

Abstract
Vaccine adjuvants are essential to drive a protective immune response in cases where vaccine antigens are weakly immunogenic, where vaccine antigen is limited, or where an increase in potency is needed for a specific population, such as the elderly. To discover novel vaccine adjuvants, we used a high-throughput screen (HTS) designed to identify small-molecule agonists of the RIG-I-like receptor (RLR) pathway leading to interferon regulatory factor 3 (IRF3) activation. RLRs are a group of cytosolic pattern-recognition receptors that are essential for the recognition of viral nucleic acids during infection. Upon binding of viral nucleic acid ligands, the RLRs become activated and signal to transcription factors, including IRF3, to initiate an innate immune transcriptional program to control virus infection. Among our HTS hits were a series of benzothiazole compounds from which we designed the lead analog, KIN1148. KIN1148 induced dose-dependent IRF3 nuclear translocation and specific activation of IRF3-responsive promoters. Prime-boost immunization of mice with a suboptimal dose of a monovalent pandemic influenza split virus H1N1 A/California/07/2009 vaccine plus KIN1148 protected against a lethal challenge with mouse-adapted influenza virus (A/California/04/2009) and induced an influenza virus-specific IL-10 and Th2 response by T cells derived from lung and lung-draining lymph nodes. Prime-boost immunization with vaccine plus KIN1148, but not prime immunization alone, induced antibodies capable of inhibiting influenza virus hemagglutinin and neutralizing viral infectivity. Nevertheless, a single immunization with vaccine plus KIN1148 provided increased protection over vaccine alone and reduced viral load in the lungs after challenge. These findings suggest that protection was at least partially mediated by a cellular immune component and that the induction of Th2 and immunoregulatory cytokines by a KIN1148-adjuvanted vaccine may be particularly beneficial for ameliorating the immunopathogenesis that is associated with influenza viruses.
Keywords: Adjuvant, Influenza, IRF3, Vaccine
1. Introduction
Seasonal influenza infections are a significant public health problem that cause severe illness and death especially in high risk populations such as children younger than 5 years and individuals older than 65 [1,2]. The vast majority of influenza immunizations are performed with inactivated virus (split vaccines) in which the virus is disrupted by detergent [3]. The effectiveness of split vaccines is modest in the general population and even lower in elderly patients [4]. In addition, it is difficult to quickly manufacture an adequate supply of inactivated vaccine during an outbreak, prompting the need for dose-sparing strategies. To address these limitations, and the increasing demand for influenza split vaccines, a variety of targeted vaccine adjuvant approaches are being investigated [3].
Our adjuvant strategy is to identify and develop small-molecule immunomodulators that target pattern-recognition receptor (PRR) signaling pathways to enhance adaptive responses to vaccine antigens. Many RNA viruses, including influenza A virus, are recognized by RIG-I-like receptors (RLRs), a family of cytosolic PRRs [5]. RLR activation induces downstream phosphorylation of IRF3, which then dimerizes and translocates into the nucleus where it induces the expression of interferon-stimulated genes (ISG) and cytokines through promoters containing IRF3-binding sites [6]. Dysregulation or disruption of RLR signaling by viruses results in impaired innate and adaptive immune responses [7]. An effective vaccine adjuvant strategy for these RNA viruses should initiate adaptive responses through the RLR pathway and IRF3 activation.
In mouse models, the use of nucleic acid RLR pathway agonists during influenza vaccination is a promising approach to induce IRF3 and a protective immune response [8,9]. Here, we describe the properties of KIN1148, an IRF3-activating small-molecule adjuvant. Immunization of mice with a suboptimal dose of a pandemic influenza virus split vaccine plus KIN1148 provided protection against a lethal challenge with a mouse-adapted influenza virus (H1N1 A/California/04/09). Increased protection over vaccine alone appears to be due in part to the ability of KIN1148 to promote a balanced immune response that may reduce infection-related tissue damage.
2. Materials and methods
2.1. Reporter cell lines, high-throughput screening, and hit validation assays
The construction of reporter cell lines, high-throughput screening, and hit validation assays were carried out essentially as described previously [10]. Briefly, Huh 7 cells were stably transfected with a luciferase promoter construct driven by the interferon-β (IFN-β), ISG56, or β-actin promoter and grown under antibiotic selection. Screening was done on a 47,000-member small-molecule diversity library (Life Chemicals). Confirmation of initial hits was performed in triplicate. Nuclear translocation of IRF3 was determined using PH5CH8 cells [11]. IRF3-dependent gene expression was measured using PHCH8 cells treated with compound (10 μM in 0.5% DMSO). Expression of IRF3-dependent genes and GAPDH housekeeping gene was quantified using Taq-man qPCR probes (Applied Biosystems). IP-10 secretion by PMA-differentiated THP-1 cells (40 ng/ml PMA for 24 h) in response to 10 μM compound was determined 18 h after compound treatment using an IP-10 luminex kit.
2.2. Viruses
Pandemic influenza virus A/California/07/2009 (pH1N1) (NYMC X-179A, CDC ID#2009713114) was used to generate the split vaccine. Mouse-adapted influenza virus H1N1 A/California/04/2009 (MA-CA04), provided by Richard J. Webby, St. Jude Children’s Research Hospital, was used as the challenge strain. A plaque assay on Madin-Darby canine kidney (MDCK) cells was used to determine viral titers.
2.3. Vaccine, KIN1148, and formulation
The split vaccine was prepared by Virapur (San Diego, CA). Virus was produced in allantoic fluid from Certified Specific Pathogen Free eggs and purified by sucrose density gradient banding. Virions were inactivated with β-propiolactone, treated with Triton-X100 and additionally purified by 10-kDa ultrafiltration and dialysis in HEPES-buffered saline. The split vaccine had a total protein concentration of 454 μg/ml and a hemagglutinin (HA) concentration of 81.2 μg/ml. The vaccine had an HA titer of 1024 U/ml and an endotoxin level of 9.5 EU/ml.
KIN1148 was formulated in liposomes containing phosphatidylcholine, pegylated phosphatidylethanol and cholesterol in phosphate-buffered saline (PBS) by 6 h ultrasonication. The final liposomes prior to mixing with vaccine contained KIN1148 (5 mg/ml) and phospholipids (40 mg/ml) and were delivered intramuscularly (KIN1148 at 50 μg and phospholipid at 400 μg per dose). Blank liposomes were prepared without KIN1148 as vehicle control. KIN1148 liposome and blank liposome formulations were stable for at least 4 months at 4 °C and were used for in vivo experiments within a month after preparation.
