophages were investigated by quantitative PCR and flow cytometry. Interestingly, macrophages displayed changes in multiple transcriptional programs only under 4T1ADV cell stimulation, including downregulation of M1 markers (CD86, iNOS, IL-1β, and IL-6) and upregulation of M2 markers (CD206, Arg-1, and IL-10) (Figures S5A and S5B). Additionally, the degree of macrophage polarization was affected by changing the ratio of 4T1ADV cells to macrophages (Figure S5C).
To evaluate the effect of Tα1 on the phenotypic reprogramming of macrophages, we incubated macrophages with Tα1, 4T1ADV cells, or 4T1ADV cells with Tα1 (4T1ADV & Tα1) (Figure 5A). As expected, increased downregulation of M1 markers (CD86, iNOS, and IL-1β) and reduced expression of upregulated M2 markers (CD206, Arg-1, and IL-10) were observed in the macrophages stimulated by 4T1ADV & Tα1 (Figures 5B–5D). Notably, 4T1ADV & Tα1 could not increase the expression of IL-6 in macrophages (Figure 5D), but this finding is not unexpected, as many studies have shown the ability of IL-6 to drive tumor progression and evade immune responses.33,34 Furthermore, we did not detect significant differences in phagocytosis and Ki67 expression levels of macrophages across all treatment groups (Figure S5E). Similar results were obtained in the THP-1 macrophages and the MDA-MB-231 cell line (Figures S5F–S5H). Based on these results, we further confirmed this phenomenon by intraperitoneally injecting 6% starch broth into BALB/c mice to recruit macrophages to better mimic the role of ADV and Tα1 in the TME (Figure S5I). Intraperitoneal injection of 4T1ADV cells increased the expression of M2 markers in peritoneal macrophages but decreased the expression of M2 markers and increased the expression of M1 markers in peritoneal macrophages after Tα1 intervention (Figure S5J).
Figure 5.

ADV-infected tumor cells induce M2 polarization, and Tα1 can reverse the phenotype of macrophages
(A) Experimental schematic of cell coculture from Figures 5B–5D: Macrophages (RAW264.7 cells) were stimulated by different interventions (Tα1, 4T1ADV cells, or 4T1ADV cells with Tα1) for 18 h, followed by analysis of macrophage polarization (n = 3 biological replicates).
(B) IL-1β and IL-10 concentrations in the supernatants were measured by ELISA (n = 3 biological replicates).
(C) Expression of CD86 and CD206 in macrophages was measured by flow cytometry (FCM) (n = 3 biological replicates).
(D) The expression (left) of iNOS, IL-1β, IL-6, Arg-1, and IL-10 in macrophages was measured by qPCR (n = 3 biological replicates and 3 technical replicates), and the mRNA expression level of iNOS, IL-1β, IL-6, Arg-1, and IL-10 was showed as heatmap (right).
(E) PCA plot of macrophages after different treatments.
(F) Heatmap of DEGs in all macrophage groups.
(G) KEGG pathway enrichment analysis of DEGs between 4T1ADV-treated macrophages and control group.
(H) KEGG pathway enrichment analysis of DEGs between 4T1ADV with Tα1-treated macrophages and 4T1ADV-treated macrophages.
(I) Volcano plot showing the DEGs between 4T1ADV-treated macrophages and 4T1ADV with Tα1-treated macrophages. DEGs with an absolute log-transformed fold change >1 and a p value < 0.05 (determined by two-sided Wilcoxon rank-sum test) were considered significant.
(J) Representative plots and percentages of CD4+ Foxp3+ T cells (top) and IFN-γ+CD8+ T cells (bottom) from cell coculture system (n = 3 biological replicates).
(K) CD4+ T cells and CD8+ T cells were respectively cocultured with macrophages, or conditioned medium of macrophages or stimulated with Tα1. Expression of Foxp3 in CD4+ T cells (top) and IFN-γ in CD8+ T cells (bottom) was measured by FCM (n = 3 biological replicates).
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
We next sought to understand the mechanisms by which ADV and Tα1 regulate macrophages. First, we performed RNA sequencing (RNA-seq) on 4T1 cells treated with ADV and ADV combined with Tα1. Although ADV infection can impact 4T1 gene signature, these differentially expressed genes (DEGs) do not appear to be significantly associated with macrophage phenotype or function (Figure S5K). Additionally, analysis of RNA-seq data from 4T1 cells showed that the transcriptomes of ADV-treated 4T1 cells were almost identical to those of 4T1 cells treated with ADV combined with Tα1 (Figure S5L). Then, we performed RNA-seq of macrophages after different stimulation. Principal components analysis (PCA) and the heatmap of differential genes showed that macrophages cocultured with 4T1ADV cells showed a substantial shift in transcriptomic levels, which was partially reversed by the intervention of Tα1 (Figures 5E and 5F). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of DEGs showed that cytokine-cytokine receptor interaction and PI3K/Akt signaling pathway were significantly enriched in macrophages treated with 4T1ADV cells (Figure 5G). Activation of the PI3K/Akt pathway is critical in restricting pro-inflammatory responses in TLR-stimulated macrophages and has been considered as a negative regulator of TLR and nuclear factor κB signaling in macrophages.35 In contrast, we observed that TLR signaling pathway and NOD-like receptor (NLR) signaling pathway were significantly enriched in 4T1ADV & Tα1-treated macrophages compared to 4T1ADV-treated macrophages. TLRs and NLRs play vital roles in regulating the pro-inflammatory responses of macrophages.36 Furthermore, IFN-stimulated genes (Ifit1bl1, Ifit3, Ifi206, and Ifi208) and function genes (CXCL10, CCL5, IL33, and SLAMF7) were significantly upregulated in 4T1ADV & Tα1-treated macrophages compared to 4T1ADV-treated macrophages (Figure 5I). These results suggested that ADV-infected tumor cells and/or Tα1 may regulate macrophages through the PI3K/Akt pathway and TLR signaling pathway, respectively.
Next, we investigated the effects of different interventions (including 4T1 cells, ADV, 4T1ADV cells, and Tα1) on the Tregs, but there was no difference across all treatment groups (Figure S5M). Some previous studies have shown that macrophages can promote Foxp3 expression in CD4+ T cells.37 To test the hypothesis that ADV combined with Tα1 affected Tregs through macrophages, we stimulated macrophages with different interventions to alter the phenotype of macrophages, which were then co-cultured with CD4+ T cells. In fact, macrophages stimulated by 4T1ADV cells increased Foxp3 expression in CD4+ T cells in the coculture system, but Foxp3 expression levels were reversed when macrophages stimulated with 4T1ADV+Tα1 were cocultured with CD4+ T cells (Figure 5J). To test whether macrophages can produce this effect in a way independent of cell contact, CD4+ T cells were respectively cocultured with macrophages (M-CTRL, M-4T1ADV, and M-4T1ADV + Tα1), or conditioned medium of macrophages (M-4T1ADV-CM, and M-4T1ADV + Tα1-CM) or conditioned medium of macrophages with added Tα1 (M-4T1ADV-CM+Tα1). The results showed that the conditioned medium had similar effects to macrophages, but the conditioned medium added to the Tα1 (M-4T1ADV-CM+Tα1) did not change the number of CD4+Foxp3+ T cells (Figure 5K). Moreover, macrophages stimulated by 4T1ADV cells reduced the proportion of IFN-γ+ CD8+ T cells, and IFN-γ+ CD8+ T cells increased when Tα1 intervened (Figure 5J). Macrophage conditioned medium also had similar effects on CD8+ T cells (Figure 5K). From these experiments, we conclude that there is an “immune response pathway” during ADV combined with Tα1 therapy: first, TAM phenotype is reprogrammed by ADV-infected 4T1 cells (and Tα1), and then CD4+ T and CD8+ T cells are affected by TAMs. Finally, CD8+ T cells play the major role of antitumor immune response.
