


Abstract
Human immunodeficiency virus (HIV), like all retroviruses, requires a cellular tRNA as a primer for initiation of reverse transcription. In a previous study, we demonstrated that an HIV-1 with a primer binding site complementary to yeast tRNAPhe (psHIV-Phe) was not infectious unless yeast tRNAPhe was supplied in trans. This unique in vivo complementation system has now been used to define the elements of the tRNA required for HIV-1 replication. Mutant tRNAPhe with deletions in TΨC stem–loop, anticodon stem–loop or D stem–loop of the tRNA were generated and assessed for the capacity to rescue psHIV-Phe. Mutant tRNAPhe with disrupted TΨC stem–loop did not rescue psHIV-Phe. In contrast, a mutant tRNAPhe without the D stem–loop was fully functional for the rescue. The tRNA anticodon stem–loop region was found to be important for efficient complementation. The results of our studies demonstrate for the first time the importance of specific structural and sequence elements of the tRNA primer for HIV-1 reverse transcription and define new targets for interruption of HIV-1 replication.
INTRODUCTION
Retroviruses use cellular tRNA as a primer to initiate reverse transcription (1). tRNALys3 is the natural primer for HIV-1 and related lentiviruses. In HIV virions, the tRNA is bound to an 18-nucleotide (nt) region of the viral genome designated as the primer binding site (PBS), which is complementary to the 3′-terminal nucleotides of the tRNA. Studies from this laboratory, and others, have shown that the complementarity between the PBS and the tRNA 3′ terminus is a major determinant for the selection of a tRNA primer in HIV-1 (2–5). Changing the HIV-1 PBS to be complementary to tRNAs other than tRNALys3 resulted in viruses which used the alternative tRNAs as primers in reverse transcription. However, these viruses rapidly reverted to use tRNALys3 following in vitro culture unless an accompanying region in U5, the A-loop, was also mutated (5–9). Even with optimized A-loop-PBS combinations in the viral genome though, not all tRNAs were stably used by viruses following in vitro culture (5), highlighting the complexity of the interactions between HIV-1 genome and the tRNA primer.
Most in vivo studies on HIV-1 RNA–tRNA interactions have been focused on understanding the sequence elements within the HIV-1 genome required for the selection and use of the tRNA primer. Although in vitro experiments exploring the interactions of tRNA with HIV-1 reverse transcriptase (RT) or with viral RNA have been reported (10–14), little is known about the sequence and structural requirements for a tRNA to be selected and used in HIV-1 reverse transcription in vivo. Studies on the elements of the tRNA primer important for yeast retrotransposon Ty reverse transcription have taken advantage of techniques to genetically manipulate yeast cells (15,16). A system has also been described for murine leukemia virus which used a genetically engineered tRNA to complement a defective provirus (17). We recently established an in vivo complementation system to allow identification of specific elements within the tRNA that are important for the primer selection and use in HIV-1 reverse transcription (18). The replication of a defective HIV-1 with a PBS complementary to yeast tRNAPhe, which previous studies have shown to be non-infectious (19), was restored when yeast tRNAPhe was provided during virus production.
Yeast tRNAPhe is an ideal candidate to examine the role of specific regions of tRNA structure in primer selection because its three-dimensional structure is well-characterized (20). The cloverleaf base-pairing yeast tRNAPhe sequence folds into an L-shaped molecule, which is universal among tRNAs. An L-shaped tRNA can be divided into two main domains: the ‘top half’, a minihelix motif consisting of the acceptor stem and TΨC stem–loop, and the ‘bottom half’, including the anticodon stem–loop, D stem–loop and variable loop. In this study, a series of yeast tRNAPhe mutants were made and tested for their capacity to restore psHIV-Phe virus infectivity. We found that while the TΨC stem–loop and anticodon stem–loop of yeast tRNAPhe are essential for complementation, the tRNAPhe D stem–loop structure was not required. The results of these studies are discussed with respect to the elements of the tRNA required for primer selection and use and potential application in designing new therapeutics to disrupt this step in HIV-1 reverse transcription.
MATERIALS AND METHODS
Tissue culture and cell lines
293T cells and HeLa H1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco BRL) with 10% fetal calf serum (FCS) and 1% antibiotic–antimycotic (penicillin/streptomycin/amphotericin cocktail; Gibco BRL) in a 37°C incubator supplied with 5% CO2.