2.4. Immunization, challenge, and cytokine profiling
Experiments using animals were conducted in accordance with IACUC-approved institutional guidelines. Female C57BL/6N mice (6–8 weeks old) were obtained from Charles River laboratories. Mice were anesthetized with isoflurane and immunized intramuscularly with 50 μl (3.3 μg total protein corresponding to 0.6 μg HA with and 0.069 EU) of vaccine, split between two 25-μl injections, one in each gastrocnemius muscle. The suboptimal dose of vaccine (3.3 μg) was determined by vaccinating with descending amounts of split-vaccine alone until reaching suboptimal protection from lethal challenge (>25%). All vaccination studies were performed using the same dose and batch of the Virapur split vaccine. For prime and prime/boost immunization, mice were immunized either once (prime) or twice 21 days apart. Twenty-one days after the receiving the final vaccine dose, mice were challenged intranasally with 10× the LD50 (2500 plaque-forming units/mouse, lethal virus dose that causes the death of 50% of the naïve mouse population inoculated with virus) of mouse-adapted influenza virus. Mice were euthanized humanely by carbon dioxide asphyxiation if displaying weight loss ⩾30% of starting weight or when displaying symptoms of severe disease including hunched posture, irregular breathing, pillory erection, and reduced activity. For serology, mice were anesthetized with isoflurane and whole blood (200 μl) was collected retro-orbitally for the processing of serum. For cytokine profiling, T cells from lung and lung-draining lymph nodes were stimulated for 18 h with split vaccine (1 μg/ml), CD4 epitope peptide MA-CA/04 NP260–283 (5 μM), CD8 epitope peptide MA-CA/04 NP366–374 (5 μM), or concanavalin A (Con A 5 μg/ml). Cytokines in culture supernatants were then measured using a multiplex ELISA (Quansys). Cytokine experiments were performed in triplicate. Bone marrow-derived dendritic cells were added as antigen-presenting cells to the lymph node T cell assays.
2.5. Split vaccine-specific IgG ELISA
ELISA plates were coated with the split vaccine at 1 μg of HA per ml of PBS. Plates were blocked with 5% BSA in PBS plus 0.1% Tween-20 (PBST). Mouse sera were added at the indicated sera dilutions (1:1000/ 1:10,000) in duplicate to plates, and allowed to bind to antigen for 1.5 h. Mouse antibodies were detected with biotinylated goat anti-Mouse IgG antibodies (Southern Biotech) at 1:5000. Streptavidin-HRP (Thermo Scientific) was then added at 1:20,000 and incubated for 30 min. Plates were washed, developed for 2 min with TMB substrate (Thermo Scientific), and stopped with 1 M H3PO4. Individual well optical density (OD) was measured at 450 nm.
2.6. Hemagglutination inhibition (HAI) and virus microneutralization (VMN)
HAI assays were performed using 0.5% fresh whole turkey blood according to WHO guidelines [12]. For VMN assays, serum was heat-inactivated at 56 °C for 45 min and diluted twofold in infection media. Virus was diluted in infection media and added to diluted serum at a multiplicity of infection of 0.15 and incubated for 90 min in a humidified incubator at 37 °C. The virus and serum mixture was then transferred onto MDCK cells and incubated at 37 °C for 6 h. Cells were fixed with ice-cold methanol-acetone (1:1) for 10 min. Fixing solution was aspirated and cells were washed once with PBS. Plates were blocked for 1 h with 10% horse serum containing BSA (1 mg/ml) and 0.1% Triton X-100. Fluorescein isothiocyanate-labeled mouse IgG to influenza nucleoprotein (1:3000) and Hoechst dye (1:5000) were added to plates in 1% horse serum and incubated for 2 h at room temperature. Infected cell counts and nuclear staining were quantified using an ArrayScan HCS instrument. Neutralizing titers were determined as the reciprocal of highest dilution of serum resulting in 50% reduction in infection over negative control (no serum).
2.7. Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.04 for Windows (GraphPad Software, La Jolla, California; www.graphpad.com). Survival analysis was performed using the Gehan-Breslow-Wilcoxon method to give more weight to death of mice at early time points. Weight-loss curves and the IRF3 translocation data in Fig. 1B were analyzed by two-way ANOVA. Antibody data in Fig. 4 were analyzed by using one-way ANOVA (Kruskal-Wallis test) followed by Dunn’s multiple comparison test. The IP-10 results in Fig. 1D were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. P values in Figs. 3 and 6B – D were calculated by using the two-tailed unpaired Student’s t-test.
Fig. 1.
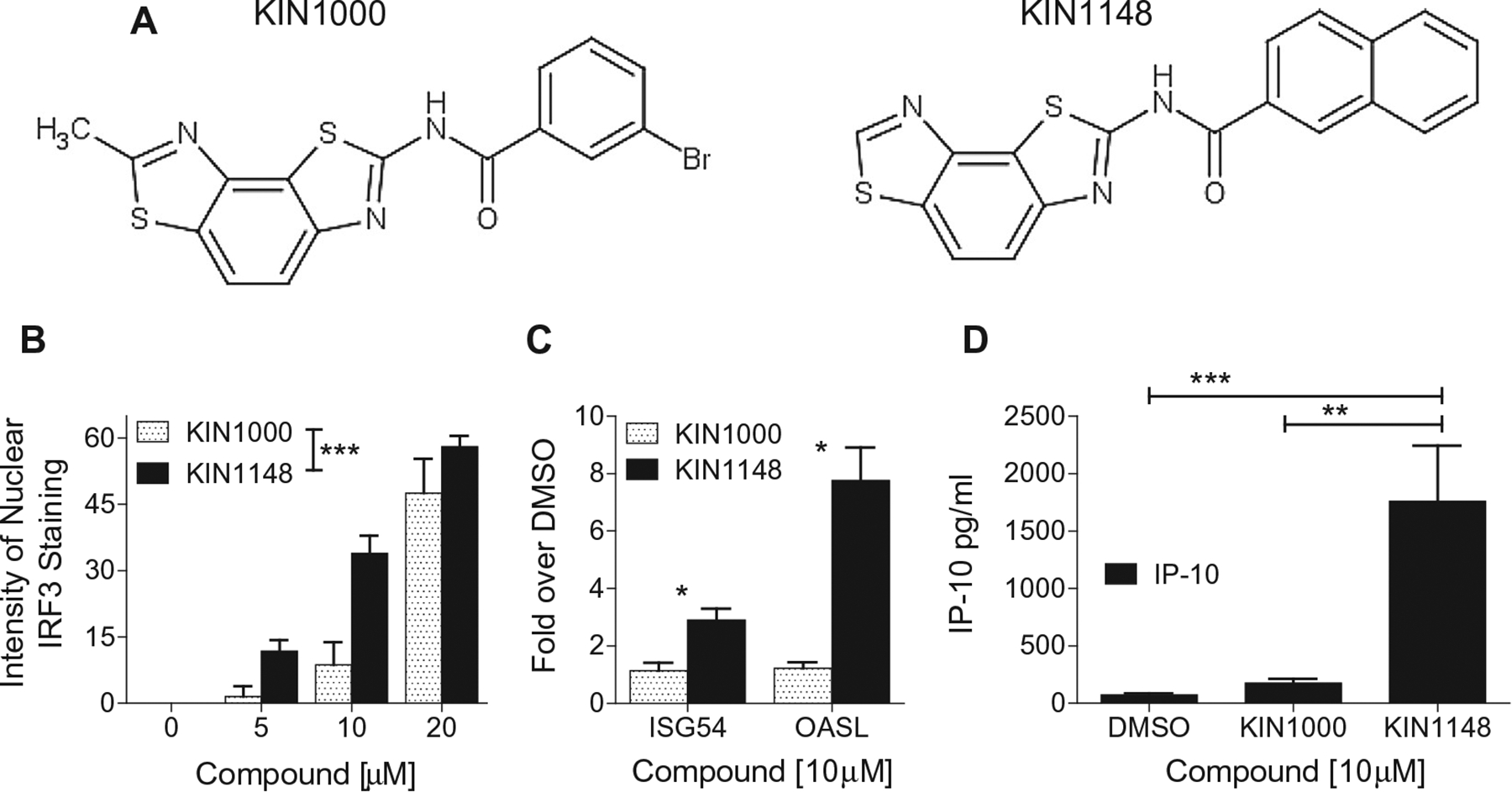
HTS hits and derivatives induce the nuclear translocation of IRF3 and IRF3-dependent gene expression. (A) Structure of KIN1000 and its derivative KIN1148. (B) KIN1000 and KIN1148 induce dose-dependent nuclear translocation of IRF3 in PH5CH8 cells. (C) KIN1148 is more potent than KIN1000 as an activator of IRF3-dependent ISG54 and OASL expression by PH5CH8. (D) KIN1148 induces the production of IP-10 by PMA activated THP-1 cells. Experiments in were performed in duplicates for (B, C) and triplicates (C). Data are representative of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Fig. 4.