The Tα1-armed recombinant adenovirus was constructed and characterized
To maximize the function of Tα1 in reprogramming macrophages within the TME during ADV treatment, we engineered ADV with genetic modification and constructed a recombinant adenovirus expressing Tα1, which was termed ADVTα1 (Figure 6A). First, we detected the secretion of Tα1 in the supernatants from the ADVTα1-infected 4T1 cells by western blotting (Figure 6B). Next, we evaluated the oncolytic ability and replicative capacity of ADVTα1. We infected 4T1, MDA-MB-231, CT26, HCT116, H22, HepG2, and MET-5A cells with ADV or ADVTα1, and then measured the oncolytic ability using CCK-8 assays and measured the replicative capacity using TCID50 assays. ADV and ADVTα1 presented similar selective oncolytic ability and replicative capacity in tumor cells (Figures 6C, 6D, S6A, and S6B). Compared with the control, both ADV and ADVTα1 effectively induced early and late apoptosis (and/or necrosis) of tumor cells (Figure 6E). We also observed EGFP expression of the two adenoviruses in multiple cell lines (Figure 6F). Finally, we tested whether the ADVTα1-infected 231 cells could have a similar effect on the polarization of macrophages as 231ADV and Tα1 (Figure 6G). Interestingly, we found the upregulation of M1 markers (CD86, iNOS, and IL-1β) and the downregulation of M2 markers (CD206, Arg-1, and IL-10) in macrophages after stimulation by ADVTα1-infected MDA-MB-231 cells (Figures 6G–6K). Furthermore, we did not detect significant differences in phagocytosis and Ki67 expression levels of macrophages across all treatment groups. (Figures 6I and S6I). Similar results were obtained in the RAW264.7 macrophages and 4T1 cell line (Figures S6C–S6H).
Figure 6.

Construction and characterization of the recombinant adenovirus ADVTα1
(A) Schematic diagram of a recombinant adenovirus expressing Tα1, ΔE1 contains E1B55K-deletion. The expression of EGFP and Tα1 is driven by inserting CMV and EF-1α promoters in the E1 region, respectively.
(B) 4T1 cells were infected with ADV or ADVTα1 (MOI = 2) for 48 h, and the secretion of Tα1 was confirmed by western blotting.
(C) 4T1 cells and MDA-MB-231 cells were infected with ADVTα1 or ADV at different MOIs, and cell viability was measured 48 h later by CCK-8 assay (n = 3 biological replicates).
(D) 4T1 cells and MDA-MB-231 cells were infected with ADVTα1 or ADV at an MOI of 1. The virus titers were measured by TCID50 assay at different time points after infection (n = 3 biological replicates).
(E) Infection of 4T1 cells with ADV or ADVTα1 (MOI = 10) for 24 h. Early apoptotic cells were confirmed as Annexin-V+/PI− cells, and late apoptotic (and/or necrotic) cells were confirmed as Annexin-V+PI+ cells (n = 3 biological replicates).
(F) 4T1 cells, CT26 cells, H22 cells, MDA-MB-231 cells, HCT116 cells, HepG2 cells, and MET-5A cells were infected with ADVTα1 or ADV at an MOI of 1. The expression of EGFP in the cells was measured by FCM.
(G–K) Macrophages (THP-1 cells) were cocultured with 231ADV (ADV), 231ADV cells with Tα1 (ADV + Tα1), or ADVTα1-infected MDA-MB-231 cells (ADVTα1), followed by analysis of macrophage polarization. (F) Schematic representation of experimental design. (H) Expression of CD86 (top) and CD206 (bottom) in macrophages was measured by FCM (n = 3 biological replicates). (I) Expression of Ki67 was measured by FCM in CD86+ macrophages or CD206+ macrophages (n = 3 biological replicates). (J) Expression of iNOS, IL-1β, IL-6, Arg-1, and IL-10 in macrophages was measured by qPCR (n = 3 biological replicates and 3 technical replicates). (K) The mRNA expression level of iNOS, IL-1β, IL-6, Arg-1, and IL-10 (from Figure 6J) was showed as heatmap.
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Taken together, these results indicate that Tα1 gene insertion does not impair the replicative or oncolytic capabilities of the virus and that Tα1 is functional in ADVTα1-infected cells in vitro.
ADVTα1 effectively recruits and activates CD8+ T cells to control tumor progression
We used the 4T1 model and H22 model to investigate the antitumor activity of ADVTα1 in vivo; ADVTα1 significantly suppressed tumor growth and prolonged survival in mice, and its antitumor efficacy was no less than that of ADV combined with Tα1 (Figures 7A–7D, S7A–S7D). Then, we further addressed whether ADVTα1 produced immune responses similar to those produced by ADV combined with Tα1 by regulating macrophage polarization. We observed that ADVTα1 had comparable effects on reprogramming TAMs, CD25+Foxp3+ Tregs, and CD3+CD8+ T cells in the 4T1 model (Figures 7E–7H). Moreover, we found that the expression of CD25, CD69, and IFN-γ was highest in the CD8+ T cells of the ADVTα1 mice, indicating that ADVTα1 treatment could effectively promote the activation of CD8+ T cells (Figure 7I).
Figure 7.

The antitumor activity of ADVTα1 is mediated by macrophages and CD8+ T cells, and ADVTα1 can reprogram the TME toward a more beneficial state for antitumor immunity
(A) Experimental schematic of mice from Figures 7B–1I: 4T1 tumor-bearing WT mice were administered different immunotherapies or vehicle control (PBS) starting on day 10 when the tumor volume reached approximately 50–100 mm3. TILs from the tumor were assessed by flow cytometry on day 24; s.c., subcutaneous; i.t., intra-tumoral.
(B–D) 4T1 tumor-bearing WT mice were administered different immunotherapies or vehicle control (PBS) starting on day 10 when the tumor reached approximately 50–100 mm3 in volume (n = 10 biological replicates). (B) Tumor growth. (C) Body weight. (D) Survival.
