

Abstract

Inherently limited by poor bioavailability, antiviral agent tenofovir (TFV) is administered to people living with HIV in prodrug form. However, current prodrugs are prematurely metabolized, compromising access to HIV-infected cells and inducing toxicity. Inspired by lipid conjugate TFV exalidex (TXL), we recently disclosed TXL analogs with potent activity and robust hepatic stability in vitro, as well as attractive oral PK profiles in vivo. In parallel, we discovered the equipotent and equally stable hexadecylthiopropyl (HTP) derivative of TXL (2a). Reported herein are the synthetic and bioanalytic efforts that led to potent, safe, and hepatically stable HTP derivatives. While HTP analog 16h showed the most attractive PK profile in mice (55% F) discrepancies in translating in vitro cell-based results to in vivo PK data, for certain prodrugs, indicated that further in vitro/in vivo optimization is required for continued advancement of this program.
Introduction
Very little perusing of scientific literature is required to determine how clinically impactful nucleoside analogs have been with regard to antiviral treatment.1−6 Among the commercially successful nucleoside therapies, acyclic nucleoside phosphonates (ANPs, Figure 1) have achieved considerable acclaim due to their broad-spectrum antiviral activity, low drug resistance, controllable toxicological profile, and availability in highly potent oral prodrug formats.3,7,8 Mechanistically speaking, the therapeutic function of ANPs requires transformation into an active metabolite via two phosphorylation events catalyzed by cytosolic enzymes, such as nucleotide kinase and nucleoside diphosphate kinase (Figure 1A). The activated species then serves as an inhibitor/substrate for either pathogenic RNA and/or DNA polymerases. The primary advantages of ANPs over their cyclic nucleoside and nucleotide brethren stems from the ingenious insertion of a phosphonate moiety, which is not only metabolically stable to premature hydrolytic processing by physiologically abundant phosphatase enzymes, but also enables ANP drugs to bypass the first phosphorylation step toward activation, which is the bottleneck for many synthetic nucleoside therapeutics.7−9 However, the phosphonate component, which is negatively charged at physiological pH, limits the permeability of ANP drugs into virally infected cells and impedes oral bioavailability. Fortunately, various prodrug strategies have been successfully applied to ANPs to overcome these limitations.10−15
Figure 1.

Conversion of ANPs to their active metabolite via enzyme-catalyzed phosphorylation by nucleoside kinases (A) and representative structures for clinically relevant ANPs (B). NMPK, nucleoside monophosphate kinase; NDPK, nucleoside diphosphate kinase.
Tenofovir (TFV), a crucial ANP drug in the antiviral toolbox of medicinal chemists and medical practitioners, has a rich history of prodrug development.3,12−16 Therapeutically, TFV is a 2′,3′-dideoxyadenine analog that elicits broad-spectrum antiviral activity against human immunodeficiency virus (HIV),16−18 hepatitis B virus (HBV),19,20 and herpes simplex virus type-2 (HSV-2).21 Nevertheless, the uncapped phosphonate functionality of TFV imparts undesirable pharmacokinetic (PK) properties and poor oral bioavailability in clinical models, necessitating the creation of TFV prodrugs. As a result, the two most prevalent scaffolds that emerged in the race to improve TFV’s therapeutic potential and oral absorption are the phosphonate diester prodrug, tenofovir disoproxil fumarate (TDF),11,17,22 and the phosphonamidate prodrug, tenofovir alafenamide (TAF).23 Both prodrugs can be found in an assortment of commercially approved combination antiviral therapies used today as the backbones of anti-HIV therapeutics.2,15
The intrigue with TDF and TAF goes far beyond the simple masking of anionic character imposed by the TFV phosphonate moiety and generating a more lipophilic compound with amplified intestinal permeability. These prodrugs depend on unique enzyme-mediated cleavage mechanisms to physiologically distribute parent TFV, each with advantages and disadvantages (Figure 2).15 For example, TDF strategically relies upon esterases to release a TFV payload into virally infected cells. Through the installation of isopropyloxycarbonyloxymethyl (POC) esters on TFV, TDF provides a significantly higher oral bioavailability (i.e., 25–40% depending on food intake) versus the parent drug.10,11 However, due to the ubiquitous nature of esterase enzymes, TDF undergoes premature cleavage in the liver and plasma and elicits adverse side-effects over long-term use. Some TDF-induced toxic events, include: (1) liver function alteration through the accumulation of TFV-diphosphate (TFV-DP) in hepatocytes,22 (2) nephrotoxicity promoted by TFV renal tubular efflux,24,25 and (3) bone mineral density depletion driven by osteoclast dysfunction from TFV aggregation.26 Contrary to TDF, TAF possesses a phosphoramidate-based masking group (ProTide technology), which depends upon intracellular cleavage mechanisms for TFV release.27 Specifically, TAF is a substrate for the lysosomal serine protease, cathepsin A,28 and the serine hydrolase, carboxylesterase I (CES1), located in the cytoplasm and endoplasmic reticulum.29 The ProTide prodrug approach not only provides increased intracellular levels of TFV in target cells but imparts higher metabolic stability in plasma. As a result, TAF confers significantly reduced nephrotoxicity and TFV plasma exposure in comparison to TDF.15,30 Nevertheless, the distribution profile of TAF is severely limited to the liver (i.e., 65% of oral dose undergoes hepatic processing in dogs) since considerable levels of cathepsin A and carboxylesterase I are found in hepatocytes.27−29,31,32 Though TAF was repurposed as an HBV treatment in 2016,33 ProTide-based prodrugs do not mechanistically provide the ideal distribution profile required to tackle a more organ diffuse viral target, such as HIV. Furthermore, these agents necessitate strict adherence to once daily oral treatment schedules to prevent viral rebound, the onset of drug resistance, and progression to AIDS. And this essential strict adherence is directly compromised by deleterious drug-induced side effects like renal toxicity and bone mineral density depletion.
Figure 2.
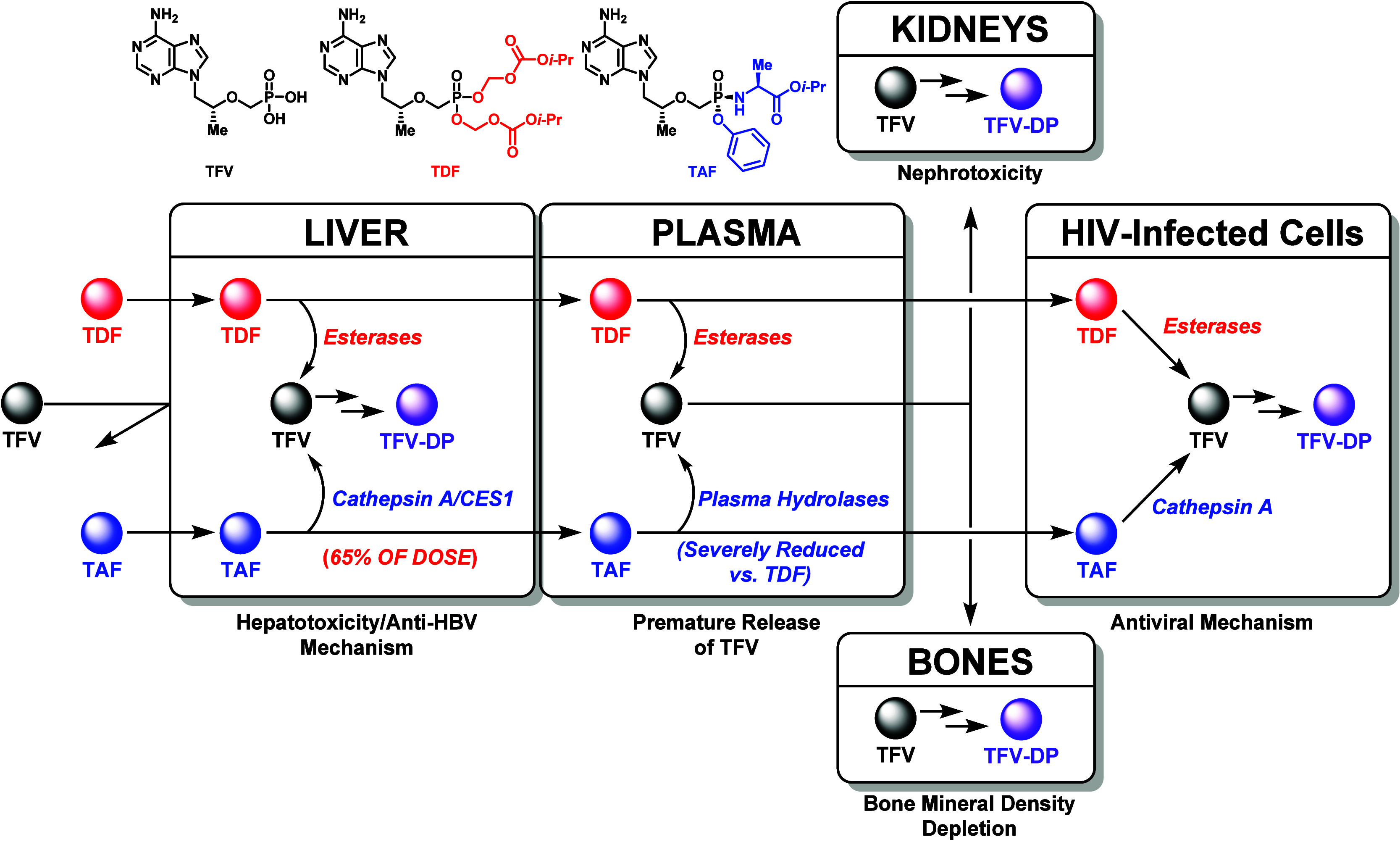
Mechanism of action and metabolic profile of TFV prodrugs in vivo.
In order to mitigate the long-term side-effects associated with POC-based prodrugs, such as TDF, Hostetler and co-workers developed a lipid-based strategy inspired by lysoglycerophospholipids (Figure 3).13 After years of modifications, the Hostetler group introduced the key hexadecyloxypropyl (HDP) moiety, which was installed on ANP cidofovir to create brincidofovir (CMX-001)34 and on TFV to generate TFV exalidex (TXL, CMX-157).35 Mechanistically, the prodrugs are predominantly cleaved intracellularly by membrane-associated enzyme, especially phospholipase C (PLC) and sphingomyelinase.13 By targeting these intracellular catabolic processes, the prodrugs exhibited favorable PK properties and safety profiles in relation to TDF (i.e., increased plasma stability and abated nephrotoxicity). However, just like TAF, the HDP scaffold undergoes significant metabolism in the liver since the terminal methyl group found on the lipid component is susceptible to cytochrome P450 (CYP450)-mediated ω-oxidation.13,36−38
Figure 3.

Hostetler and co-worker’s lipid-based strategy to enhance the oral absorption and reduce the toxicity of nucleoside drugs.
Despite the limitation with HDP-derived prodrugs, our lab seized upon an opportunity to improve the pharmacological properties of TXL and developed novel analogs capable of resisting ω-oxidation in vitro and in vivo.39,40 More specifically, by functionalizing the lipid terminus with chemical moieties designed to resist CYP450-mediated oxidation, we discovered the ω-CF3 prodrug 1, which displayed similar in vitro antiviral activity and cytotoxicity, improved human liver microsome (HLM) stability, and enhanced exposure levels and prodrug t1/2 in mice (Figure 4) compared to TXL. During these studies, we also discovered that the HDP component could be replaced with a hexadecylthiopropyl (HTP) group, resulting in compound 2a that showed comparable HIV activity (IC50 = 10 nM) and HLM metabolic half-life (t1/2 = 42 min) to TXL. Though the cytotoxic potential of 2a (CC50 = 50 μM) was 2-fold greater than TXL (CC50 = 98 μM), the molecule still exhibited an impressive therapeutic index of approximately 5000. Considering that substituting the HDP oxygen atom with a sulfur atom presents certain chemical and biological advantages (e.g., ease of nucleophilic reactions under mild basic conditions and potentially higher cellular permeability due to increased lipophilicity),41,42 we saw 2a as a great starting point for developing potent TXL analogs with elevated biological distribution and robust drug safety profiles. Additionally, since CYP450-mediated ω-oxidation of a lipid substrate is mechanistically controlled by a variety of factors, including steric effects, electronegativity, and bond dissociation energies,38,43−47 we used the HTP platform to further explore the prodrug terminal motif. Herein, we outline the synthesis and systematic pharmacological optimization of these HTP-derived TFV prodrugs, with detailed investigations of sulfur position, lipid chain length, and newly installed CYP450-resistant terminal motifs. Several of these compounds displayed HLM stability that far exceeded TXL, while maintaining potent anti-HIV activity and safe cellular cytotoxicity levels in vitro. Additionally, one prodrug demonstrated 2-fold greater oral bioavailability than TXL in vivo.
Figure 4.