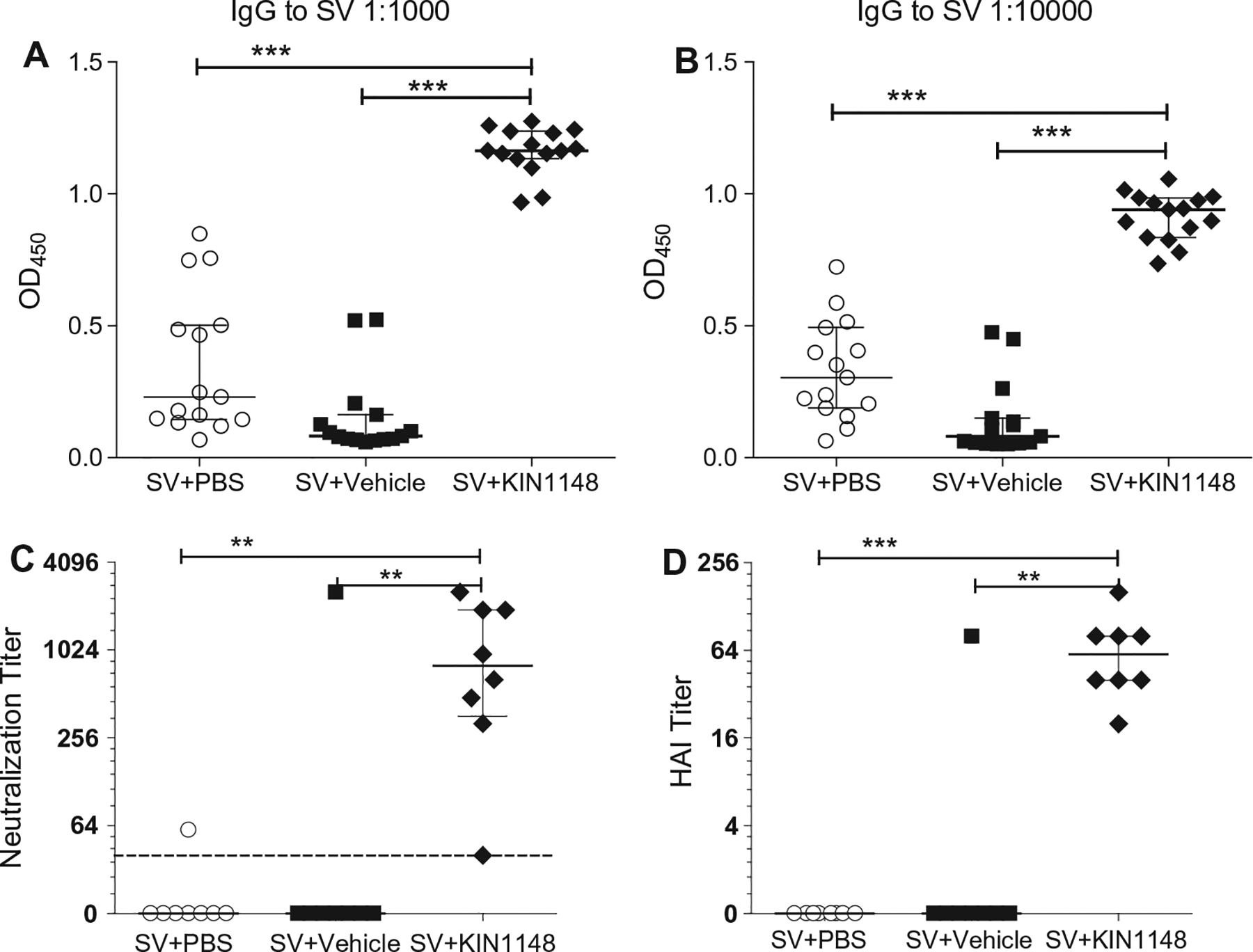
Prime-boost immunization with vaccine plus KIN1148 induces a strong humoral response. Groups of C57BL/6N mice were immunized twice, 21 days apart, with vaccine plus PBS, vaccine plus vehicle, or vaccine plus KIN1148. Mice were bled 18 days after prime and 14 days after boost and sera was used for determination of IgG, neutralizing antibody, or HAI titer. (A) IgG titers at day 18 (post prime). (B) IgG titers at day 35 (14 days post boost). (C) Neutralizing antibody titer at day 35. (D) HAI titer at day 35. Data shown as individual mice with median (horizontal line) and interquartile range (error bar) of the respective groups (n = 15, A/B; n = 8 C/D). One-way ANOVA (Kruskal-Wallis test) followed by Dunn’s multiple comparison test was performed for statistical analysis. **P < 0.01, ***P < 0.001. Data are representative of three individual experiments.
Fig. 3.