(E–I) Representative plots (left), percentages (top), and total cells normalized to g (gram) tumor tissue (bottom) of TILs. (E) “M1-like” macrophages, (F) “M2-like” macrophages, (G) CD25+Foxp3+ Tregs, and (H) CD3+CD8+ T cells within the TME of mice. (I) Representative plots and percentages of CD25+ CD8+ T cells, CD69+ CD8+ T cells, and IFN-γ+ CD8+ T cells within the TME of mice (n = 6 biological replicates).
(J–M) 4T1 tumor-bearing mice were administered different immunotherapies and treated with intraperitoneal injections of anti-CSF1R or anti-CD8a; s.c., subcutaneous; i.t., intra-tumoral; i.p, intra-peritoneal. (J) Schematic diagram of the timeline of the antibody depletion experiment in the 4T1 model. (K) Tumor growth. (L) Body weight. (M) Survival (n = 10 biological replicates).
(N–Q) Tumor-bearing NCG mice were intravenously injected with human peripheral blood mononuclear cells on day 1, and then mice were administered different immunotherapies; s.c., subcutaneous; i.t., intra-tumoral; i.v., intravenous. (N) Schematic representation of experimental design and treatment timeline. (O) Tumor growth. (P) Body weight. (Q) Survival (n = 5 biological replicates).
(R–U) The PDX tumor cells were injected into the fourth mammary fat pads of NCG mice, and these tumor-bearing mice were intravenously injected with human peripheral blood mononuclear cells on day 1, followed by different immunotherapies starting on day 10; s.c., subcutaneous; i.t., intra-tumoral; i.v., intravenous. (R) Schematic representation of experimental design and treatment timeline. (S) Tumor growth. (T) Body weight. (U) Survival (n = 5 biological replicates).
The data are shown as the means ± SD. NS, no significant difference; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next, we used antibody depletion to determine the importance of macrophages and CD8+ T cells during ADVTα1 treatment. 4T1 tumor growth inhibition was sharply diminished by the depletion of macrophages and/or CD8+ T cells (Figures 7J–7M). To further explore whether ADVTα1 treatment can inhibit metastasis, we used triple-negative 4T1 orthotopic mammary carcinoma that will have spontaneous lung metastasis. ADVTα1 not only inhibited the primary tumor growth but also reduced the formation of metastasis 4T1 foci in the lung on day 24 and 35 (Figures S7E–S7G). In addition, ADVTα1 displayed broad antitumor efficacy with suppressed tumor growth and prolonged survival in the humanized TNBC (triple-negative breast cancer) model and the HCC model (Figures 7N–7Q and S7H–S7K). More importantly, ADVTα1 treatment also inhibited tumor growth in the patient-derived xenograft (PDX) TNBC model (Figures 7R–7U).
In summary, the recombinant oncolytic adenovirus ADVTα1 can effectively orchestrate the reprogramming of TAMs and activate CD8+ T cells to exert superior antitumor activity in vivo.
Discussion
In this study, we have found that the induction of an immunosuppressive feedback loop by ADV diminishes antitumor immunity. More specifically, ADV-infected tumor cells stimulate macrophages to polarize toward an “M2-like” pro-tumoral phenotype, resulting in an expansion of Tregs within the TME. To compensate for the shortcomings of ADV treatment, we attempt to reverse the M2 phenotype of macrophages and reduce Treg numbers in the TME during ADV treatment through the intervention of Tα1, and the results have demonstrated that the strategy is feasible. Additionally, based on the superior antitumor efficacy of ADV and Tα1 combined therapy, we also construct a recombinant oncolytic adenovirus expressing Tα1, which could effectively reprogram the TME into a state more beneficial to antitumor immunity.
OVs are an attractive immunotherapy because of their ability to reprogram the TME and activate antitumor immune responses.21,22 However, the existence of immunosuppressive feedback loops induced by OVs during therapy has been poorly studied. Major players in immune suppression are Tregs, “M2-like” TAMs, and innate tumor-associated suppressive myeloid cells.38,39 In this study, we have demonstrated that ADV treatment could effectively transform the tumor from “cold” to “hot,” while a significantly increased number of “M2-like” pro-tumoral TAMs and Tregs in the TME is induced by ADV treatment at all time points assessed in the 4T1 and H22 model. In addition, Tα1 demonstrates a strong immunomodulatory capacity that reprogrammed the phenotype of macrophages in both mouse and human cell models in vitro. With respect to the relationship between viruses and macrophages, previous studies have reported that WT adenovirus-infected tumor cell corpses exhibit cell contact-dependent repression of macrophage inflammatory response.9 In this work, we further identify that ADV treatment results in increased “M2-like” pro-tumoral TAMs and Treg infiltration within the TME.
TAMs in the TME can be roughly divided into two groups, namely, classically activated “M1-like” antitumoral macrophages and alternatively activated “M2-like” pro-tumoral macrophages.40 Classical “M1-like” polarization has been defined as the expression of CD80, CD86, MHCII, and iNOS, which is related to the tumoricidal function of TAMs. M1-like TAMs can phagocytose cancer cells and produce proinflammatory cytokines to activate CD8+ T cells to coordinate antitumor immunity. In contrast, “M2-like” TAMs are characterized by high expression of CD206, VEGF, CD163, Arg-1, and IL-10 and recruit Tregs to suppress antitumor immunity.32,41 Moreover, IL-10+ TAMs have been found to be associated with immunosuppressive TMEs, where IL-10+ TAMs lead to cytotoxic CD8+ T cell dysfunction and promote immune evasion.42,43 In this research, we have found that in vitro, direct contact of ADV-infected tumor cells with macrophages results in high IL-10 expression in macrophages.
Many therapeutic strategies for targeting TAMs have been developed, and the reprogramming strategy is undoubtedly the best choice compared with other therapies, which can not only retain the phagocytic function and antigen presentation of macrophages but also enhance their potential immune stimulatory function.24 Tα1, a thymus-derived peptide, is a therapeutic immunomodulator for the adjuvant treatment of infections, malignant diseases, and efferocytotic macrophage polarization.20,26,44 In our study, Tα1 reprograms “M2-like” pro-tumoral macrophages to “M1-like” antitumoral macrophages and reduces Treg infiltration in the TME during ADV treatment. Additionally, the combination therapy shows superior antitumor effects in a variety of solid tumors compared with monotherapy and results in complete elimination of tumors and long-term immunological memory in H22 tumor-bearing mice. Therefore, we also construct a recombinant adenovirus that introduces all the elements of combination therapy and is functional, with no less antitumor efficacy than combination therapy, and at the same time has a lighter administration burden.
Tα1 has been approved in more than 35 countries for the treatment of chronic hepatitis B and C.26 Tα1 is not recommended in oncolytic adenovirus therapy because it can induce antiviral immune responses, while we do not find that Tα1 affects the oncolytic ability and replicative capacity of ADVTα1. Of note, Tα1 alone delays tumor growth and prolongs survival to a certain extent in a variety of solid tumor models, but no changes in the phenotypic markers of macrophages and CD4+ T cells are observed following treatment with Tα1 alone, either in vitro or in vivo. There is no significant difference between the subcutaneous injection of Tα1 alone and treatment with PBS in immunodeficient nude mice, indicating that the antitumor effect of Tα1 alone may be mediated by T cells.
