

SUMMARY
In 1992, the Food and Drug Administration (FDA) instituted the accelerated approval regulations that allow drugs or biologics for serious conditions that fill an unmet medical need to be approved on the basis of a surrogate endpoint or an intermediate clinical endpoint. The current definition of a serious condition includes chronic disabling conditions, such as osteoarthritis (OA), and thereby provides expanded opportunities for the use of biomarkers for regulatory approval of drugs for OA. The use of surrogates or intermediate clinical endpoints for initial regulatory approval of a drug or biologic requires confirmation in a post-marketing study of a drug effect on a clinically relevant outcome, such as on how a patient feels, functions or survives. Current FDA guidance requires that the post-marketing approval (PMA) study be ongoing during the time of initial drug approval. This white paper arose out of the need to brainstorm trial designs that might be suitable for PMA of drugs initially approved, on the basis of a surrogate or intermediate clinical endpoint, for treatment of OA to alter disease progression, abnormal function or pathological changes in the morphology of the joint. In this white paper we define the concept and regulations regarding accelerated approval and propose two major study design scenarios for PMA trials in OA. The long-term goal is to discuss and refine these designs in consultation with regulatory agencies in order to facilitate development of drugs to fill the large unmet need in OA.
Keywords: Biomarker, Drug development, Trial design, Serious condition, Randomized clinical trial
Introduction
Drugs are traditionally approved in the United States (US) based upon data from adequate and well-controlled trials demonstrating the clinical benefit related to patient symptoms, function or survival and potential harms of the therapy. In 1992, the Food and Drug Administration (FDA) instituted the accelerated approval regulations that allowed drugs or biologics for serious conditions that fill an unmet medical need to be approved on the basis of a surrogate endpoint or an “intermediate clinical endpoint”1,2. A surrogate endpoint used for accelerated approval is a marker – a laboratory measurement, radiographic image, physical sign or other measure – that is thought to predict clinical benefit but is not itself a measure of clinical benefit3. An intermediate clinical endpoint is a measure of a therapeutic effect other than irreversible morbidity or mortality (for all definitions including a summary of biomarker nomenclature, see Supplementary Text and Supplementary Table 1). In 2012, Congress codified these FDA regulations in the Food and Drug Administration Safety Innovations Act (FDASIA); Section 901 of FDASIA amends the Federal Food, Drug, and Cosmetic Act to recognize that the FDA can base accelerated approval for drugs or biologics for serious conditions that fill an unmet medical need on whether the drug has an effect on a surrogate endpoint or an intermediate clinical endpoint4. In these cases, the surrogate or intermediate endpoints used are those believed to reasonably likely to predict patient-reported outcomes (PROs) of interest or overall survival.
An increasing use of biomarkers in drug development has now been encouraged by the 21st Century Cures Act5. The FDA has recently explained that in addition to morbidity and mortality risk, a serious condition includes progressive disability as defined in a 2014 guidance document:
a disease or condition associated with morbidity that has a substantial impact on day-to-day functioning. Short-lived and self-limiting morbidity will usually not be sufficient, but the morbidity need not be irreversible if it is persistent or recurrent. Whether a disease or condition is serious is a matter of clinical judgment, based on its impact on such factors as survival, day-to-day functioning, or the likelihood that the disease, if left untreated, will progress from a less severe condition to a more serious one3.
This expanded definition provides expanded opportunities for the use of biomarkers for regulatory approval of drugs for chronic disabling conditions such as osteoarthritis (OA)4. The 21st Century Cures Act also provides for a process of accelerated approval for regenerative medicine therapies such as cell therapy, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products5. Furthermore, the same Act also provides a possible framework for utilizing real-world evidence to provide support for the clinical relevance of an approved therapy based on a surrogate measure.
The accelerated approval pathway differs from the traditional OA trial paradigm for demonstrating a delay in structural progression as embodied in a former 1999 FDA draft guidance on OA6. The former guidance acknowledged that it is possible that certain classes of products may slow joint space narrowing without concomitantly affecting symptoms. Curiously, this now defunct FDA draft guidance stated that a demonstration of a purely structural endpoint, namely improvement of the radiograph compared to baseline that reflects new or regrown cartilage, “would be convincing and require no formal parallel evidence of improvement in clinical outcomes”6. It is generally believed that this emphasis on radiographs has hampered development of disease modifying OA drugs (DMOADs) due to inherent limitations of radiographs including: their lack of sensitivity to joint tissue changes; in contrast to MRI, their inability to report on the state of the whole joint organ (they reflect bone changes only and secondarily and inaccurately articular cartilage as “loss of joint space”); and their slowness to change7. Of note, the prior FDA draft guidance allowed for the possibility of claims related to delay in time to joint surgery6; this outcome, described below, has potential merit for post-marketing studies.
OA as a serious disease
Many patients with OA clearly suffer from a serious disease; the progressive disability observed in some of these patients is associated with reduced mobility and increased risk for death (as discussed further in an OARSI white paper presented to the FDA December 1, 20168). Gratifyingly, the FDA acknowledged, in their latest guidance document9, that “OA can be a serious disease with an unmet medical need for therapies that modify the underlying pathophysiology of the disease and potentially change its natural course to prevent long-term disability.” This formal recognition of OA as a serious disease supports the potential use of surrogate endpoints for regulatory approval of a drug or biologic under FDA’s accelerated approval regulations1,2. However, the use of a biomarker or surrogate endpoint for regulatory approval of drugs for OA poses two challenges: 1) selection of appropriate surrogate endpoints, and 2) appropriate designs for post-marketing confirmatory studies. The first challenge, establishment of appropriate imaging and/or biochemical biomarkers as intermediate or surrogate endpoints in OA trials, is ongoing in the Foundation for NIH OA Biomarkers Consortium initiative, now in phase 2 (for a discussion of criteria for surrogacy see Supplementary Text and Supplementary Tables 2 and 3). The aim of this document is to address the second challenge of developing confirmatory trial designs in consultation with regulatory agencies.