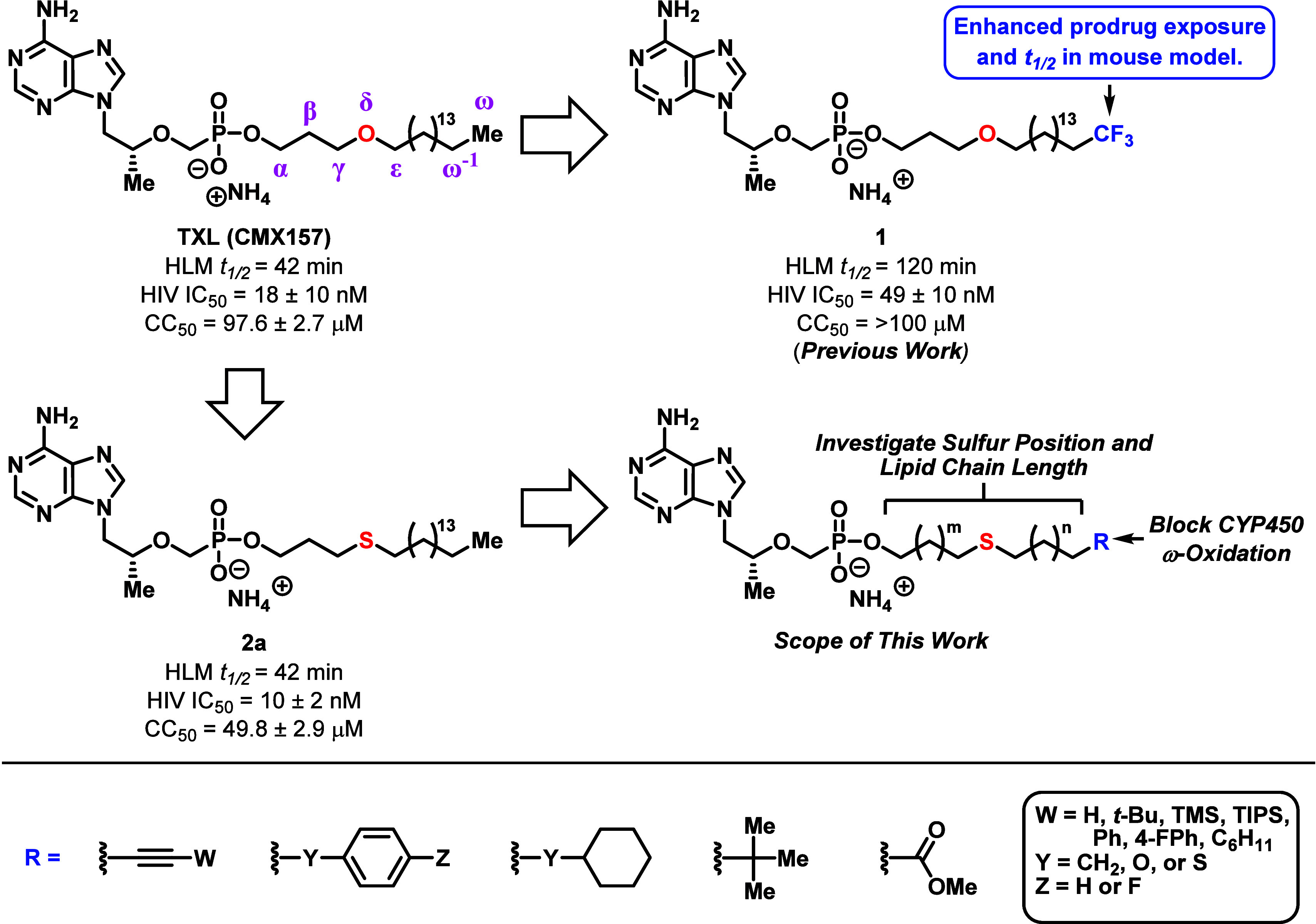
Summary of the pharmacological properties and drug design parameters for our HDP- and HTP-derived prodrugs. The scope of this study entails the optimization of HTP-based scaffolds through systematic investigation of sulfur position, lipid chain length, and newly integrated terminal motifs designed to resist CYP450-mediated ω-oxidation.
Results and Discussion
Organic Synthesis
Lipid analogs with modifications to sulfur position and chain length were synthesized utilizing the same two-step reaction sequence (Scheme 1). First, mercaptoalcohols (3) or alkyl thiols (6) were efficiently alkylated with the corresponding alkyl bromides (4) or bromo alcohols (5) respectively in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or cesium carbonate (Cs2CO3). Next, the resulting thioether-containing lipid alcohols 7a–j were conjugated to TFV using N,N′-dicyclohexylcarbodiimide (DCC) in combination with triethylamine (Et3N) and 4-(dimethylamino)pyridine (DMAP) to produce the final compounds 2a–j in low to moderate yields (30–69%). As shown in Scheme 1, these thioether-containing TXL derivatives, along with the remaining analogs described in our report, were obtained as ammonium salts due to the required use of ammonium hydroxide (NH4OH) as an additive for column chromatography.
Scheme 1. Synthesis of HTP-Derived TFV Prodrugs with Modified Sulfur Atom Position and Chain Length.
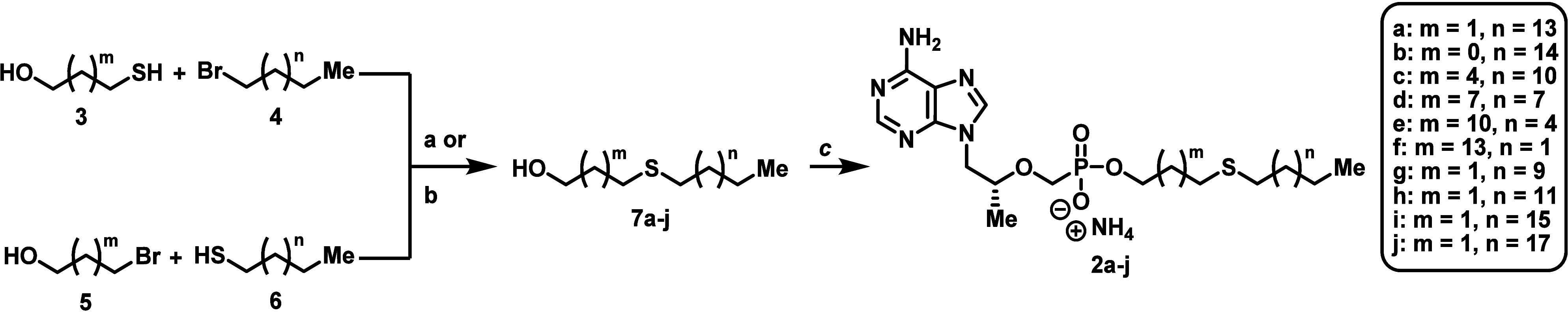
Reagents and conditions: (a) CsCO3, DMF, rt, 16 h, 92%; (b) DBU, DMF, rt to 60–65 °C, 1.5–4 h, 76 → 95%; (c) TFV, DCC, Et3N, DMAP, DMF or NMP, 100 or 105 °C, 18–24 h, 30–69%.
With regards to our target ω-functionalized lipid prodrugs (15b–h and 16e–h), the majority were constructed by starting with tetrahydropyranyl (THP)-protected bromo alcohols of varying chain length (8, Scheme 2). (Alkynyloxy)tetrahydropyran series 9 was then generated in high yield (84–94%) through a sequential treatment of commercially available asymmetrically functionalized acetylenes with n-butyllithium (n-BuLi) followed by nucleophilic substitution on bromide compounds 8. Next, alkynols 11a, 11c–f, and 11h were acquired by exposing the corresponding THP-protected alkynes (9) to standard deprotection conditions with a catalytic amount of p-toluenesulfonic acid (p-TsOH) in MeOH. Alternatively, alkynol 11g was directly synthesized using conventional Sonogashira coupling between 11-dodecyn-1-ol (10) and 1-fluoro-4-iodobenzene with a moderate 62% yield. Specific alkynols (11e–h) were then reduced with palladium on carbon (Pd/C) in either MeOH or EtOAc to generate lipid alcohols 12e–h. Even with solvents and catalysts known to slow hydrogenations, the silyl groups of compounds 11a, 11c, and 11d did not survive alkyne reduction toward the corresponding saturated lipids (results not shown). The collection of alkynols (11a and 11c–h) and alcohols (12e–h) were subsequently converted to thioether-containing lipid alcohols 13a, 13c–h, and 14e–h through an efficient two-step reaction sequence involving alcohol mesylation followed by nucleophilic displacement with 3-mercaptopropanol in the presence of DBU (68–92% yield over two steps). Trimethylsilyl (TMS) acetylene 13a was further processed to terminal alkyne 13b by simple tetrabutylammonium fluoride (TBAF)-mediated silyl deprotection. Lastly, the newly synthesized thioether lipids were coupled to TFV using the DCC procedure outlined above to produce final compounds 15b–h and 16e–h.
Scheme 2. Synthesis of ω-Functionalized HTP-Derived TFV Prodrugs Featuring Alkynyl- and Alkyl-Based Terminal Motifs.
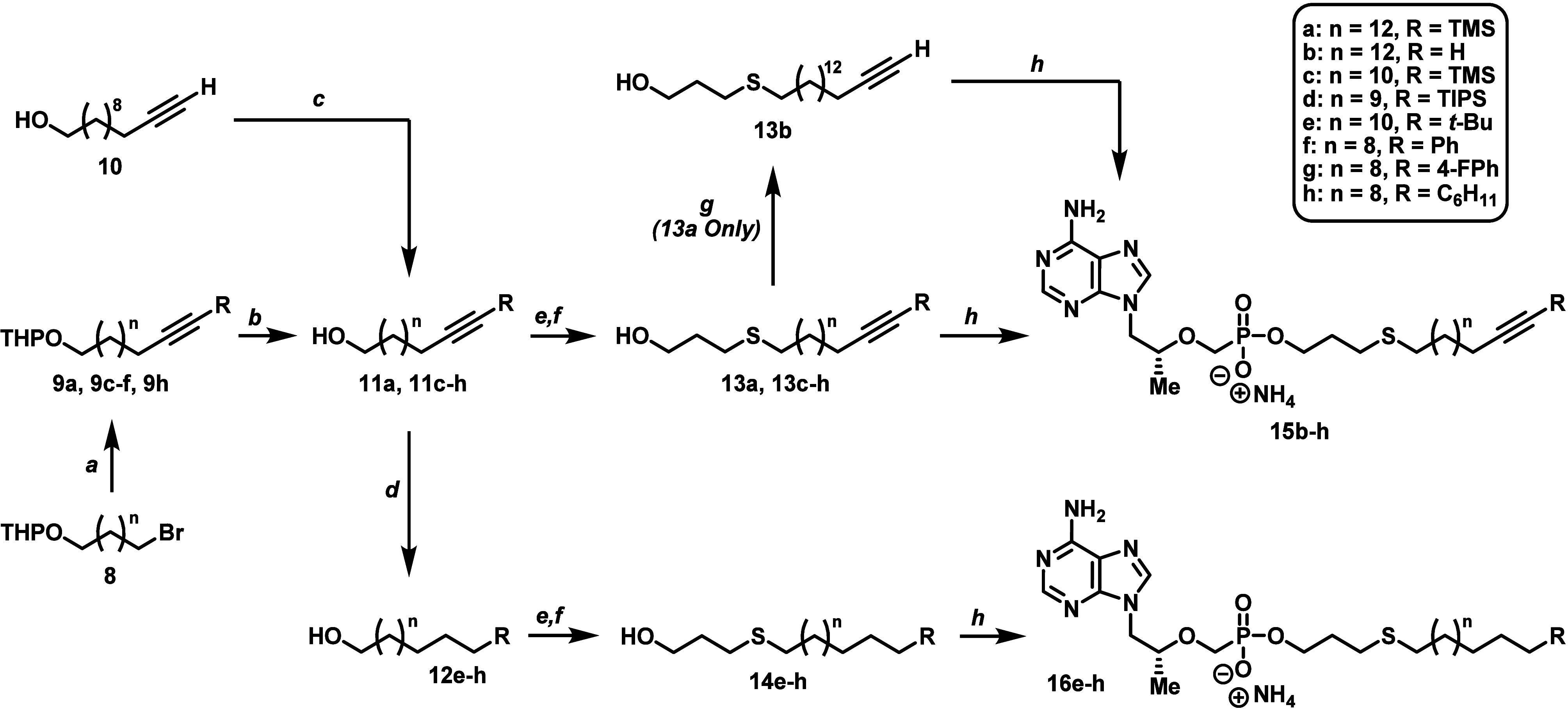
Reagents and conditions: (a) substituted acetylene, n-BuLi, HMPA, THF, −78 to −40 °C, 1 h then −40 °C to rt, 5–24 h, 84–94%; (b) p-TsOH·H2O, MeOH, rt, overnight, 69–91%; (c) 1-fluoro-4-iodobenzene, Pd(PPh3)2Cl2, CuI, Et3N, THF, rt to 70 °C, overnight, 62%; (d) 10% Pd/C, H2, MeOH or EtOAc, rt, 3–16 h, 55–83%; has MsCl, Et3N, DCM, 0 °C to rt, 45 min; (f) 3-mercaptopropanol, DBU, DMF, rt to 60–65 °C, 1.5–5 h, 68–92% over two steps; (g) TBAF, THF, rt, 45 min, 87%; (h) TFV, DCC, Et3N, DMAP, NMP, 100 °C, 18–24 h, 35–61%.
The synthesis for the remaining batch of designed thioether-containing TXL derivatives (23a–e, Scheme 3) started with 11-bromo-1-undecanol (17) or methyl 16-bromohexadecanoate (21). Phenyl ether derivatives 19a and 19c were first constructed using a standard Mitsunobu reaction between 17 and the corresponding phenols in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosphine (PPh3). On the other hand, cyclohexyl ether 19d was more difficult to produce, requiring a sequential alcohol triflation of 17(48) followed by a nucleophilic substitution with lithium cyclohexanolate under cold conditions. Next, the thioether-containing lipid alcohols 22a, 22c, and 22d were efficiently synthesized (i.e., 84–95% yield) by a simple nucleophilic substitution between bromide series 19 and 3-mercaptopropanol with Cs2CO3 as base. This reaction was also used to generate thiobenzene 20, which was carried forward to lipid 22b according to the two-step mesylation/nucleophilic displacement outlined in Scheme 2. Additionally, ester moiety 22e was obtained in a similar manner to 22a, 22c, and 22d by starting with bromide 21 and using DBU as the base for 3-mercaptopropanol deprotonation. Ultimately, lipid alcohols 22a–e were coupled to TFV to yield the final set of prodrugs analyzed for this study (23a–e).
Scheme 3. Synthesis of ω-Functionalized HTP-Derived TFV Prodrugs with Alternative Phenyl and Ester Terminal Motifs.
