

Abstract
Cardiovascular diseases (CVDs) represent a major concern for human health worldwide. Emergencies in this field include wide repertories of studies dealing primarily with CVD prevention. In addition to dietary habits and lifestyles, medical knowledge is fully needed to improve public educational programs toward cardiovascular risk factors and to enrich the endowment of pharmaceutical options and therapies to address CVDs, particularly for ischemic damage due to an impairment in the endothelial–myocardial relationship. Because ozone is a stimulator of the endothelial nitric oxide synthase/nitric oxide pathway, ozone therapy has been widely demonstrated to have the ability to counteract endothelial-cardiac disorders, providing a novel straightforward opportunity to reduce the impact of CVDs, including atrial fibrillation. In this review, we attempt to establish a state-of-the-art method for the use of ozone in CVD, suggesting that future remarks be addressed to provide fundamental insights into this issue. The purpose of this study was to highlight the role of ozone in the adjunctive medical treatment of cardiovascular pathologies such as acute myocardial infarction due to ischemic disorders.
Keywords: acute myocardial infarction, atrial fibrillation, autohemotherapy, ischemia, ozone therapy, ozone, SIOOT, stroke
Introduction
Cardiovascular disease (CVD) represents one of the leading causes of death worldwide, and the Global Burden of Disease study estimated that at least 17.8 million deaths by the last five years are associated with CVD.1,2 Among these cases, at least 50% were ischemic heart disease (IHD) and 35% were strokes.1 Therefore, IHD has been the most common cause of death in Europe since the 1980s.1,2,3,4 In Italy, the prevalence of CVD has been estimated to be twofold higher than the global estimated prevalence (12.9% vs. 6.6%); however, when evaluations were corrected for age confounders, estimates were similar (6.2% vs. 6.3%).4 The major burden of CVD is represented by IHD, with a prevalence estimated to be 3.6% in Italy and 1.7% in an age-standardized evaluation, a value representing a crucial health alarm, as doubling the corresponding global estimation (1.7%).4
IHD is a generic term used for a group of closely related syndromes that result from myocardial ischemia, usually an imbalance between the supply (perfusion) and the cardiac demand for oxygenated blood. In 90% of cases, myocardial ischemia is caused by a reduction in the blood flow of the coronary artery, which is due mainly, yet not exclusively, to atherosclerosis.5,6,7 The conventional belief about IHD pathogenesis includes the causative participation of an obstructive plaque that inhibits or reduces blood flow throughout the coronary artery and causes myocardial ischemia.8 To date, any coronary artery disease (CAD) has been fairly defined by the exclusive presence of obstruction due to atherosclerotic plaques.9 Nevertheless, in recent years, major attention has been given to the ability of the coronary artery to regulate critical stenosis in blood flow via myogenic and metabolic factors.7
In the case of atherosclerotic plaque-mediated obstruction, over 70% of the luminal cross-sectional area is involved, thus reducing the coronary diameter by approximately 50%, but in this case, an increase in the proximal resistance leads to a reduction in the distal coronary perfusion pressure and elicits an autoregulation mechanism that is able to maintain the basal coronary blood flow.8 Obviously, this should lead to a nonsymptomatic situation only at rest, whereas insufficient blood flow is associated with high oxygen demand upon physical exercise.7
Atherosclerotic lesions, underlying IHD, in most cases have a slow and silent progression, which can last even decades, and often, their origin can be traced back to childhood or adolescence.10,11
Typically, IHD is present as an acute form, often with unstable angina, heart attack, myocardial stroke and sudden cardiac death, when an atheromatous plaque undergoes an unpredictable and abrupt transformation and, owing to subsequent surface erosion, ulceration, fissuring, rupture, etc., it loses its stability, potentially becoming life-threatening for the patient.12,13 The exposure of the plaque content to the blood affects a series of reactions that lead to the formation of a mural thrombus,14 which obstructs, in a more or less marked way, the coronary artery and can detach and enter the circulation in the form of an occlusive thrombus that blocks the flow through the coronary artery.7,8,15,16
To better elucidate the etiopathogenesis of IHD, even coronary microcirculation should be considered. The existence of the coronary tree of arterioles with a diameter ranging from 50 μm to 200 μm represents a microcirculation barrier, which encompasses approximately 60% of coronary artery resistance.17 In recent years, coronary microcirculation abnormalities, which have been investigated as a leading cause of IHD, have been collectively described under the umbrella term of coronary microvascular dysfunction and related to causative factors of type II myocardial infarction.18
In fact, coronary microvascular dysfunction causes an increase in coronary flow resistance, leading to reduced perfusion pressure and subsequently to myocardial ischemia.19 Some studies have reported that shear stress impairment exacerbates endothelial dysfunction, particularly by enhancing thrombus formation in the epicardial arteries.20,21 When chronic stenosis of the coronary artery occurs, arteriole inward remodeling and rarefaction lead to an increased vasoconstriction response to endothelin (because of the loss of endothelin B receptor-mediated vasodilation) and a blunted myogenic response.22,23,24
The most important form of IHD is myocardial infarction (AMI), commonly called “heart stroke,” which is the leading cause of death in industrialized countries.25,26 The risk of developing AMI increases with increasing age and in the presence of predisposing factors to atherosclerosis. The global mortality rate in the first year from the onset of AMI is approximately 30% (including patients who fail to reach the hospital); subsequently, among survivors, the mortality rate is 3–4% per year.25,26 The most common pathogenetic evidence concerning myocardial infarction is that AMI is caused by a coronary occlusion that leads to a loss of myocardial perfusion with consequent functional, biochemical and morphological alterations. Ischemia is considered when, on the basis of its extent and duration, it can cause necrosis in the anatomical region of the affected artery, especially at the subendocardial level.7,8
The occurrence of a cardiac stroke (infarction) is the final result of a complex milieu of triggering factors, which also encompass the strategic role of the microvasculature and coronary endothelia. The role of endothelial physiology in IHD may be particularly crucial in deepening IHD pathogenesis and preventing strokes from myocardial ischemia.27,28 Therefore, it is crucial to focus on the role of endothelia in IHD to understand the activity of oxygen–ozone therapy in these districts.