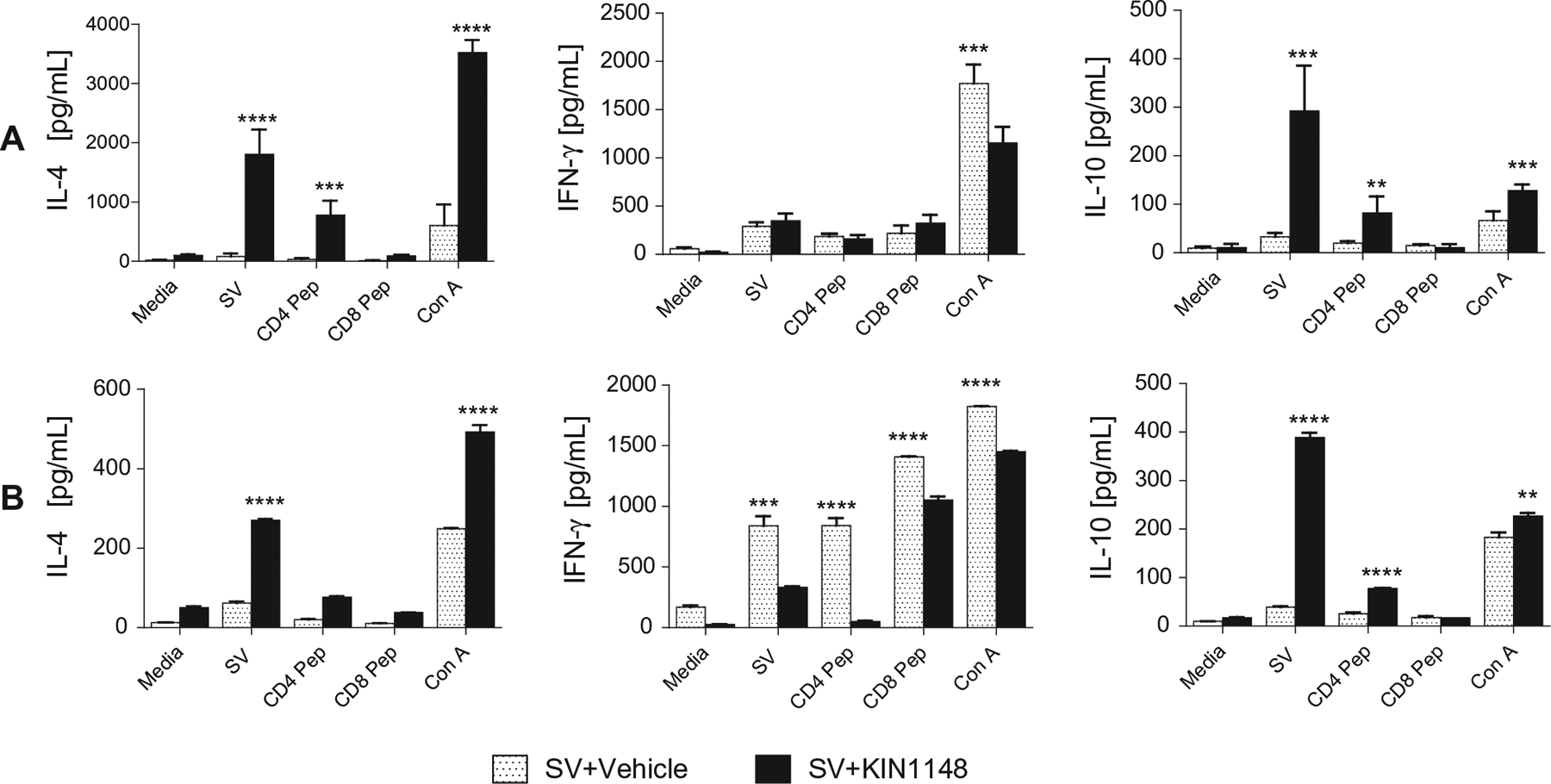
Prime-boost immunization with vaccine plus KIN1148 enhances influenza virus-specific Th2 and immunoregulatory responses in response to viral challenge. Groups of C57BL/6N mice were immunized twice, 21 days apart, with vaccine plus vehicle (n = 4), or vaccine plus KIN1148 (n = 8). Mice were then challenged with 10× the LD50 of mouse-adapted influenza virus A/California/04/2009 21 days after boost. T cells from lung and lung-draining lymph nodes (collected on day 7 after challenge) were stimulated for 18 h with split vaccine (SV), CD4 epitope peptide MA-CA/04 NP260–283 (CD4 Pep), CD8 epitope peptide MA-CA/04 NP366–374 (CD8 Pep), or concanavalin A (Con A). Cytokines in culture supernatants were then measured using a multiplex ELISA. (A) T cells derived from lungs were evaluated from individual mice. Bars represent the mean and SEM of KIN1148/vaccine (n = 8 mice) and vehicle/vaccine (n = 4 mice) groups. (B) Cells derived from lung-draining lymph nodes were evaluated as separate pools derived from mice receiving KIN1148/vaccine (n = 8 mice) or vehicle/vaccine (n = 4 mice). Stimulations were performed in triplicate with bars representing the mean and STD. Student’s two-tailed t-test was performed to compare the respective antigen-specific T cell responses from SV/vehicle control and SV/KIN1148 groups. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Fig. 6.
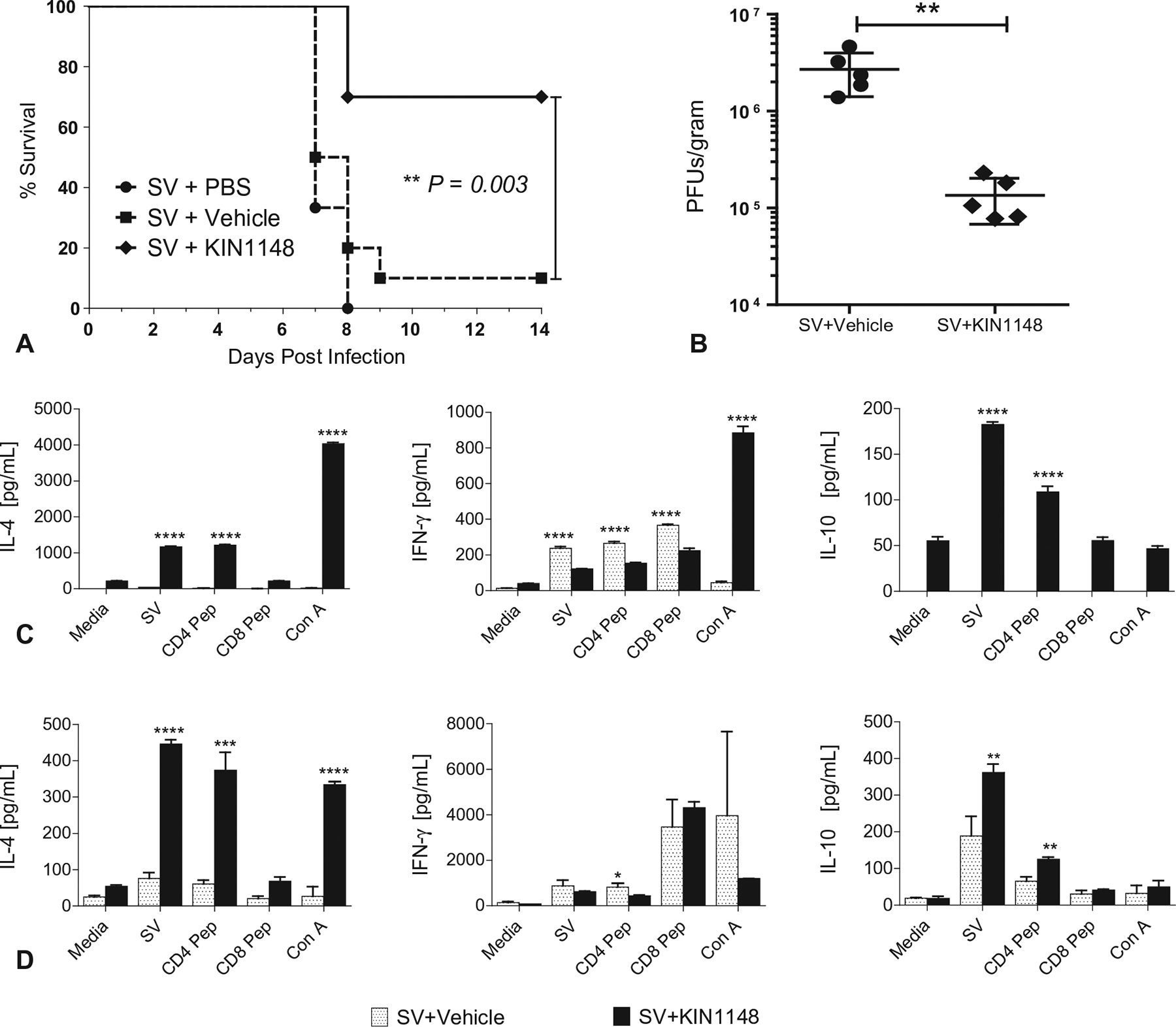
Prime immunization alone with vaccine plus KIN1148 protects mice against lethal H1N1 challenge. Groups of C57BL/6N mice were immunized twice, 21 days apart, with vaccine plus PBS, vaccine plus vehicle, or vaccine plus KIN1148. (A) Survival (10 mice per group). (B) Viral load in lungs 3 days post challenge (5 mice per group). (C/D) Cytokine response of T cells from (C) lung and (D) lung-draining lymph nodes (5 mice per group). T cells from lung and lung-draining lymph nodes (collected on day 7 after challenge) were stimulated for 18 h as described in the legend to Fig. 3. Cytokines in culture supernatants were then measured using a multiplex ELISA. T cells derived from lungs were evaluated from individual mice. Bars represent the mean and SEM of KIN1148/vaccine and vehicle/vaccine groups. Cells derived from lung-draining lymph nodes were pooled by group. Stimulations were performed in triplicate with bars representing the mean and STD. Student’s two-tailed t-test was performed to compare the respective antigen-specific T cell responses from SV/vehicle control and SV/KIN1148 groups. *P < 0.05 **P < 0.01, ***P < 0.001, ****P < 0.0001 Data represent the results from at least two independent experiments.
3. Results
3.1. Identification of small-molecule agonists of the RLR pathway