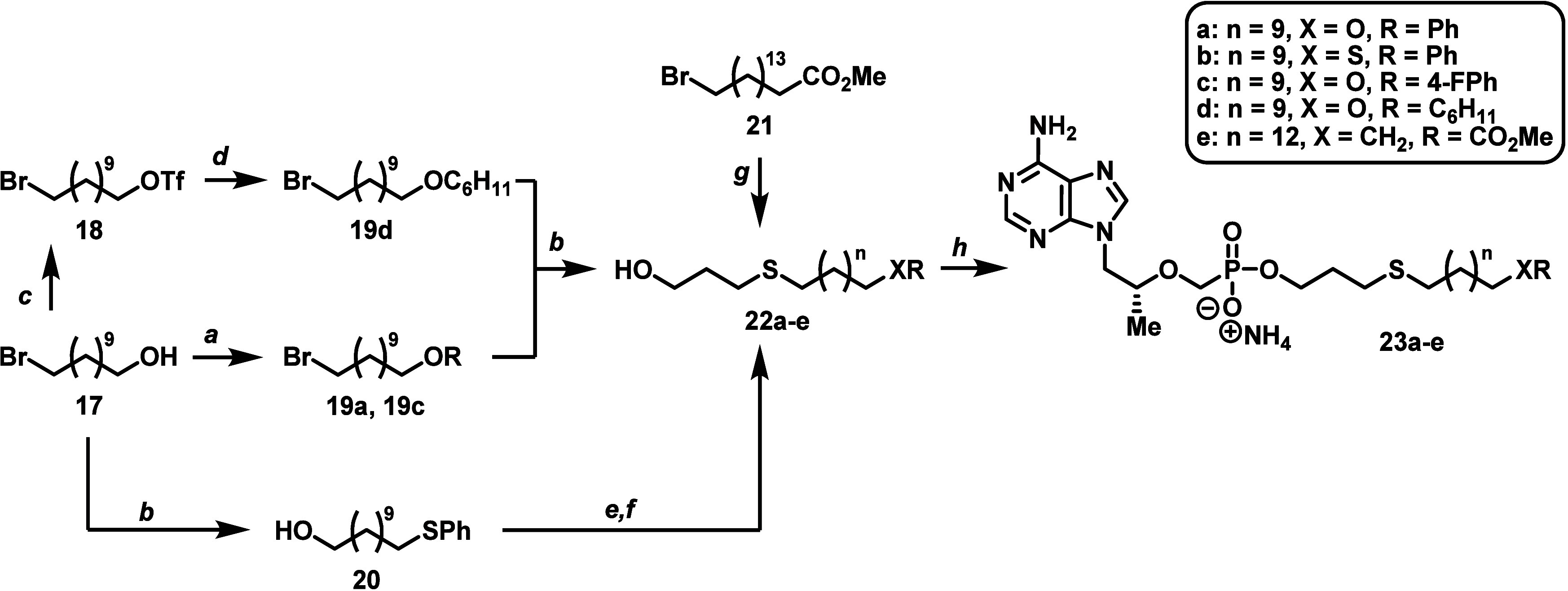
Reagents and conditions: (a) phenol, DIAD, PPh3, THF, 0 °C to rt, overnight, 70–77%; (b) thiol, CsCO3, DMF, rt, 1–16 h, 84–95%; (c) triflic anhydride, 2,6-lutidine, DCM, −78 °C, 30 min, 61%; (d) cyclohexanol, n-BuLi, HMPA, THF, −78 to 0 °C, 1 h then −78 to 0 °C, 5 h, 74%; (e) MsCl, Et3N, DCM, 0 °C to rt, 45 min; (f) 3-mercaptopropanol, DBU, DMF, rt to 60 °C, 3 h, 68% over two steps; (g) 3-mercaptopropanol, DBU, DMF, rt to 60 °C, 2 h, 88% over two steps; (h) TFV, DCC, Et3N, DMAP, DMF or NMP, 100 or 105 °C, 18–24 h, 32–54%.
In Vitro Antiviral Potency and Metabolic Stability Evaluation for HTP-Derived Prodrugs
All newly synthesized HTP-derived TFV prodrugs (2a–j, 15b–h, 16e–h, and 23a-e) underwent a preliminary pharmacological screen in vitro to ascertain HIV activity (IC50), cellular cytotoxicity (CC50), and HLM t1/2. The initial biological screen was conducted using assays and procedures our lab has previously reported (see Supporting Information).39,40 In brief, the metabolic t1/2 for each final compound was deduced in HLM (1 mg/mL) with the assistance of an LC-MS/MS to evaluate the rate of degradation. Second, HIV IC50 and cellular CC50 were determined with a pseudoviral, whole (HEK293T) cell, nonreplicative HIV assay, using the HIV-1 subtype C strain MJ4 (refer to Experimental Methods). To account for the unique amphiphilic properties of TXL and the novel HTP-based prodrugs, all compounds were formulated in a human serum albumin (HSA) solution (5:1 drug:protein). Consequently, the HIV potency for our designed prodrugs depended on several factors, including: (1) association/dissociation with HSA, (2) cell permeability, and (3) PLC kinetics for lipid cleavage.39 As the parent compound (TFV) is identical in all cases, activity differences between prodrugs hinged on disruptions or amplifications to these processes.
Considering that sulfur is chemically and biologically distinct from oxygen (i.e., lower electronegativity, larger atomic size, susceptibility to oxidation, etc.),41,42 a systematic structural evaluation of the novel HTP-based TFV prodrugs was necessary. Initially, we synthesized a series of lipid analogs with alterations in sulfur atom position and chain length to analyze how these changes could impact the HIV activity, metabolic stability, and cellular cytotoxicity for this compound class (Table 1). The results suggest that the more sterically or electronically accessible the sulfur atom is toward CYP450-mediatiated oxidation, the lower the metabolic stability of our prodrugs.38 Compound 2a displays an identical HLM stability profile to TXL (t1/2 = 42 min), which is notable due to the thioether’s position, which may provide steric or electronic protection against CYP450 enzymes. We were interested in defining which lipid positions the sulfur atom could occupy without negatively influencing the prodrug’s pharmacological properties. Additionally, since thioethers are generally more lipophilic than their ether counterparts, the lipid chain length threshold for HIV potency and cytotoxicity, which depend on compound solubility, cell permeability, and rate of PLC cleavage, could be severely influenced.13,34,35,39,40 Thus, we first aimed to identify the optimal thioether placement and linear atom count for our lipid prodrugs.
Table 1. In Vitro HIV (MJ4) Activity and HLM Stability Profiles of HTP-Derived TFV Prodrugs with Varied Sulfur Position and Chain Length.
| compound | lipid motif | linear atom # | HLM t1/2 (min)a [% remaining]b | HIV (MJ4) IC50 (μM)a | CC50 (μM)a | therapeutic indexc |
|---|---|---|---|---|---|---|
| TXL | C3H6–O-C16H33 | 20 | 41.6 ± 9.0d | 0.018 ± 0.010d | 97.6 ± 2.7 | 5420 |
| 2a | C3H6–S-C16H33 | 20 | 41.7 ± 2.3 | 0.010 ± 0.002d | 49.8 ± 2.9 | 4980 |
| 2b | C2H4–S-C17H35 | 20 | 66.1 ± 2.6 | 0.004 ± 0.003 | 28.6 ± 3.7 | 7150 |
| 2c | C6H12–S-C13H27 | 20 | 66.2 ± 3.0 | 0.073 ± 0.011 | >100 | >1370 |
| 2d | C9H18–S-C10H21 | 20 | 13.3 ± 0.5 | 0.058 ± 0.009 | >100 | >1720 |
| 2e | C12H24–S-C7H15 | 20 | 20.6 ± 0.1 | 0.338 ± 0.004 | >100 | >296 |
| 2f | C15H6–S-C4H9 | 20 | 19.7 ± 0.003 | 0.302 ± 0.062 | >100 | >331 |
| 2g | C3H6–S-C12H25 | 16 | 11.3 ± 0.2 | 0.583 ± 0.188 | >100 | >532 |
| 2h | C3H6–S-C14H29 | 18 | 15.6 ± 0.1 | 0.007 ± 0.006 | >100 | >16,700 |
| 2i | C3H6–S-C18H37 | 22 | >120 [>95%] | n.d. | n.d. | n.d. |
| 2j | C3H6–S-C20H41 | 24 | >120 [>95%] | n.d. | n.d. | n.d. |
n = 2.
Relative percent remaining of prodrug at 120 min. Linear atom # does not count H or F.
Therapeutic index = CC50/HIV (MJ4) IC50.
n = 4.
As illustrated with compounds 2a–f in Table 1, thioether placement on the lipid prodrugs clearly affected all studied in vitro parameters. Situating the thioether closer to the phosphonate functionality, as in compound 2b, produced slight improvements in both HIV activity (IC50 = 4 nM) and HLM stability (t1/2 = 66 min), but resulted in an estimated 2-fold and 4-fold increase in cytotoxicity (CC50 = 29 μM) over 2a and TXL, respectively. Despite the elevation in cellular toxicity, 2b maintained a therapeutic index of over 7000 and presented another starting scaffold for prodrug modification. On the other hand, the data for compounds 2c–f indicated that installing the thioether further down the lipid chain severely reduced HIV activity (IC50 = 60–340 nM) and/or HLM tolerance (t1/2 = 13–21 min). While compound 2c possessed a metabolic stability comparable to 2b (t1/2 = 66 min), the prodrug showed an 18-fold decrease in HIV potency (IC50 = 73 nM), providing a threshold for thioether placement on our HTP-derived analogs. On the other hand, compounds 2d–f demonstrated drastic declines in both antiviral activity and HLM t1/2, despite possessing better cytotoxicity profiles over 2a and 2b (CC50 > 100 μM). Ultimately, the results observed with 2d–f support our initial hypothesis that the HLM stability for HTP-derived prodrugs depends on the accessibility of the sulfur atom toward CYP450-mediated oxidation. Furthermore, in terms of the intracellular availability of 2c–f to inhibit HIV activity, further study is required to pinpoint how the thioether position mechanistically disrupts HSA binding, cell permeability, and/or PLC cleavage. When taking Table 1 into account, 2a and 2b revealed that the γ and δ positions on the lipid chain (see Figure 4) are pharmacologically optimal for the thioether. Though 2b is a potential starting point for future drug development, we decided to move forward with prodrug 2a for structural consistency with TXL and our previously reported analogs.39,40
As anticipated, compounds 2a and 2g–j, with lipid moieties ranging from 16 to 24 atoms, demonstrated the significant impact chain length has on both HIV activity and HLM stability, following the pattern observed for alkoxyalkyl-based TFV prodrugs reported previously (Table 1).13,34,35,39,40 For example, while compound 2h (18 atoms) maintained high antiviral potency (IC50 = 7 nM) in relation to 2a (20 atoms), the shorter lipid accelerated HLM metabolism (t1/2 = 15.6 min). When the prodrug was further truncated, as in 2g (16 atoms), dramatic loses in both HIV activity (IC50 = 583 nM) and HLM stability (t1/2 = 11.3 min) were observed. Conversely, extension of the lipid moiety beyond 20 atoms (2i and 2j) imparted considerable resistance to CYP450-mediated oxidation (t1/2 > 120 min with 95% prodrug remaining). Unfortunately, 2i and 2j could not be tested for antiviral potency and cell cytotoxicity because of poor solubility in the assay medium, despite formulation with HSA. Nevertheless, due to the promising metabolic profile of 2i and 2j, our lab is continuing to analyze TXL analogs with chain lengths beyond 20 atoms and will disclose a report in the future. However, for the sake of this study, 2a remained as the most pharmacologically attractive starting point for ω-functionalization. Hence, with one exception (refer to 23e), we focused on prodrugs containing lipid moieties with a linear atom count of 20 atoms.
Upon discovering the most pharmacologically attractive starting point for our thioether scaffolds, we moved toward installing ω-functionalized lipids on TFV to block CYP450-mediated oxidation at the terminal position. Apart from chain length45 and bond-dissociation energy considerations,39,43 previous structure–activity relationship (SAR) studies on fatty acid hydroxylases (i.e., CYP450 isoform family 4 enzymes, CYP-4) have obstructed ω-oxidation of an unbranched lipid substrate through the strategic installation of steric bulk (cycloalkanes or aromatic rings)44 or large sized atoms (bromo or iodo)47 at the terminus. Generally, the observed steric control occurs due to the structural characteristics of the CYP-4 catalytic site, which possesses a narrow, sterically restricted channel near the heme moiety to allow for regioselective oxidation of the ω position on lipids (Figure 5).49−53 However, depending on the functional group properties at the ω-position, CYP-4 enzymes will favorably engage in ω–1 hydroxylation.38 Consequently, despite blocking the ω-position with chemical moieties known to resist CYP450 metabolism, prodrug decomposition could potentially occur at the ω–1-site. Therefore, motivated by these studies, we performed a systematic pharmacological analysis on a variety of alkynyl, phenyl, silyl, and bulky alkyl terminal groups to assess how to fully limit CYP450-mediated ω-oxidation without attenuating HIV potency and impacting drug safety (Tables 2 and 3).
Figure 5.

Proposed binding mode for substrates of CYP-4 enzymes, using compound 2a as an example. AA+ = Positively Charged Amino Acid.
Table 2. In Vitro HIV (MJ4) Activity and HLM Stability Profiles of ω-Functionalized HTP-Derived TFV Prodrugs Featuring Acetylenic Terminal Groups.
| compound | lipid motif | linear atom # | HLM t1/2 (min)a [% remaining]b | HIV (MJ4) IC50 (μM)a | CC50 (μM)a | therapeutic indexc |
|---|---|---|---|---|---|---|
| TXL | C3H6–O-C16H33 | 20 | 41.6 ± 9.0d | 0.018 ± 0.010d | 97.6 ± 2.7 | 5420 |
| 2a | C3H6–S-C16H33 | 20 | 41.7 ± 2.3 | 0.010 ± 0.002d | 49.8 ± 2.9 | 4980 |
| 15b | C3H6–S-C14H28–C≡CH | 20 | 95.8 ± 1.8 | 0.028 ± 0.0002 | >100 | >3570 |
| 15c | C3H6–S-C12H24–C≡CTMS | 20 | >120 [82%] | 0.024 ± 0.0001 | >100 | >4170 |
| 15d | C3H6–S-C11H22–C≡CTIPS | 20 | >120 [>95%] | 0.265 ± 0.069d | 59.3 ± 2.4 | 224 |
| 15e | C3H6–S-C12H24–C≡Ct-Bu | 20 | >120 [71%] | 0.194 ± 0.044c | >100 | >516 |
| 15f | C3H6–S-C10H20-C≡CPh | 20 | 71.3 ± 1.1 | 0.008 ± 0.0004 | >100 | >12,500 |
| 15g | C3H6–S-C10H20-C≡C4-FPh | 20 | >120 [67%] | 0.010 ± 0.002 | >100 | >10,000 |
| 15h | C3H6–S-C10H20-C≡CC6H11 | 20 | >120 [70%] | 0.013 ± 0.002 | >100 | >7580 |
n = 2.
Relative percent remaining of prodrug at 120 min. Linear atom # does not count H or F.
Therapeutic index = CC50/HIV (MJ4) IC50.
n = 4.