Finally, atrial fibrillation (AF) is considered another comorbid factor in AMI pathogenesis.29 AF is the most common type of sustained arrhythmia found in clinical practice. His prevalence in the general population was reported to be just under 1%: 0.95% in the study by North American AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA)30 and 0.87% in the Scottish study by Murphy et al.31 In two more publications related to data collected in the United States, the prevalence was higher: 1.12% and 2.5%, with a net increase compared with previous years.32,33 This trend would confirm forecasts of a 2–3-fold increase in the number of affected patients in the United States AF by the year 2050 (from the current 2.5–6 million to 6–15 million).30,33 The prevalence of AF increases with increasing age. In the ATRIA1 study, the prevalence was 0.1% in subjects aged < 55 years and 9% in those aged > 80 years; in the Framingham study, the prevalence progressively increased from 0.5% in the age group between 50 and 59 years to 1.8% between 60 and 69 years, 4.8% between 70 and 79 years and 8.8% between 80 and 89 years,34 and 70% of patients with AF were over 65 years, with a median age of 75 years.35 The prevalence is slightly higher in men than in women in all age groups (on average, 1.1% vs. 0.8%).35 Finally, more recent epidemiology on AF and insights into the use of stem cells for AF are included in this introduction survey.36,37
The pathogenetic link between AF and ischemic damage, leading to AMI, shares many markers with IHD,38 such as inflammation39 and platelet activation,40 where some insightful biomarkers, such as prostaglandin-F2-alpha, beta-thromboglobulin, P-selectin and soluble CD40L, are elevated during AF.40 In this review, we highlight the role of ozone in the treatment of ischemic myocardial infarction and other cardiovascular pathologies. The survey included a literature search within PubMed, Embase, Web of Science, and Scopus with the term search “ozone & cardiovascular disease” from 2001 to date.
The endothelial involvement in the ischemic heart diseases and the role of ozone
CAD, which leads to myocardial ischemia or, more generally, IHD, is also caused by disorders in endothelial function.41 Endothelial dysfunction is one of the leading causes exacerbating myocardial ischemia in the daily activities of individuals at risk for IHD.42,43 Therefore, coronary artery endothelia, as well as microcirculating endothelia, may represent fundamental targets for therapy and preventive approaches against IHD and myocardial ischemic stroke.44 Cardioprotection targeting endothelia is a leading matter of debate in the context of IHD.
Endothelial health is paramount for myocardial contraction, as the endothelium contributes to improving the sensitivity of myofilaments to calcium45 but, in particular, protects the microvasculature upon ischemia/reperfusion (I/R) injury,46,47 preventing myocardial infarction.
In this context, mitochondrial dysfunction is a frequent hallmark of I/R injury in the heart and is often driven via an unfolded protein response (UPRmt).48 A recent gene expression investigation revealed that the UPRmt partially inhibits oxidative stress in myocardial tissue by upregulating the expression of mitochondrion-localized antioxidant proteins but also by promoting the upregulation of mitochondrial fusion-related genes and mitochondrial biogenesis in the reperfused heart, triggering FUN14 domain-containing 1-induced mitophagy to restore functional mitochondria during I/R injury.49 Factors able to elicit mild mitochondrial stress in the context of I/R injury, such as the lipid peroxide 4-hydroxynonenal (4-HNE), may overcome the stress response and lead to full recovery from I/R injury.50
The role of mitochondria in the healthy physiology of endothelia to prevent I/R injury and, subsequently, the risk of myocardial ischemic stroke is paramount.51 The vicious shortcut leading to heart stroke includes disorders in microvascular perfusion, endothelial dysfunction leading to platelet/endothelial impairment and platelet activation and ultimately cardiomyocyte apoptosis caused by additional ischemic events during reperfusion.51,52
Endothelia contain fewer mitochondria than cardiomyocytes do; therefore, the effect of ischemic injury and the consequent increase in reactive oxygen species (ROS) is particularly stringent in this tissue, which is much more prone to undergo cellular senescence and apoptosis once ROS signals exceed the survival threshold.51,53
Moreover, even the role of nitric oxide (NO) must be considered.54 NO is widely known to play a protective role in I/R injury,55 and the bioavailability of endothelial NO is dramatically reduced upon myocardial infarction and heart failure.56
A recent study by Ajamieh et al.57 The authors used ozone oxidative preconditioning by rectal insufflation (1 mg/kg) on adult male Wistar rats before experimental induction of I/R injury and reported that ozone pretreatment exerted a protective effect on I/R-induced liver damage, increased the synthesis of endothelial NO and reduced the effect of N-ω-nitro-L-arginine methyl ester (NAME) on endothelial NO synthase (eNOS) and inducible nitric oxide synthase activity.57 Specifically, the rats were classified into six groups: the sham-operated (control), NAME, I/R, ozone + I/R, ozone + NAME + I/R and NAME + I/R groups. The animals underwent surgical intervention involving laparotomy and surgical manipulation, which included the right vs. the left hepatic arteria and vein, without inducing ischemia; intravenous injection of 10 mg/kg NAME (groups B, E, and F); and 90 minutes of right hepatic lobe ischemia following 90 minutes of reperfusion and 15 ozone treatments with 50 μg/mL O3.57 Ozone reduces the impact of ischemia and the worsening effect of NO and ameliorates the oxidative stress response.57
Medical ozone, which is used at low physiological dosages to treat numerous disorders,58 can target many fundamental components in the prevention or therapy of IHDs and myocardial infarction, including cardiac arrhythmias.
Di Filippo et al.59 reported the antiarrhythmic effect of ozone on experimental animals; however, to the best of our knowledge, no clinical investigations on ozone and AF have been reported. Sprague–Dawley rats were anesthetized and subjected to three kinds of treatments (three groups), namely, saline (control), aconitine 15 μg/kg (intravenous injection), 1.5% KCl (intravenous injection) and lidocaine, to induce two kinds of arrhythmias, i.e., sodium (aconitine) and potassium (KCl)-dependent arrhythmia.59 When rats were treated with 100 to 300 μg/kg O3, doses ranging from 150 to 300 μg/kg O3 greatly reduced ventricular fibrillation, ventricular tachycardia and mortality caused by reperfusion, whereas a dose of 100 μg/kg O3 was ineffective.59
From a clinical point of view, the role of oxygen ozone therapy in patients suffering from IHD or affected by myocardial infarction was initially presumptive on the basis of several tests on laboratory animals, which were performed in Italy since 1991, when Lettieri and colleagues successfully treated a cohort of patients with AMI with oxygen-ozone autohemotherapy, reporting evidence on the use of ozone, the mechanism of which they investigated many years later in laboratory animals.
In 2010, Di Filippo’s group60 subjected Sprague–Dawley rats to 25 minutes of occlusion of the left descending coronary artery, followed by 2 hours of reperfusion, whereas 30 minutes before ischemia, three different doses of an oxygen/ozone mixture, i.e., 100, 150 and 300 μg/mL ozone, were insufflated in the test group. The biomarkers used were myocardial infarct size and immunochemistry for endothelial progenitor cells (EPCs).60 A dose of 150 μg/mL ozone in the O2/O3 mixture reduced the infarct size by 35% (P < 0.01) and increased eNOS, contributing to the upregulation of EPCs.60
The purported positive use of medical ozone in IHD and other CVDs may lie in the ability of ozone to act in a hormetic way61 on fundamental targets, such as mitochondrial turnover and biogenesis and NO release, in addition to promoting the proliferation of EPCs and a cardiomyocyte stem repair phenotype,60 an issue we address further in the manuscript.