Prior precedents of approvals under Subpart H regulations
Accelerated approval is relatively common in some therapeutic areas such as cancer and HIV. For example, between December 11, 1992 and May 31, 2017, under the accelerated approval authority, the FDA approved 64 products (53 new molecular entities) for 93 new indications related to hematologic and non-hematologic malignancies10. The FDA approved most of these drugs on the basis of response rates, such as evidence that the drug shrinks tumors, because tumor shrinkage is considered reasonably likely to predict a real clinical benefit, such as survival. In addition to response rate, other intermediate endpoints used to support accelerated approval of oncologic drugs include time-to-event endpoints such as progression-free survival or time-to-progression, disease-free survival or recurrence-free survival.
Many antiretroviral drugs were approved to treat HIV/AIDS based initially on the surrogate endpoint of an increase in CD4 cells, and later, a decrease in HIV-RNA (viral load). With more experience (including subsequent drug approvals), the FDA concluded that treatment-induced decreases in HIV-RNA levels were highly predictive of clinical benefit, and determined that measurement of HIV-RNA could serve as a clinical endpoint in trials designed to support either accelerated or traditional approvals. The FDA’s position has further evolved and under current guidance, traditional approval can be the initial approval for all antiretroviral drugs, with the duration of viral load reductions dependent on the population studied11.
To date, there have been a moderate number of accelerated drug approvals for serious diseases besides cancer and HIV12 (see Table I); these provide insights into possible study designs and endpoints for use in OA trials. For instance, drug development in other disease indications with fewer patients, such as non-alcoholic steatohepatitis (NASH), already involves both larger pivotal studies as currently undertaken for OA and implementation of the Subpart H approval process. Lessons learned from the surrogate endpoints in NASH, and how they later translate into modifications of PROs may benefit the OA field (for a discussion of development hurdles in OA compared to other diseases and for a summary of representative studies related to NASH, osteoporosis, type II diabetes and OA, see Supplementary Table 4).
Table I.
Accelerated approvals based on intermediate clinical endpoints (top) or biomarker surrogate endpoints (bottom)
| Drug | Indication | Date of approval | Accelerated approval/confirmatory study |
|---|---|---|---|
|
| |||
| Accelerated approval based on an intermediate clinical endpoint. | |||
| Betaseron | For use in ambulatory patients with relapsing-remitting multiple sclerosis. | 7/23/1993 | Accelerated approval based on the rate and extent of exacerbations of multiple sclerosis (intermediate clinical endpoint, although the size of the treatment effect was small); and improvements in MRI-measured lesion area (surrogate). Confirmatory study: 4—6 year study using disability as measured by the Kurtzke Expanded Disability Status Scale (EDSS); plus correlation of MR imaging with clinical endpoints. |
| Remicade | Treatment of moderately to severely active Crohn’s disease. | 8/24/1998 | Accelerated approval based on “clinical response,” defined as a reduction from baseline in the Crohn’s Disease Activity Index (CDAI) of at least 70 at 4 weeks. CDAI is a research tool used to quantify the status of patients with Crohn’s disease that includes a combination of clinical features (number of stools, abdominal pain, well-being, abdominal mass and other clinical features) in addition to quantitative measures such as amount of anti-diarrheal drug use, hematocrit and body weight. Confirmatory study: Maintaining a sustained clinical outcome (“clinical response” at week 30 and “clinical remission” at week 54) in patients with moderate to severely active Crohn’s disease. |
| Remodulin | Treatment for pulmonary arterial hypertension. | 5/21/2002 | Accelerated approval based on a combined exercise (6-minute walk test/Borg score) analysis. Confirmatory study: Time to first occurrence of death, hospitalization for complications of hypertension or other clear evidence of deterioration. |
| Tysabri | For the treatment of patients with relapsing forms of multiple sclerosis. | 11/23/2004 | Accelerated approval based on a large therapeutic effect on relapse rate through approximately 13 months of treatment. Confirmatory study: Continue the existing trials into the post-marketing period to confirm the durability of the observed effect at 2 years. |
| Makena | To reduce the risk of preterm birth. | 2/3/2011 | Accelerated approval based on a demonstration of delay in delivery. Confirmatory studies: Post-marketing studies to demonstrate improved long-term postnatal outcomes. |
| Accelerated approval based on a biomarker as a surrogate. | |||
| Priftin | Treatment of pulmonary tuberculosis. | 6/22/1998 | Accelerated approval based on sputum culture status at 6 months. Confirmatory study: Negative sputum culture up to 2 years post-treatment. |
| Synercid | Treatment of patients with infections associated with vancomycin-resistant Enterococcus faecium (VREF) bacteremia. | 9/21/1999 | Accelerated approval based on a laboratory measurement of bacteria in the blood. Confirmatory study: Clinical resolution of infection. |
| Celebrex | To reduce the number ofadenomatous colorectal polyps in familial adenomatous polyposis (FAP), as an adjunct to usual care. | 12/23/1999 | Accelerated approval based on the % change in the number of colorectal adenomas. Confirmatory study: Reduction in the incidence of FAP-related events (e.g., polypectomy, surgery, cancer, desmoids, death). The sponsor did not demonstrate the link between polyp number and onset of colonic cancer after the allotted time allowed to produce these data; thus, this indication and dose were removed from the label. |
| Sirturo | Combination therapy in adults with pulmonary multi-drug resistant tuberculosis (MDR-TB). | 12/28/2012 | Accelerated approval based on sputum culture status at 6 months. Confirmatory study: Resolution of pulmonary tuberculosis. |
| Ferriprox | Treatment of patients with transfusional iron overload due to thalassemia syndromes. | 9/9/2015 | Accelerated approval based on a decrease in iron stores for patients with iron overload caused by thalassemia. Confirmatory study: Decrease in transfusion-related adverse events caused by iron overload in the body. |
Proposed study designs for OA
Presently, it is expected that prospects for regulatory approval of a DMOAD will require large numbers of patients and potentially long periods of observation to discern whether improvement in signs and symptoms follows structural benefit, particularly if applying therapies to unselected patient populations rather than to trial candidates with specific OA phenotypes and/or high risk of progression13. It is difficult to power trials for both symptom improvement as well as potential structural change at the same time. Currently, PROs used in OA trials, although not so costly, are potentially subject to large placebo effects. The OMERACT-OARSI responder criteria, based on PROs from non-steroidal trials of at least 6 weeks duration, require sample sizes of ~100 patients per study arm14. In contrast, structural outcomes require long periods of observation. Adequate powering of a trial for structural outcomes is anticipated to require fewer patients and shorter observation periods using MRI compared with radiography due to the greater sensitivity of MRI imaging outcomes15. It is hoped that the use of imaging and/or biochemical markers during DMOAD trials could provide early indications of a potential treatment related effect on structure. Initial approval on the basis of a surrogate could allow for marketing of a product and the acquisition of revenue to facilitate funding of the necessary post-marketing confirmation trials with PRO endpoints and/or joint survival assessments to verify and describe its clinical benefit, required under FDA’s accelerated approval regulations, when there is uncertainty as to the relation of the surrogate endpoint to clinical benefit, or of the observed clinical benefit to ultimate outcome1,2.