DNA plasmids
The construction of plasmid psHIV-Phe was described in detail in our previous study (18). Briefly, psHIV-Phe contains a defective HIV-1 proviral genome in which the env gene was deleted and replaced by a drug-resistant gene gpt (xanthine-guanosine phosphoribosyl transferase). The PBS region of this provirus was mutated to be complementary to the 3′-terminal 18-nt of yeast tRNAPhe. Plasmid pLGRNL used to express vesicular stomatitis virus G glycoprotein (VSV-G) has been described previously (21).
In vitro transcription
Plasmid p67YF0 which contains the yeast tRNAPhe gene was kindly provided by Dr O. C. Uhlenbeck (22). p67YF0 contains a T7 promoter directly adjacent to the yeast tRNAPhe gene and a BstNI restriction site at the 3′ end of the gene. ‘Run off’ transcription of the BstNI-digested p67YF0 gives a 76-nt RNA identical to the unmodified yeast tRNAPhe. All transcription reactions were carried out using a commercial in vitro transcription kit (Gibco BRL), following the instructions of the manufacturer.
To generate tRNAPhe mutants, cDNA fragments starting with sequence containing a T7 promoter (5′-CTGCAGTAATACGACTCACTATA) followed by the exact coding sequence for one of the following yeast tRNAPhe mutants were made: the 35mer, 45mer, 46mer, 47mer, 55mer, 62mer, 62-loop and 62-stem (see Figs 2A, 3A and 4A for the nucleotide sequences). These cDNA fragments were generated by annealing the plus-strand and the complementary minus-strand synthetic DNA oligomers at 94°C and then slowly cooling down to room temperature. Each of the cDNA fragments was used as a template to generate the encoded yeast tRNAPhe mutant by in vitro transcription.
Figure 2.
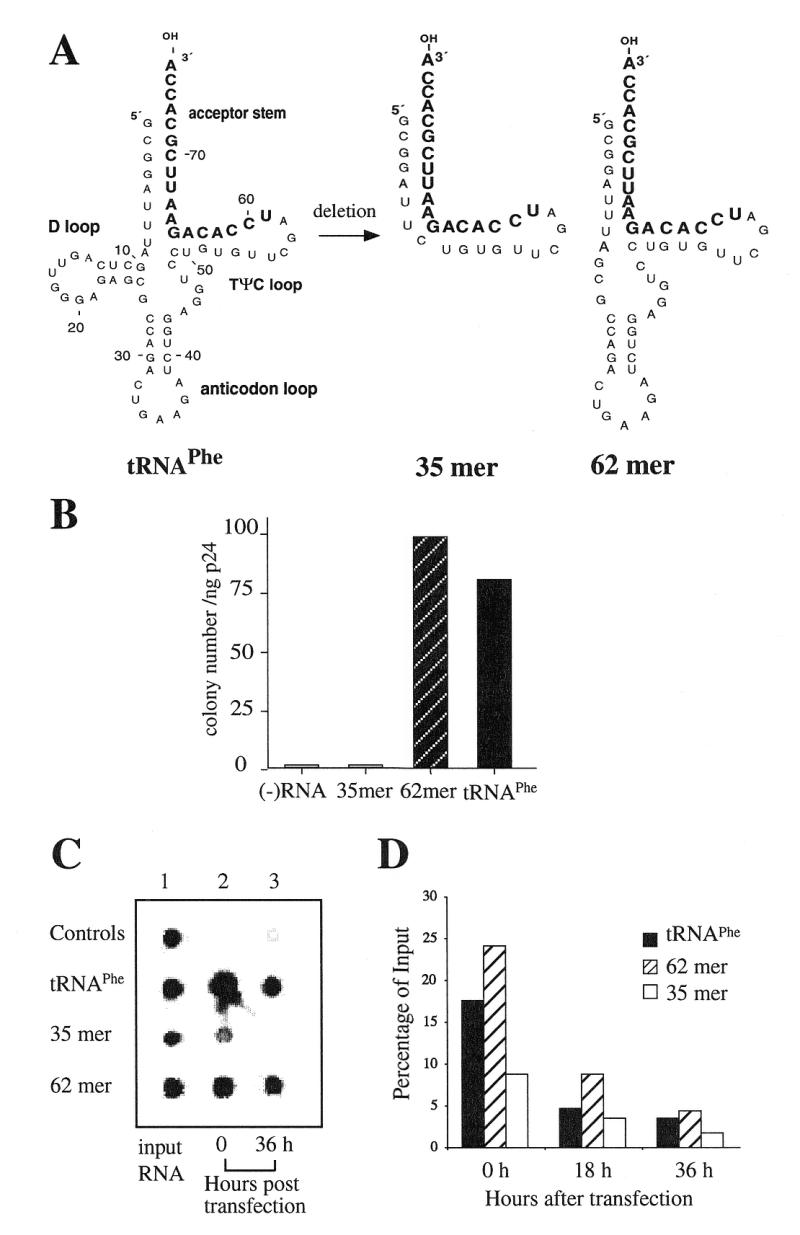
The capacity of the yeast tRNAPhe deletion mutants to restore psHIV-Phe infectivity. (A) The RNA nucleotide sequences of yeast tRNAPhe, mutant 35mer and 62mer. The 3′-terminal 18-nt complementary to the PBS of psHIV-Phe are in bold. (B) The numbers of drug-resistant colonies derived from the infection of psHIV-Phe pseudoviruses generated by cotransfecting psHIV-Phe provirus and VSV-G expression plasmid (pLGRNL) along with wild-type yeast tRNAPhe, 35mer or 62mer. psHIV-Phe virus without tRNA complementation served as a negative control. (C) The stability of biotin-labeled tRNAPhe mutants following cotransfection. Biotin-labeled yeast tRNAPhe or mutants were cotransfected with psHIV-Phe provirus and pLGRNL into 293T cells by incubating the transfection mixtures with the cells for 8–10 h. At 0 or 36 h after cotransfection, total RNAs were isolated from the transfected cells, spotted on a Nylon membrane and visualized using a biotin luminescent