Oxygen-ozone therapy in cardiovascular disorders: the evidence
Bocci et al.62 wondered if ozone could be useful in improving cardiovascular disorders. Recent evidence should support the hypothesis that ozone is able to improve cardiovascular disorders. A recent study by Martínez-Sánchez et al.63 reported the effective use of ozone in 53 patients with CAD previously treated with an antithrombotic therapy protocol. In this randomized controlled trial, 27 patients were treated with only antithrombotic therapy (Aspirin®, acetyl-salicylic acid (125 mg) and Ateromixol®, policosanol (5 mg), once a day for 20 days), and 26 patients were treated with the same antithrombotic protocol plus 40 μg/mL via rectal insufflation of 3% ozone in 200 mL of an oxygen–ozone calibrated mixture.63 Patients undergoing the ozone protocol showed that ozone enhanced the antithrombotic effect of acetyl salicylate with policosanol, improving the antioxidant status of patients and ameliorating their clinical status, although they reported mild platelet activation, which was attributed to the ozone effect on platelet adenosine receptors.63,64 Bocci et al.65 reported in past reports that the use of heparin, instead of calcium chelators such as sodium citrate, prior to oxygen-ozone autohemotherapy may cause platelet aggregation rather than inhibiting platelet activation, whereas oxygen-ozone autohemotherapy with 50 μg/mL O3 in the presence of sodium citrate as an anticoagulant did not induce spontaneous or adenosine-induced platelet aggregation.66 This raises the fundamental concern that protocols in ozone therapy must be particularly tailored and standardized to achieve the best outcome and prevent any adverse effects.
The ability of ozone to elicit eNOS and promote the recruitment of EPCs is a landmark of the ability of this oxygen allotrope to intervene in restoring healthy cardiovascular physiology.60 Ozone triggers NO from endothelia via a mechanism enhanced by NO precursors such as L-arginine and blunted by eNOS inhibitors such as NAME.67 This should lead to the assessment that ozone, and its lipoperoxide mediators, act mainly on the microvasculature district to achieve positive outcomes in patients with myocardial disorders.
In peripheral occlusive arterial disease, oxygen–ozone autohemotherapy ameliorated both the rheological properties of circulating blood and oxygen release to tissues.68,69,70 In Giunta et al.’s clinical trial,68 27 patients with peripheral occlusive arterial disease, clinical stage II−III (La Fontaine criteria), underwent major oxygen-ozone autohemotherapy. Ozone treatment reduced blood viscosity (P < 0.01) and fibrinogen, whereas hematocrit and 2,3-diphosphoglycerate did not change significantly.68 Protocols should often be debated to achieve the best performance possible. The use of ozone therapy to treat cardiovascular and myocardial disorders needs to be reappraised and debated at a consensus conference to tailor the most correct and suitable protocol for these pathologies.
In heart failure with a reduced ejection fraction, Buyuklu and coworkers71 treated 40 patients undergoing conventional heart failure therapy with three major additional treatments of 20–50 μg/mL oxygen-ozone autohemotherapy and one minor treatment with 20–40 μg/mL intramuscular O3 for 5 weeks, matching the results with 40 patients treated with only heart failure therapy as controls. In this study, ozone increased the plasma levels of antioxidant enzymes, i.e., superoxide dismutase, catalase, glutathione and glutathione peroxidase.71 The left ventricular end-systolic and diastolic volumes were significantly reduced by ozone treatment, whereas the left ventricular ejection fraction (LVEF) was only modestly increased but not significantly (P = 0.567). Furthermore, ozone treatment reduced plasma NO and malonyl-dialdehyde, in addition to the heart failure marker N-terminal pro-brain natriuretic peptide.71 The 6-minute walk distance exercise, performed by patients undergoing ozone therapy, was notably improved.71
The complex pattern of results from this study suggests that a sound and rigorous protocol of oxygen–ozone therapy for IHD, CAD and general CVDs is particularly crucial.
Pandolfi and colleagues72 reported, in a significant case report, that a 76-year critically ill patient suffering from IHD with some episodic occurrences of myocardial infarction improved his LVEF under 33% for 14 months upon 40 μg/mL ozone treatment, despite several medical treatments and coronary angioplasty, to an LVEF of 50%, only after two oxygen–ozone autohemotherapy sessions on a weekly basis, according to protocols from the Italian Society of Oxygen Ozone Therapy.
Cecilio Cespedes-Suarez et al.73 succeeded in increasing the LVEF in a cohort of six patients with Class II–III chronic heart failure from 33% to 50%, following a protocol of ozone rectal insufflation arranged in four weeks, each with 5 sessions and with constantly 5 μg/mL weekly increasing dosages of ozone from 25 to 40 μg/mL. In Cespedes-Suarez et al.’s study,73 all twelve patients underwent conventional heart failure therapy, whereas only a cohort of six patients were additionally treated with ozone. In this group, 66.7% of patients had class III heart failure, whereas following ozone therapy, patients were no longer classified as class III heart failure; however, 16.6% were class I heart failure, and 83.3% were class II heart failure, which improved their clinical stage.73,74 Furthermore, ozone-treated patients presented 50% lower levels of the heart failure marker natriuretic peptide N-terminal pro-brain natriuretic peptide.73 In this research study, the authors used rectal ozone via insufflation via a 20-session protocol and increasing concentrations every 5 sessions, starting from 25 μg/mL in 200 mL to 40 μg/mL in 300 mL.73
The promising action of ozone on endothelia may be a possible reason for the myocardial physiology in response to ozone therapy. For example, the role of NO signaling in heart failure with preserved ejection fraction (HFpEF) is crucial74, and in HFpEF, the function of endothelia may be deranged, as shown recently by comparisons of serum NO-derived metabolites in heart failure patients with a reduced ejection fraction and HFpEF patients.75 If NO is a stringent factor in heart failure, the role of ozone should be particularly tuned to the most functional protocol available thus far. NO clearly has a beneficial effect on heart failure, improving left ventricular function.76,77 The activity of ozone, as well as its lipid mediators, such as the electrophile 4-HNE, on endothelia indirectly affects full myocardial physiology. The role of 4-HNE is particularly intriguing. This lipid hydroperoxide byproduct, which ozone may form by the oxidation of lipids with ω-6 fatty acids, is particularly abundant in the vascular district, where it exerts a hormetic effect on the arterial lining of the endothelium and smooth musculature, depending on the fine balance between the rate of lipid peroxidation by ROS and the scavenging action of 4-HNE adducts by glutathione-S-transferases.78 Therefore, 4-HNE represents an outstanding signaling molecule involved in the interplay between inflammation, immune surveillance, the stress response, mitochondrial biogenesis and cell turnover, all of which are functional districts where the hormetic activity of ozone is clearly demonstrable.61,79
Role of ozone oncell precursors to prevent cardiac ischemic stroke and treat cardiovascular diseases
Cell therapy is a leading, straightforward and modern approach to repair tissue damage following AMI and allowing myocardial functionality to restore health status.80
As cardiologists know in depth, despite some similarities, HFpEF (with an ejection fraction ≥ 50%) is quite different from heart failure with a reduced ejection fraction (ejection fraction < 50%), therefore representing a challenging cardiovascular syndrome because it is particularly refractory to medicines and medical therapies.81,82,83,84 In addition to sodium glucose cotransporter 2 inhibitors and the promising effect of ozone along with conventional therapy,85 improvements in morbidity or mortality due to HFpEF seem to be overcome by the use of cardiosphere-derived cells, a type of stromal/progenitor cells that are distinct from c-kit-positive cells but that exhibit clonogenicity and multilineage potential and are active via indirect mechanisms.80 Myocardial cell therapy can potentially have beneficial effects, but long-term studies on its safety and efficacy are important. In particular, with regard to its effectiveness, it is necessary to identify its weaknesses and try to address this issue in further studies.