Generally, T cells play a key role in antitumor immunotherapy.45,46 In our study, compared with ADV alone, ADV combined with Tα1 and ADVTα1 significantly increase the number of tumor-infiltrating T cells in the TME. The expression of CD8+ T cell activation markers (CD25, CD69, and IFN-γ) is highest after treated with ADVTα1, which is mainly underpinned by the ability of ADVTα1 to effectively reprogram TAMs to secrete proinflammatory cytokines, thereby activating CD8+ T cells and reducing the number of Tregs in the TME. In addition, we have demonstrated that macrophages and CD8+ T cells were indispensable in the antitumor immune responses induced by combination therapy and ADVTα1. For the ADV and ADVTα1 usage, we confirm their replication and lytic effect in murine and human cell lines, and we validate their antitumor effect in various mouse model and peripheral blood mononuclear cell-humanized solid tumor models.
In conclusion, ADV-infected tumor cells induce macrophages to adopt an “M2-like” pro-tumoral phenotype in a cell contact-dependent manner, and the intervention of Tα1 can effectively orchestrate macrophage reprogramming, thereby enhancing T cell-mediated antitumor responses. Our study provides inspiration for oncolytic viral combinatorial therapeutic strategies and the design of oncolytic engineered viruses. In addition, our findings have translational potential given that (1) adenovirus is one of the most frequently employed OVs in immunotherapy, (2) Tα1 has been clinically approved in over 30 countries worldwide as an immunomodulator, (3) the antitumor activity of the combination therapy and ADVTα1 has been shown in multiple murine and humanized tumor models,4,26,47 and (4) the ability of ADV & Tα1 and ADVTα1 to regulate macrophages is applicable to human cell lines.
Limitations of the study
There are several limitations to our study. No patient samples have been obtained from existing oncolytic adenovirus clinical trials to demonstrate an immune feedback loop mediated by OV treatment, which is critical for clinical translational prospects. In addition, it is undefined whether the antigen-presenting function of macrophages changes after different treatments. Finally, although we have found that ADV-infected tumor cells and additional Tα1 may regulate macrophage function to affect antitumor immune responses through the PI3K/Akt pathway and TLR signaling pathway, the precise mechanism by which ADV-infected tumor cells cause M2 polarization in TAMs and Tα1 reprograms TAMs during ADV treatment also remains to be investigated and should be the focus of future studies.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Junhua Wu (wujunhua@nju.edu.cn).
Materials availability
Recombinant adenoviruses generated in this study are available from the lead contact upon request.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Acknowledgments
We thank Research Center for Basic Medical Science of Nanjing University Medical School for the technical support.
The research was supported by the Shandong Provincial Laboratory Project (SYS202202), the National Natural Science Foundation of China (81972888 and 82272819), the Research Project of Jinan Microecological Biomedicine Shandong Laboratory (JNL-202219B, JNL-202204A, and JNL-2023017D), the Primary Research and Development Plan of Jiangsu Province (BE2022840), and the Open Project of Chinese Materia Medica First-Class Discipline of Nanjing University of Chinese Medicine (2020YLXK007).
Author contributions
Conceptualization, J.W., C.J., and K.L.; methodology, K.L., L.K., J.W., and C.J.; investigation, K.L., L.K., H.C., Q.X., Y.Z., and C.G.; visualization, K.L. and L.K.; funding acquisition, J.W., C.J., and X.G.; project administration, J.W. and C.J.; supervision, J.W. and C.J.; writing – original draft, K.L. and J.W.; writing – review and editing, J.W., C.J., and K.L.
Declaration of interests
K.L., J.W., C.J., L.K., H.C., X.G., and Q.X. are named as inventors on China patent application (no. 2024110168233), which is related to this work. Jinan Microecological Biomedicine Shandong Laboratory and Nanjing University are the applicants of this patent application. X.G. and Q.X. are employees of Jinan Microecological Biomedicine Shandong Laboratory. J.W. and C.J. are employees of Nanjing University.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC anti-mouse CD45.1 Antibody | BioLegend | Cat#110714; RRID:AB_313503 |
| FITC anti-mouse CD3 Antibody | BioLegend | Cat#100204; RRID:AB_312661 |
| PE anti-mouse CD4 Antibody | BioLegend | Cat#100408; RRID:AB_312693 |
| FITC anti-mouse CD4 Antibody | BioLegend | Cat#100406; RRID:AB_312691 |
| PerCP/Cyanine5.5 anti-mouse CD8a Antibody | BioLegend | Cat#100734; RRID:AB_2075238 |
| FITC anti-mouse CD11b Antibody | BioLegend | Cat#101206; RRID:AB_312789 |
| PE anti-mouse CD86 Antibody | BioLegend | Cat#159203; RRID:AB_2832567 |
| PE/Cyanine7 anti-mouse CD11c Antibody | BioLegend | Cat#117318; RRID:AB_493568 |
| PE anti-mouse IFN-γ Antibody | BioLegend | Cat#163504; RRID:AB_2890730 |
| PE anti-mouse FOXP3 Antibody | BioLegend | Cat#126404; RRID:AB_1089117 |
| PE/Cyanine7 anti-mouse F4/80 Antibody | BioLegend | Cat#123114; RRID:AB_893478 |
| PE/Cyanine7 anti-mouse CD69 Antibody | BioLegend | Cat#104512; RRID:AB_493564 |
| PerCP/Cyanine5.5 anti-mouse CD206 (MMR) Antibody | BioLegend | Cat#141716; RRID:AB_2561992 |
| PE anti-mouse CD49b Antibody | BioLegend | Cat#103506; RRID:AB_313029 |
| PerCP/Cyanine5.5 anti-mouse CD25 Antibody | BioLegend | Cat#101912; RRID:AB_10613643 |
| PE/Cyanine7 anti-mouse/human Ki67 Antibody | BioLegend | Cat#151217; RRID:AB_2910305 |
| PerCP/Cyanine5.5 anti-human CD206 (MMR) Antibody | BioLegend | Cat#321122; RRID:AB_10899411 |
| PE anti-human CD86 Antibody | BioLegend | Cat#381010; RRID:AB_3083375 |
| InVivoMAb anti-mouse CD8a | BioXCell | Cat#BE0061; RRID:AB_1125541 |
| InVivoMAb anti-mouse CSF1R(CD115) | BioXCell | Cat#BE0213; RRID:AB_2687699 |
| 6x-His Tag Monoclonal Antibody | Invitrogen | Cat#4E3D10H2/E3; RRID:AB_2536841 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, HRP | Invitrogen | Cat#31430; RRID:AB_228307 |
| Biological samples | ||
| Human PBMC | Blood from the healthy donor | N/A |
| Human TNBC tumor | Tumor sample from the TNBC patients in Nanjing Drum Tower Hospital | N/A |