detection kit. Biotin-labeled tRNAPhe was spotted directly on the membrane as a positive control (control 1). The cotransfection of unlabeled tRNAPhe with psHIV-Phe and pLGRNL served as a negative control (control 2); cells incubated with biotin-labeled tRNAPhe, psHIV-Phe and pLGRNL in the absence of transfection reagent were also included as a control (control 3). The RNA stocks used for cotransfection were diluted and then spotted on the membrane as controls (labeled as input RNA). (D) Stability of 35S-labeled tRNAPhe mutants following cotransfection. Similar experiments as described in (C) were carried out with 35S-labeled tRNAs in place of the biotin-labeled tRNAs. Nucleic acids were precipitated from the cells at 0, 18 or 36 h after cotransfection and collected to determine radioactivity by counting. The counts obtained at 0 h (time 0 corresponds to 8 h after the incubation of transfection mixtures with cells), 18 and 36 h were divided by the input amounts for each sample. The values presented represent percentages of the input amount.
Figure 3.

Complementation of psHIV-Phe virus by cotransfected yeast tRNAPhe mutants. (A) The sequence of four yeast tRNAPhe mutants generated by further deletion within the 62mer. The 3′-terminal 18-nt that are complementary to the PBS of psHIV-Phe are in bold. (B) The numbers of drug-resistant colonies derived from infection of the complemented pseudoviruses; psHIV-Phe virus without complementation was used as a control. (C) The stability of the tRNAPhe mutants following cotransfection. The biotin-labeled yeast tRNAPhe or mutants cotransfected with psHIV-Phe and pLGRNL were recovered from the transfected cells and analyzed as described in Figure 2C.
Figure 4.
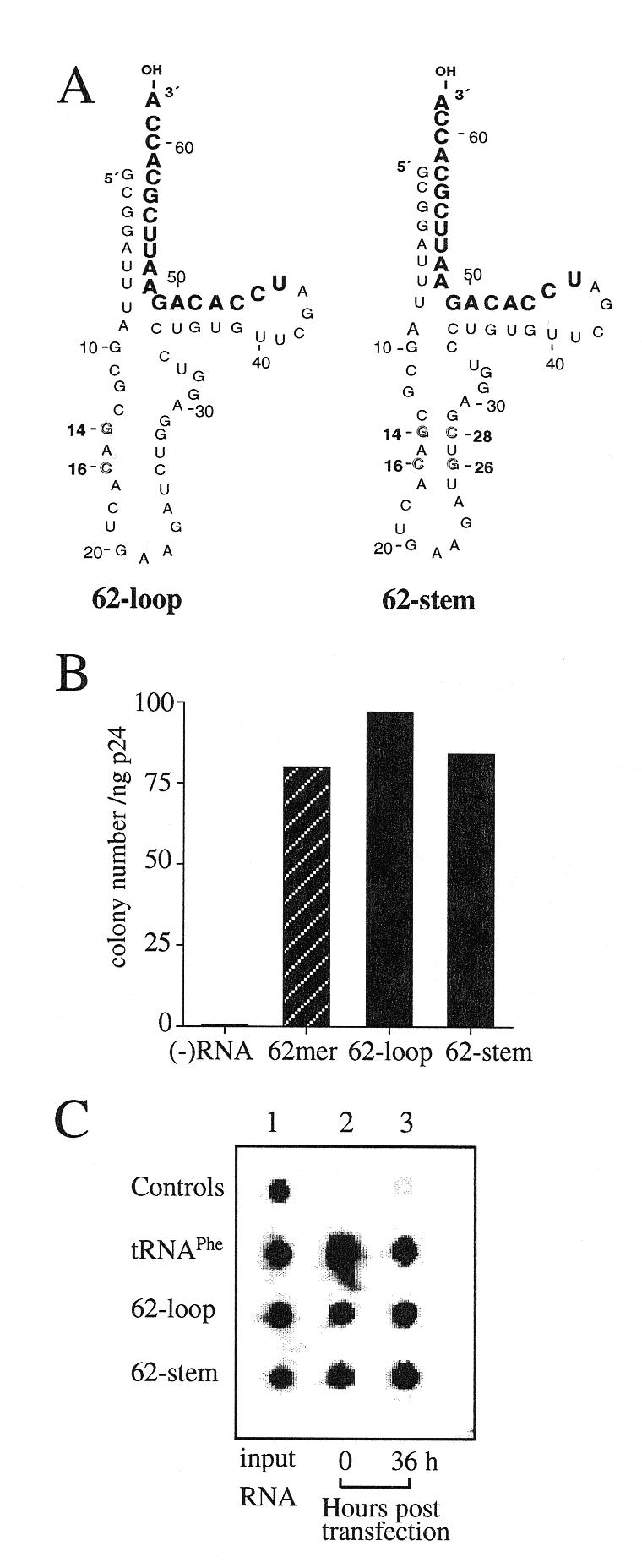
Comparison of the tRNAPhe mutants with different structures in the anticodon stem–loop region. (A) Two mutants were generated by altering nucleotides in the anticodon stem of mutant 62mer. The C14-G16 to G14-C16 (outlined) change in the 62-loop was predicted to disrupt the anticodon stem. The 62-stem was generated by introducing mutations (C26-G28 to G26-C28) (outlined) into mutant 62-loop to restore the complementarity between the two strands. (B) Drug-resistant colonies derived from the infection of psHIV-Phe virus complemented by 62-loop or 62-stem; psHIV-Phe virus without tRNA complementation was used as a control. (C) Stability of the tRNAPhe mutants following cotransfection. See Figure 2C for description of the procedure.
RNA synthesis