The following criteria must be met by post-marketing confirmatory studies to prove clinical relevance:
Post-marketing studies must be adequate and well-controlled;
Although the FDA does not mandate that the post-marketing approval (PMA) study is necessarily conducted in the original trial population, it may be more efficient and cost-effective to conduct the trial in the same population used to assess the effect on surrogate or intermediate clinical endpoints because new patient identification and recruitment would be unnecessary and it would also be possible to evaluate the durability of the treatment response.
- If a true controlled study is required post-marketing, it could be a challenge to maintain patients on placebo for long periods of time once the drug is conditionally approved and clinically available. To overcome this challenge,
- It would be possible to use rescue therapy for OA symptoms.
- As an alternative to a placebo controlled randomized clinical trial (RCT), the study could be designed to compare high vs low doses of the active drug without a placebo arm.
- As an alternative to a placebo controlled RCT, the study could be designed to compare high vs low doses of the drug to an approved active comparator.
Both adverse and beneficial outcomes can and should be monitored post-marketing.
Study design proposals – one size does not fit all
As described below, there are several different drugs under development with different mechanisms of action. Ultimately, post-marketing studies are based on an interaction and negotiations with FDA/EMA that will not be the same for all mechanisms of action, as one size clearly does not fit all. Current guidance requires that the PMA study must be ongoing during the time of initial approval. For the purposes of DMOAD indications, we propose two major study design scenarios (Figs. 1 and 2) and describe variations on these designs and the drug profile categories (Table II) to which they might apply. These trials involve an initial Phase 3 trial period of up to 2 years with collection of surrogate and PRO outcomes with approval based on the surrogate. In both cases, the subsequent phase of the trial follows the same or different patients over an additional period of time (to be determined based on the anticipated time to a treatment effect on a clinical endpoint) with collection of PRO outcomes or some measure of joint survival.
Fig. 1. Scenario 1 – prospective trial continuation.
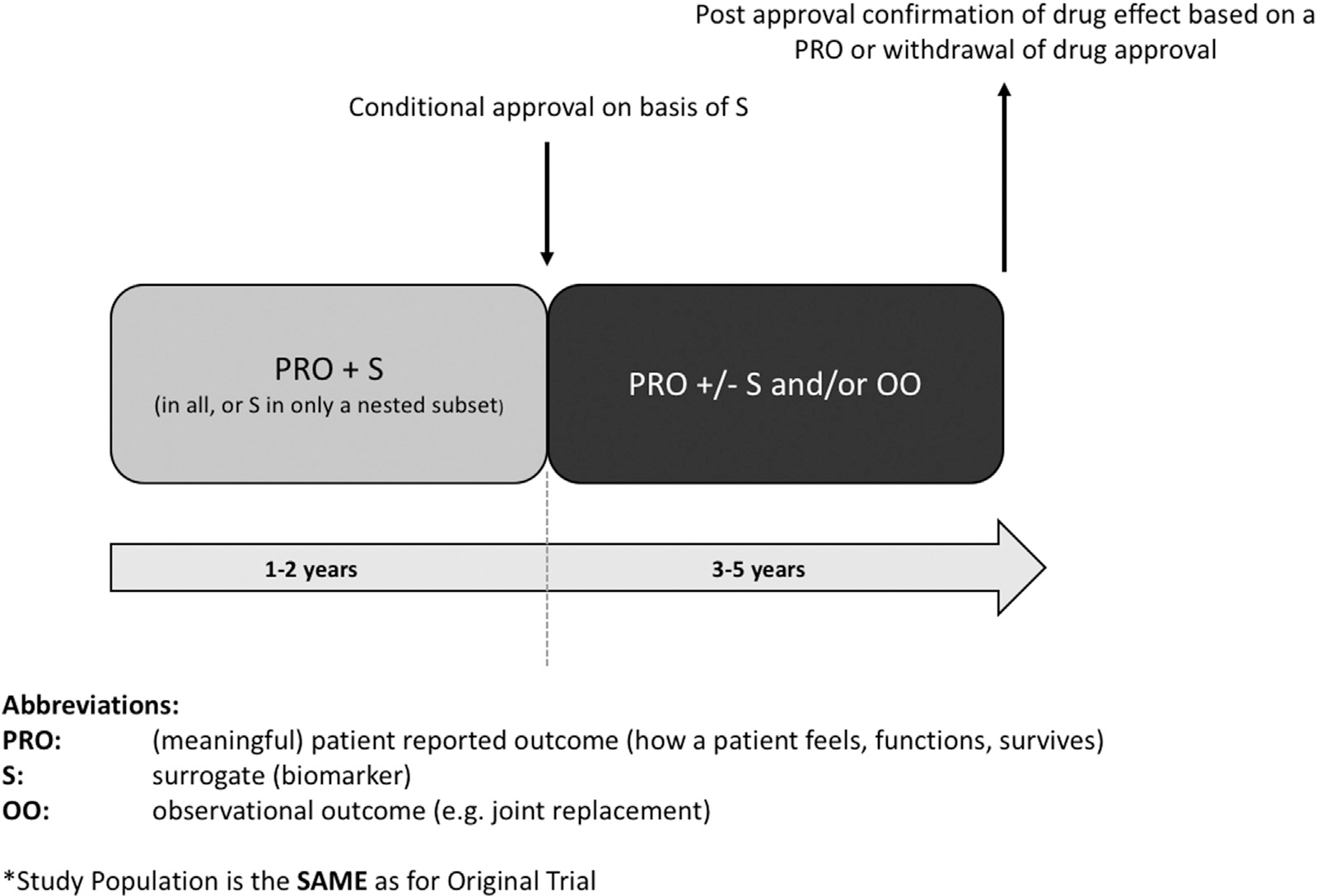
PMA study design scenario 1 represents the continuation, post-approval, of the Phase 3 double blind, placebo controlled trial. The PMA study population contains the same patients as the original trial. Clinically relevant endpoints might be the time-to-event of joint replacement surgeries or clinically relevant symptomatic worsening or whichever is first.