Table 3. In Vitro HIV (MJ4) Activity and HLM Stability Profiles of ω-Functionalized HTP-Derived TFV Prodrugs Featuring Alkyl- or Heteroatom-Linked Terminal Groups.
| compound | lipid motif | linear atom # | HLM t1/2 (min)a [% remaining]b | HIV (MJ4) IC50 (μM)a | CC50 (μM)a | therapeutic indexc |
|---|---|---|---|---|---|---|
| TXL | C3H6–O-C16H33 | 20 | 41.6 ± 9.0d | 0.018 ± 0.010d | 97.6 ± 2.7 | 5420 |
| 2a | C3H6–S-C16H33 | 20 | 41.7 ± 2.3 | 0.010 ± 0.002d | 49.8 ± 2.9 | 4980 |
| 16e | C3H6–S-C14H28-t-Bu | 20 | >120 [>95%] | 0.174 ± 0.045d | 24.0 ± 1.4 | 138 |
| 16f | C3H6–S-C12H24–Ph | 20 | 68.4 ± 0.1 | 0.008 ± 0.001 | >100 | >12,500 |
| 23a | C3H6–S-C11H22–OPh | 20 | 8.4 ± 0.1 | 0.009 ± 0.004 | >100 | >11,100 |
| 23b | C3H6–S-C11H22–SPh | 20 | 7.9 ± 0.2 | 0.050 ± 0.008 | >100 | >2000 |
| 16g | C3H6–S-C12H24-4-FPh | 20 | >120 [69%] | 0.012 ± 0.002 | >100 | >8065 |
| 23c | C3H6–S-C11H22–O4-FPh | 20 | 49.7 ± 0.03 | 0.118 ± 0.014e | >100 | >594 |
| 16h | C3H6–S-C12H24–C6H11 | 20 | >120 [85%] | 0.008 ± 0.002 | 47.5 ± 8.7 | 6250 |
| 23d | C3H6–S-C11H22–OC6H11 | 20 | 47.9 ± 0.4 | 0.184 ± 0.023d | >100 | >542 |
| 23e | C3H6–S-C15H30-CO2Me | 22 | 5.5 ± 0.3 | 1.420 ± 0.675 | >100 | >70 |
n = 2.
Relative percent remaining of prodrug at 120 min. Linear atom # does not count H or F.
Therapeutic index = CC50/HIV (MJ4) IC50.
n = 4.
n = 3.
Our previous report indicated that installation of substituted acetylenic functionality at the lipid terminus generally produced pharmacologically beneficial results in vitro and in vivo,39 and thus we initially focused on applying the same concept to the HTP-derived prodrugs (Table 2). Acetylene 15b and TMS acetylene 15c exhibited improvements in HLM stability (t1/2 > 95 min) and cell cytotoxicity (CC50 > 100 μM) while maintaining relative antiviral potency (IC50 = 24–28 nM) compared with TXL and 2a. Contrarily, despite demonstrating significant resistance to metabolic oxidation (t1/2 > 120 min with >95% prodrug remaining), triisopropylsilyl (TIPS) acetylene 15d displayed very poor HIV activity with an IC50 value of 265 nM. More surprisingly, we observed similar results from tert-butyl (t-Bu) acetylene 15e (IC50 = 196 nM, t1/2 > 120 min with 71% prodrug remaining). While the chemical distinction between TIPS and TMS acetylenic substitution at the terminus was obvious, we were intrigued by the drastic impact to antiviral activity seen with the t-Bu group. Therefore, we conducted further studies on 15e in HepaRG cells (data presented below) to better characterize this SAR. Moving forward, we were pleased to see that phenylacetylene 15f showed HIV potency proportional to 2a (IC50 = 8 nM) with a slight boost in HLM stability (t1/2 = 71 min) and appreciable reduction in cytotoxicity (CC50 > 100 μM). By capping a potential metabolic hotspot on the phenyl ring of 15f with a fluorine atom, we generated prodrug 15g, which not only performed well against HIV (IC50 = 10 nM) but also exhibited a desirable metabolic stability (t1/2 > 120 min with 67% prodrug remaining) and cytotoxicity (CC50 > 100 μM) profile relative to TXL and 2a. Moreover, upon strategically replacing the phenyl ring with its fully saturated counterpart, we created the cyclohexyl-derived compound 15h that was just as potent, resistant to metabolic oxidation, and safe to cells as 15g (IC50 = 13 nM, t1/2 > 120 min with 67% prodrug remaining, CC50 > 100 μM). Ultimately, based on the data in Table 2, we thought compounds 15c, 15g, and 15h met our initial screening criteria for PK analysis. However, during the discovery process, we observed qualitatively by eye (i.e., white-to-yellow solid transition) and by LC–MS (i.e., rising impurity, m/z suggests amination) that these acetylenic prodrugs can undergo spontaneous decomposition while being stored at −20 °C (refer to Figure S52 for example). Considering that our TFV prodrugs were synthesized as ammonium salts, it is possible that acetylenic degradation occurs via a slow hydroamination of the terminal triple bond. However, additional experiments are required to fully characterize this process, and salt screens must be conducted to assess the relationship between counterion properties and prodrug stability. Because the rest of the compounds reported herein were quite bench-stable, and since we do not seek to further develop these acetylenic prodrugs, no further data on these apparently unstable analogs were collected. In contrast, we sought to progress forward by avoiding the triple bond entirely.
As shown with compounds 16e–16h in Table 3, our acetylene reduction strategy produced similar trends across all in vitro parameters studied. For example, with exception to an appreciable boost in cytotoxicity (CC50 = 24 μM), t-Bu analog 16e demonstrated very poor antiviral activity (IC50 = 177 nM) with high metabolic stability (t1/2 > 120 min with 95% prodrug remaining) akin to 15e. Additionally, phenyl compound 16f exhibited a comparable profile to 15f, with proportional HIV potency to 2a (IC50 = 8 nM), a slight increase in HLM stability (t1/2 = 68 min) and low cytotoxic potential (CC50 > 100 μM). By addressing the metabolic hotspots on the phenyl ring terminus of 16f, we generated our lead compounds 4-fluorophenyl 16g and cyclohexyl 16h, which were both effective against HIV activity (IC50 = 8–12 nM) and resistant to CYP450-mediated oxidation (t1/2 > 120 min) in HLM. Though the cytotoxic potential of 16h was greater than other prodrugs in the series (CC50 = 48 μM), the compound possessed an attractive therapeutic index of 6250.
In addition to the ω-functionalized lipids described above, we also sought to install a fatty acid ester (23e) to mimic more naturally occurring substrates. Regrettably, this strategy negatively impacted all studied parameters presumably due to the metabolic instability and increased polarity imparted by the ester moiety. Next, in efforts to simplify prodrug synthesis and take advantage of the promising pharmacological behavior of the phenyl and cyclohexyl terminal group insertions found in 16f–h, we constructed ethers 23a, 23c, and 23d and thioether 23b. While 23a showed antiviral activity (IC50 = 9 nM) and cellular cytotoxicity (CC50 > 100 μM) on par with 15f and 16f, the compound’s HLM stability was severely impacted by the phenoxy terminal group (t1/2 = 8 min). In principle, the electronic properties of the phenoxy terminus of 23a could enable the prodrug to bind more favorably to the heme complexes of CYP450 enzymes than phenylacetylene 15f and phenyl 16f to accelerate aromatic ring oxidation, which could explain the 8-fold shorter t1/2 in HLM versus its hydrocarbon counterparts. Unsurprisingly, thioether 23b also exhibited very low metabolic stability, presumably on account of the exposed sulfur atom in proximity to the lipid terminus (refer to Table 1). Interestingly, relative to the acetylenic- and alkyl-based prodrugs described above, we observed minimal improvements in HLM stability (t1/2 = 48–50 min) when mitigating potential metabolic hotspots found in compounds 23a and 23b with 4-fluorophenyl (23c) and cyclohexyl (23d) substitutions. While speculative, this could suggest that dealkylation/dearylation could be a major route of metabolism for this prodrug class. Furthermore, we were intrigued by the drastic decrease in antiviral activity observed with compounds 23c and 23d, which gave IC50 values of 118 and 184 nM, respectively. While the electronic effects induced by the 4-fluorophenyl ether of 23c could potentially disrupt HSA binding, cell permeability, and/or PLC kinetics versus 15g and 16g, the results for 23d are not as easy to rationalize. One thought is that the cyclohexyl ether terminus of 23d could interfere with the lipid membrane mechanics required for cell permeability13 and/or PLC cleavage. However, further studies are certainly required to pinpoint which step or steps of the TFV prodrug processing pathway (i.e., prodrug cellular uptake and/or cleavage) are most perturbed by these structural modifications.
In Vivo Pharmacokinetics for Compounds 2a, 16g, and 16h
Considering the data presented in Tables 2 and 3, our final candidates for in vivo studies (C57BL/6 mice) were 16g and 16h with 2a as a control compound against TXL. Prior to submission, these prodrugs were evaluated in mouse liver microsomes (MLMs) and human and mouse plasma in vitro (Table 4) to assess whether the thioether linker would impart a distinct metabolic profile from our previously reported TXL analogs in mice.39 Just as with TXL, 2a demonstrates a much higher metabolic stability in MLM (t1/2 > 120 min), most likely due to species differences in prodrug metabolizing CYP450 enzymes required for ω-oxidation. Additionally, 2a was completely stable in human and mouse plasma (t1/2 > 240 min with 95% prodrug remaining at 240 min). Furthermore, similar trends were observed across human and mouse matrices for 16g and 16h, with exception to 16h showing minimal decomposition in mouse plasma over the studied time course (71% prodrug remaining after 240 min). Since rodent plasma presents distinct esterase and hydrolase activity profiles compared to human plasma, 16h decomposition within the mouse matrix may have emerged from an enzyme isoform not found in humans.54 Nevertheless, we moved forward with our PK analysis of these three prodrugs.
Table 4. In Vitro Stability Profiles for 2a, 16g, and 16h in Human and Mouse Liver Microsomes and Plasma.
| compound | lipid motif | HLM t1/2 (min)a [% remaining]b | MLM t1/2 (min)a [% remaining]b | human plasma t1/2 (min)a [% remaining]c | mouse plasma t1/2 (min)a [% remaining]c |
|---|---|---|---|---|---|
| 2a | C3H6–S-C16H33 | 41.7 ± 2.3 | >120 [>95%] | >240 [>95%] | >240 [>95%] |
| 16g | C3H6–S-C12H24-4-FPh | >120 [69%] | >120 [88%] | >240 [>95%] | >240 [>95%] |
| 16h | C3H6–S-C12H24–C6H11 | >120 [85%] | >120 [>95%] | >240 [>95%] | >240 [71%]d |
n = 2.
Relative percent remaining of prodrug at 120 min.
Relative percent remaining of prodrug at 240 min.
Low LC–MS/MS signal intensity seen throughout assay time course.
To compare the in vivo PK profiles to TXL and our previously reported drug lead 1 (Tables S19 and S26),39 one group of male C57BL/6 mice were administered a single oral dose (p.o., 10 mg/kg) of 2a, 16g, or 16h (refer to Experimental Methods). Additionally, a separate group of mice were given a single intravenous dose (i.v., 3 mg/kg) of each prodrug to obtain plasma clearance (Cl), volume of distribution (Vd), and oral bioavailability data. Blood was sampled at 8 time points over 8 h (i.v.) or 24 h (p.o.) postdose, and plasma was isolated via centrifugation. Tissues (liver, kidney, and brain) were also harvested and homogenized at 3 time points over 24 h after oral dose. The concentrations of prodrug and TFV metabolite in each of these samples was then quantified using LC–MS/MS methodology.
After oral dosing, compound 16g achieved the highest hepatic exposure levels, as indicated by Cmax and AUC0–24h, followed by 16h, which demonstrated greater than a 4-fold elevated AUC0–24h relative to 2a (Figure 6A, Table 5). However, while TXL exhibited a comparable mouse liver profile to 2a, analog 1 (Figure 4) achieved higher Cmax (8710 ng/mL) and AUC0–24h (109,000 h·ng/mL) values than each of the HTP-derived lipid prodrugs.39 Regrettably, as signified by TFV AUC0–24h, compounds 16g and 16h delivered significantly higher levels (greater than 2-fold) of TFV metabolite to the liver relative to 2a, TXL, and 1 (TFV AUC0–24h = 23000–29000 h·ng/mL). Additionally, while 16g and 16h achieved liver prodrug to TFV AUC0–24h ratios that were 4- and 2-fold higher than TXL respectively, analog 1 solely displayed a ratio greater than 1 (Table S26). Considering that 16g and 16h were optimized against CYP450-mediated ω-oxidation alongside 1, one potential explanation for the unexpected increase in hepatic TFV levels is that these two prodrugs could be effectively metabolized by phase I and/or II enzymes absent in mouse and human liver microsome matrices. Alternatively, seeing as all prodrugs studied released some degree of TFV in the liver, metabolite exposure may significantly depend on the compound’s ability to permeate species-specific hepatocytes and engage in PLC cleavage.55−57 Evidence for the latter was observed during in vitro cellular uptake and stability experiments conducted with HepaRG cells, which simulate primary human hepatocytes (results outlined below).
Figure 6.

Mouse plasma and liver pharmacokinetic profiles of HTP-derived analogs of TXL. Male C57BL/6 mice (n = 3 per time point) were administered a single oral dose (10 mg/kg) of TFV prodrug using 90:10 olive oil:EtOH as a vehicle. Plasma, liver, and kidney levels of prodrug and TFV were quantified using LC–MS/MS. Liver (A), plasma (B), and kidney (C) concentrations of TFV prodrug and TFV metabolite are plotted together. Data represent the mean concentration at each time point ± SEM. The figures are generated with GraphPad Prism v9.