Synthetic RNA oligonucleotides 35mer, 62mer, 55mer, 45mer, 47mer, 46mer and 62-UUU (see Figs 2A, 3A and 5), generated using an Applied Biosystems 392 DNA/RNA synthesizer, were provided by Dr S. Hajduk (University of Alabama at Birmingham). The synthetic oligonucleotides were gel-purified prior to use in the complementation system. All RNAs were quantitated by UV absorption and by gel electrophoresis and ethidium bromide staining. The latter was followed by the use of Alpha-Imager (Alpha Innotech), which achieved quantitation through comparing the density of each RNA band with the standards.
Figure 5.

Complementation of psHIV-Phe by a tRNAPhe mutant with the anticodon of tRNALys3. (A) The anticodon of tRNAPhe (GAA) was substituted with the anticodon of tRNALys3 (UUU, in bold) in the 62mer (Fig. 2A) to generate mutant 62-UUU. (B) The numbers of drug-resistant colonies derived from the infection of psHIV-Phe virus rescued by 62-UUU, 62mer or tRNAPhe.
Transfection and selection of drug-resistant cell lines
The procedure used for complementation has been previously described (18,23). Briefly, lipofectin (Gibco BRL) was used to cotransfect plasmid DNA (1 µg) and RNA oligonucleotides (2 µg) into 293T cells. At 36 h post-transfection, the supernatants of transfected cells were collected and filtered through a 0.45-µm pore-size filter (Nalgene). Virus production was monitored by measuring the p24 antigen levels using a commercial ELISA kit (Coulter Laboratories). Different dilutions of viruses produced by cotransfections were used to infect HeLa H1 cells. At 2 h after infection, the cells were washed once and fresh DMEM with 10% FCS and 1% antibiotics was added. At 24 h after infection, the medium was replaced with the selection medium [DMEM containing 10% FCS, 1% antibiotics, 20 mM HEPES (pH 7.5), 250 µg/ml xanthine and 50 µg/ml mycophenolic acid]. This medium was changed every 2–3 days until colonies of drug-resistant cells were formed (10–12 days). Cell colonies were washed with phosphate buffered saline and stained with crystal violet solution (0.2% crystal violet, 25% isopropanol and 5% acetic acid). The numbers of the drug-resistant colonies presented are representative of three independent experiments. For analysis of integrated proviral primer binding site, cell colonies were isolated and expanded in selection medium; chromosomal DNA was isolated using a Wizard genomic DNA purification kit (Promega). The U5-PBS region of integrated provirus in the isolated chromosomal DNA was amplified by PCR with the following primers: primer 1 (5′-TAGACCAGATCTGAGCCTGGGAGCTC-3′; nt 13–48), and primer 2 (5′-CTCCTTCTAGCCTCCGCTAGTC-3′, complementary to nt 331–310). The PCR-amplified fragments were cloned into pGEM-T-Easy vector (Promega) and the resultant plasmid DNAs were amplified and prepared for sequence analysis.