| Chemicals, proteins, and recombinant proteins | ||
| Dulbecco’s modified Eagle’s medium (DMEM) | Invitrogen | Cat#10564011 |
| Fetal Bovine Serum (FBS) | Invitrogen | Cat#A4766801 |
| Streptomycin/penicillin | Invitrogen | Cat#15140122 |
| Opti-MEM | Invitrogen | Cat#31985070 |
| Roswell Park Memorial Institute (RPMI) 1640 | Invitrogen | Cat#21875034 |
| 293 Pro | BasalMedia | Cat#F431166 |
| Foxp3 fixation/permeabilization buffer | eBioscience | Cat#00-5523-00 |
| Permeabilization kit | eBioscience | Cat#00-5523-00 |
| DNAse I | Roche | Cat#10104159001 |
| Collagenase A | Roche | Cat#10103586001 |
| Lipofectamine 2000 | Invitrogen | Cat#11668019 |
| GeltrexTM Basement Membrane Matrix | Invitrogen | Cat#A1413202 |
| Perteinase K Solution | Invitrogen | Cat#25530049 |
| Kananmycin | Invitrogen | Cat#11-815-024 |
| Ampicillin | Bioreagents | Cat#BP1760-25 |
| Critical commercial assays | ||
| HiScript Reverse Transcriptase reagent kit | Vanzyme | Cat#R101-02 |
| HiScript II One Step RT-PCR Kit | Vanzyme | Cat#P611-01 |
| Adeno-XTM rapid titer kit | Clontech | Cat#632250 |
| CD4+ T cell isolation kit, mouse | Selleck | Cat#B90000 |
| CD8+ T cell isolation kit, mouse | Selleck | Cat#B90011 |
| Mouse IL-1β ELISA kit | Elabscience | Cat#E-EL-M0046c |
| Human IL-1β ELISA kit | Elabscience | Cat#E-EL-H0149 |
| Mouse IL-10 ELISA kit | Elabscience | Cat#E-EL-H0149c |
| Human IL-10 ELISA kit | Elabscience | Cat#E-EL-H6154 |
| Deposited data | ||
| 4T1 cells RNA sequencing | GEO database | GEO: GSE271046 |
| Macrophages RNA sequencing | GEO database | GEO: GSE271202 |
| Experimental models: Cell lines | ||
| 4T1 cells | ATCC | Cat#CRL-2537 |
| CT26 cells | ATCC | Cat#CRL-2638 |
| H22 cells | This paper | N/A |
| B16-F10 | ATCC | Cat#CRL-6475 |
| MDA-MB-231 cells | ATCC | Cat#CRM-HTB-26 |
| HCT116 cells | ATCC | Cat#CCL-247EMT |
| HepG2 cells | ATCC | Cat#HB-8065 |
| MET-5A cells | ATCC | Cat#CRL-9444 |
| THP-1 cells | ATCC | Cat#TIB-202 |
| HEK 293T cells | ATCC | Cat#CRL-1573 |
| RAW264.7 cells | ATCC | Cat#TIB-71 |
| Experimental models: Organisms/strains | ||
| Mouse: BALB/c | GemPharmatech | SN#000651 |
| Mouse: Outbred athymic nude | GemPharmatech | SN#007850 |
| Mouse: C57BL/6J | GemPharmatech | SN#000664 |
| Mouse: CD11c-dtr B6.FVB-1700016L21RikTg(Itgax−HBEGF/EGFP)57Lan/J |
Jackson Laboratory | SN#004509 |
| Mouse: NCG mice NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt |
GemPharmatech | SN#T001475 |
| DH5α | Vanzyme | Cat# C502-02 |
| GFP+Escherichia coli | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 for primers | N/A | N/A |
| Recombinant DNA | ||
| pAd-DEST | This paper | N/A |
| pAd-Tα1 | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 9 | N/A | N/A |
| FlowJo 10.7.1 | Tree Star | N/A |
| Adobe Illustrator | Adobe | N/A |
| BioRender | BioRender | N/A |
Experimental model and study participant details
Mice
Four-to 8-week-old mice were used for the experiments. WT BALB/c mice, WT C57BL/6J mice, immunodeficient BALB/c-nude mice, CD11c-dtr mice (Jackson Laboratory, B6.FVB-1700016L21RikTg(Itgax−HBEGF/EGFP)57Lan/J) and NCG mice (GemPharmatech, NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) were housed under specific pathogen-free conditions at a temperature of ∼18°C–24°C with water and food and kept on 12-h light/dark cycles. All animal experiments were approved by the Ethics Committee of The Affiliated Drum Tower Hospital, Medical School of Nanjing University.
Cell lines
The mouse breast cancer cell line 4T1, human breast cancer cell line MDA-MB-231, mouse colorectal cancer cell line CT26, human colorectal cancer cell line CT116, human HCC HepG2 cells, mouse macrophage-like cell line RAW264.7 and human embryonic kidney 293T cells were cultured in DMEM with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin, and mouse HCC H22 cells and THP-1 cells were cultured in RPMI 1640 medium. All cells were incubated at 37°C with 5% CO2.
Method details
Construction of recombinant adenovirus
As described in a previous study,10 the adenovirus shuttle plasmid encoding the Tα1 domains of PTMA was purchased from Sino Biological. For the secretion and detection of Tα1, the IL-2 signal peptide (MYRMQLLSCIALSLALVTNS) was designed upstream of the Tα1 sequence, and the His-tag (HHHHHH) was designed downstream of the Tα1 sequence. The ENTR plasmid was further recombinant with the pAd-DEST human adenovirus type 5 backbone to generate a recombinant adenovirus expression vector. After digestion with the restriction enzyme PacI, the recombinant adenovirus was generated and amplified in 293T cells. The recombinant adenoviruses were purified by sucrose gradient ultracentrifugation and titrated using the Adeno-X rapid titer kit according to the manufacturer’s instructions.
Viral oncolysis and replication
A total of 2×103 4T1, CT26, and H22 cells were seeded into 96-well plates and cultured overnight prior to treatment with different adenoviruses for 48 h. Cell viability was evaluated by CCK-8 assays. The expression of EGFP in cells infected with adenoviruses was measured by flow cytometry. The TCID50 assay was used to calculate the replication of the adenoviruses, Briefly, cells were seeded into 24-well plates and infected with different adenoviruses. The medium containing viruses was removed, and fresh medium was added 2 h after infection. Cells were harvested at serial time points as indicated (24, 48, 72, and 96 h after infection). Then, the adenovirus titers were measured by TCID50 assay.
Western blot
First, 4T1 cells were seeded into 6-well plates and infected with ADV or ADVTα1 at an MOI of 2 for 48 h. Supernatants from infected cells were harvested and mixed with loading buffer. The following experimental steps of Western blotting were performed according to previously described procedures.48 The antibodies used were anti-6x-His tag monoclonal (Invitrogen, 4E3D10H2/E3) and goat anti-mouse IgG (H + L) (Invitrogen, 31430).
qRT-PCR
Total cellular RNA was collected and then reverse transcribed to cDNA, mRNA expression levels were analyzed using SYBR Green PCR master mix and an ABI QuantStudio 5 real-time PCR system (Thermo Fisher Scientific) and normalized to GAPDH expression. Primer sequences are listed in Table S1.