The AViiD high-throughput screening platform [10] was used to identify small molecules that activate RLR signaling. In this platform, Huh7 reporter cell lines containing the ISG54, ISG56, or IFN-β promoters, all IRF3-responsive genes triggered through the RLR pathway, were used to screen a diversity library of over 47,000 small-molecule compounds. The benzobisthiazole compound KIN1000 was identified as a hit molecule from the library, and 418 subsequent analogs of this compound, including KIN1148, were designed, synthesized (Fig. 1A) and screened in the IRF3-translocation assay. Of these compounds, 136 were validated in the IRF3 translocation assay and were then further analyzed for the induction of IRF3-dependent gene expression and cytokine/chemokine production by differentiated THP-1 cells. Both KIN1000 and KIN1148 induced the dose-dependent nuclear translocation of IRF3 in PH5CH8 cells (Fig. 1B). KIN1148 induced IRF3 translocation at lower concentrations than did KIN1000, and also elicited greater induction of endogenous IRF3-dependent ISG54 and OASL expression by PH5CH8 cells (Fig. 1C). In addition, PMA-differentiated THP-1 cells produced significant higher levels of the IRF3-dependent chemokine IP-10 than produced by THP-1 cells stimulated with KIN1000 (Fig. 1D). KIN1148 was selected for further characterization in vaccine adjuvant studies based on additional screens using human primary cells (manuscript in preparation) and the ability to formulate the compound in liposomes.
3.2. Prime-boost immunization with a sub-optimal dose of split influenza virus vaccine plus KIN1148 provides a protective immune response
We assessed the potential of KIN1148 to adjuvant a pandemic human influenza virus pH1N1 A/California/07/2009 split vaccine by immunizing mice with a suboptimal dose of the vaccine plus PBS, vaccine plus vehicle (blank liposomes), or vaccine plus KIN1148 liposomes. Mice were immunized on day 0 (prime), again on day 21 (boost), and then challenged with 10× the LD50 of mouse-adapted influenza virus H1N1 A/California/04/2009 (MA-CA04) at day 42 (21 days after boost). Mice were observed for 14 days after challenge, and weight loss and symptoms of disease were recorded. All mice that received vaccine plus KIN1148 survived lethal challenge (Fig. 2A) and lost only an average of 5% of their starting weight during the study (Fig. 2B). In contrast, the median survival of mice receiving vaccine alone was 8 days and the mice experienced significant weight loss post-challenge, with the majority dropping below 25% of starting weight. Overall, prime-boost vaccination with a suboptimal dose of vaccine plus KIN1148 resulted in protection from subsequent challenge with live virus.
Fig. 2.
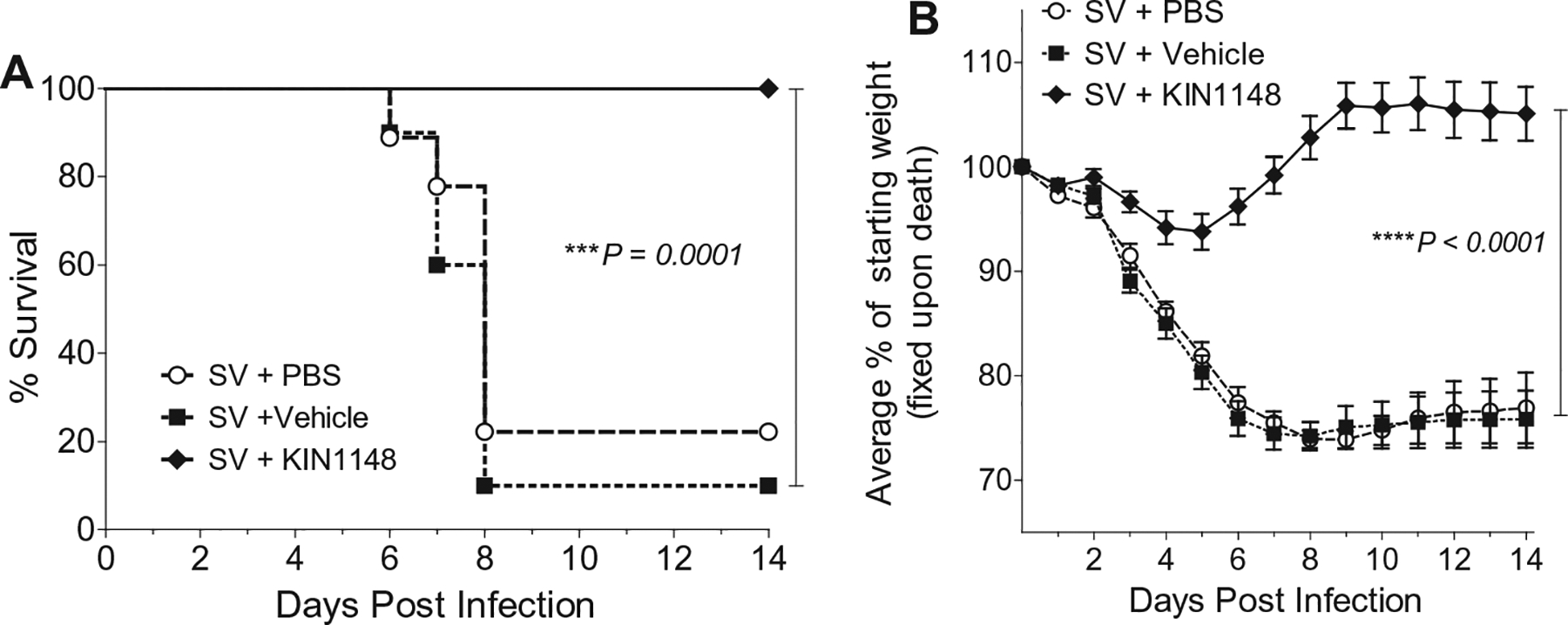
Prime-boost immunization with vaccine plus KIN1148 protects against lethal H1N1 challenge. Groups of 10 C57BL/6N mice were immunized twice, 21 days apart, with vaccine plus PBS, vaccine plus vehicle, or vaccine plus KIN1148. The mice were then challenged with 10× the LD50 of mouse-adapted influenza virus A/California/04/2009 21 days after boost. Mice were observed for 14 days after challenge (A) Survival. (B) Weight loss. Data are representative of two independent experiments. ***P < 0.001, ****P < 0.0001.