Biotinylation or radiolabeling of tRNAs
The biotin RNA labeling mix (Boehringer Mannheim) was used in the in vitro transcription as substrates to generate biotin-labeled (biotinylated) RNA oligonucleotides, following the instructions of the manufacturer. 35S-labeled tRNAs were generated using the MEGAshortscript transcription kit (Ambion) as instructed by the manufacturer, with the exception that the final concentration of UTP was changed from 7.5 to 0.75 mM (one-tenth of other NTPs) and 10 mCi/ml [35S]UTP stock (ICN) was also added to achieve a final concentration of 1 mCi/ml in the reaction. The transcripts were purified by phenol/chloroform extraction and ethanol precipitation.
Two micrograms of biotin-labeled or 35S-labeled tRNAs was cotransfected into 293T cells with psHIV-Phe provirus and plasmid pLGRNL. At specified time points the transfected cells were washed with phosphate buffered saline, incubated with RNase A (10 µg/ml in PBS solution) for 20 min, and then washed again three times with phosphate buffered saline. Subsequently, total intracellular RNAs of the cells transfected with biotin-labeled tRNAs were isolated using TRI REAGENT (Molecular Research Center). One percent of the total RNAs were spotted on positively-charged Nylon membrane (Boehringer Mannheim) followed by the use of a biotin luminescent detection kit (Boehringer Mannheim). The biotin-labeled tRNA was detected with streptavidin coupled to alkaline phosphatase (AP) and was visualized by adding the chemiluminescent AP-substrate CSPD. The cells transfected with 35S-labeled tRNAs were harvested by trypsinization and lysed in 10% trichloroacetic acid solution; the precipitated nucleic acids were collected on a 0.45 µm membrane filter (Gelman Science). The dried filters were placed in scintillation fluor (Research Products International) and radioactivity was determined by counting.
RESULTS
In a previous study, we described a defective HIV-1 provirus with a PBS complementary to the 3′-terminal 18-nt of yeast tRNAPhe, designated as psHIV-Phe, which contains a drug-resistant gene, xanthine-guanosine phosphoribosyl transferase (gpt), in place of the deleted env gene (Fig. 1) (18). Pseudoviruses derived from cotransfection of psHIV-Phe provirus and the plasmid encoding VSV-G protein were non-infectious as measured by the ability to induce resistance to mycophenolic acid. Yeast tRNAPhe added into the cotransfection restored the infectivity of the pseudovirus in a dose-dependent fashion.
Figure 1.

psHIV-Phe proviral genome. The defective psHIV-Phe proviral genome with a PBS complementary to yeast tRNAPhe is depicted. The gene encoding xanthine-guanosine phosphoribosyl transferase (gpt) under the control of SV40 early promoter was substituted for the env gene of HIV. The 18-nt sequence of the PBS complementary to yeast tRNAPhe is referred to as PBSPhe. To generate pseudoviruses, this plasmid was co-transfected with the plasmid encoding VSV-G protein either with or without tRNA. Successful infection of cells with the pseudoviruses confers resistance to mycophenolic acid.
Dissection of the tRNA required for complementation of psHIV-Phe
Since little is known about the region of the tRNA required for in vivo primer selection and use in reverse transcription, our strategy was to generate deletion mutants of tRNAPhe. We first tested two tRNAPhe deletion mutants, a 35mer and 62mer (Fig. 2A). The 35mer is the minihelix of yeast tRNAPhe, consisting of the acceptor stem and TΨC stem–loop. The 62mer represents a yeast tRNAPhe without the D stem–loop. Unless otherwise specified, yeast tRNAPhe mutants were generated by direct RNA synthesis, followed by cotransfection with psHIV-Phe provirus and the plasmid encoding VSV-G protein (pLGRNL); the resultant pseudoviruses were normalized for p24 and used to infect HeLa cells, followed by selection for resistance to mycophenolic acid. A low number of drug-resistant colonies were recovered from the infection of psHIV-Phe virus complemented by 35mer, which was not greater than the background levels of the assay (Fig. 2B). In contrast, the 62mer restored the psHIV-Phe virus infectivity with a similar efficiency as the wild-type yeast tRNAPhe (Fig. 2B). Similar results were found with a 62mer generated from in vitro transcription, indicating that the ability of the RNA to function in the assay did not depend on the method of RNA generation (data not shown).