Fig. 2. Scenario 2 – separate PMA study.
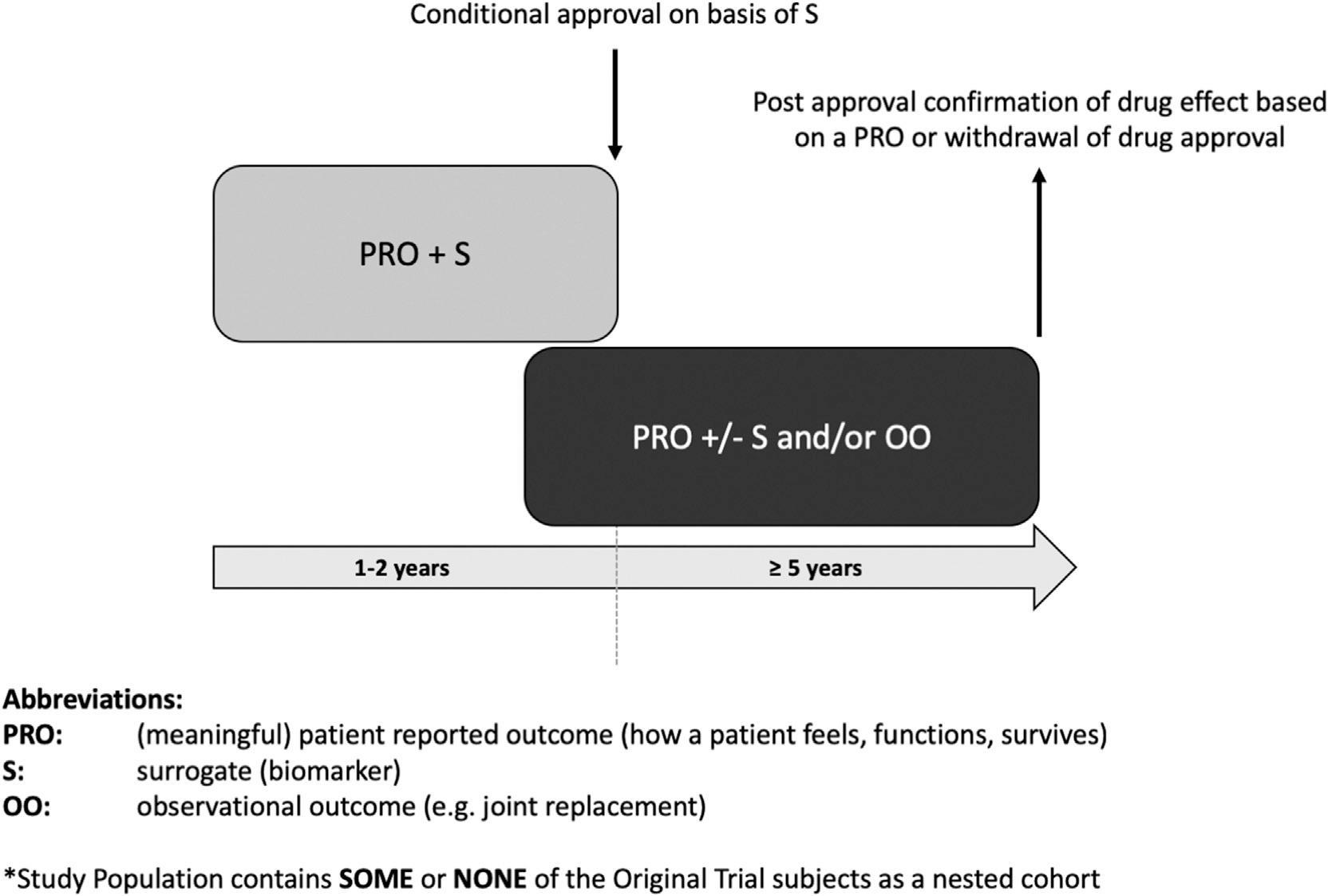
Study design scenario 2 represents a PMA study that might be conducted as a separate study from the phase 3 trial. The PMA study population contains some or none of the original phase 3 trial subjects as a nested cohort. All patients may be on active (high vs low dose) treatment in the PMA study and followed for rates of OA progression. As for scenario 1, clinically relevant endpoints might be the time-to-event of joint replacement surgeries or clinically relevant symptomatic worsening or whichever is first.
Table II.