ELISA
The supernatants of RAW264.7 cells and THP-1 cells stimulated with different interventions were collected (1000 ×g, 20 min, 4°C) respectively, and then concentrations of human and/or mouse IL-1β and IL-10 were detected by related ELISA kits (Elabsicence) according to the manufacturer’s instructions.
In vitro macrophage coculture experiment
4T1 cells and MDA-MB-231 cells were infected with ADV or ADVTα1 at 37°C for 48 h. ADV-infected tumor cells (4T1ADV; 231ADV), ADVTα1-infected tumor cells (ADVTα1) and ADV-infected tumor cells were preincubated with Tα1 (4T1ADV & Tα1; 231ADV & Tα1) for 30 min. Afterward, RAW264.7 macrophages were cocultured with 4T1ADV cells at ratio of 10:1 (THP-1 macrophages were cocultured with 231ADV cells at a ratio of 10:1). The medium containing adenoviruses and tumor cells was removed, and fresh medium was added 2 h after incubation. After 18 h, the macrophages and their supernatants were collected and ready for investigating changes in the phenotypic and proliferative markers of macrophages.
In vitro CD4 T cell and CD8 T cell stimulation with macrophage (culture supernatant)
CD4 T cells and CD8 T cells derived from spleen of WT mice were purified with CD4+ T cell isolation kit (Selleck, 90000) and CD8+ T cell isolation kit (Selleck, B90011). As described in the previous methods, macrophages stimulated with different interventions and their culture supernatants (conditioned medium) were harvested. CD4+ T cells and CD8+ T cells were respectively cocultured with macrophages (M-CTRL, M-Tα1, M-4T1ADV, and M-4T1ADV +Tα1), or conditioned medium of macrophages (M-CTRL, M-4T1ADV-CM, and M-4T1ADV +Tα1-CM) or conditioned medium of macrophages with added Tα1 (M-4T1ADV-CM+ Tα1). After 48 h later, the percentages of CD4+Foxp3+ T cells and IFN-γ+CD8+ T cells in the coculture system were assessed by flow cytometry.
Escherichia coli phagocytosis experiment
As described in the previous methods, macrophages were stimulated with different interventions. Macrophages (RAW264.7 cells and/or THP-1 cells) were cocultured with GFP+ Escherichia coli, and were harvested after 3 h. Then, macrophages were washed three times with PBS to remove the GFP+ Escherichia coli, and the EGFP levels in macrophages were measured by flow cytometry to assess phagocytosis efficiency. Macrophages from different intervention groups were mixed together to be used as a control to exclude interference from adenovirus EGFP fluorescence.
Flow cytometry
Samples were run on a FACSCaliber cytometer (BD) and Beckman Coulter Cytoflex S and analyzed with FlowJo 10. For the apoptosis analysis, tumor cells were incubated with viruses (MOI 10 or 20). After 24 h, cells were collected and stained with Annexin-V/PI for 20 min. For the immune cells in the TME and in the spleen, tissues from mice were collected and filtered to make single-cell suspensions by digestion with collagenase IV (50 μg/mL) for 1 h at 37°C. For the peritoneal macrophages, cells were collected by peritoneal lavage solution centrifugation at 1000 rpm for 5 min. For the polarization of macrophages in vitro, cells were collected at the indicated time points after different treatments. The harvested cells were stained with different antibodies. To exclude dead cells, 4′,6-diamidino-2-phenylindole (DAPI) were added shortly before analysis or sorting. Alternatively, fixable viability dye eFluor520 or LIVE/DEAD Near IR Fixable Stain was used to determine live cells. Fluorescent antibodies recognizing murine CD45-APC, CD3-FITC, CD4-PE, IFN-γ-PE, FoxP3-PE, CD11b-FITC, F4/80-PE/Cy7, CD86-PE, CD206-PerCP/Cy5.5, CD8-PerCP/Cy5.5, CD69-PE/Cy7, CD49b-PE, CD25-PerCP/Cy5.5, CD11c-PE/Cy7, and Ki67-PE/Cy7 were used in this assay and acquired from BioLegend.
Cell gating strategies: lymphocytes (FSC-H and SSC-H), single cells (SSC-A and SSC-H), CD45+ cells (CD45+ gated single cells), CD3+ T cells (CD3+ gated CD45+ cells), CD4+ T cells (CD4+ gated CD45+ cells), CD8+ T cells (CD8+ gated CD3+ cells), activated CD8+ T cells (CD25+CD8+, or CD69+CD8+, or IFN-γ+CD8+ gated CD3+ CD8+ cells), Tregs (CD25+FoxP3+ gated CD4+ T cells), macrophages (CD11b+F4/80+ gated CD45+ cells), “M1-like” macrophages (CD86+ gated macrophages), “M2-like” macrophages (CD206+ gated macrophages), DCs (CD11c+CD86+ gated CD45+ cells), and NK cells (CD3−CD49b+ gated CD45+ cells).
RNA sequencing
4T1 cells were treated with different interventions, including ADV, ADV combined with Tα1, or vehicle control at 37°C for 48 h. Macrophages were co-cultured with 4T1 cells after different stimulation for 18 h, and then macrophages were collected to perform transcriptome RNA-seq analysis. Total RNA of these samples was extracted using TRIzol (Vazyme) and stored at −80°C. Transcriptome analysis of these samples was performed using the Illumina NovaSeq 6000 sequencing platform (Majorbio). These data are available at the GEO accession number GSE271046 (4T1 cells) and GSE271202 (macrophages).
In vivo experiment
Subcutaneous tumor model
Exponentially growing 4T1, CT26, H22, or B16-F10 cells were harvested and then injected subcutaneously into the flanks of mice. Tumor volume was calculated using the formula (length×width2×0.5). Once the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups. Then, 0.1 mL of PBS, ADV or ADVTα1 (2 × 108 PFU) was injected intratumorally every other day for a total of three times, and 0.25 mg/kg Tα1 was injected subcutaneously once daily into the peritumoral site.49 Tumor volume was measured three times per week during the treatment. For survival experiments, mice were sacrificed when the tumor volume reached ≥1500 mm3. For flow cytometry analysis of tumor and/or spleen, mice were euthanized at related time points.
Rechallenge model and bilateral tumor bearing model
For the rechallenge model, the cured mice administered immunotherapies were injected subcutaneously with 5 × 106 H22 cells on day 0. For the bilateral tumor bearing model, the cured mice with immunological memory were rechallenged with 5 × 106 H22 cells and the opposing flank was injected subcutaneously with 5×105 4T1 cells on day 90 after the first rechallenge, naive mice were used as a control. Mice were sacrificed when the tumor volume reached ≥1500 mm3.
4T1 orthotopic tumor model
4T1 cells were injected into the fourth mammary fat pads of NCG mice on day 0. When the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups and treated with immunotherapies. Mice were euthanized and lung tissue samples of 4T1 tumor-bearing mice were collected on day 24 (n = 6 mice/group) and day 35 (n = 6 mice/group) to assess the antitumor effects of ADV combined with Tα1 and ADVTα1.