The enormous bulk of knowledge acquired thus far suggests that to overcome the problems of cell therapy, a more complex approach is needed, one that involves more innate biological mechanisms and the other that enhances current strategies. In addition, researchers need approaches that associate the use of stem cells with mechanisms that increase the body’s ability to recall cells and allow their insertion and survival in the damaged area. Homing approaches and engineering are essential to allow stem cells to perform their functions in damaged tissue, particularly when systemically infused. Hypoxic stimuli, which characterize ischemic preconditioning, have been proposed as a therapeutic strategy to improve the survival and engraftment of stem cells.86,87
Chacko and colleagues86 reported that rat mesenchymal stem cells (MSCs) under hypoxia for 24 and 48 hours optimally expressed proteins essential for survival and engineering of the cells in the infarcted heart. The authors used cryopreserved primary rat MSCs, which were positive for CD29 and CD54 and were isolated from the bone marrow of adult Fisher rats treated with hypoxic preconditions (0.5% O2) for 24, 48 and 72 hours. Hypoxic preconditions reduced apoptosis due to severe hypoxia and enhanced the ability of MSCs to differentiate into cardiomyocytes or endothelial precursors.86 The authors concluded that the results obtained provided indications of possible methods for the preparation of MSCs capable of enhancing the effectiveness of in vivo cellular therapy.86
In 2005, Tang and colleagues87 reported in an in vivo mouse model that MSCs engineered to overexpress, in a hypoxic context, the heme oxygenase-1 (HO-1) gene, one of the effectors activated during the late phase of ischemic preconditioning, presented increased survival and viability in the ischemic heart. Other research groups have shown that the cardioprotection conferred by ischemic preconditioning (IPC) is also linked to the mobilization of stem cells and to the production of cardioprotective cytokines that enhance recovery following damage from I/R and chronic ischemia.88,89 During both the early and late stages of IPC, stem cells are mobilized from the bone marrow to the site of damage via the bloodstream, which is likely attracted by the secretion of cytokines and other chemoattractive substances from the damaged organ.
Ii et al.88 using an in vivo mouse model, reported that one classical preconditioning ischemic stimulus could recruit cells into myocardium medullary-derived EPCs within 3 hours. They “imported” cardioprotective mediators, such as inducible nitric oxide synthase and eNOS, in the area of damage.88
Furthermore, when volunteers are subjected to repeated tests for 1 month with cycles of I/R in the arm (remote ischemic preconditioning), an increased level of circulating EPCs and improved endothelial function have been observed in humans,90 but the contribution of EPCs to the improvement of endothelial function has not yet been fully clarified. In this study, upper limb ischemia (by cuff inflation of over 200 mmHg lasting 5 minutes) was performed 6 times a day within 1 month in 30 young healthy men to induce the late phase of IPC and investigate its effect on endothelial function.90 The IPC stimulus increased plasma vascular endothelial growth factor, circulating levels of EPCs and the forearm blood flow response to acetylcholine, suggesting that repetition of late IPC enhances endothelia-mediated vasodilation by increasing NO and EPCs.90
Furthermore, Kamota et al.89 demonstrated that remote IPCs had beneficial effects in an in vivo mouse model. In fact, researchers have reported a reduction in the size of the heart attack and preservation of the ejection fraction of the left ventricle. During the early stage, the serum levels of vascular endothelial growth factor and SDF-1α increase, but during the late phase, there is an increase in the peripheral levels of CD34+ and CD34+/flk– stem cells and an accumulation of bone marrow stem cells in the ischemic risk zone.89 In addition, the use of antibodies directed against stem cells abolished cardioprotective effects.89
Gyöngyösi and colleagues91 showed, in a model of a heart attack in an animal of large size (pig), that the IPC was able to promote mobilization and the recruitment of MSCs and heart stem cells from the circulating sector to the infarcted area and border areas. MSCs seemed to head faster into damaged areas. In addition, during the early phase of IPC, they reported an increase in the levels of some circulating cytokines and growth factors, including vascular endothelial growth factor. These studies have potential clinical implications for cardiac cell therapy, as they suggest that ischemic preconditioning, as well as strategies such as the administration of exogenous cytokines,82 can promote the mobilization and integration of stem cells in ischemic areas.
Di Filippo and colleagues60 reported that oxygen-ozone therapy has a myocardial protective effect against infarction by triggering endothelial expression of eNOS and NO release and by inducing the activity of EPCs and CD34- and CD117/c-kit-positive cells.
Ozone promotes progenitor clonogenetic cells, which are able to repair myocardial damage; however, its activity is broader than its well-known antioxidant and anti-inflammatory effects. EPCs represent a new, encouraging frontier in the improvement of cardiac stress and damage and cardiovascular problems.92 The mechanism by which ozone can regulate the fate and turnover of EPCs may be driven by the same eNOS, which ozone and 4-HNE are able to target. When eNOS is uncoupled, as occurs during diabetes, it produces ROS instead of NO, causing endothelial dysfunction and impairing both the mobilization and function of EPCs, whose biology is controlled by eNOS.93 Under ischemic conditions, pleiotrophin induces angiogenesis in vivo but recruits not only endothelial cells but also EPCs,94 including NO.95 Interestingly, the activity of pleiotrophin, a heparin-binding growth factor, is concurrent with that of the initial pro-oxidant action of ozone and 4-HNE or lipoperoxides, as pleiotrophin is involved both in the formation of ROS as signaling molecules and in being a target of ROS in EPC migration,96 even inducing the transdifferentiation of monocytes into endothelial cells.97 This intriguing ozone-pleiotrophin-ROS-EPC circuit, which also involves eNOS and NO, may be the fundamental hallmark of how ozone can induce EPCs and actively participate in myocardial repair and the recovery of healthy endothelial function in the cardiovascular system.