The complementation of psHIV-Phe requires the tRNA present in the cells to be selected as primer through an as yet undefined mechanism, and positioned at the PBS. To further investigate the differences between 35mer and 62mer with respect to complementation, the stability of the RNAs was analyzed following transfection. Biotin-labeled wild-type tRNAPhe, 35mer and 62mer were generated by in vitro transcription, and cotransfected with psHIV-Phe and pLGRNL. The cotransfections were carried out by first incubating nucleic acids–lipofectin mixtures with the cells for 8 h. After the 8-h incubation, growth medium was added to the cells; we designated this as time point 0 h. At time point 0 or 36 h, the transfected cells were incubated with RNase A and washed extensively to eliminate any untransfected tRNA that might remain outside of the cells (Q.Yu and C.D.Morrow, unpublished results); total cellular RNAs were then isolated from the transfected cells and spotted on a membrane for the detection of biotin-labeled tRNA (Fig. 2C). Our analysis revealed that after 8-h incubation with the cells (time point 0 h), less 35mer was detected than wild-type tRNAPhe or the 62mer. The 62mer had a similar stability profile as the wild-type tRNAPhe, at least up to 36 h after cotransfection, which corresponds to the time for harvesting pseudoviruses for infections (Fig. 2C). The results from this analysis suggested that the 35mer might be less stable than the 62mer following transfection. To further address this possibility, we repeated the above experiments with 35S-labeled tRNAPhe, 35mer and 62mer (Fig. 2D). All three RNAs were detected at 36 h post-transfection at levels 2–5% of the amounts used in transfection. However, at 0 h time point there was approximately twice as much 62mer and wild-type tRNAPhe as the 35mer, again suggesting the 35mer is less stable following transfection. Given the differences in stability between the 62mer and 35mer, we have not pursued the analysis of the 35mer to complement psHIV-Phe. Instead, elements within the 62mer were further analyzed to explore the role of different regions of the tRNA in complementation. A 47mer containing partial deletion of the TΨC stem–loop was generated (Fig. 3A). The acceptor stem and anticodon stem–loop were predicted to remain intact in the 47mer by the RNA secondary-structure modeling program M-fold (24–26). Although the 47mer remained stable after cotransfection, the RNA had a very low capacity to rescue psHIV-Phe virus (Fig. 3B and C), indicating the importance of an intact TΨC stem–loop for complementation.
Role of the anticodon stem–loop region of the tRNA in primer selection and use in reverse transcription
To address the role of the anticodon stem–loop and variable loop of the tRNA in primer selection and use in HIV-1, three new tRNAPhe mutants based on the 62mer were generated and tested in our complementation system: the 55mer (the anticodon loop was deleted), 45mer (both anticodon stem and loop were deleted) and 46mer (the anticodon stem and variable loop were deleted) (Fig. 3A). Upon infection of the cells with the psHIV-Phe pseudoviruses complemented by each of these mutants, we recovered fewer drug-resistant colonies compared to the virus complemented by 62mer, although the values obtained for 55mer and 45mer were always above background (Fig. 3B). The lower complementation capacity of these mutants was not due to reduced stability compared to the wild-type tRNAPhe (Fig. 3C). To confirm the use of yeast tRNAPhe mutants as primers for reverse transcription by the rescued psHIV-Phe viruses, the PBS sequences of integrated psHIV-Phe proviruses were analyzed. Chromosomal DNA was isolated from cell clones derived from the infection of psHIV-Phe virus complemented by the 62mer, 55mer or 45mer. The proviral U5-PBS region in chromosomal DNA was amplified by PCR, followed by subcloning and sequence analysis. The results showed that all the PBS sequences recovered were complementary to the 3′-terminal 18-nt of yeast tRNAPhe. Since the PBS sequence of integrated HIV provirus reflects the actual primer used in reverse transcription, the yeast tRNAPhe mutants examined had functioned as reverse transcription primers in the rescued psHIV-Phe viruses (data not shown).