PBMC-humanized CDX model
Exponentially growing MDA-MB-231 and/or HepG2 cells were harvested and then injected subcutaneously into the flanks of NCG mice (GemPharmatech, NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) on day 0. PBMCs (it’s obtained from human peripheral blood) were inoculated into the mice on day 1. When the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups. Then, 0.1 mL of PBS, ADV or ADVTα1 (2 × 108 PFU) was injected intratumorally every other day for a total of three times, and 0.25 mg/kg Tα1 was injected subcutaneously once daily into the peritumoral site. Tumor volume was measured three times per week during the treatment. For survival experiments, mice were sacrificed when the tumor volume reached ≥1500 mm3.
PBMC-humanized PDX model
The TNBC tumor cells were resuspended in 50% volume Matrigel (Invitrogen, A1413202) and were injected into the fourth mammary fat pads of NCG mice on day 0. PBMCs (it’s obtained from human peripheral blood) were inoculated into the mice on day 1. When the tumor volume reached approximately 50–100 mm3, the mice were randomly divided into different groups. Then, 0.1 mL of PBS, ADV or ADVTα1 (2 × 108 PFU) was injected intratumorally every other day for a total of three times, and 0.25 mg/kg Tα1 was injected subcutaneously once daily into the peritumoral site. Tumor volume was measured every other day.
For the generation of PDX model, the breast tumor of patients diagnosed with TNBC was maintained on ice and brought to the laboratory within 1 h, after which they were harvested and dissociated into single cells and/or organoids by mechanical mincing and digestion. Cells were filtered through a 70 μm sterile filter. A total of 5×106 viable tumor cells were resuspended in 50% volume Matrigel and injected into the fourth mammary fat pads of NCG mice. When tumors reach about 1000 mm3, they were harvested and dissociated into single cells as previously described. The initial tumor (which reaches a volume of 1000 mm3 in NCG mice) is termed ‘passage 0’ (P0), and passages continued to be tracked with each generation. The oncolytic adenovirus (ADV+Tα1 and ADVTα1) treatment of PDX model used mice at passage 3.
In vivo immune cell depletion
For macrophage and CD8+ T cell depletion, BALB/c mice were intraperitoneally injected with 500 μg of anti-CD8a (Bioxcell, West Lebanon, NH, USA) or anti-CSF1R (Bioxcell, West Lebanon, NH, USA) every other day for a total of three times at related time points (n = 10 mice/group). For confirming the cell depletion, tumors from the mice injected with anti-CD8a or anti-CSF1R were harvested and flow cytometry confirmed the anti- CD8a and anti- CSF1R depletion effects at related time points.
For dendritic cell depletion, CD11c-dtr mice were administered 100 ng of diphtheria toxin the day before tumor injection (n = 6 mice/group). For confirming the cell depletion, tumors from the mice injected with diphtheria toxin were harvested and flow cytometry confirmed the depletion effects on day 10.
Quantification and statistical analysis
The statistical significance of differences in multiple groups was analyzed using GraphPad Prism by one-way ANOVA. The survival of tumor-bearing mice was analyzed using the Kaplan‒Meier method with the log rank test. Student’s t test or paired t test was used to compare two independent or matched groups. Data distribution was assumed to be normal, but this was not formally tested. Data are shown as the mean ± SD (NS, no significant differences; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Published: October 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101751.
Contributor Information
Xiaosong Gu, Email: nervegu@ntu.edu.cn.
Chunping Jiang, Email: chunpingjiang@163.com.
Junhua Wu, Email: wujunhua@nju.edu.cn.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Sanmamed M.F., Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2019;176:677. doi: 10.1016/j.cell.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Liu C., Yang M., Zhang D., Chen M., Zhu D. Clinical cancer immunotherapy: Current progress and prospects. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.961805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma R., Li Z., Chiocca E.A., Caligiuri M.A., Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. 2023;9:122–139. doi: 10.1016/j.trecan.2022.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2016;15:660. doi: 10.1038/nrd.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achard C., Surendran A., Wedge M.E., Ungerechts G., Bell J., Ilkow C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine. 2018;31:17–24. doi: 10.1016/j.ebiom.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macedo N., Miller D.M., Haq R., Kaufman H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan A.C., Bagley S.J., Wen P.Y., Lim M., Platten M., Colman H., Ashley D.M., Wick W., Chang S.M., Galanis E., et al. Systematic review of combinations of targeted or immunotherapy in advanced solid tumors. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radke J.R., Grigera F., Ucker D.S., Cook J.L. Adenovirus E1B 19-kilodalton protein modulates innate immunity through apoptotic mimicry. J. Virol. 2014;88:2658–2669. doi: 10.1128/JVI.02372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Zhang H., Wei M., Mou T., Shi T., Ma Y., Cai X., Li Y., Dong J., Wei J. Recombinant Adenovirus Expressing a Soluble Fusion Protein PD-1/CD137L Subverts the Suppression of CD8(+) T Cells in HCC. Mol. Ther. 2019;27:1906–1918. doi: 10.1016/j.ymthe.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding L., Gao Q., Xu Z., Cai L., Chen S., Zhang X., Cao P., Chen G. An Inter-Supplementary Biohybrid System Based on Natural Killer Cells for the Combinational Immunotherapy and Virotherapy of Cancer. Adv. Sci. 2022;9 doi: 10.1002/advs.202103470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv P., Liu X., Chen X., Liu C., Zhang Y., Chu C., Wang J., Wang X., Chen X., Liu G. Genetically Engineered Cell Membrane Nanovesicles for Oncolytic Adenovirus Delivery: A Versatile Platform for Cancer Virotherapy. Nano Lett. 2019;19:2993–3001. doi: 10.1021/acs.nanolett.9b00145. [DOI] [PubMed] [Google Scholar]
- 13.Chen J., Gao P., Yuan S., Li R., Ni A., Chu L., Ding L., Sun Y., Liu X.Y., Duan Y. Oncolytic Adenovirus Complexes Coated with Lipids and Calcium Phosphate for Cancer Gene Therapy. ACS Nano. 2016;10:11548–11560. doi: 10.1021/acsnano.6b06182. [DOI] [PubMed] [Google Scholar]
- 14.Cerullo V., Pesonen S., Diaconu I., Escutenaire S., Arstila P.T., Ugolini M., Nokisalmi P., Raki M., Laasonen L., Särkioja M., et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010;70:4297–4309. doi: 10.1158/0008-5472.CAN-09-3567. [DOI] [PubMed] [Google Scholar]