OA general drug profile categories
| Drug profile | Description of profile | Expectations | Type of approval | Challenge |
|---|---|---|---|---|
|
| ||||
| The Pure-Anticatabolic-Profile | • A drug candidate that demonstrates statistical difference on structure (less worsening compared to placebo) but fails to demonstrate symptomatic and/or functional benefit in a phase 3 trial. | • It might be expected that the structural difference to placebo will result in clinical benefit in longer trials e.g., by less worsening on symptoms and/or function or by delaying joint replacements. The profile is similar to a protease blocker without immediate direct effects on symptoms and/or function. | • Accelerated approval on the basis of an OA progression surrogate endpoint • Post-marketing trial to confirm benefit on signs/symptoms |
• Risk of post-marketing withdrawal of regulatory approval for drug if it fails to show benefit for signs/ symptoms |
| The Pure-Anabolic-Profile | • A drug candidate that demonstrates statistical difference on structure by increasing cartilage but fails to demonstrate symptomatic and/or functional benefit in a phase 3 trial. | • It might be expected that the structural difference to placebo will result in clinical benefit in longer trials e.g., by less worsening on symptoms and/or function or by delaying joint replacements. The profile is similar to a growth factor without direct effects on symptoms and/or function. | • Based on former draft FDA guidance, demonstration of new or regrowth of cartilage would be convincing and require no formal parallel evidence of improvement in clinical outcomes • Alternatively could pursue accelerated approval on the basis of a surrogate endpoint • Post-marketing trial to confirm benefit on signs/symptoms |
• Need to show, for instance by specialized imaging, that growth of cartilage is functional matrix rather than cartilage swelling • Risk of post-marketing withdrawal of regulatory approval for drug if it fails to show benefit for signs/symptoms |
| Pain-Lowering-Anticatabolic-Profile | • A drug candidate that demonstrates durable symptomatic and/or functional benefit in a phase 3 trial, but does not achieve statistical difference or the MCID on a radiographic structural endpoint. | • The structural endpoint might have failed because of a short trial duration (1 or 2 years only). The profile is similar to a NSAID after phase 3. | • Traditional approval for signs/symptoms indication • A structure indication may be achieved concurrent with signs/symptoms indication on the basis of a surrogate, such as MRI feature, especially if linked to legacy or other data demonstrating its clinical meaningfulness and/or relation to reduced joint replacement • Alternatively, post-marketing study to determine DMOAD effect. |
• Cost of drug based on signs/symptom benefit; • If DMOAD effect shown subsequent to clinical availability of drug, difficulty later changing cost to get return on additional investment required to show DMOAD effect |
| Pain-Lowering-Anabolic-Profile | • A drug candidate that demonstrates durable symptomatic and/or functional benefit in a phase 3 trial but does not achieve statistical difference on a structural endpoint despite anabolic properties. | • The structural endpoint might have failed because of short trial duration of 1 or 2 years only. The profile is similar to a growth factor with some direct effects on symptoms and/or function. | • Traditional approval for signs/symptoms indication. • A structure indication may be achieved concurrent with signs/symptoms indication on the basis of a surrogate, such as MRI feature, especially if linked to legacy or other data demonstrating its clinical meaningfulness and/or relation to reduced joint replacement • Alternatively, post-marketing study to determine DMOAD effect with possible addition of DMOAD indication. |
• Cost of drug based on signs/symptom benefit; • If DMOAD effect shown subsequent to clinical availability of drug, difficulty later changing cost to get return on additional investment required to show DMOAD effect |
MCID = minimal clinical important difference; DMOAD = disease modifying OA drug; NSAID = non-steroidal anti-inflammatory drug.
For both scenarios, it is important to note that the consideration to pursue either one of these strategies could be predicated upon the failure, or likelihood of failure, to attain a treatment effect on a clinically relevant and validated PRO. When the PRO is not achievable in the short-term, an accelerated (conditional) approval is sought on the basis of a surrogate endpoint likely to predict clinical benefit in a longer study.
Alternatively, attainment of a treatment effect on a PRO could result in traditional regulatory approval for signs and symptoms indications with subsequent pursuit of a DMOAD approval with a PMA study to demonstrate disease modification. This poses clear challenges and potentially acts as a disincentive to pursuing long term studies for a DMOAD indication (see Table II) because the cost setting for the drug will be dictated by the signs and symptoms indication (and not a DMOAD indication) that may not ultimately provide enough return on investment to cover the added costs of the research necessary to achieve a DMOAD indication. It would also be difficult to imagine a marketed drug increasing in price when and if a DMOAD indication is granted, again acting as a disincentive to pursuing the necessary long-term studies once the drug cost has been set. It will be necessary to consult with regulatory authorities to determine whether simultaneous approval of a drug could be granted on the basis of benefit on signs and symptoms (traditional approval) concurrent with approval on the basis of an expected DMOAD effect (for instance based on a surrogate (S) that predicts slowing of OA progression), with subsequent longer term study with an observational outcome such as reduced joint replacement rate (time-to-event) of replacement surgeries, or slowing of radiographic OA.
Joint failure endpoints for “time to failure” determination might include a predefined increase in pain, a predetermined and clinically important amount of change in MRI features associated with OA progression and/or joint failure, total joint replacement for OA, a predefined decline in function or a combination of any of these endpoints.
Scenario 1 (Fig. 1): prospective trial continuation
This scenario represents the continuation, post-approval, of the Phase 3 double blind, placebo controlled trial. The PMA study population contains the same patients as the original trial. The following characteristics and possible variations on this study design are as follows:
The Surrogate (S) in the initial phase may be measured in all or only a subset of the study population (determined based on study power estimates for the S and PRO outcomes); if the surrogate involves an expensive technique, a cost savings could be envisioned by not collecting further surrogate data in the confirmatory trial period.
Inclusion of the Surrogate (e.g., MRI) in the PMA study is optional; it is however potentially important to show that the change in the surrogate in the pre-approval study is linked to a PRO or observational outcome and this may need to be shown in the same patients (important point for discussion with regulatory authorities).
Continue all patients on initial drug allocation into the PMA trial until a failure threshold is achieved; this could allow crossover of placebo treated patients to active agent or exit from trial; for placebo patients transitioned to active treatment, their failure to ‘catch up’ to patients treated with active agent for the entire study duration (throughout the pre-approval and PMA study) would be evidence of drug efficacy and a persistent treatment effect on the disease course; failure threshold(s) would have to be defined in advance (for instance based on a certain amount of rescue medication use, or attainment of a threshold level of pain or disability).
An endpoint might be the time-to-event of joint replacement for OA or clinically relevant symptomatic worsening or whichever is first (see discussion below).
Scenario 2 (Fig. 2): separate PMA study
There are circumstances in which the phase 3 study could be amended to be a PMA study, especially if the demonstration of symptomatic and/or functional benefit is needed and the prolongation of a placebo controlled study for 1 or 2 years might be appropriate (scenario 1). Other profiles may need to demonstrate an effect on structure or even joint survival which might be more appropriate in a study population which is enriched for progressors. In this case, the PMA study might be conducted as a separate study as in this scenario 2. A combination of the two scenarios is possible as well. The following characteristics and possible variations on this study design are as follows:
The PMA study population is different than the population in the original trial (although some patients may be the same).
Inclusion criteria in the PMA study might be different from the pre-approval or pre-registrational trial.
All patients may be on active (high vs low dose) treatment in the PMA study and followed for rates of OA progression; such a design would facilitate retention of the maximal number of patients as no one would be on placebo once the agent is approved and available clinically/commercially; greater numbers of individuals retained during the PMA trial would provide a larger patient population to monitor for adverse effects.