Table 5. Mouse Liver Pharmacokinetic Properties of Top HTP-Derived TFV Prodrugs After Oral Dose (10 mg/kg).
| PK parameter (p.o.) | 2a (CH3) | 16g (4-FPh) | 16h (C6H11) |
|---|---|---|---|
| Tmax (h) | 1.00 | 1.00 | 4.00 |
| Cmax (ng/mL) | 1250 | 5330 | 2150 |
| AUC0–24h (h·ng/mL) | 6450 | 63,600 | 27,300 |
| TFVTmax (h) | 4.00 | 24.0 | 24.0 |
| TFVCmax (ng/mL) | 1440 | 3980 | 3900 |
| TFV AUC0–24h (h·ng/mL) | 29,100 | 72,500 | 66,600 |
| Prodrug AUC0–24h/TFV AUC0–24h | 0.222 | 0.877 | 0.411 |
Interestingly, following i.v. (Figures S43, S46, and S49, Table 6) and p.o. (Figure 6B, Table 7) administration, compound 16h achieved the best plasma exposure levels among the thioether-lipid derivatives, as illustrated by the reported Cmax and AUC0–24h. In fact, after oral dosing, prodrug 16h demonstrated a plasma Cmax (195 ng/mL) that surpassed all tested lipid moieties (Table S19) and a greater than 3-fold improvement in AUC0–24h (1070 h·ng/mL) over TXL, 2a, and 16g. Moreover, the plasma AUC0–24h value of 16h was attractively comparable to that of our previous drug lead 1 (1280 h·ng/mL) and resulted in an impressive oral bioavailability (%F) of 54.9%. Surprisingly, compound 16g showed a similar oral bioavailability to 2a (overall %F of 20%), despite 16g exhibiting significantly higher hepatic levels than 2a and 16h. Additionally notable was that the plasma exposure profile of TXL outperformed 16g in oral experiments, raising questions about the PLC and membrane diffusion kinetics of 16g in mouse hepatocytes. Next, prodrug 16h displayed the lowest Cl (5.15 L/h/kg) and Vd (22.8 L/kg) of the HTP series in correlation with the significantly higher plasma exposure levels observed after oral administration. Analog 16h also exhibited a plasma t1/2 (3.07 h) comparable to TXL (2.92 h) and 2a (3.49 h), presumably due to compound’s propensity to engage with the liver and kidneys (refer to Table 8). Although 16g exhibited the longest p.o. plasma t1/2 (4.19 h) among the three congeners, surpassing TXL but being 2-fold shorter than that of compound 1 (8.66 h), relatively poor linear regression (0.65 ≤ R2 ≤ 0.77) of first-order kinetic plots suggest that 2a and 16g undergo nonlinear pharmacokinetic elimination just like TXL (refer to Supporting Information). As t1/2 and Vd values are calculated from the slopes (k) of these linear fits, these parameters should be considered approximations as opposed to absolute values. In contrast, 16h and 1 appear to nicely undergo first-order pharmacokinetic elimination (0.94 ≤ R2 ≤ 0.96), thereby placing higher confidence on t1/2 and Vd values for these compounds. Nevertheless, further studies are necessary to elucidate which prodrug properties are responsible for providing the distinct plasma PK profile observed with 16h relative to 2a and 16g. Based on our initial in vitro stability evaluation of 16h in mouse plasma, the prodrug’s poor matrix solubility or increased protein binding compared to the other HTP derivatives may play a role.
Table 6. Mouse Plasma Pharmacokinetic Properties of Top HTP-Derived TFV Prodrugs After Intravenous Dose (3 mg/kg).
| PK parameter (p.o.) | 2a (CH3) | 16g (4-FPh) | 16h (C6H11) |
|---|---|---|---|
| Tmax (h) | 0.10 | 0.10 | 0.10 |
| Cmax (ng/mL) | 2060 | 863 | 2380 |
| AUC0–8h (h·ng/mL) | 340 | 373 | 583 |
| k | 0.467 | 0.406 | 0.574 |
| R2 | 0.85 | 0.93 | 0.91 |
| t1/2 (h) | 1.49 | 1.71 | 1.21 |
| Cl(L/h/kg) | 8.82 | 8.05 | 5.15 |
| Vd(L/kg) | 18.9 | 19.9 | 8.97 |
Table 7. Mouse Plasma Pharmacokinetic Properties of Top HTP-Derived TFV Prodrugs After Oral Dose (10 mg/kg).
| PK parameter (p.o.) | 2a (CH3) | 16g (4-FPh) | 16h (C6H11) |
|---|---|---|---|
| Tmax (h) | 1.00 | 1.00 | 2.00 |
| Cmax (ng/mL) | 73.3 | 58.6 | 195 |
| AUC0–24h (h·ng/mL) | 219 | 260 | 1070 |
| %F | 19.3 | 20.9 | 54.9 |
| k | 0.199 | 0.165 | 0.226 |
| R2 | 0.65 | 0.77 | 0.94 |
| t1/2 (h) | 3.49 | 4.19 | 3.07 |
| Cl(L/h/kg) | 8.82 | 8.05 | 5.15 |
| Vd(L/kg) | 44.4 | 48.7 | 22.8 |
| TFVTmax (h) | 4.00 | 4.00 | 4.00 |
| TFVCmax (ng/mL) | 39.4 | 47.4 | 47.8 |
| TFV AUC0–24h (h·ng/mL) | 657 | 635 | 740 |
| Prodrug AUC0–24h/TFV AUC0–24h | 0.333 | 0.409 | 1.44 |
Table 8. Mouse Kidney and Brain Pharmacokinetic Properties of Top HTP-Derived TFV Prodrugs After Oral Dose (10 mg/kg).
| tissue | PK parameter (p.o.) | 2a (CH3) | 16g (4-FPh) | 16h (C6H11) |
|---|---|---|---|---|
| kidney | Tmax (h) | 1.00 | 4.00 | 4.00 |
| Cmax (ng/mL) | 581 | 602 | 795 | |
| AUC0–24h (h·ng/mL) | 5740 | 7650 | 10,800 | |
| TFVTmax (h) | 4.00 | 24.0 | 24.0 | |
| TFVCmax (ng/mL) | 695 | 1420 | 1420 | |
| TFV AUC0–24h (h·ng/mL) | 14,600 | 23,800 | 22,100 | |
| Prodrug AUC0–24h/TFV AUC0–24h | 0.393 | 0.322 | 0.488 | |
| brain | Tmax (h) | 1.00 | 4.00 | 1.00 |
| Cmax (ng/mL) | 43.8 | 5.74 | 16.2 | |
| AUC0–24h (h·ng/mL) | 69.0 | 13.5 | 36.8 | |
| TFVTmax (h) | ||||
| TFVCmax (ng/mL) | ||||
| TFV AUC0–24h (h·ng/mL) |
Furthermore, despite 16h delivering the highest plasma TFV Cmax and AUC0–24h after oral dose, the ratio of 16h AUC0–24h to TFV AUC0–24h was most attractive compared to 2a and 16g, both of which exhibited ratios below 1 (Table 7). However, TXL and 1 delivered lower plasma TFV Cmax and AUC0–24h values and demonstrated higher AUC0–24h ratios than each of the HTP-derived lipid prodrugs evaluated herein. Once more, considering that 2a, 16g, and 16h displayed high in vitro mouse plasma stability (t1/2 > 4 h), the mechanism by which each prodrug released such a high degree of TFV in vivo (5-fold greater AUC0–24h versus TXL) remains unclear. Additionally, while the hepatic TFV levels generated from 16g and 16h may have led to increased plasma exposure in relation to TXL and 1, the same does not apply to 2a, which possessed a similar hepatic profile to TXL.
Moreover, we ventured to further elucidate the distribution profile for the 3 HTP-derived lipid prodrugs by specifically analyzing the kidney and brain, which are relevant due to TFV-mediated nephrotoxicity and HIV reservoirs in the CNS (Figure 6C, Table 8). After oral administration, compound 16h achieved the highest renal exposure levels, followed by 16g and 2a. Interestingly, while 16g demonstrated an 8-fold higher AUC0–24h in the liver versus kidney, 16h and 2a displayed significantly lower liver to kidney AUC0–24h ratios. Considering that glomerular filtration in the kidneys is negatively impacted by protein binding, lipophilicity, and molecular size and HDP prodrugs historically exhibit very low renal uptake,13 the prodrug exposure levels for 16h and 2a were unexpected. Additionally, based on the liver and plasma PK results outlined above, prodrugs 16g and 16h unsurprisingly delivered the highest TFV Cmax and AUC0–24h, whereas 2a yielded the lowest renal exposure levels of TFV. Regrettably, all compounds displayed prodrug to TFV AUC0–24h ratios below 1, posing nephrotoxic implications over long-term use. Finally, in contrast to exposure levels in liver and kidney, the 3 HTP-derived lipid prodrugs achieved very low prodrug Cmax and AUC0–24h values in brain tissue, and none of them delivered detectable quantities of TFV, at least not at the time points evaluated. It is worth noting however that we did not measure levels of TFV monophosphate or diphosphate, and therefore, additional experiments are required to draw strong conclusions about CNS penetration. Overall, 2a achieved the highest prodrug exposure levels, followed by 16h then 16g. Based on these results, we are currently conducting experiments in brain homogenate to evaluate whether HDP and HTP prodrug strategies have utility for targeting latent HIV in the CNS.
In Vitro Cellular Uptake and Stability Evaluation For Select HTP-Derived Prodrugs In HepaRG Cells
Between the in vitro HLM (Tables 1–4) and in vivo mouse liver PK (Tables 5 and S26) data sets presented above, we discovered certain discrepancies in the pharmacological profiles of our HTP-derived TFV prodrugs that warranted further investigation. For example, compound 16g unexpectedly demonstrated a 2-fold higher in vivo hepatic AUC0–24h than 16h, despite the 4-fluorophenyl lipid moiety potentially having an order of magnitude lower lipophilicity versus its cyclohexyl counterpart according to predictions (Table S41). Furthermore, while HDP and trifluoromethyl lipid derivatives are anticipated to possess similar clogP values, analog 1 displayed a nearly 22-fold increase in hepatic AUC0–24 in vivo over TXL. Lastly, contrary to our optimization efforts in HLM in vitro, compounds 16g and 16h exhibited 3-fold greater TFV AUC0–24h levels compared to TXL and prodrug 1. Such inconsistencies pose challenges for the translation of in vitro data obtained in human matrices toward mouse PK models. Hence, we utilized an in vitro HepaRG cell-based assay to evaluate the pharmacological behavior of our best prodrugs 16g and 16h, along with TXL, 1, and 2a as control compounds, in a more metabolically complete, human-related system. Additionally, we spiked the cells with prodrugs 15e and 16e to provide insight into the relationship between cellular drug delivery and sterics at the lipid terminus. Plated HepaRG cells were employed for this purpose since they retain many of the characteristics found in primary human hepatocytes without compromising CYP450 activity, including non-CYP450 and phase II enzymes, active transport mechanisms, tight cellular junctions, and fatty acid catabolic pathways.58−60 Naturally, due the metabolic complexity of HepaRG cells, we specifically analyzed prodrug cellular uptake and stability, as represented by Cmax and intrinsic clearance (Clint,u) accordingly (Figure 7 and Table 9). Furthermore, the in vitro parameters described in Table 9 were obtained using identical compound formulation (5:1 drug:HSA) and LC-MS/MS method development as outlined above (refer to Experimental Methods).
Figure 7.

HepaRG cellular uptake profiles of select HTP-derived analogs of TXL. Data represent the mean concentration at each time point ± SD. The figure was generated with GraphPad Prism v9.
Table 9. In Vitro HepaRG Uptake and Stability Profiles For Select Lipid-Based TFV Prodrugs.
| compound | lipid motif | HepaRG Cmax (ng/mL)a,b | HepaRG t1/2 (min)a | HepaRG Clint,u (μL/min/million cells)c |
|---|---|---|---|---|
| TXL | C3H6–O-C16H33 | 397.1 ± 1.4 | 44.3 ± 0.5 | 28.5 |
| 1 | C3H6–O-C15H31–CF3 | 337.4 ± 1.0 | 42.9 ± 0.5 | 29.4 |
| 2a | C3H6–S-C16H33 | 828.0 ± 0.2 | 89.4 ± 0.4 | 14.1 |
| 15e | C3H6–S-C12H24–C≡Ct-Bu | 242.0 ± 5.5 | 66.6 ± 5.0 | 18.9 |
| 16e | C3H6–S-C14H28-t-Bu | 1100.0 ± 63.6 | 123.1 ± 3.0 | 10.2 |
| 16g | C3H6–S-C12H24-4-FPh | 209.8 ± 0.2 | 46.7 ± 0.3 | 27.0 |
| 16h | C3H6–S-C12H24–C6H11 | 1412.4 ± 1.5 | 71.0 ± 0.4 | 17.8 |
n = 2.
Cmax value that was recorded after 3 h incubation.
Clint,u value was calculated from cellular t1/2 (refer to Experimental Methods for equation). Linear atom # does not count H or F.
Contrary to the mouse hepatocyte data described above,39 TXL and compound 1 demonstrated comparable HepaRG uptake with Cmax values of 397 ng/mL and 337 ng/mL after 3 h incubation, respectively. Moreover, despite displaying a substantially longer t1/2 in HLM and a 25-fold higher prodrug to TFV AUC0–24h ratio in mouse liver, prodrug 1 showed a nearly identical rate of cellular clearance to TXL (t1/2 = 43–44 min, Clint,u = ∼ 29 μL/min/million cells). These results suggest drastic differences in PLC cleavage and/or membrane diffusion kinetics across cell lines. Next, prodrug 2a exhibited greater than 2-fold higher cellular uptake (Cmax = 828 ng/mL) relative to TXL and 1, indicating that HepaRG permeability improves with increasing lipophilicity. Further evidence for this trend was observed with cyclohexyl derivative 16h demonstrating an impressive HepaRG Cmax value of 1410 ng/mL, followed by t-Bu moiety 16e as the next best permeating prodrug (Cmax = 1100 ng/mL). Consequently, due to the heightened cellular prodrug levels observed with 2a, 16e, and 16h, these compounds displayed lower Clint,u values (10–18 μL/min/million cells) than TXL and 1. However, despite delivering the largest drug payload into HepaRG cells, compound 16h exhibited a faster rate of clearance than both 2a and 16e, potentially suggesting that the cyclohexyl lipid moiety is a more favorable PLC substrate under these biological conditions. On the other hand, prodrug 16e markedly showed the slowest rate of cellular clearance (t1/2 = 123 min, Clint,u = 10.2 μL/min/million cells), indicating that the t-Bu lipid derivative undergoes inefficient cell processing and may resist cleavage to TFV. Interestingly, in direct contrast to mouse liver PK experiments, compound 16g displayed the lowest cellular uptake with a Cmax value of 210 ng/mL, a nearly 4-fold and 7-fold reduction compared to 2a and 16h, respectively. Hence, due to the decreased cellular prodrug levels, analog 16g demonstrated a clearance rate analogous to TXL and 1 (t1/2 = 47 min, Clint,u = 27 μL/min/million cells). Lastly, t-Bu acetylene 15e showed a greater than 4-fold decrease in cell uptake (Cmax = 242 ng/mL) versus counterpart 16e, hinting at a relationship between structural conformation at the lipid terminus and HepaRG membrane diffusion efficiency that requires further study. Additionally notable is that although 15e exhibited nearly a 6-fold lower cellular uptake efficiency compared to 16h, these compounds displayed nearly equivalent rates of cellular clearance (t1/2 = 67–71 min, Clint,u = 18–19 μL/min/million cells). Akin to 16e, this result suggests that our t-Bu terminally capped lipids are poorly processed by cells.
Overall, we discovered key data set differences while evaluating lipid-based TFV prodrugs with in vitro HepaRG assays and in vivo mouse liver PK experiments. Particularly, drastic variances in the pharmacological parameters of TXL, 1, 2a, 16g, and 16h raise questions about how well data obtained from in vitro human-related systems translates to in vivo mouse models. We are currently analyzing the behavior of these prodrug constructs in a series of species-specific hepatocytes and PLC isozymes that will be reported in the future. Nonetheless, on a preliminary level, the compilation of in vitro and in vivo results presented here demonstrates the significance of fatty acid catabolic pathways and membrane diffusion kinetics in each cell line have on prodrug delivery, cleavage, and distribution for HIV treatment.