- 15.Dias J.D., Hemminki O., Diaconu I., Hirvinen M., Bonetti A., Guse K., Escutenaire S., Kanerva A., Pesonen S., Löskog A., et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19:988–998. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- 16.Jiang H., Rivera-Molina Y., Gomez-Manzano C., Clise-Dwyer K., Bover L., Vence L.M., Yuan Y., Lang F.F., Toniatti C., Hossain M.B., Fueyo J. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017;77:3894–3907. doi: 10.1158/0008-5472.CAN-17-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y.H., Liu S.H., Hao F.R., Zhang Y.H. Recombinant adenovirus p53 combined with radiotherapy improves efficacy and safety in the treatment of head and neck lymphoma. Cancer Biomarkers. 2018;23:213–220. doi: 10.3233/CBM-181286. [DOI] [PubMed] [Google Scholar]
- 18.Serafino A., Pierimarchi P., Pica F., Andreola F., Gaziano R., Moroni N., Zonfrillo M., Sinibaldi-Vallebona P., Garaci E. Thymosin alpha1 as a stimulatory agent of innate cell-mediated immune response. Ann. N. Y. Acad. Sci. 2012;1270:13–20. doi: 10.1111/j.1749-6632.2012.06707.x. [DOI] [PubMed] [Google Scholar]
- 19.Mandaliti W., Nepravishta R., Pica F., Vallebona P.S., Garaci E., Paci M. Potential mechanism of thymosin-alpha1-membrane interactions leading to pleiotropy: experimental evidence and hypotheses. Expet Opin. Biol. Ther. 2018;18:33–42. doi: 10.1080/14712598.2018.1456527. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y.T., Wang X.R., Yan C., Huang F., Zhang Y., Liu X., Wen Z.F., Sun X.T., Zhang Y., Chen Y.Q., et al. Thymosin alpha-1 Reverses M2 Polarization of Tumor-Associated Macrophages during Efferocytosis. Cancer Res. 2022;82:1991–2002. doi: 10.1158/0008-5472.CAN-21-4260. [DOI] [PubMed] [Google Scholar]
- 21.Ilkow C.S., Marguerie M., Batenchuk C., Mayer J., Ben Neriah D., Cousineau S., Falls T., Jennings V.A., Boileau M., Bellamy D., et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 22.Dyer A., Baugh R., Chia S.L., Frost S., Iris, Jacobus E.J., Khalique H., Pokrovska T.D., Scott E.M., Taverner W.K., et al. Turning cold tumours hot: oncolytic virotherapy gets up close and personal with other therapeutics at the 11th Oncolytic Virus Conference. Cancer Gene Ther. 2019;26:59–73. doi: 10.1038/s41417-018-0042-1. [DOI] [PubMed] [Google Scholar]
- 23.Munir M.T., Kay M.K., Kang M.H., Rahman M.M., Al-Harrasi A., Choudhury M., Moustaid-Moussa N., Hussain F., Rahman S.M. Tumor-Associated Macrophages as Multifaceted Regulators of Breast Tumor Growth. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22126526. ARTN 6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang X., Wang J., Lu D., Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Targeted Ther. 2021;6:75. doi: 10.1038/s41392-021-00484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wing J.B., Tanaka A., Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity. 2019;50:302–316. doi: 10.1016/j.immuni.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 26.King R., Tuthill C. Immune Modulation with Thymosin Alpha 1 Treatment. Vitam. Horm. 2016;102:151–178. doi: 10.1016/bs.vh.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Li J., Cheng Y., Zhang X., Zheng L., Han Z., Li P., Xiao Y., Zhang Q., Wang F. The in vivo immunomodulatory and synergistic anti-tumor activity of thymosin alpha1-thymopentin fusion peptide and its binding to TLR2. Cancer Lett. 2013;337:237–247. doi: 10.1016/j.canlet.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Russell S.J., Peng K.W., Bell J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robert-Guroff M. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 2007;18:546–556. doi: 10.1016/j.copbio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gholamin S., Mitra S.S., Feroze A.H., Liu J., Kahn S.A., Zhang M., Esparza R., Richard C., Ramaswamy V., Remke M., et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaf2968. [DOI] [PubMed] [Google Scholar]
- 31.Lecoultre M., Dutoit V., Walker P.R. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christofides A., Strauss L., Yeo A., Cao C., Charest A., Boussiotis V.A. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 2022;23:1148–1156. doi: 10.1038/s41590-022-01267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter C.A., Jones S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015;16:448–457. doi: 10.1038/ni.3153. [DOI] [PubMed] [Google Scholar]
- 34.Unver N., McAllister F. IL-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 2018;41:10–17. doi: 10.1016/j.cytogfr.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukao T., Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/S1471-4906(03)00139-X. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Q., Tang B., Zhang C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct Tar. 2022;7:182. doi: 10.1038/s41392-022-01036-5. ARTN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W. TGF-β Regulation of T Cells. Annu. Rev. Immunol. 2023;41:483–512. doi: 10.1146/annurev-immunol-101921-045939. [DOI] [PubMed] [Google Scholar]
- 38.Ugel S., De Sanctis F., Mandruzzato S., Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Invest. 2015;125:3365–3376. doi: 10.1172/Jci80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saha D., Martuza R.L., Rabkin S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell. 2017;32:253–267.e5. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jayasingam S.D., Citartan M., Thang T.H., Mat Zin A.A., Ang K.C., Ch'ng E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019;9:1512. doi: 10.3389/fonc.2019.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 42.Xu Y., Zeng H., Jin K., Liu Z., Zhu Y., Xu L., Wang Z., Chang Y., Xu J. Immunosuppressive tumor-associated macrophages expressing interlukin-10 conferred poor prognosis and therapeutic vulnerability in patients with muscle-invasive bladder cancer. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salmaninejad A., Valilou S.F., Soltani A., Ahmadi S., Abarghan Y.J., Rosengren R.J., Sahebkar A. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell. Oncol. 2019;42:591–608. doi: 10.1007/s13402-019-00453-z. [DOI] [PubMed] [Google Scholar]
- 44.Camerini R., Garaci E. Historical review of thymosin alpha 1 in infectious diseases. Expet Opin. Biol. Ther. 2015;15 Suppl 1:S117–S127. doi: 10.1517/14712598.2015.1033393. [DOI] [PubMed] [Google Scholar]
- 45.O'Donnell J.S., Teng M.W.L., Smyth M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019;16:151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 46.Kishton R.J., Sukumar M., Restifo N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metabol. 2017;26:94–109. doi: 10.1016/j.cmet.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuthill C., Rios I., McBeath R. Thymosin alpha 1: past clinical experience and future promise. Ann. N. Y. Acad. Sci. 2010;1194:130–135. doi: 10.1111/j.1749-6632.2010.05482.x. [DOI] [PubMed] [Google Scholar]
- 48.Gao D., Zhang X., Zhang J., Cao J., Wang F. Expression of thymosin alpha1-thymopentin fusion peptide in Pichia pastoris and its characterization. Arch Pharm. Res. (Seoul) 2008;31:1471–1476. doi: 10.1007/s12272-001-2132-z. [DOI] [PubMed] [Google Scholar]
- 49.Peng R., Xu C., Zheng H., Lao X. Modified Thymosin Alpha 1 Distributes and Inhibits the Growth of Lung Cancer in Vivo. ACS Omega. 2020;5:10374–10381. doi: 10.1021/acsomega.0c00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.