An endpoint might be the time-to-event of joint replacement for OA or clinically relevant symptomatic worsening or whichever is first (see discussion below)
Use of joint replacement outcomes in post-marketing confirmatory trials
Although the ultimate proof of DMOAD activity could be demonstrated on the time-to-event (delay or elimination) of joint replacement surgery for OA, this outcome poses considerable barriers. While clinical benefit in the case of “joint survival” is clear, this outcome poses challenges due to the need for long study durations, large sample sizes and the impact of non-disease and other factors on the outcome (such as level of patient education, socioeconomic status and expectations of surgical outcomes, cost, and physician willingness to operate based on health status, comorbidities and/or age of the patient)16,17. So, although joint replacement can be considered an observational outcome, it is impacted by numerous subjective factors. Moreover, it is important to consider the treatment context in order to infer reduction in joint replacement as a benefit on structure; a reduction in joint replacement due solely to pain reduction would not be considered a reflection of a benefit on structure. The time frame for a study using a joint replacement outcome is most likely more than 5 years for the population with Kellgren/Lawrence grade 2 and 3 radiographic knee OA (7–11 years depending on the sample size)18. There are no consensus criteria guiding patient recommendations regarding replacement surgery; this results in the obvious problem of differences between countries, regions and even centers within the same region. If these differences are adequately addressed by the study design, e.g., by randomization per study center, then the time-to-event of joint replacement surgery for OA might represent a feasible primary endpoint. It will be important to discuss with regulatory authorities whether this observational outcome would fulfill the criterion for how a patient feels, function or survives for purposes of a PMA study.
Use of placebo in post-marketing confirmatory studies
The study designs may be different for the first drug to market compared to the second or subsequent drugs to market. For instance, subsequent drugs may be compared to existing drugs on the market rather than placebo, particularly if patient harm is anticipated due to placebo treatment once any effective disease treatment is available. An exception to this is evident in the osteoporosis field; even the latest drugs approved for osteoporosis were tested against true placebo treatments – this was undoubtedly facilitated by the fact that the disease is asymptomatic throughout its course until a fracture ensues – this is not the case for OA. In the rheumatoid arthritis (RA) field there are several disease modifying treatment options that could be the basis for a comparator in a drug trial but there are none in OA.
All post approval confirmatory studies must address a fundamental question: How can a patient be kept in the study if the drug is available? It is unlikely that a patient would accept the risk of randomization into the placebo or even standard of care arm once the drug is available clinically/commercially, particularly when a prolonged use of placebo in a PMA study would be anticipated. A precedent has been established in FDA guidance on RA trials for limiting the exposure of patients to placebo or ineffective therapies for a prolonged period of time (i.e., beyond 12 weeks)19. It is recommended that studies longer than 12 weeks should include an active comparator as the control or provisions for rescue treatment for patients with active disease. Procedures for enabling prolonged PMA studies could possibly maintain blinding until a study participant reaches a failure endpoint; patients on placebo could be offered active treatment at that time; patients on active treatment reaching a failure endpoint would be withdrawn from the study and considered therapeutic failures in the analysis. This scenario would require the establishment of threshold criteria for failure. Alternatively, the study could be designed to treat all patients with the active agent, comparing high vs low dose levels of the active drug without a placebo arm. This variation might be appropriate for each of the scenarios. Of note, this trial option (high vs low dose without placebo) for symptom and structure indications was embodied in the prior 1999 draft clinical trial guidance that encouraged “at least one trial showing superiority of the test product to placebo, to a lower dose of the agent, or to an active control”6. Another pragmatic option would be to offer all patients an exercise (core) treatment representing a high standard of care as “background therapy” and thereby promote their retention in the PMA study, whether on active agent or placebo treatment.
Possible outcomes for post-marketing approval study and use of real-world evidence in OA trials
In traditional trials, direct evidence of treatment benefit is derived from clinical trial effectiveness endpoints that measure survival or a meaningful aspect of how a patient feels or functions in daily life. There are four types of clinical outcomes that may support either direct or indirect evidence of a treatment benefit. The clinical outcome assessments include (see Fig. 3):
Fig. 3. Diagram of types of clinical outcomes.
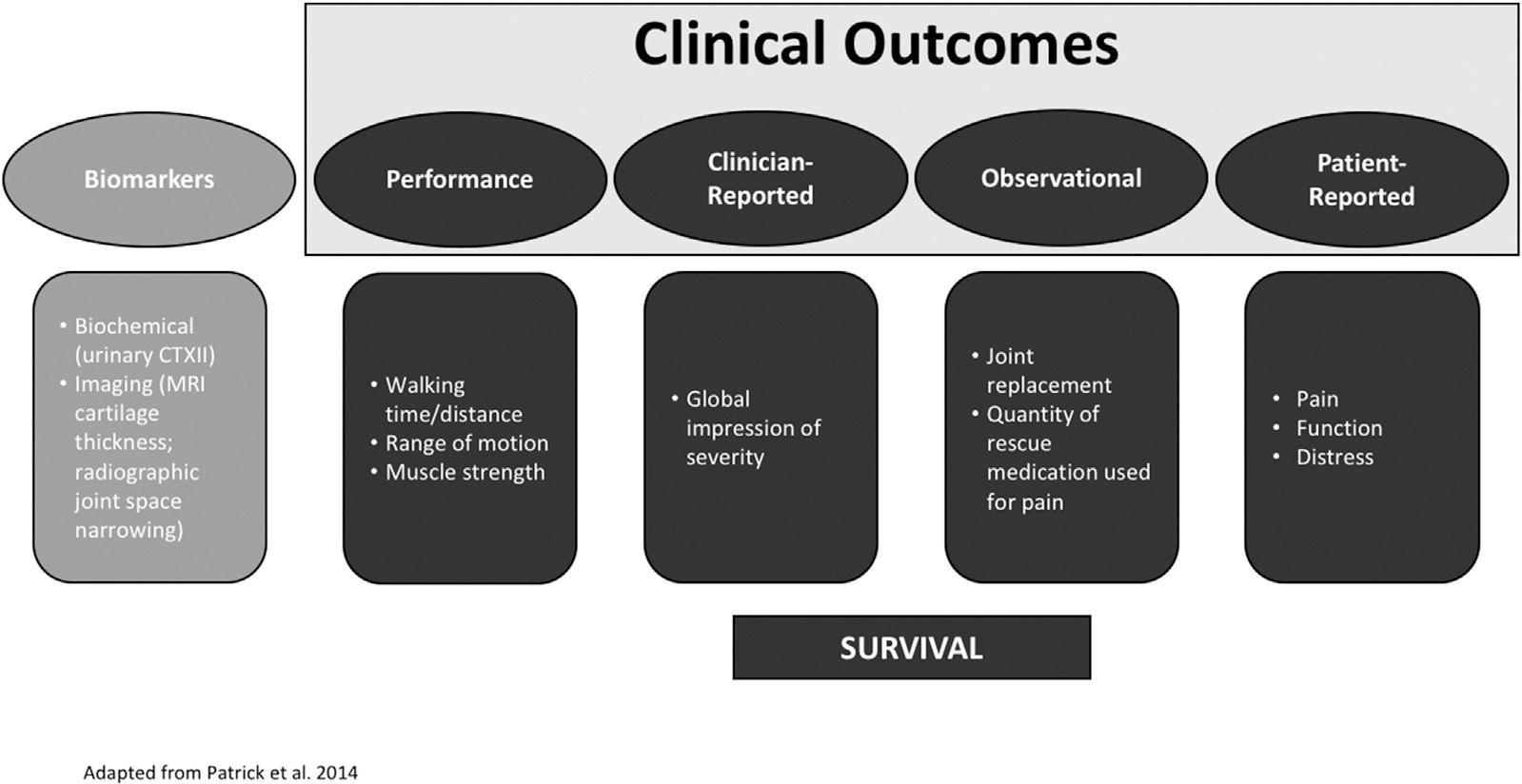
Clinical outcomes may include PROs, clinician-reported outcomes, observer-reported outcomes and performance based outcomes. The focus of this white paper is on biomarker outcomes and trials demonstrating their relationship to clinical outcomes in PMA trials. Graphic adapted from Patrick, Arbuckle, and Burke presentation at the ISPOR 17th Annual European Congress, November 11, 2014 (https://www.ispor.org/Event/GetReleasedPresentation/148).