3.3. Prime-boost immunization with vaccine plus KIN1148 promotes influenza virus-specific Th2 and functional antibody responses following challenge
To determine how prime-boost immunization with the KIN1148/split vaccine adjuvant system modulates the cellular immune response after viral challenge, we evaluated ex vivo cytokine production by T-cells from the lung and lung draining lymph nodes in response to influenza virus antigens. Seven days after challenge with MA-CA04, mice were euthanized, and lung and lung-draining lymph nodes were collected and processed into mononuclear cell suspensions. T cells were stimulated for 18 h with split vaccine or influenza virus nucleoprotein peptides comprising CD4+ and CD8+ T cell epitopes. The levels of the Th1-associated cytokine IFN-γ, the Th2-associated cytokine IL-4, and the immune-regulatory cytokine IL-10 (which down regulates the expression of Th1 cytokines) was determined in supernatants by using a multiplex ELISA. Upon antigen-specific activation with split vaccine antigens or the influenza virus nuclear protein (NP) CD4+ T cell epitope, T cells from animals immunized with split vaccine plus KIN1148 produced higher levels of IL-4 than did T cells from animals immunized with vaccine alone (Fig. 3). Notably, the IFN-γ response elicited by split vaccine antigen or the NP CD4+ peptide in lymph node-derived T cells from the KIN1148-adjuvanted split vaccine group was markedly reduced when compared to the IFN-γ production of T cells immunized with the vehicle/split vaccine control (Fig. 3B, middle panel). In addition, the split vaccine antigen and the NP CD4+ peptide epitope stimulated the production of immune-regulatory IL-10 by T cells from animals that received vaccine plus KIN1148, but not in animals immunized with vaccine alone. The production of IL-4 and IL-10 by the KIN1148-adjuvanted vaccine was specific to CD4+ T cells since the response elicited by the influenza virus peptide epitope recognized by CD8+ T cells did not induce any detectable levels of these cytokines.
To assess the influenza-specific humoral response elicited by prime-boost immunization, we determined the level of split vaccine-specific IgG antibodies in serum samples taken on day 18 (post prime) (Fig. 4A) and day 35 (14 days post boost) (Fig. 4B). Both prime and prime-boost immunizations (3.3 μg protein/0.6 μg HA per dose) with the KIN1148 split vaccine adjuvant system induced a strong and significant increase in total split vaccine-specific IgG response compared to mice receiving vaccine formulated with vehicle control. To examine the protective functionality of the generated antibody response, we determined neutralizing antibody and hemagglutinin inhibition titers (HAI) [13] to H1N1 A/California/07/09 influenza virus. At day 35, prime-boost immunization with vaccine plus KIN1148 enhanced neutralizing serum antibody levels to H1N1 A/California/07/09 (median seroconversion titer of 1:800) (Fig. 4C). Only one out of 8 mice that received prime-boost vaccine formulated in PBS or vehicle control produced any detectable neutralizing antibodies, whereas 7 out of the 8 mice that received vaccine paired with KIN1148 showed significantly higher neutralizing serum antibody levels after prime-boost vaccination when compared to the control groups. The HAI assay is the standard assay used by the FDA for measuring protection (seroconversion) in response to vaccination, and an HAI titer of 1:40 is typically used as a cutoff for efficacy in clinical studies [14]. We found that mice immunized with vaccine plus KIN1148 had a median HAI titer of 1:64 at day 35 after prime-boost vaccination (Fig. 4D). By comparison, mice immunized with vaccine alone were seronegative at day 35. The outlier mouse in the SV + vehicle group with the high HAI titer was the same mouse with the high virus neutralizing titer in Fig 4C. We were not able to detect either virus neutralizing antibodies or HAI titers on day 18 after prime immunization alone, with or without KIN1148 (data not shown). In summary, prime-boost immunization with the KIN1148/split vaccine adjuvant system elicited a protective humoral immune response.
3.4. Passive transfer of serum from mice that received prime-boost immunization with vaccine plus KIN1148 protects naïve animals from challenge
To further evaluate the functional role of antibodies generated in response to immunization with vaccine plus KIN1148, we transferred serum from vaccinated animals into naïve mice. Sera was collected from mice that had been immunized by prime or prime-boost with the KIN1148/split vaccine adjuvant system or with vehicle/split vaccine control on day 21 (prime) or day 35 (prime-boost). Pooled sera was then administered to naïve recipient mice by intraperitoneal injection of two 200-μl doses over the course of 2 days followed by challenge with a lethal dose of MA-CA04. Passive transfer of serum from animals that received only prime immunization with vaccine plus KIN1148 provided limited protection against viral challenge by extending the medium survival from 6 to 10 days. The post-challenge weight loss was significantly delayed in these animals when compared to weight loss of mice that received serum from the vehicle/split vaccine control group or naïve mice (Fig. 5A). In contrast, passive transfer of serum from animals that received prime-boost KIN1148/split vaccine immunizations provided complete protection with significantly reduced weight loss from subsequent virus challenge when compared to mice immunized with vaccine/vehicle control (Fig 5B), consistent with the protective humoral immune response observed following prime-boost immunization with the KIN1148/split vaccine adjuvant system.
Fig. 5.