Conclusions
Figure 1 summarizes the major impact of ozone in the complex milieu of CVD. The role of ozone in cardiovascular disorders has been debated for several years,62 and to date, novel biomedical data can encourage the use of ozone adjunct treatment in this field. Further insights are needed to assess the fundamental role of ozone in cardiovascular disorders, including IHD and myocardial infarction. Promising clinical and experimental studies, which need to be further implemented, should expand the debate about the correct protocols of ozone therapy to be applied in singular and defined cardiac and cardiovascular disorders to optimize clinical outcomes and provide patients with more favorable perspectives and life expectance. Ozone has been widely demonstrated to target important components of cardiac physiology, both in vitro and in vivo, and in this sense, research on oxygen-ozone therapy is still attractive and encouraging for clinical cardiology.
Figure 1.
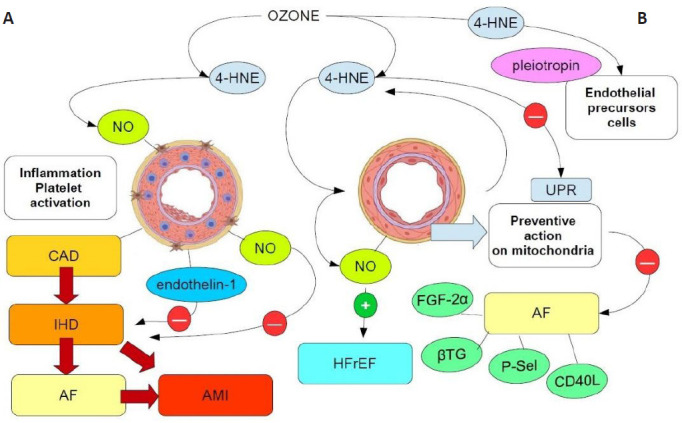
Carton shows the major functional pathways associated with the effects of ozone therapy on CVD.
The major ozone intermediate is 4-HNE, which may hormetically act on compromised arterial functions due to plaque-mediated obstruction (A) or other etiopathogenetic mechanisms involving microcirculation and arterial endothelia (B). In A, plaque-induced inflammation, platelet activation and IHD may worsen the clinical scenario, ultimately leading to AMI, despite the activation of myogenic protective factors, such as NO and endothelin-1. Ozone prevents AMI and AF mainly by inducing the endothelial production of NO and reducing inflammation and innate cell pyroptosis via 4-HNE in mitochondria (not shown). The triggering of eNOS and production of NO can reduce AF biomarkers (FGF-2α, CD40L, P-Sel, βTG, and so on), improving HFrEF and rejuvenating endothelia and myocardial tissue via the induction of EPCs by 4-HNE-induced pleiotropin (B). Red circles: inhibition; green circles: activation. 4-HNE: 4-Hydroxynonenal; AF: atrial fibrillation; AMI: acute myocardial infarction; CAD: coronary artery disease; CVD: cardiovascular disease; eNOS: endothelial nitric oxide synthase; EPC: endothelial progenitor cell; FGF-2α: fibroblast growth factor-2α; HFrEF: heart failure with reduced ejection fraction; IHD: ischemic heart disease; NO: nitric oxide; P-Sel: P-selectin; UPR: unfolded protein response; βTG: β-thromboglobulin.
Footnotes
Conflicts of interest
The authors state that they have no conflicts of interest.
Data availability statement:
No additional data are available.
References
- 1.Townsend N, Kazakiewicz D, Lucy Wright F, et al. Epidemiology of cardiovascular disease in Europe. Nat Rev Cardiol. 2022;19:133–143. doi: 10.1038/s41569-021-00607-3. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1736–1788. doi: 10.1016/S0140-6736(18)32203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panico S, Mattiello A. Epidemiology of cardiovascular diseases in women in Europe. Nutr Metab Cardiovasc Dis. 2010;20:379–385. doi: 10.1016/j.numecd.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Saglietto A, Manfredi R, Elia E, et al. Cardiovascular disease burden: Italian and global perspectives. Minerva Cardiol Angiol. 2021;69:231–240. doi: 10.23736/S2724-5683.21.05538-9. [DOI] [PubMed] [Google Scholar]
- 5.Jensen RV, Hjortbak MV, Bøtker HE. Ischemic heart disease: an update. Semin Nucl Med. 2020;50:195–207. doi: 10.1053/j.semnuclmed.2020.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Khan MA, Hashim MJ, Mustafa H, et al. Global epidemiology of ischemic heart disease: results from the Global Burden of Disease Study. Cureus. 2020;12:e9349. doi: 10.7759/cureus.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boden WE, Stone PH. Lessons learned from the ISCHEMIA trial for the management of patients with stable ischemic heart disease. Annu Rev Med. 2023;74:189–198. doi: 10.1146/annurev-med-042921-124013. [DOI] [PubMed] [Google Scholar]
- 8.Severino P, D’Amato A, Pucci M, et al. Ischemic heart disease pathophysiology paradigms overview: from plaque activation to microvascular dysfunction. Int J Mol Sci. 2020;21:8118. doi: 10.3390/ijms21218118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao C, Wang J, Tian J, Tang YD. Coronary artery disease: from mechanism to clinical practice. Adv Exp Med Biol. 2020;1177:1–36. doi: 10.1007/978-981-15-2517-9_1. [DOI] [PubMed] [Google Scholar]
- 10.Bassareo PP, O’Brien ST, Dunne E, et al. Should we be screening for ischaemic heart disease earlier in childhood? Children (Basel) 2022;9:982. doi: 10.3390/children9070982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer JF, Larsen SB, Blond K, Damsgaard CT, Bjerregaard LG, Baker JL. Associations between body mass index and height during childhood and adolescence and the risk of coronary heart disease in adulthood: A systematic review and meta-analysis. Obes Rev. 2021;22:e13276. doi: 10.1111/obr.13276. [DOI] [PubMed] [Google Scholar]
- 12.Alatorre CI, Hoogwerf BJ, Deeg MA, et al. Factors associated with stroke, myocardial infarction, ischemic heart disease, unstable angina, or mortality in patients from real world clinical practice with newly-diagnosed type 2 diabetes and early glycemic control. Curr Med Res Opin. 2018;34:337–343. doi: 10.1080/03007995.2017.1396969. [DOI] [PubMed] [Google Scholar]
- 13.Owlia M, Dodson JA, King JB, et al. Angina severity, mortality, and healthcare utilization among veterans with stable angina. J Am Heart Assoc. 2019;8:e012811. doi: 10.1161/JAHA.119.012811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prati F, Gurguglione G, Biccire F, et al. Sudden cardiac death in ischaemic heart disease: coronary thrombosis or myocardial fibrosis? Eur Heart J Suppl. 2023;25:B136–B139. doi: 10.1093/eurheartjsupp/suad093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scheitz JF, Sposato LA, Schulz-Menger J, Nolte CH, Backs J, Endres M. Stroke-heart syndrome: recent advances and challenges. J Am Heart Assoc. 2022;11:e026528. doi: 10.1161/JAHA.122.026528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frąk W, Wojtasińska A, Lisińska W, Młynarska E, Franczyk B, Rysz J. Pathophysiology of cardiovascular diseases: new insights into molecular mechanisms of atherosclerosis, arterial hypertension, and coronary artery disease. Biomedicines. 2022;10:1938. doi: 10.3390/biomedicines10081938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedele F, Severino P, Bruno N, et al. Role of ion channels in coronary microcirculation: a review of the literature. Future Cardiol. 2013;9:897–905. doi: 10.2217/fca.13.65. [DOI] [PubMed] [Google Scholar]