Conclusions
Herein, we describe the strategies, synthesis, and pharmacological assessment of an HTP-derived prodrug series of TFV (Figure 4). In consideration of the promising in vitro characteristics of 2a, we performed systematic thioether position, chain length, and lipid terminus optimizations to identify the most therapeutically effective and metabolically stable TFV prodrugs. As demonstrated in Table 1, analogs bearing thioether functionality beyond the δ position (2c–2f) or lipid chain lengths below 20 atoms (2g and 2h) exhibited considerable reductions in HLM stability and/or anti-HIV activity in vitro. Interestingly, prodrugs carrying lipids over 20 atoms (2i and 2j) showed significantly enhanced HLM resistance versus TXL and 2a but could not be properly assessed for antiviral potency due to poor solubility in assay media. Accordingly, we opted to employ a terminal group SAR study using the general lipid characteristics of 2a (i.e., 20 atom lipids with sulfur in δ position), coupled with functional groups designed to block CYP450-mediated ω-oxidation (Tables 2 and 3). While our investigation resulted in the synthesis of several prodrugs with substantially longer t1/2 in HLM than TXL and 2a, dramatic variability in anti-HIV activity was observed among structurally similar congeners (e.g., 16e vs 16h). Ultimately, compounds 16g and 16h passed our initial in vitro screening requirements, and hence were subjected to in vivo mouse PK experiments, alongside 2a as a control (Tables 5–8). Regrettably, while 16h arguably demonstrated the best PK profile in vivo with an oral bioavailability of 55% after a 10 mg/kg dose, we observed significant levels of TFV in mouse liver, plasma, and kidney for all HTP-based compounds studied. Additionally, the HTP-derived TFV prodrugs were unable to surpass our previously discovered drug lead 1 in terms of mouse plasma t1/2 and overall prodrug to TFV AUC0–24h ratio. Motivated by discrepancies found between in vitro HLM and in vivo mouse liver PK experiments, we analyzed select lipid-based TFV prodrugs in HepaRG uptake and stability assays (Table 9) to simulate an advanced human-related metabolic system. After discovering significant deviation between the in vitro and in vivo uptake and clearance profiles of our prodrugs, we determined that careful animal model selection will be necessitated for future studies. Nevertheless, the preliminary results presented in this report demonstrate that our lead 16h is significantly absorbed and prematurely processed by the liver, which is somewhat inconsistent with our optimization efforts in HLM. Thus, further prodrug optimization using more physiologically relevant systems is required to achieve more attractive distribution and stability in vivo. Toward this aim, our lab is currently experimenting with longer chain length lipids featuring elements of unsaturation that will be reported soon.
Experimental Methods
Chemistry
Automated flash column chromatography was performed using a Teledyne ISCO CombiFlash Companion system with RediSep Rf normal-phase silica gel-packed columns or RediSep Rf Gold reverse-phase C18 columns (Teledyne Isco). Melting/decomposition ranges were determined on a REACH Devices RD-MP digital melting point apparatus. Analytical thin-layer chromatography (TLC) was carried out on commercially available (Sigma) aluminum-supported (thickness: 200 μm) or glass (2.5 × 7.5 cm) silica gel plates with fluorescent indicator (F-254). Visualization of compounds on TLC plates was achieved using UV light (254 nm) and/or using ethanolic phosphomolybdic acid (PMA) or aqueous potassium permanganate (KMnO4) solutions. TLC retention factors (Rf) were determined on glass (2.5 × 7.5 cm) silica gel plates with fluorescent indicator (F-254) and calculated as the average of three replicate experiments. NMR spectra (1H, 13C, and 31P) were acquired using a Bruker Ascend 600 MHz spectrometer, a Varian INOVA 600 MHz spectrometer, a Varian INOVA 500 MHz spectrometer, a Bruker NEO 400 MHz spectrometer, a Varian INOVA 400 MHz spectrometer, or a Varian VNMR 400 MHz spectrometer (Emory University NMR Center, directed by Dr. Shaoxiong Wu). NMR samples were prepared in deuterated chloroform (CDCl3) or deuterated methanol (CD3OD) using tetramethylsilane (TMS) or residual solvent peaks (CDCl3: 1H = 7.26 ppm, 13C = 77.16 ppm; CD3OD: 1H = 3.31 ppm, 13C = 49.00 ppm; TMS: 1H = 0.00 ppm) as internal references. Alternatively, the residual chloroform peak in 1H NMR was used as an absolute reference for 31P NMR, unless otherwise specified. MestreNova software was used to process all NMR spectra. NMR data includes chemical shifts (δ) reported in ppm, multiplicities indicated as s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), td (triplet of doublets), m (multiplet), br (broad), or app (apparent), and coupling constants (J) reported in Hz. High resolution mass spectrometry (HRMS) was performed by the Emory University Mass Spectrometry Center, directed by Dr. Fred Strobel. Liquid chromatography–mass spectrometry (LC–MS) was performed on an Agilent 1200 HPLC equipped with a 6120 Quadrupole mass spectrometer (ESI) eluting with mixtures of HPLC grade MeOH and H2O or MeCN and H2O (all spiked with 0.1% HCO2H) through an analytical, reverse-phase Agilent InfinityLab Poroshell 120 EC-C8 (2.1 mm × 50 mm, 2.7 μm) column. LC-MS samples were prepared with HPLC grade MeOH at a concentration of 1 mg/mL. Final compound purity was assessed using LC-MS, and purity of all final compounds reported herein were determined to be ≥95% pure. The chemical synthesis and characterization for all compounds can be found in the Supporting Information.
Cellular Toxicity and Antiviral Activity Assays
Assessment of cytotoxicity and antiviral activity of all final compounds in vitro was conducted according to previously reported procedures.39 In summary, human embryonic kidney (HEK293T) cells maintained in high glucose (25 mM), Dulbecco’s modified Eagle’s medium (DMEM) supplemented with fetal bovine serum (10%), sodium pyruvate (1 mM), l-glutamine (2 mM), HEPES (25 mM), and gentamicin (50 μg/mL) were used for determinations. The cells were maintained in an incubator at 37 °C under 5% CO2 in a humidified atmosphere. For both cytotoxicity and antiviral screening, all compounds were evaluated in duplicate in at least two independent experiments. All final compounds were formulated with human serum albumin (HSA, 5:1 compound to HSA molar ratio) and subsequently diluted with complete DMEM to 200 μM. Fifty microliters of test compound serial dilutions (3-fold) were prepared in 96-well culture plates, after which 50 μL of HEK293T cells (2 × 104 cells/well) was added. Final test compound concentrations for toxicity assessments ranged from 100 μM to 1.7 nM. A growth medium control without test compound was included as an indicator of 100% cell viability (no cytotoxicity). Ninety-six-well plates were incubated for 48 h at 37 °C under 5% CO2 in a humidified atmosphere. Cytotoxicity was then assessed by quantifying cell viability using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI) or resazurin sodium salt (cat# R7017, Merck, Darmstadt, Germany).61 The concentration of test compound that kills 50% of cultured cells (CC50) was calculated using Microsoft Excel (Redmond, WA). Antiviral activity was then assessed using a single-cycle, nonreplicating, envelope deleted HIV pseudoviral in vitro assay.62,63 The assay relies on the introduction of the firefly luciferase gene into HEK293T cells through infection with HIV-like viral particles, containing Reverse Transcriptase (RT) and Integrase (IN) from the HIV-1 subtype C strain MJ4.64 The expression of the luciferase in the cells is directly proportional to the level of infection by the HIV-like viral particles and can be used to assess the inhibition of HIV-1 RT and IN. To evaluate anti-HIV activity, 3-fold serial dilutions of all final compounds were prepared in 50 μL over the noncytotoxic concentration range (as determined from the cytotoxicity screens described above) in 96-well culture plates. HEK293T cells (2 × 104 cells/well) and HIV-like viral particles, standardized to produce a luminescence signal of 1 × 106 relative light units (RLUs) in the growth medium only control, were combined, and 50 μL was added to the wells containing serially diluted test compounds. A growth medium control without test compound was included as a reference for 100% viral activity (no inhibition). Ninety-six-well plates were incubated for 48 h at 37 °C under 5% CO2 in a humidified atmosphere. The expression of luciferase was subsequently quantified by adding 100 μL of the Bright-Glo Luciferase Assay substrate (Promega, Madison, WI) to each well of the 96-well plates. After incubation at rt for 3 min, luminescence was quantified on the GloMax Explorer Multimode Microplate Reader (Promega, Madison, WI). The concentration of each compound required to inhibit viral activity by 50% (IC50) was calculated using Microsoft Excel (Redmond, WA).
Metabolic Stability Assays
Liver Microsome Stability
Human liver microsomes (HLMs, 20 mg/mL) and CD-1 mouse liver microsomes (MLMs, 20 mg/mL) were purchased from Xenotech. NADPH was purchased from Sigma-Aldrich and prepared in 10 mM stock solutions of distilled H2O (Invitrogen UltraPure). Verapamil and diphenhydramine were both purchased from Sigma-Aldrich and served as positive controls for HLM and MLM stability, respectively. Test compounds and positive controls were initially dissolved in MeOH to make 10 mM stock solutions. Sample solutions were then further diluted in 70/30 MeOH/H2O to 500 μM. Next, the reactions were prepared by mixing human or mouse liver microsomes (55 μL) with potassium phosphate buffer (100 mM, 928 μL) in 1.5 mL Eppendorf tubes. The test compounds (6.6 μL of 500 μM solution) were subsequently added to the suspensions, and the reaction mixtures were incubated at 37 °C for 5 min. Afterward, the liver microsome reactions were initiated with 110 μL of 10 mM NADPH and further incubated at 37 °C for the designated time course of the study. This procedure provided experiments with a final volume of 1100 μL (<0.2% organic solvent content), a concentration for HLMs and MLMs of 1 mg/mL, and a final test compound concentration of 3 μM. Aliquots (100 μL) were removed from each reaction mixture in duplicate at 0, 15, 30, 60, and 120 min time intervals and quenched with 100 μL of cold internal standard solution (ISTD, 2 μM 7-ethoxy-d5-coumarin in MeOH). Quenched aliquots were then centrifuged at 12,500 g for 5–10 min, and the resulting supernatant solutions were withdrawn and placed in LC–MS vials to be analyzed by LC–MS/MS (Agilent G6460C QQQ MS coupled with an Infinity II 1260 HPLC). Each test compound was run in tandem with positive and negative control experiments for quality assurance. Positive control reactions were conducted at a final volume of 550 μL for a single run of each time point. Lastly, the negative control experiment was conducted with test compounds and liver microsomes in the absence of NADPH (150 μL) and analyzed at the 120 min time point.
Plasma Stability
Human plasma (lithium heparin (LiHep) mixed, gender pooled, 0.2 μm filtered) and mouse plasma (BALB/C, LiHep mixed, male pooled, 0.2 μm filtered) were purchased from BioIVT. Procaine (Sigma-Aldrich) served as a positive control for both human and mouse plasma experiments. Test compounds and positive controls were initially dissolved in MeOH to make 10 mM stock solutions. The solutions of test and control compounds were then further diluted in 70/30 MeOH/H2O to 500 μM. Next, the human or mouse plasma (994 μL) was aliquoted into 1.5 mL Eppendorf tubes with duplicates (reactions A and B) being prepared for each compound. The plasma was then incubated at 37 °C for 10 min. Afterward, the reaction was initiated by the addition of test compound (6 μL of 500 μM solution) and further incubated at 37 °C for the designated time course of the study. This procedure provided duplicate experiments with a final volume of 1000 μL (<0.2% organic solvent content) and a final test compound concentration of 3 μM. Aliquots (100 μL) were removed from each reaction mixture at 0, 30, 60, 120, and 240 min time intervals and quenched with 150 μL of cold ISTD solution (2 μM 7-ethoxy-d5-coumarin in MeOH). Quenched aliquots were then centrifuged at 15,000g for 30–45 min, and the resulting supernatant solutions (∼70 μL) were withdrawn and placed in LC–MS vials to be analyzed by LC–MS/MS (Agilent G6460C QQQ MS coupled with an Infinity II 1260 HPLC). Each test compound was run in tandem with positive and negative control experiments for quality assurance. The positive control reaction was conducted at a final volume of 1000 μL for a single run of each time point. Finally, the negative control experiment was conducted with test compounds in Dulbecco’s phosphate buffered saline without calcium and magnesium (DPBS, Fisher Scientific, 143 μL) and analyzed at the 240 min time point.
Data Analysis
For both LM and plasma stability assays, each data point was analyzed in duplicate using in-between blank washes to avoid carry over and to equilibrate the column for the subsequent runs. Averages of these duplicate for individual compounds at each time point were then normalized to the data at 0 min, representing 100% test compound remaining or 0% metabolism. Half-lives (t1/2) were calculated by plotting ln of % test compound remaining versus time and performing linear regression to determine slope. Slope = −k and t1/2 = 0.693/k for first-order kinetics (Figures S1–S34).