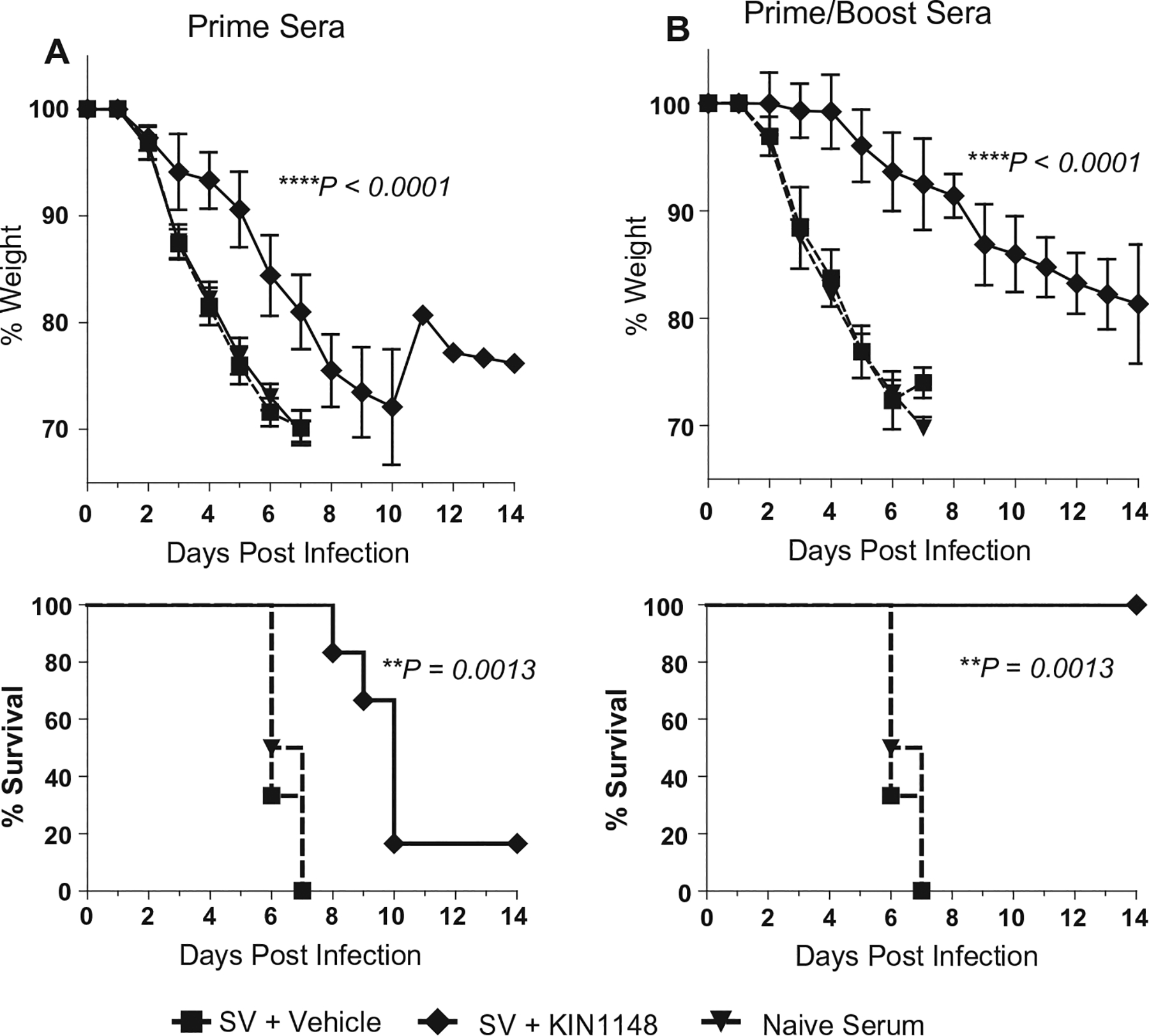
Passive transfer of serum from mice immunized with vaccine plus KIN1148 protects mice against lethal H1N1 challenge. Immune passive transfer sera was generated from mice immunized either once (prime), or twice (prime/boost), 21 days apart, with vaccine plus vehicle, or vaccine plus KIN1148. Groups of 6 C57BL/6N mice received 200 μl of pooled immune sera by intraperitoneal injection on day −2 and day −1 prior to challenge. Mice were then challenged with 10× the LD50 dose of mouse-adapted influenza virus A/California/04/2009 and observed for 14 days after challenge. (A) Passive transfer of prime sera. (B) Passive transfer of prime-boost sera. **P < 0.01, ****P < 0.0001.
3.5. Prime immunization with the KIN1148/split vaccine adjuvant system mediates protection against viral challenge
Next we determined whether mice receiving a single dose of vaccine plus KIN1148 were protected from virus challenge. Mice were immunized with vaccine alone or vaccine plus KIN1148 and challenged with 10× LD50 dose of MA-CA04 twenty-one days after immunization. Mice primed with vaccine plus KIN1148 displayed significant improvement in survival when compared with groups of mice immunized with vaccine formulated with PBS or vehicle control (Fig. 6A). In addition, prime immunization with KIN1148-adjuvanted vaccine reduced the viral burden in the lungs 3 days after viral challenge. Compared with animals immunized with vaccine controls, mice in the KIN1148/split vaccine group had a 20-fold reduction in viral titers (Fig. 6B). Finally, prime immunization enhanced the production of IL-4 and IL-10 after challenge by influenza-specific T cells from lung (Fig. 6C) and lung-draining lymph nodes (Fig. 6D). Upon antigen-specific ex vivo activation with split vaccine antigen or the NP CD4+ T cell epitope, T cells-derived from animals immunized with KIN1148/split vaccine produced higher levels of IL-10 and IL-4 than did T cells from animals that received vaccine formulated with vehicle control (Fig. 6C and D).
Taken together, our data indicate that prime immunization with the KIN1148/split vaccine adjuvant system provides protection against viral challenge and reduces the viral burden in the lungs. The protection appears partially mediated by a cellular immune component inducing the Th2 cytokine IL-4 and the immunoregulatory anti-inflammatory cytokine IL-10. This balanced immune response may contribute to controlling the inflammation induced by the remaining viral burden in the lungs.
4. Discussion
Various studies show that activation of RLR signaling during vaccination is a promising approach to adjuvant influenza virus vaccines. Mice immunized with a Sendai virus-derived RNA co-delivered with a suboptimal dose of influenza virus vaccine showed enhanced humoral responses to influenza virus as well as protective immunity to challenge [8]. Additionally, mice co-immunized with the RIG-I ligand 5′ppp-dsRNA developed robust HAI titers, enhanced T follicular helper cell responses, and protective immunity with a dose-sparing amount of vaccine [9].
We identified KIN1148 as a small-molecule agonist of the RLR pathway that activates IRF3. Unlike nucleic acid RLR agonists, our small-molecule agonist can be optimized for efficacy, reactogenicity and delivery [15,16]. KIN1148 is a lipophilic small molecule compound with a cLogP of 4.76 and limited solubility in aqueous solutions. The incorporation of KIN1148 liposomes increased the adjuvant activity of the compound over KIN1148 formulated in PBS (data not shown). This effect is likely mediated by the homogenous dispersion of KIN1148 liposomes and split vaccine and the ability to deliver a higher compound dose. In contrast to nucleic acid RIG-I ligands, the KIN1148 adjuvant system does not require transfection reagents or the integration into DNA vaccines for in vivo adjuvant activity [8,17].