- 18.Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction (2018) J Am Coll Cardiol. 2018;72:2231–2264. doi: 10.1016/j.jacc.2018.08.1038. [DOI] [PubMed] [Google Scholar]
- 19.Crea F, Montone RA. Pathophysiology of coronary microvascular dysfunction. Vascul Pharmacol. 2023;153:107239. doi: 10.1016/j.vph.2023.107239. [DOI] [PubMed] [Google Scholar]
- 20.Lerman A, Holmes DR, Herrmann J, Gersh BJ. Microcirculatory dysfunction in ST-elevation myocardial infarction: cause, consequence, or both? Eur Heart J. 2007;28:788–797. doi: 10.1093/eurheartj/ehl501. [DOI] [PubMed] [Google Scholar]
- 21.Berry C, Duncker DJ. Coronary microvascular disease: the next frontier for cardiovascular research. Cardiovasc Res. 2020;116:737–740. doi: 10.1093/cvr/cvaa035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbieta Caceres VH, Lin J, Zhu XY, et al. Early experimental hypertension preserves the myocardial microvasculature but aggravates cardiac injury distal to chronic coronary artery obstruction. Am J Physiol Heart Circ Physiol. 2011;300:H693–701. doi: 10.1152/ajpheart.00516.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorop O, Merkus D, de Beer VJ, et al. Functional and structural adaptations of coronary microvessels distal to a chronic coronary artery stenosis. Circ Res. 2008;102:795–803. doi: 10.1161/CIRCRESAHA.108.172528. [DOI] [PubMed] [Google Scholar]
- 24.Duncker DJ, Koller A, Merkus D, Canty JM., Jr Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis. 2015;57:409–422. doi: 10.1016/j.pcad.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Virani SS, Alonso A, Aparicio HJ, et al. Heart Disease and Stroke Statistics-2021 update: a report from the American Heart Association. Circulation. 2021;143:e254–e743. doi: 10.1161/CIR.0000000000000950. [DOI] [PubMed] [Google Scholar]
- 26.Tsao CW, Aday AW, Almarzooq ZI, et al. Heart Disease and Stroke Statistics-2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052. [DOI] [PubMed] [Google Scholar]
- 27.Oikonomou E, Siasos G, Tsigkou V, et al. Coronary artery disease and endothelial dysfunction: novel diagnostic and therapeutic approaches. Curr Med Chem. 2020;27:1052–1080. doi: 10.2174/0929867326666190830103219. [DOI] [PubMed] [Google Scholar]
- 28.Medina-Leyte DJ, Zepeda-García O, Domínguez-Pérez M, González-Garrido A, Villarreal-Molina T, Jacobo-Albavera L. Endothelial dysfunction, inflammation and coronary artery disease: potential biomarkers and promising therapeutical approaches. Int J Mol Sci. 2021;22:3850. doi: 10.3390/ijms22083850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Violi F, Soliman EZ, Pignatelli P, Pastori D. Atrial fibrillation and myocardial infarction: a systematic review and appraisal of pathophysiologic mechanisms. J Am Heart Assoc. 2016;5:e003347. doi: 10.1161/JAHA.116.003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA. 2001;285:2370–2375. doi: 10.1001/jama.285.18.2370. [DOI] [PubMed] [Google Scholar]
- 31.Murphy NF, Simpson CR, Jhund PS, et al. A national survey of the prevalence, incidence, primary care burden and treatment of atrial fibrillation in Scotland. Heart. 2007;93:606–612. doi: 10.1136/hrt.2006.107573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol. 2009;104:1534–1539. doi: 10.1016/j.amjcard.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 33.Miyasaka Y, Barnes ME, Gersh BJ, et al. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation. 2006;114:119–125. doi: 10.1161/CIRCULATIONAHA.105.595140. [DOI] [PubMed] [Google Scholar]
- 34.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–988. doi: 10.1161/01.str.22.8.983. [DOI] [PubMed] [Google Scholar]
- 35.Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med. 1995;155:469–473. [PubMed] [Google Scholar]
- 36.Kornej J, Börschel CS, Benjamin EJ, Schnabel RB. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ Res. 2020;127:4–20. doi: 10.1161/CIRCRESAHA.120.316340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leowattana W, Leowattana T, Leowattana P. Human-induced pluripotent stem cell-atrial-specific cardiomyocytes and atrial fibrillation. World J Clin Cases. 2022;10:9588–9601. doi: 10.12998/wjcc.v10.i27.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Shetry M, Mahfouz R, Frere AF, Abdeldayem M. The interplay between atrial fibrillation and acute myocardial infarction. Br J Hosp Med (Lond) 2021;82:1–9. doi: 10.12968/hmed.2020.0584. [DOI] [PubMed] [Google Scholar]
- 39.Pignatelli P, Pastori D, Carnevale R, et al. Serum NOX2 and urinary isoprostanes predict vascular events in patients with atrial fibrillation. Thromb Haemost. 2015;113:617–624. doi: 10.1160/TH14-07-0571. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Sun K, Zhao R, et al. Inflammatory biomarkers of coronary heart disease. Front Biosci (Schol Ed) 2018;10:185–196. doi: 10.2741/s508. [DOI] [PubMed] [Google Scholar]
- 41.Bockus L, Kim F. Coronary endothelial dysfunction: from pathogenesis to clinical implications. Open Heart. 2022;9:e002200. doi: 10.1136/openhrt-2022-002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allbritton-King JD, García-Cardeña G. Endothelial cell dysfunction in cardiac disease: driver or consequence? Front Cell Dev Biol. 2023;11:1278166. doi: 10.3389/fcell.2023.1278166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimabukuro M. L-Arginine, Nitric Oxide, and Endothelial Dysfunction Underlying Atherosclerotic Cardiovascular Disease (ASCVD) J Atheroscler Thromb. 2023;30:1311–1312. doi: 10.5551/jat.ED235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrera-Zelada N, Zuñiga-Cuevas U, Ramirez-Reyes A, Lavandero S, Riquelme JA. Targeting the endothelium to achieve cardioprotection. Front Pharmacol. 2021;12:636134. doi: 10.3389/fphar.2021.636134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen X, Tan Z, Zhong X, et al. Endocardial endothelium is a key determinant of force-frequency relationship in rat ventricular myocardium. J Appl Physiol (1985) 2013;115:383–393. doi: 10.1152/japplphysiol.01415.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res. 2016;8:765–777. [PMC free article] [PubMed] [Google Scholar]