To further delineate the features of anticodon stem–loop structure for the selection and use of the tRNA in reverse transcription, two additional mutants were designed. In the mutant 62-loop, C14-G16 to G14-C16 change disrupts the anticodon stem structure; in the second mutant, designated as 62-stem, a C26-G28 to G26-C28 change in addition to the C14-G16 to G14-C16 change was introduced to restore the complementarity between the two strands of the anticodon stem (Fig. 4A). The disruption of the anticodon stem–loop structure in the mutant 62-loop and the restoration of this structure in the mutant 62-stem were predicted by the M-fold program (24–26). These mutant RNAs were generated by in vitro transcription and analyzed for their capacity to complement psHIV-Phe virus. Surprisingly, both mutant 62-loop and 62-stem showed a similar capacity in rescuing psHIV-Phe virus as the 62mer (Fig. 4B); and no difference was detected between the stability of the mutants and the wild-type tRNAPhe following cotransfection (Fig. 4C). Taken together, the results of our analysis established that the anticodon region is essential for primer selection and use in HIV-1 reverse transcription, whereas the native anticodon stem–loop structure per se is not required.
The anticodon of tRNALys3 has been shown to interact with the A-loop in the U5 region of HIV-1 RNA (11). Although previous studies have shown that certain combinations of A-loop-PBS resulted in viruses that stably maintained a PBS complementary to an alternative tRNA (e.g., tRNAHis or tRNAMet), other A-loop-PBS mutations were not stable (e.g. tRNAIle) (5). Based on the results of this and other studies, we believe that the U5-PBS interaction with tRNA is complex, and most probably depends upon the formation of specific RNA structures in the U5. As an alternative to mutating the A-loop region of U5 to correspond to the anticodon of tRNAPhe, we generated a tRNAPhe mutant (62-UUU) in which the anticodon was altered to that of tRNALys3 (GAA to UUU) (Fig. 5A). This mutant should take advantage of the wild-type A-loop structure of psHIV-Phe genome for initiation of reverse transcription. Surprisingly, 62-UUU rescued the virus with a similar efficiency, as determined by colony numbers, as the 62mer with a tRNAPhe anticodon (GAA) (Fig. 5B). Thus, for the single-round infection analysis with tRNAPhe, the complementarity with A-loop and PBS did not result in more efficient virus rescue. This result is consistent with other A-loop-PBS combinations (i.e., tRNAMet) but is different from that found for tRNAHis (Q.Yu and C.D.Morrow, unpublished results).
DISCUSSION
Elements of the tRNA essential for primer selection and use in HIV-1 reverse transcription were delineated by using the yeast tRNAPhe mutants to complement the replication of a defective HIV-1 virus, psHIV-Phe. Analysis of tRNAPhe mutants revealed that while the TΨC stem–loop region was essential for complementation, the tRNA without the D stem–loop region was fully functional for complementation. Deletion of the anticodon stem and/or loop severely reduced the complementation capacity of the tRNA; however, mutations to disrupt or promote the anticodon stem–loop structure did not affect complementation (summarized in Fig. 6).
Figure 6.

Summary of the effect of tRNAPhe mutations on primer selection and use in HIV-1 reverse transcription. The tertiary or ‘L-shape’ representation of yeast tRNAPhe is depicted. Regions of the tRNA important for complementing the psHIV-Phe virus are indicated by triangles. The region not required for virus complementation is labeled with an x. Circles indicate the positions of the point mutations affecting the secondary structure of the anticodon stem while not interfering with the virus complementation.
The results of our experiments provide the first insights into what tRNA sequence and structures are required for the selection and use of the tRNA primer for HIV-1 reverse transcription. Our experimental approach is unique because an in vivo, rather than an in vitro system was used, which necessitates the tRNA to be selected from the intracellular milieu and positioned at the PBS in addition to being functional for initiation and completion of reverse transcription. In an in vitro system, reverse transcription can be accomplished with an RNA primer of 18 nt, indicating that the complementarity between the PBS and a primer is sufficient for the use of the primer in reverse transcription (27). Our studies revealed that the requirement for primer selection and use in vivo is more complex than the simple complementarity between the potential primer and the PBS of HIV-1. We initially tested the minihelix of tRNAPhe (35mer) consisting of the acceptor stem and TΨC stem–loop. The tRNA minihelix motif has been proposed to be the oldest element of the tRNA (28) and was used by a primitive RT encoded by the Mauriceville retroplasmid to initiate cDNA synthesis (29). However, the 35mer failed to rescue psHIV-Phe. A complication with the interpretation of this experiment was the difference in stability of 35mer compared to the wild-type tRNAPhe following cotransfection. However, tRNAPhe mutants containing the minihelix and additional elements (45mer, 46mer and 55mer) which exhibited similar stability as tRNAPhe, failed to rescue psHIV-Phe virus. Collectively, elements in the minihelix (35mer) were probably not sufficient for the virus complementation. Our results do suggest that the minihelix contains essential elements for psHIV-Phe complementation. One of these elements would be the TΨC stem–loop as demonstrated by the incapability of a tRNAPhe mutant (47mer) to rescue psHIV-Phe. Previous studies have suggested that the TΨC stem–loop of tRNALys3 interacts with HIV-1 RT in vitro (14); the TΨC stem–loop was also proposed to interact with the U5 sequence of HIV-1 (1). These potential interactions may also occur between yeast tRNAPhe and psHIV-Phe, since the TΨC loop sequence is highly conserved among tRNAs. Further experiments will be required to address this issue.