Prime-boost immunization with the KIN1148/split vaccine adjuvant system elicited protective antibodies and induced IL-10 and a Th2 response in the lung and lung-draining lymph nodes after viral challenge. Similarly, DMXAA, a potent IRF3-dependent inducer of type I IFN, also induces a Th2 response to a split influenza virus vaccine [18].
Although Th2 immune responses, particularly eosinophilia, have the potential to elicit allergic responses [19,20], recent research into Th2 immunity at mucosal surfaces has revealed plurality within the Th2 response [21–23]. Th2 immune responses may fall into two categories of outcome: anti-inflammatory (tissue protective or reparative) or pro-inflammatory (tissue damaging mast cell degranulation, asthma, allergy, and inflammation). The observations of influenza antigen-specific IL-10-producing T cell populations in lungs and lung-draining lymph nodes obtained from mice after challenge implies that immunization with vaccine plus KIN1148 promotes a balanced immune response that dampens influenza virus-induced immunopathology. Our results encourage the preclinical development of small-molecule RLR agonists as novel immunomodulatory adjuvants for vaccines against RNA viruses. In addition, the use of KIN1148-adjuvanted vaccines for pathogenic human and avian influenza viruses, where the excessive inflammatory responses caused by these viruses might be controlled by the induction of a tissue-protective Th2 regulatory immune response will be explored.
Acknowledgments
We thank Christopher B. Fox (Infectious Disease Research Institute, Seattle, Washington) for manufacturing the KIN1148 liposomes and Marcus J. Korth for assistance in preparing the manuscript for publication.
Financial support
This work was funded by the National Institute of Allergy and Infectious Disease, National Institutes of Health, under contract numbers HHSN272200900035C and HHSN272201323C and by Public Health Service grants R01AI104002 and U19AI083019.
Footnotes
Conflict of interest
PP, JG, MW, MG, SI, and KB own equity shares in Kineta, Inc.
References
- [1].World Health Organization. Influenza fact sheet; 2016. http://www.who.int/mediacentre/factsheets/fs211/en/ [accessed December 21, 2016]. [Google Scholar]
- [2].Thompson WW, Weintraub E, Dhankhar P, Cheng P-Y, Brammer L, Meltzer MI, et al. Estimates of US influenza-associated deaths made using four different methods. Influenza Other Respir Viruses 2009;3:37–49. 10.1111/j.1750-2659.2009.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Del Giudice G, Rappuoli R. Inactivated and adjuvanted influenza vaccines. Curr Top Microbiol Immunol 2015;386:151–80. 10.1007/82_2014_406. [DOI] [PubMed] [Google Scholar]
- [4].Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 2012;12:36–44. 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- [5].Kell AM, Gale M. RIG-I in RNA virus recognition. Virology 2015;479–480:110–21. 10.1016/j.virol.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Loo Y-M, Gale M Immune signaling by RIG-I-like receptors. Immunity 2011;34:680–92. 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Errett JS, Gale M. Emerging complexity and new roles for the RIG-I-like receptors in innate antiviral immunity. Virol Sin 2015;30:163–73. 10.1007/s12250-015-3604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Martínez-Gil L, Goff PH, Hai R, García-Sastre A, Shaw ML, Palese P. A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J Virol 2013;87:1290–300. 10.1128/JVI.02338-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kulkarni RR, Rasheed MAU, Bhaumik SK, Ranjan P, Cao W, Davis C, et al. Activation of the RIG-I pathway during influenza vaccination enhances the germinal center reaction, promotes T follicular helper cell induction, and provides a dose-sparing effect and protective immunity. J Virol 2014;88:13990–4001. 10.1128/JVI.02273-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bedard KM, Wang ML, Proll SC, Loo Y-M, Katze MG, Gale M, et al. Isoflavone agonists of IRF-3 dependent signaling have antiviral activity against RNA viruses. J Virol 2012;86:7334–44. 10.1128/JVI.06867-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sumpter R, Loo Y-M, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 2005;79:2689–99. 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].World Health Organization. WHO manual on animal influenza diagnosis and surveillance. http://www.who.int/csr/resources/publications/influenza/en/whocdscsrncs20025rev.pdf [accessed December 21, 2016].
- [13].Veguilla V, Hancock K, Schiffer J, Gargiullo P, Lu X, Aranio D, et al. Sensitivity and specificity of serologic assays for detection of human infection with 2009 pandemic H1N1 virus in U.S. populations. J Clin Microbiol 2011;49:2210–5. 10.1128/JCM.00229-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].FDA. Guidance for industry: clinical data needed to support the licensure of seasonal inactivated influenza vaccines; 2014.
- [15].Flower DR. Systematic identification of small molecule adjuvants. Expert Opin Drug Discov 2012;7:807–17. 10.1517/17460441.2012.699958. [DOI] [PubMed] [Google Scholar]
- [16].Wu TY-H, Singh M, Miller AT, De Gregorio E, Doro F, D’Oro U, et al. Rational design of small molecules as vaccine adjuvants. Sci Transl Med 2014;6:263ra160. 10.1126/scitranslmed.300998. [DOI] [PubMed] [Google Scholar]
- [17].Luke JM, Simon GG, Söderholm J, Errett JS, August JT, Gale M, et al. Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J Virol 2011;85:1370–83. 10.1128/JVI.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tang CK, Aoshi T, Jounai N, Ito J, Ohata K, Kobiyama K, et al. The chemotherapeutic agent DMXAA as a unique IRF3-dependent type-2 vaccine adjuvant. PLoS ONE 2013;8:e60038. 10.1371/journal.pone.0060038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Harker J, Bukreyev A, Collins PL, Wang B, Openshaw PJM, Tregoning JS. Virally delivered cytokines alter the immune response to future lung infections. J Virol 2007;81:13105–11. 10.1128/JVI.01544-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Minne A, Jaworska J, Gerhold K, Ahrens B, Avagyan A, Vanbever R, et al. Intranasal delivery of whole influenza vaccine prevents subsequent allergen-induced sensitization and airway hyper-reactivity in mice. Clin Exp Allergy J Br Soc Allergy Clin Immunol 2007;37:1250–8. 10.1111/j.1365-2222.2007.02767.x. [DOI] [PubMed] [Google Scholar]
- [21].Kimoto T, Mizuno D, Takei T, Kunimi T, Ono S, Sakai S, et al. Intranasal influenza vaccination using a new synthetic mucosal adjuvant SF-10: induction of potent local and systemic immunity with balanced Th1 and Th2 responses. Influenza Other Respir Viruses 2013;7:1218–26. 10.1111/irv.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pulendran B, Artis D. New paradigms in type 2 immunity. Science 2012;337:431–5. 10.1126/science.1221064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog 2011;7:e1002003. 10.1371/journal.ppat.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]