- 47.Devan AE, Umpierre D, Harrison ML, et al. Endothelial ischemia-reperfusion injury in humans: association with age and habitual exercise. Am J Physiol Heart Circ Physiol. 2011;300:H813–819. doi: 10.1152/ajpheart.00845.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu MX, Ma XF, Niu X, Fan GB, Li Y. Mitochondrial unfolded protein response in ischemia-reperfusion injury. Brain Res. 2022;1797:148116. doi: 10.1016/j.brainres.2022.148116. [DOI] [PubMed] [Google Scholar]
- 49.Ji H, Wang J, Muid D, Song W, Jiang Y, Zhou H. FUNDC1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury. Cell Signal. 2022;92:110249. doi: 10.1016/j.cellsig.2022.110249. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Henderson GI, Freeman GL. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. J Mol Cell Cardiol. 2001;33:1919–1927. doi: 10.1006/jmcc.2001.1454. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;23:299–314. doi: 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- 52.Cong L, Bai Y, Guo Z. The crosstalk among autophagy, apoptosis, and pyroptosis in cardiovascular disease. Front Cardiovasc Med. 2022;9:997469. doi: 10.3389/fcvm.2022.997469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Toan S. Pathological roles of mitochondrial oxidative stress and mitochondrial dynamics in cardiac microvascular ischemia/reperfusion injury. Biomolecules. 2020;10:85. doi: 10.3390/biom10010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weerateerangkul P, Chattipakorn S, Chattipakorn N. Roles of the nitric oxide signaling pathway in cardiac ischemic preconditioning against myocardial ischemia-reperfusion injury. Med Sci Monit. 2011;17:RA44–52. doi: 10.12659/MSM.881385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navati MS, Lucas A, Liong C, et al. Reducing ischemia/reperfusion injury by the targeted delivery of nitric oxide from magnetic-field-induced localization of S-nitrosothiol-coated paramagnetic nanoparticles. ACS Appl Bio Mater. 2019;2:2907–2919. doi: 10.1021/acsabm.9b00282. [DOI] [PubMed] [Google Scholar]
- 56.Landmesser U, Engberding N, Bahlmann FH, et al. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–1939. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 57.Ajamieh HH, Menéndez S, Martínez-Sánchez G, et al. Effects of ozone oxidative preconditioning on nitric oxide generation and cellular redox balance in a rat model of hepatic ischaemia-reperfusion. Liver Int. 2004;24:55–62. doi: 10.1111/j.1478-3231.2004.00885.x. [DOI] [PubMed] [Google Scholar]
- 58.Elvis AM, Ekta JS. Ozone therapy: A clinical review. J Nat Sci Biol Med. 2011;2:66–70. doi: 10.4103/0976-9668.82319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di Filippo C, Cervone C, Rossi C, et al. Antiarrhythmic effect of acute oxygen-ozone administration to rats. Eur J Pharmacol. 2010;629:89–95. doi: 10.1016/j.ejphar.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 60.Di Filippo C, Luongo M, Marfella R, et al. Oxygen/ozone protects the heart from acute myocardial infarction through local increase of eNOS activity and endothelial progenitor cells recruitment. Naunyn Schmiedebergs Arch Pharmacol. 2010;382:287–291. doi: 10.1007/s00210-010-0545-2. [DOI] [PubMed] [Google Scholar]
- 61.Franzini M, Valdenassi L, Pandolfi S, et al. The biological activity of medical ozone in the hormetic range and the role of full expertise professionals. Front Public Health. 2022;10:979076. doi: 10.3389/fpubh.2022.979076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bocci V, Travagli V, Zanardi I. May oxygen-ozone therapy improves cardiovascular disorders? Cardiovasc Hematol Disord Drug Targets. 2009;9:78–85. doi: 10.2174/187152909788488681. [DOI] [PubMed] [Google Scholar]
- 63.Martínez-Sánchez G, Delgado-Roche L, Díaz-Batista A, Pérez-Davison G, Re L. Effects of ozone therapy on haemostatic and oxidative stress index in coronary artery disease. Eur J Pharmacol. 2012;691:156–162. doi: 10.1016/j.ejphar.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 64.León Fernández OS, Ajamieh HH, Berlanga J, et al. Ozone oxidative preconditioning is mediated by A1 adenosine receptors in a rat model of liver ischemia/ reperfusion. Transpl Int. 2008;21:39–48. doi: 10.1111/j.1432-2277.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- 65.Bocci V, Valacchi G, Rossi R, et al. Studies on the biological effects of ozone: 9. Effects of ozone on human platelets. Platelets. 1999;10:110–116. doi: 10.1080/09537109976167. [DOI] [PubMed] [Google Scholar]
- 66.Tylicki L, Lizakowski S, Biedunkiewicz B, et al. Platelet function unaffected by ozonated autohaemotherapy in chronically haemodialysed patients. Blood Coagul Fibrinolysis. 2004;15:619–622. doi: 10.1097/00001721-200410000-00014. [DOI] [PubMed] [Google Scholar]
- 67.Valacchi G, Bocci V. Studies on the biological effects of ozone: 11. Release of factors from human endothelial cells. Mediators Inflamm. 2000;9:271–276. doi: 10.1080/09629350020027573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giunta R, Coppola A, Luongo C, et al. Ozonized autohemotransfusion improves hemorheological parameters and oxygen delivery to tissues in patients with peripheral occlusive arterial disease. Ann Hematol. 2001;80:745–748. doi: 10.1007/s002770100377. [DOI] [PubMed] [Google Scholar]
- 69.Turczyński B, Sroczyński J, Antoszewski Z, et al. Ozone therapy and viscosity of blood and plasma, distance of intermittent claudication and certain biochemical components in patients with diabetes type II and ischemia of the lower extremities. Pol Tyg Lek. 1991;46:708–710. [PubMed] [Google Scholar]
- 70.Verrazzo G, Coppola L, Luongo C, et al. Hyperbaric oxygen, oxygen-ozone therapy, and rheologic parameters of blood in patients with peripheral occlusive arterial disease. Undersea Hyperb Med. 1995;22:17–22. [PubMed] [Google Scholar]
- 71.Buyuklu M, Kandemir FM, Set T, et al. Beneficial effects of ozone therapy on oxidative stress, cardiac functions and clinical findings in patients with heart failure reduced ejection fraction. Cardiovasc Toxicol. 2017;17:426–433. doi: 10.1007/s12012-017-9400-8. [DOI] [PubMed] [Google Scholar]