PRO measures (objectively reported symptoms and function, such as provided by WOMAC or KOOS scores in OA, that could lead to the derivation of a time-to-event of clinically relevant symptomatic worsening);
Clinician-reported outcome measures (ratings based on specific professional training such as physician global assessment);
Observer-Reported outcome measures (items assessing directly reportable behavior without interpretation or interference such as total joint replacement and quantity of rescue medication used for pain);
Performance outcome measures (objectively measured function such as 6 min walk test).
The 21st Century Cures Act includes a provision for post-approval studies to include clinical evidence, clinical studies, patient registries, or other sources of real-world evidence, such as electronic health records, collection of larger confirmatory datasets or post-approval monitoring of all patients treated prior to approval of the therapy5. An electronic medical record based assessment of effectiveness could show paradoxically negative results because of biased loss to follow up (patients return for care when they are faring poorly and stay home when they are doing well).
For drugs that are approved on the basis of a PRO, a sponsor might seek to add efficacy indications to the label of an already approved drug based on endpoints relevant to payers and/or patients using confirmatory studies. Endpoints for these confirmatory studies might be derived from real-world evidence. As described in a white paper by Berger et al.20, for chronic obstructive pulmonary disease for example, a sponsor may wish to generate real-world evidence supporting indications of reduced exacerbation-related hospitalizations or improved quality of life – endpoints more readily useful in clinical decision-making and coverage decisions than the endpoint of forced expiratory volume in one minute (FEV1) used for initial drug approval. Because these endpoints may be measured using real-world data with good validity and reliability and would be captured in the same indicated population, they could lend themselves to a rigorous observational study design that harnesses electronic health records and claims. Alternatively, treated patients in a PMA study might be compared to a standard of care cohort or to historical databases.
Types of real-world evidence that could be derived from electronic health records that might be used to monitor status of OA patients include amount and strength/dose of real world rescue medication use [acetaminophen, non-steroidal anti-inflammatory drug (NSAIDs), opioids]; disease exacerbation (disease ‘relapse’) as measured by use of an intra-articular therapy, disease failure as measured by a total joint replacement, and all-cause mortality (based on knowledge that the natural history of OA, under the current treatment paradigm, increases mortality). Blinding may not be necessary when mortality is used as an endpoint in a confirmatory trial because bias may be less likely. Given the increased prevalence and incidence of diabetes in individuals with lower limb arthritis, with a large proportion (37–46%) attributable to walking disability21, the incidence or worsening of diabetes and step counts or mobility data (made increasingly available through use of wearable devices) are examples of the types of real-world data that could contribute to a real-world efficacy indication for a DMOAD.
Some questions for regulatory consultation
Do the two study design paradigms capture the majority of variation possible and feasible in OA?
How can patients be retained long-term in PMA studies for purposes of demonstrating benefit on signs and symptoms of OA?
Is it necessary to link the PRO in the confirmatory study to the biomarker (surrogate) in the initial approval study? Such a linkage is of course of high interest for potential DMOADs with similar modes of action. However, the clinical benefit of the drug is the matter of paramount importance for the confirmatory trial as opposed to retrospective justification of the surrogate.
Is it feasible to use real world evidence for the post-approval study? The study has to be well-controlled, which can be interpreted that a randomization procedure might be required. However, a comparison of treatments known to have substantial placebo effects, such as intra-articular therapy compared to standard of care, might result in an imbalanced comparison with respect to the placebo-related contextual effects.
Can function (both patient reported and/or measured) be used as a primary outcome in a PMA? Can PRO-function and objectively measured function have lower placebo response rates and higher treatment effects than PRO pain in OA trials?
Given the known interaction of pain and function, can mobile health technology be used in OA trials to provide objective function outcomes for trial purposes? The “work in the garden” problem is the phenomenon whereby pain reduction can result in function enhancement and increased physical activity resulting in an apparent overall minimal improvement in pain. Objective monitoring of function and possibly subjective PRO function could unmask a benefit on signs and symptoms of a drug under these circumstances.
Can slowing of pain worsening by a pre-specified clinically relevant amount be used to support a claim of slowing of OA progression?
Can a time-to-event study based on joint survival (time to joint replacement) provide ultimate proof of DMOAD activity and be used as a design option for confirmatory PMA trials?
Can the placebo treated study participants be switched to active drug in the post-marketing study?
Other disease fields cross placebo to active treatment during the confirmatory study phase with failure to catch up as the metric of success.
How will OA clinical trial guidance change when MRI measures are qualified as predictors of long-term patient benefits in delaying or preventing the progression to disability or joint replacement related to OA?