HepaRG Cell Uptake and Metabolic Stability
The HepaRG cell line (HPRGC10, Thermo Fisher Scientific) was used to evaluate the cellular permeability and metabolic stability for select final compounds in vitro. The cells were initially thawed with working media composed of HepaRG Thaw, Plate, & General Purpose Medium Supplement (HPRG670), Williams Medium E (100 mL), and GlutaMax (1 mL), all purchased from Thermo Fisher Scientific. Next, the cells were seeded in sterile collagen-coated 24-well plates (0.55 × 106 cells/well) and maintained in an incubator at 37 °C under 5% CO2 in a humidified atmosphere for 24 h. After the thawing medium was aspirated from each well, the cells were cultured for an additional 6 days with working media consisting of HepaRG Maintenance/Metabolism Medium Supplement (HPRG720), Williams Medium E (500 mL), and GlutaMax (1 mL), all purchased from Thermo Fisher Scientific. Upon completing the 7-day culture period, HepaRG cells were plated in enough wells to allow for duplicate analysis of prodrugs in two independent cellular uptake or metabolic stability experiments. All studied compounds were formulated with human serum albumin (HSA, 5:1 compound to HSA molar ratio) and subsequently diluted to a 20 μM final concentration with assay medium composed of serum-free INVITROGRO HI Medium and TORPEDO Antibiotic Mix purchased from BioIVT. For cellular uptake determinations, the maintenance/metabolism medium was first aspirated from each well, and then the plated cells were spiked with 1.0 mL of test compound stock solution. Subsequently, the plate was incubated at 37 °C under 5% CO2 environment over the desired time course (seven data points/compound). Blank wells not treated with prodrug were included in assay runs as controls and initial time points (0 min). At each time point, the test compound solution was removed from the appropriate wells, and the plated cells were washed with 1.0 mL of DPBS twice. The HepaRG cells were then extracted with 500 μL of cold ISTD solution (1 μM 7-ethoxy-d5-coumarin in 70/30 MeOH/H2O), and the quenched aliquots were centrifuged at 15,000 g for 10 min at 4 °C. The resulting supernatant solutions were withdrawn and placed in LC–MS vials to be analyzed by LC–MS/MS (Agilent G6460C QQQ MS coupled with an Infinity II 1260 HPLC). After obtaining prodrug Cmax and Tmax values from the cellular uptake assay, HepaRG metabolic stability was then assessed. Following the initial 7-day culture procedure, the maintenance/metabolism medium was aspirated from each well, and the plated cells were spiked with 1.0 mL of test compound stock solution. Next, the plate was incubated at 37 °C under 5% CO2 atmosphere until the calculated Tmax was reached (3 h). The test compound solution was then removed from each well and replaced with untreated dilution medium (i.e., serum-free INVITROGRO HI Medium and TORPEDO Antibiotic Mix), initializing the stability determination. The HepaRG plate was once more incubated at 37 °C under 5% CO2 atmosphere over the desired time course (nine data points/compound), and sample preparation and analysis were handled in identical fashion to cellular uptake experiments.
Data Analysis
Concentrations of lipid-based TFV prodrugs were determined using a fit-for-purpose LC-MS/MS method (see Tables S2–S8). Analyses of these data were conducted at Emory University according to the following specifications. Cmax represents the mean observed maximum concentration achieved by prodrug in HepaRG cells. Similarly, Tmax represents the average time at which Cmax was observed. Metabolic half-lives (t1/2) were then calculated using methodology from LM and plasma stability assays described above (Figures S36–S42). The unscaled intrinsic clearance was determined using the equation: Clint,u = (0.693/t1/2) × (1000/0.55) μL/min/million cells.
Mouse Pharmacokinetic Experiments (Conducted by Sai Life Sciences)
Test System
Healthy male C57BL/6 mice (8–12 weeks old) weighing 20–30 g were procured from Global, India. Three mice were housed in each cage. Temperature and humidity were maintained at 22 ± 3 °C and 30–70%, respectively, and illumination was controlled to provide for 12 h light and 12 h dark cycles. Temperature and humidity were recorded by an autocontrolled data logger system. All animals were provided a laboratory rodent diet (Envigo Research private Ltd., Hyderabad). Reverse osmosis H2O treated with ultraviolet light was provided ad libitum.
Study Design
Male mice were divided into two groups (n = 18 total, n = 9 per treatment group, n = 3 per time point, sparse sampling). One group was intravenously administered a solution of 2a, 16g, and 16h in 10% PEG-300, 10% Solutol HS-15, and 80% saline at 3 mg/kg with a dosing volume of 5 mL/kg. The second group was administered a solution of 2a, 16g, and 16h in 10% EtOH and 90% olive oil at 10 mg/kg via oral gavage with a dosing volume of 10 mL/kg.
Sample Collection
Blood samples (approximately 60 μL) were collected from retro-orbital plexus under light isoflurane anesthesia such that the samples were obtained predose, as well as at eight time points post dose: 0.1, 0.25, 0.5, 1, 2, 4, 6, and 8 (i.v.) and 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h (p.o.). Immediately after blood collection, plasma samples were harvested by centrifugation at 4000 rpm and 4 °C for 10 min and stored at −70 °C until LC–MS/MS analysis. Following blood collection from p.o. group, liver, kidney, and brain samples were collected (n = 3 per treatment group per time point) at 1, 4, and 24 h post dose. After isolation, tissue samples were rinsed three times with ice-cold saline and dried with blotting paper. Next, the tissue samples were homogenized using ice-cold PBS (pH = 7.4), and the resulting homogenates were stored below −70 °C until LC-MS/MS analysis. The total homogenate volume was 3 times the brain weight and 5 times the liver and kidney weight.
Data Analysis
Concentrations of 2a, 16g, and 16h were determined using a fit-for-purpose LC–MS/MS method by Sai Life Sciences (see Tables S9–S40). Analyses of these data were conducted at Emory University according to the following specifications. Cmax represents the mean observed maximum concentration achieved by prodrug (2a, 16g, and 16h) and common metabolite TFV in plasma or tissue. Similarly, Tmax represents the average time at which Cmax was observed. Areas under the concentration–time curves (AUC0–8h and AUC0–24h) were calculated via the linear trapezoidal rule65 using GraphPad Prism v9. Furthermore, terminal elimination rate constants k were determined using linear regression (slope = −k) of the terminal portion of semilog-transformed concentration–time curves (Figures S44, S45, S47, S48, S50, and S51). Half-lives (t1/2) were then estimated using the first-order kinetics equation: t1/2 = 0.693/k. After i.v. or p.o. dosing, plasma clearance was calculated using the equations: Cli.v. = Vd × k = Dosei.v./AUC0–8h and Clp.o. = Vd × k = Dose/AUC0–24h. The oral bioavailability for each prodrug was calculated using the equation: %F = [(AUC0–24h × Dose(i.v.))/(AUC0–8h × Dose(p.o.))] × 100.
Ethical Declaration
These studies were performed with approval from the Institutional Animal Ethics Committee (IAEC) in accordance with the requirements of and the guidelines provided by The Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA, India), as published in The Gazette of India, December 15, 1998.
Acknowledgments
The authors would like to thank the NIH Center for AIDS Research (CFAR) for partially funding this work through a developmental grant (P30AI050409). Partial funding for this research also came from the NIH National Institute of Allergy and Infectious Diseases (NIAID, R01-AI128837). Funding for the in vitro toxicity and antiviral activity assays was received from the South African National Health Laboratories Services (NHLS) Research Trust. The authors would additionally like to acknowledge Dr. Shaoxiong Wu and the Emory University NMR Center, Dr. Fred Strobel and the Emory University Mass Spectrometry Center, and the Emory University Comprehensive Glycomics Core Facility for experimental support. Finally, the authors specially thank the Emory University CFAR network and Dr. Colleen S. Kraft for facilitating internal collaboration.
Glossary
Abbreviations
- ANP
acyclic nucleoside phosphonate
- AUC
area under the curve
- cART
combination antiretroviral therapies
- CES1
carboxyesterase I
- Cl
clearance
- CYP450
cytochrome P450
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCC
N,N′-dicyclohexylcarbodiimide
- DCM
dichloromethane
- DIAD
diisopropyl azodicarboxylate
- DMAP
4-(dimethylamino)pyridine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DNA
DNA
- DPBS
Dulbecco’s phosphate buffered saline
- HBV
hepatitis B virus
- HDP
hexadecyloxypropyl
- HIV
human immunodeficiency virus
- HLM
human liver microsome
- HMPA
hexamethylphosphoramide
- HPLC
high performance liquid chromatography
- HSA
human serum albumin
- HSV
herpes simplex virus
- HTP
hexadecylthiopropyl
- ISTD
internal standard
- LC–MS
liquid chromatography–mass spectrometry
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- MLM
mouse liver microsome
- NADPH
nicotinamide adenine dinucleotide phosphate
- NMP
N-methylpyrrolidone
- Ph
phenyl
- POC
isopropyloxycarbonyloxymethyl
- PK
pharmacokinetics
- PLA1
phospholipase A1
- PLA2
phospholipase A2
- PLC
phospholipase C
- PLD
phospholipase D
- PMA
phosphomolybdic acid
- Rf
retention factors
- SAR
structure–activity relationship
- SD
standard deviation
- SEM
standard error of the mean
- TAF
tenofovir alafenamide
- TBAF
tetrabutylammonium fluoride
- TDF
tenofovir disoproxil fumarate
- TFV
tenofovir
- TFV-DP
tenofovir diphosphate
- THF
tetrahydrofuran
- THP
tetrahydropyranyl
- TLC
thin-layer chromatography
- TMS
trimethylsilyl
- TIPS
triisopropylsilyl
- TXL
tenofovir exalidex
- Vd
volume of distribution
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01510.
Protocols for and data from in vitro bioassays (metabolic stability and cellular uptake and stability assays), mouse PK experiments, synthetic procedures for all intermediates and final compounds, 1H and 13C NMRs for thioether-containing lipids and prodrugs, and LC–MS traces of all compounds tested in vivo (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the Development of Nucleoside and Nucleotide Analogues for Cancer and Viral Diseases. Nat. Rev. Drug Discovery 2013, 12, 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Groaz E.; De Jonghe S. Overview of Biologically Active Nucleoside Phosphonates. Front. Chem. 2020, 8, 616863 10.3389/fchem.2020.616863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seley-Radtke K. L.; Yates M. K. The Evolution of Nucleoside Analogue Antivirals: A Review for Chemists and Non-Chemists. Part 1: Early Structural Modifications to the Nucleoside Scaffold. Antiviral Res. 2018, 154, 66–86. 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates M. K.; Seley-Radtke K. L. The Evolution of Antiviral Nucleoside Analogues: A Review for Chemists and Non-Chemists. Part II: Complex Modifications to the Nucleoside Scaffold. Antiviral Res. 2019, 162, 5–21. 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh D.; Vijayakumar B. G.; Kannan T. Advances in Nucleoside and Nucleotide Analogues in Tackling Human Immunodeficiency Virus and Hepatitis Virus Infections. ChemMedChem. 2021, 16, 1403–1419. 10.1002/cmdc.202000849. [DOI] [PubMed] [Google Scholar]
- Kataev V. E.; Garifullin B. F. Antiviral Nucleoside Analogs. Chem. Heterocycl. Compd. (NY) 2021, 57, 326–341. 10.1007/s10593-021-02912-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E.; Holý A. Acyclic Nucleoside Phosphonates: A Key Class of Antiviral Drugs. Nat. Rev. Drug Discovery 2005, 4, 928–940. 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- Holý A. Antiviral Acyclic Nucleoside Phosphonates Structure Activity Studies. Antiviral Res. 2006, 71, 248–253. 10.1016/j.antiviral.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Johansson N. G.; Eriksson S. Structure-Activity Relationships for Phosphorylation of Nucleoside Analogs to Monophosphates by Nucleoside Kinases. Acta Biochim. Polym. 1996, 43, 143–160. 10.18388/abp.1996_4573. [DOI] [PubMed] [Google Scholar]
- Cundy K. C.; Sueoka C.; Lynch G. R.; Griffin L.; Lee W. A.; Shaw J. P. Pharmacokinetics and Bioavailability of the Anti-Human Immunodeficiency Virus Nucleotide Analog 9-[(R)-2-(Phosphonomethoxy)propyl]adenine (PMPA) in Dogs. Antimicrob. Agents Chemother. 1998, 42, 687–690. 10.1128/AAC.42.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney B. P.; Flaherty J. F.; Shah J. Tenofovir Disoproxil Fumarate: Clinical Pharmacology and Pharmacokinetics. Clin. Pharmacokinet. 2004, 43, 595–612. 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- Peterson L. W.; McKenna C. E. Prodrug Approaches to Improving the Oral Absorption of Antiviral Nucleotide Analogues. Expert Opin. Drug Delivery 2009, 6, 405–420. 10.1517/17425240902824808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler K. Y. Alkoxyalkyl Prodrugs of Acyclic Nucleoside Phosphonates Enhance Oral Antiviral Activity and Reduce Toxicity: Current State of the Art. Antiviral Res. 2009, 82, A84–98. 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradere U.; Garnier-Amblard E. C.; Coats S. J.; Amblard F.; Schinazi R. F. Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev. 2014, 114, 9154–9218. 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassner C.; Bradley N.; Lee Y. A Review and Clinical Understanding of Tenofovir: Tenofovir Disoproxil Fumarate versus Tenofovir Alafenamide. J. Int. Assoc. Provid. AIDS Care 2020, 19, 2325958220919231 10.1177/2325958220919231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Clinical Potential of the Acyclic Nucleoside Phosphonates Cidofovir, Adefovir, and Tenofovir in Treatment of DNA Virus and Retrovirus Infections. Clin. Microbiol. Rev. 2003, 16, 569–596. 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H. B.; Stone E. A.; Piacenti F. J. Tenofovir Disoproxil Fumarate: A Nucleotide Reverse Transcriptase Inhibitor for the Treatment of HIV Infection. Clin. Ther. 2002, 24, 1515–1548. 10.1016/S0149-2918(02)80058-3. [DOI] [PubMed] [Google Scholar]