- 72.Pandolfi S, Zammitti A, Franzini M, et al. Effects of oxygen ozone therapy on cardiac function in a patient with a prior myocardial infarction. Ozone Ther. 2017;2:6745. [Google Scholar]
- 73.Cecilio Cespedes-Suarez J, Conde-Perez P, Calunga-Fernandez JL, et al. Study of Left Ventricle Ejection Fraction and Natriuretic Peptides in patients with heart failure treated with systemic medical ozone. J Ozone Ther. 2021;5:69–78. [Google Scholar]
- 74.Greene SJ, Butler J, Spertus JA, et al. Comparison of New York Heart Association class and patient-reported outcomes for heart failure with reduced ejection fraction. JAMA Cardiol. 2021;6:522–531. doi: 10.1001/jamacardio.2021.0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cruz L, Ryan JJ. Nitric oxide signaling in heart failure with preserved ejection fraction. JACC Basic Transl Sci. 2017;2:341–343. doi: 10.1016/j.jacbts.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rassaf T, Poll LW, Brouzos P, et al. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699–1705. doi: 10.1093/eurheartj/ehl096. [DOI] [PubMed] [Google Scholar]
- 77.Koul K, LaPenna K, Li Z, et al. Nitric oxide therapy attenuates heart failure with preserved ejection fraction. FASEB J. 2022;36:R2429. [Google Scholar]
- 78.Chapple SJ, Cheng X, Mann GE. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013;1:319–331. doi: 10.1016/j.redox.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bocci VA, Zanardi I, Travagli V. Ozone acting on human blood yields a hormetic dose-response relationship. J Transl Med. 2011;9:66. doi: 10.1186/1479-5876-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Couto G, Mesquita T, Wu X, et al. Cell therapy attenuates endothelial dysfunction in hypertensive rats with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2022;323:H892–H903. doi: 10.1152/ajpheart.00287.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kelly JP, Mentz RJ, Mebazaa A, et al. Patient selection in heart failure with preserved ejection fraction clinical trials. J Am Coll Cardiol. 2015;65:1668–1682. doi: 10.1016/j.jacc.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mentz RJ, Kelly JP, von Lueder TG, et al. Noncardiac comorbidities in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. 2014;64:2281–2293. doi: 10.1016/j.jacc.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scantlebury DC, Borlaug BA. Why are women more likely than men to develop heart failure with preserved ejection fraction? Curr Opin Cardiol. 2011;26:562–568. doi: 10.1097/HCO.0b013e32834b7faf. [DOI] [PubMed] [Google Scholar]
- 84.Tschöpe C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep. 2014;11:436–444. doi: 10.1007/s11897-014-0219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anker SD, Butler J, Filippatos G, et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med. 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038. [DOI] [PubMed] [Google Scholar]
- 86.Chacko SM, Ahmed S, Selvendiran K, Kuppusamy ML, Khan M, Kuppusamy P. Hypoxic preconditioning induces the expression of prosurvival and proangiogenic markers in mesenchymal stem cells. Am J Physiol Cell Physiol. 2010;299:C1562–1570. doi: 10.1152/ajpcell.00221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang YL, Tang Y, Zhang YC, Qian K, Shen L, Phillips MI. Improved graft mesenchymal stem cell survival in ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. J Am Coll Cardiol. 2005;46:1339–1350. doi: 10.1016/j.jacc.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 88.Ii M, Nishimura H, Iwakura A, et al. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported” nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 89.Kamota T, Li TS, Morikage N, et al. Ischemic pre-conditioning enhances the mobilization and recruitment of bone marrow stem cells to protect against ischemia/reperfusion injury in the late phase. J Am Coll Cardiol. 2009;53:1814–1822. doi: 10.1016/j.jacc.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 90.Kimura M, Ueda K, Goto C, et al. Repetition of ischemic preconditioning augments endothelium-dependent vasodilation in humans: role of endothelium-derived nitric oxide and endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2007;27:1403–1410. doi: 10.1161/ATVBAHA.107.143578. [DOI] [PubMed] [Google Scholar]
- 91.Gyöngyösi M, Posa A, Pavo N, et al. Differential effect of ischaemic preconditioning on mobilisation and recruitment of haematopoietic and mesenchymal stem cells in porcine myocardial ischaemia-reperfusion. Thromb Haemost. 2010;104:376–384. doi: 10.1160/TH09-08-0558. [DOI] [PubMed] [Google Scholar]
- 92.Edwards N, Langford-Smith AWW, Wilkinson FL, Alexander MY. Endothelial progenitor cells: new targets for therapeutics for inflammatory conditions with high cardiovascular risk. Front Med (Lausanne) 2018;5:200. doi: 10.3389/fmed.2018.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thum T, Fraccarollo D, Schultheiss M, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–674. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- 94.Heiss C, Wong ML, Block VI, et al. Pleiotrophin induces nitric oxide dependent migration of endothelial progenitor cells. J Cell Physiol. 2008;215:366–373. doi: 10.1002/jcp.21313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heiss C, Schanz A, Amabile N, et al. Nitric oxide synthase expression and functional response to nitric oxide are both important modulators of circulating angiogenic cell response to angiogenic stimuli. Arterioscler Thromb Vasc Biol. 2010;30:2212–2218. doi: 10.1161/ATVBAHA.110.211581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsirmoula S, Lamprou M, Hatziapostolou M, Kieffer N, Papadimitriou E. Pleiotrophin-induced endothelial cell migration is regulated by xanthine oxidase-mediated generation of reactive oxygen species. Microvasc Res. 2015;98:74–81. doi: 10.1016/j.mvr.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 97.Sharifi BG, Zeng Z, Wang L, et al. Pleiotrophin induces transdifferentiation of monocytes into functional endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1273–1280. doi: 10.1161/01.ATV.0000222017.05085.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.