Supplementary Material
Acknowledgments
We wish to sincerely thank Valorie Thompson (Innovations Consulting Group LLC) for her excellent administrative assistance and all the members of the phase 2 working group of the OARSI Precompetitive Consortium in OA (listed in Supplementary Text).
Footnotes
Conflicts of interest
No author has any conflicts of interest related specifically to this work. Relevant financial activities outside the submitted work during the prior 36 months are as follows:
V Kraus received personal fees from Novartis, Flexion Therapeutics, TissueGene, GlaxoSmithKline, 23andME, Sanofi;
L Simon has no conflicts of interest related to this work but as a drug development consultant, he has consulted with multiple companies regarding trial designs and outcome measures including PROs, clinician-reported outcomes, and functional measures; consulting fees have been received from Eupraxia, Asahi, Samumed, Metabolex, Flexion, EMDSerono, Talagen, Tigenix, Genzyme.
CaloSyn, Horizon, Pozen, Analgesic Solutions, Bayer, Durect, Sanofi, JRX Biopharm;
JN Katz received a grant through his institution from Samumed; T Neogi received personal fees from EMD Merck Serono and Novartis;
DJ Hunter received personal fees from Merck Serono, TLC Bio and TissueGene;
A Guermazi received personal fees from BICL, LLC, Pfizer, GE, AstraZeneca, TissueGene, Roche, Galapagos and Merck Serono;
MA Karsdal is Chief Executive Officer of Nordic Bioscience specializing in biomarkers.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.joca.2018.11.002.
References
- 1.Food and Drug Administration. In: Services HaH, Ed. Subpart H – Accelerated Approval of New Drugs for Serious or Life-threatening Illnesses, 2017. [Google Scholar]
- 2.Food and Drug Administration. In: Services HaH, Ed. Subpart E – Accelerated Approval of Biological Products for Serious or Life-threatening Illnesses, 2017. [Google Scholar]
- 3.Food and Drug Administration. Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics, 2014. [Google Scholar]
- 4.Food and Drug Administration Safety and Innovation Act 2012. Public Law No. 112–144, 901, 126 Stat 993, 1083.
- 5.Public Law No. 114–255, 130 Stat 103321st Century Cures Act 2016.
- 6.Food and Drug Administration. Guidance for Industry: Clinical Development Programs for Drugs, Devices and Biological Products Intended for the Treatment of OA. Rockville: U.S. Department of Health and Human Services; 1999. [Google Scholar]
- 7.Guermazi A, Roemer FW, Felson DT, Brandt KD. Motion for debate: osteoarthritis clinical trials have not identified efficacious therapies because traditional imaging outcome measures are inadequate. Arthritis Rheum 2013;65(11):2748–58. [DOI] [PubMed] [Google Scholar]
- 8.March L, Cross M, Lo C, Arden N, Gates L, Leyland K, et al. Osteoarthritis: a Serious Disease. Osteoarthritis Research Society International; 2016. [Google Scholar]
- 9.Food and Drug Administration. Osteoarthritis: Structural Endpoints for the Development of Drugs, Devices, and Biological Products for Treatment Guidance for Industry 2018. [Google Scholar]
- 10.Beaver JA, Howie LJ, Pelosof L, Kim T, Liu J, Goldberg KB, et al. A 25-year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: a review. JAMA Oncol 2018;1(2673837). [DOI] [PubMed] [Google Scholar]
- 11.Amur S, LaVange L, Zineh I, Buckman-Garner S, Woodcock J. Biomarker qualification: toward a multiple stakeholder framework for biomarker development, regulatory acceptance, and utilization. Clin Pharmacol Ther 2015;98(1):34–46, 10.1002/cpt.136 (Epub 2015 Jun 6). [DOI] [PubMed] [Google Scholar]
- 12.Sasinowski F, Varond A. FDA’s flexibility in subpart H approvals: assessing quantum of effectiveness evidence. Food Drug Law J 2016;71(1):135–200. [PubMed] [Google Scholar]
- 13.Karsdal MA, Michaelis M, Ladel C, Siebuhr AS, Bihlet AR, Andersen JR, et al. Disease-modifying treatments for osteoarthritis (DMOADs) of the knee and hip: lessons learned from failures and opportunities for the future. Osteoarthritis Cartilage 2016;24(12):2013–21. [DOI] [PubMed] [Google Scholar]
- 14.Pham T, Van Der Heijde D, Lassere M, Altman RD, Anderson JJ, Bellamy N, et al. Outcome variables for osteoarthritis clinical trials: the OMERACT-OARSI set of responder criteria. J Rheumatol 2003;30(7):1648–54. [PubMed] [Google Scholar]
- 15.Buck R, Hellio Le Graverand-Gastineau M-P, Wirth C, Eckstein F . Efficacy trials for knee cartilage change may achieve reasonable treatment goals in <12 months and sample size <200/arm. Osteoarthritis Cartilage 2014;22:S69. [Google Scholar]
- 16.Kwoh CK, Vina ER, Cloonan YK, Hannon MJ, Boudreau RM, Ibrahim SA. Determinants of patient preferences for total knee replacement: African-Americans and whites. Arthritis Res Ther 2015;17:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wetterholm M, Turkiewicz A, Stigmar K, Hubertsson J, Englund M. The rate of joint replacement in osteoarthritis depends on the patient’s socioeconomic status. Acta Orthop 2016;87(3):245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwoh C, Guehring H, Hannon M, Aydemir A. Clinical relevance of structural measures in knee osteoarthritis: baseline values and change from baseline discriminate patients subsequently receiving knee replacement [abstract]. Arthritis Rheumatol 2017;69(Suppl 10). Abstract 1207. [Google Scholar]
- 19.Food and Drug Administration. Guidance for Industry, Rheumatoid Arthritis: Developing Drug Products for Treatment. In: 2013. Rockville, MD. [Google Scholar]
- 20.Berger M, Daniel G, Frank K, Hernandez A, McClellan M, Okun S, et al. , for the Duke-Margolis Center for Health Policy. A Framework for Regulatory Use of Real-world Evidence; 2017. [Google Scholar]
- 21.Kendzerska T, King L, Lipscombe L, Croxford R, Stanaitis I, Hawker G. The impact of hip and knee osteoarthritis on the subsequent risk of incident diabetes: a population-based cohort study. Diabetologia 2018;61:2290–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.