- Gallant J. E.; DeJesus E.; Arribas J. R.; Pozniak A. L.; Gazzard B.; Campo R. E.; Lu B.; McColl D.; Chuck S.; Enejosa J.; Toole J. J.; Cheng A. K. Tenofovir DF, Emtricitabine, and Efavirenz vs. Zidovudine, Lamivudine, and Efavirenz for HIV. N. Engl. J. Med. 2006, 354, 251–260. 10.1056/NEJMoa051871. [DOI] [PubMed] [Google Scholar]
- Delaney W. E. T.; Ray A. S.; Yang H.; Qi X.; Xiong S.; Zhu Y.; Miller M. D. Intracellular Metabolism and In Vitro Activity of Tenofovir Against Hepatitis B Virus. Antimicrob. Agents Chemother. 2006, 50, 2471–2477. 10.1128/AAC.00138-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying C.; De Clercq E.; Nicholson W.; Furman P.; Neyts J. Inhibition of the Replication of the DNA Polymerase M550V Mutation Variant of Human Hepatitis B Virus by Adefovir, Tenofovir, L-FMAU, DAPD, Penciclovir and Lobucavir. J. Viral Hepat. 2000, 7, 161–165. 10.1046/j.1365-2893.2000.00210.x. [DOI] [PubMed] [Google Scholar]
- Andrei G.; Lisco A.; Vanpouille C.; Introini A.; Balestra E.; van den Oord J.; Cihlar T.; Perno C. F.; Snoeck R.; Margolis L.; Balzarini J. Topical Tenofovir, a Microbicide Effective against HIV, Inhibits Herpes Simplex Virus-2 Replication. Cell Host Microbe 2011, 10, 379–389. 10.1016/j.chom.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H. H.; Stock H.; Rausch L.; Bunin D.; Wang A.; Brill S.; Gow J.; Mirsalis J. C. Tenofovir Disoproxil Fumarate: Toxicity, Toxicokinetics, and Toxicogenomics Analysis After 13 Weeks of Oral Administration in Mice. Int. J. Toxicol. 2015, 34, 4–10. 10.1177/1091581814565669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A. S.; Fordyce M. W.; Hitchcock M. J. Tenofovir Alafenamide: A Novel Prodrug of Tenofovir for the Treatment of Human Immunodeficiency Virus. Antiviral Res. 2016, 125, 63–70. 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Ray A. S.; Cihlar T.; Robinson K. L.; Tong L.; Vela J. E.; Fuller M. D.; Wieman L. M.; Eisenberg E. J.; Rhodes G. R. Mechanism of Active Renal Tubular Efflux of Tenofovir. Antimicrob. Agents Chemother. 2006, 50, 3297–3304. 10.1128/AAC.00251-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Fernandez B.; Montoya-Ferrer A.; Sanz A. B.; Sanchez-Niño M. D.; Izquierdo M. C.; Poveda J.; Sainz-Prestel V.; Ortiz-Martin N.; Parra-Rodriguez A.; Selgas R.; Ruiz-Ortega M.; Egido J.; Ortiz A. Tenofovir Nephrotoxicity: 2011 Update. AIDS Res. Treat. 2011, 2011, 354908 10.1155/2011/354908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P. M.; Cotter A. G. Tenofovir and Bone Health. Curr. Opin. HIV AIDS 2016, 11, 326–332. 10.1097/COH.0000000000000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehellou Y.; Rattan H. S.; Balzarini J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018, 61, 2211–2226. 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkus G.; Wang R.; Liu X.; Kutty N.; MacArthur H.; Cihlar T.; Gibbs C.; Swaminathan S.; Lee W.; McDermott M. Cathepsin A is the Major Hydrolase Catalyzing the Intracellular Hydrolysis of the Antiretroviral Nucleotide Phosphonoamidate Prodrugs GS-7340 and GS-9131. Antimicrob. Agents Chemother. 2007, 51, 543–550. 10.1128/AAC.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami E.; Tolstykh T.; Bao H.; Niu C.; Steuer H. M.; Bao D.; Chang W.; Espiritu C.; Bansal S.; Lam A. M.; Otto M. J.; Sofia M. J.; Furman P. A. Mechanism of Activation of PSI-7851 and its Diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Lu X.; Yang X.; Xu N. The Efficacy and Safety of Tenofovir Alafenamide Versus Tenofovir Disoproxil Fumarate in Antiretroviral Regimens for HIV-1 Therapy: Meta-Analysis. Medicine (Baltimore) 2016, 95, e5146 10.1097/MD.0000000000005146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babusis D.; Phan T. K.; Lee W. A.; Watkins W. J.; Ray A. S. Mechanism for Effective Lymphoid Cell and Tissue Loading Following Oral Administration of Nucleotide Prodrug GS-7340. Mol. Pharmaceutics 2013, 10, 459–466. 10.1021/mp3002045. [DOI] [PubMed] [Google Scholar]
- Murakami E.; Wang T.; Park Y.; Hao J.; Lepist E. I.; Babusis D.; Ray A. S. Implications of Efficient Hepatic Delivery by Tenofovir Alafenamide (GS-7340) for Hepatitis B Virus Therapy. Antimicrob. Agents Chemother. 2015, 59, 3563–3569. 10.1128/AAC.00128-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Kean L. M.; Egelund E. F.; Jourjy J. Tenofovir Alafenamide for the Treatment of Chronic Hepatitis B Monoinfection. Pharmacotherapy 2018, 38, 1051–1057. 10.1002/phar.2174. [DOI] [PubMed] [Google Scholar]
- Beadle J. R.; Hartline C.; Aldern K. A.; Rodriguez N.; Harden E.; Kern E. R.; Hostetler K. Y. Alkoxyalkyl Esters of Cidofovir and Cyclic Cidofovir Exhibit Multiple-Log Enhancement of Antiviral Activity Against Cytomegalovirus and Herpesvirus Replication In Vitro. Antimicrob. Agents Chemother. 2002, 46, 2381–2386. 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter G. R.; Almond M. R.; Trost L. C.; Lampert B. M.; Neyts J.; Clercq E. D.; Korba B. E.; Aldern K. A.; Beadle J. R.; Hostetler K. Y. Evaluation of Hexadecyloxypropyl-9-R-[2-(Phosphonomethoxy)Propyl]-Adenine, CMX157, as a Potential Treatment for Human Immunodeficiency Virus Type 1 and Hepatitis B Virus Infections. Antimicrob. Agents Chemother. 2007, 51, 4538–4538. 10.1128/AAC.01375-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier R.; Trost L.; Tippin T.; Lampert B.; Robertson A.; Foster S.; Rose M.; Painter W.; O’Mahony R.; Almond M.; Painter G. Development of CMX001 for the Treatment of Poxvirus Infections. Viruses 2010, 2, 2740–2762. 10.3390/v2122740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanwandee T.; Chatsiricharoenkul S.; Thongsawat S.; Sukeepaisarnjaroen W.; Tangkijvanich P.; Komolmit P.; Avihingsanon A.; Piratvisuth T.; Sunthi P.; Mahasanprasert T.; Matkovits T.; Conover M.; Cobb J.; Canizares C.; Sullivan-Bolyai J. Pharmacokinetics, Safety and Antiviral Activity of CMX157, a Novel Prodrug of Tenofovir, Administered as Ascending Multiple Doses to Healthy Volunteers and Hepatitis B Virus-Infected Subjects. J. Hepatol. 2017, 66, S24–S25. 10.1016/S0168-8278(17)30311-2. [DOI] [Google Scholar]
- Johnston J. B.; Ouellet H.; Podust L. M.; Ortiz de Montellano P. R. Structural Control of Cytochrome P450-Catalyzed ω-Hydroxylation. Arch. Biochem. Biophys. 2011, 507, 86–94. 10.1016/j.abb.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribut N.; D’Erasmo M.; Dasari M.; Giesler K. E.; Iskandar S.; Sharma S. K.; Bartsch P. W.; Raghuram A.; Bushnev A.; Hwang S. S.; Burton S. L.; Derdeyn C. A.; Basson A. E.; Liotta D. C.; Miller E. J. ω-Functionalized Lipid Prodrugs of HIV NtRTI Tenofovir with Enhanced Pharmacokinetic Properties. J. Med. Chem. 2021, 64, 12917–12937. 10.1021/acs.jmedchem.1c01083. [DOI] [PubMed] [Google Scholar]
- Toti K. S.; Pribut N.; D’Erasmo M.; Dasari M.; Sharma S. K.; Bartsch P. W.; Burton S. L.; Gold H. B.; Bushnev A.; Derdeyn C. A.; Basson A. E.; Liotta D. C.; Miller E. J. Expanding the Toolbox of Metabolically Stable Lipid Prodrug Strategies. Front. Pharmacol. 2023, 13, 1083284 10.3389/fphar.2022.1083284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M.; Tang B.; Liang S. H.; Jiang X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top Med. Chem. 2016, 16, 1200–1216. 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa M.; Winum J. Y. The Importance of Sulfur-Containing Motifs in Drug Design and Discovery. Expert Opin. Drug Discovery 2022, 17, 501–512. 10.1080/17460441.2022.2044783. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R.; Reich N. O. Specific Inactivation of Hepatic Fatty Acid Hydroxylases by Acetylenic Fatty Acids. J. Biol. Chem. 1984, 259, 4136–4141. 10.1016/S0021-9258(17)43020-1. [DOI] [PubMed] [Google Scholar]
- Bambal R. B.; Hanzlik R. P. Effects of Steric Bulk and Conformational Rigidity on Fatty Acid Omega Hydroxylation by a Cytochrome P450 4A1 Fusion Protein. Arch. Biochem. Biophys. 1996, 334, 59–66. 10.1006/abbi.1996.0429. [DOI] [PubMed] [Google Scholar]
- Hoch U.; Zhang Z.; Kroetz D. L.; Ortiz de Montellano P. R. Structural Determination of the Substrate Specificities and Regioselectivities of the Rat and Human Fatty Acid Omega-Hydroxylases. Arch. Biochem. Biophys. 2000, 373, 63–71. 10.1006/abbi.1999.1504. [DOI] [PubMed] [Google Scholar]
- Hoch U.; Falck J. R.; de Montellano P. R. Molecular Basis for the Omega-Regiospecificity of the CYP4A2 and CYP4A3 Fatty Acid Hydroxylases. J. Biol. Chem. 2000, 275 (35), 26952–26958. 10.1016/S0021-9258(19)61465-1. [DOI] [PubMed] [Google Scholar]
- He X.; Cryle M. J.; De Voss J. J.; Ortiz de Montellano P. R. Calibration of the Channel That Determines the ω-Hydroxylation Regiospecificity of Cytochrome P4504A1: Catalytic Oxidation of 12 Halododecanoic Acids. J. Biol. Chem. 2005, 280, 22697–22705. 10.1074/jbc.M502632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang H.; Cox N.; Lalic G. Copper-Catalyzed Reduction of Alkyl Triflates and Iodides: An Efficient Method for the Deoxygenation of Primary and Secondary Alcohols. Angew. Chem., Int. Ed. 2014, 53, 752–756. 10.1002/anie.201307697. [DOI] [PubMed] [Google Scholar]
- Johnson A. L.; Edson K. Z.; Totah R. A.; Rettie A. E. Cytochrome P450 ω-Hydroxylases in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 223–262. 10.1016/bs.apha.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edson K. Z.; Rettie A. E. CYP-4 Enzymes as Potential Drug Targets: Focus on Enzyme Multiplicity, Inducers and Inhibitors, and Therapeutic Modulation of 20-Hydroxyeicosatetraenoic Acid (20-HETE) Synthase and Fatty Acid ω-Hydroxylase Activities. Curr. Top Med. Chem. 2013, 13, 1429–1440. 10.2174/15680266113139990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks E. A.; Zhang Z.; Johnson E. F.; de Montellano P. R. The Catalytic Site of Cytochrome P4504A11 (CYP4A11) and Tts L131F Mutant. J. Biol. Chem. 1998, 273, 23055–23061. 10.1074/jbc.273.36.23055. [DOI] [PubMed] [Google Scholar]
- Hsu M. H.; Baer B. R.; Rettie A. E.; Johnson E. F. The Crystal Structure of Cytochrome P450 4B1 (CYP4B1) Monooxygenase Complexed with Octane Discloses Several Structural Adaptations for ω-Hydroxylation. J. Biol. Chem. 2017, 292, 5610–5621. 10.1074/jbc.M117.775494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eksterowicz J.; Rock D. A.; Rock B. M.; Wienkers L. C.; Foti R. S. Characterization of the Active Site Properties of CYP4F12. Drug Metab. Dispos. 2014, 42, 1698–1707. 10.1124/dmd.114.059626. [DOI] [PubMed] [Google Scholar]
- Bahar F. G.; Ohura K.; Ogihara T.; Imai T. Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J. Pharm. Sci. 2012, 101, 3979–3988. 10.1002/jps.23258. [DOI] [PubMed] [Google Scholar]
- Xu G. G.; Geng Z.; Zhou X. C.; He Y. G.; He T. T.; Mei J. X.; Yang Y. J.; Liu Y. Q.; Xu C. S. Three Branches of Phospholipase C Signaling Pathway Promote Hepatocyte Growth in Rat Liver Regeneration. Genet. Mol. Res. 2015, 14, 5710–5723. 10.4238/2015.May.29.3. [DOI] [PubMed] [Google Scholar]
- Klein B. M.; Andrews J. B.; Bannan B. A.; Nazario-Toole A. E.; Jenkins T. C.; Christensen K. D.; Oprisan S. A.; Meyer-Bernstein E. L. Phospholipase C beta 4 in Mouse Hepatocytes: Rhythmic Expression and Cellular Distribution. Comp. Hepatol. 2008, 7, 8. 10.1186/1476-5926-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh P. G.; Park J. I.; Manzoli L.; Cocco L.; Peak J. C.; Katan M.; Fukami K.; Kataoka T.; Yun S.; Ryu S. H. Multiple Roles of Phosphoinositide-Specific Phospholipase C Isozymes. BMB Rep. 2008, 41, 415–434. 10.5483/BMBRep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- Lübberstedt M.; Müller-Vieira U.; Mayer M.; Biemel K. M.; Knöspel F.; Knobeloch D.; Nüssler A. K.; Gerlach J. C.; Zeilinger K. HepaRG Human Hepatic Cell Line Utility as a Surrogate for Primary Human Hepatocytes in Drug Metabolism Assessment In Vitro. J. Pharmacol. Toxicol. Methods 2011, 63, 59–68. 10.1016/j.vascn.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Guillouzo A.; Corlu A.; Aninat C.; Glaise D.; Morel F.; Guguen-Guillouzo C. The Human Hepatoma HepaRG Cells: A Highly Differentiated Model for Studies of Liver Metabolism and Toxicity of Xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. 10.1016/j.cbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Grünig D.; Szabo L.; Marbet M.; Krähenbühl S. Valproic Acid Affects Fatty Acid and Triglyceride Metabolism in HepaRG Cells Exposed to Fatty Acids by Different Mechanisms. Biochem. Pharmacol. 2020, 177, 113860 10.1016/j.bcp.2020.113860. [DOI] [PubMed] [Google Scholar]
- Riss T. L.; Moravec R. A.; Niles A. L.; Duellman S.; Benink H. A.; Worzella T. J.; Minor L.. Cell Viability Assays. In The Assay Guidance Manual; NIH, 2013; pp 355–385. [Google Scholar]
- Gupta R. K.; Kohli A.; McCormick A. L.; Towers G. J.; Pillay D.; Parry C. M. Full-Length HIV-1 Gag Determines Protease Inhibitor Susceptibility within in-Vitro Assays. AIDS 2010, 24, 1651–1655. 10.1097/QAD.0b013e3283398216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry C. M.; Kohli A.; Boinett C. J.; Towers G. J.; McCormick A. L.; Pillay D. Gag Determinants of Fitness and Drug Susceptibility in Protease Inhibitor-Resistant Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 9094–9101. 10.1128/JVI.02356-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndung’u T.; Renjifo B.; Essex M. Construction and Analysis of an Infectious Human Immunodeficiency Virus Type 1 Subtype C Molecular Clone. J. Virol. 2001, 75, 4964–4972. 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urso R.; Blardi P.; Giorgi G. A Short Introduction to Pharmacokinetics. Eur. Rev. Med. Pharmacol. Sci. 2002, 6, 33–44. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.