A surprising result was that the mutant tRNAPhe without the D stem–loop functioned as effectively as wild-type tRNAPhe in complementing the psHIV-Phe virus. The D stem–loop is essential for tRNA tertiary folding through the D loop–TΨC loop interaction (30,31). The lack of requirement for the D stem–loop for psHIV-Phe rescue strongly argues that the D stem–loop, and consequently the tRNA tertiary structure, are not essential for the tRNA primer selection and use in HIV-1 in vivo. This conclusion is consistent with the structure model of HIV-1 reverse transcription initiation complex proposed by Marquet and co-workers, which suggested that the D stem–loop of the tRNA primer protrudes out of the initiation complex and does not interact with either viral RNA or RT (11,13). Conversely, earlier studies suggested that the D stem–loop was important for HIV-1 RT–tRNALys3 binding in vitro (14,32,33). However, this interaction might be non-specific, since HIV-1 RT binds with many other tRNAs with the same efficiency (34). It is possible the suggested tRNA–RT interaction, if it occurs in vivo, may not be involved in the specific selection and use of the tRNA primer for reverse transcription. Additional experiments will be required to address this possibility.
The deletion of the anticodon stem and/or loop severely impaired the capacity of the tRNA to rescue psHIV-Phe virus, while the disruption of the anticodon stem structure by point mutations did not affect the virus rescue. Alteration of the anticodon (from GAA to UUU) also did not compromise the ability of the tRNA to rescue. Collectively, our results may imply that the anticodon stem–loop region is important in forming the correct three-dimensional structure of tRNA for primer selection, which was affected severely by the deletion of anticodon stem–loop, not by the point mutations. Alternatively, certain nucleotides within the anticodon stem–loop might be important for the viral genomic RNA–tRNA interaction. Brulé et al. have suggested that the anticodon stem of tRNALys3 interacts with the U3 region during first strand transfer of reverse transcription in vitro (10). The same region in psHIV-Phe U3 exhibits base-pairing potential with the tRNAPhe anticodon stem. Therefore, it is possible that the anticodon stem deletion mutant in our study was blocked in the strand-transfer step. Previous studies from this laboratory and others have suggested that the interaction between the anticodon of the tRNA primer and the A-loop region of HIV-1 RNA is important for primer selection (6,7,9,11). However, the role of this interaction is not fully understood, since some but not all A-loop-PBS combinations resulted in viruses that stably use the alternative tRNAs. Furthermore, even when A-loop-PBS combinations resulted in viruses which stably use the alternative tRNAs (e.g., tRNAMet and tRNAHis) as primers, the alternative A-loop may or may not enhance the virus infectivity. In a single-round infection assay, an A-loop region complementary to tRNAMet did not enhance the infectivity of the virus using tRNAMet as the primer, whereas an A-loop complementary to tRNAHis did improve the infectivity of the virus using tRNAHis as the primer (Q.Yu and C.D.Morrow, unpublished results). In this study, we addressed the question by generating a tRNAPhe mutant with the tRNALys3 anticodon to facilitate the anticodon and A-loop interaction. Surprisingly, the change of the tRNAPhe anticodon to match tRNALys3 did not result in enhanced virus rescue (Fig. 5). One explanation for this result is that the anticodon region of tRNAPhe may interact with an alternative sequence in U5. This speculative interaction might play a similar role as the interaction between the tRNALys3 anticodon and the A-loop, which could explain why the tRNAPhe mutants with the anticodon of tRNALys3 or tRNAPhe are similar in rescuing psHIV-Phe. Alternatively, our single-round complementation system may not be sensitive enough to detect the contribution of the tRNALys3 anticodon and A-loop interaction to the virus infectivity. Additional experiments will be required to address these possibilities.
In summary, the results of our studies provide the first characterization of the critical elements of the tRNA required for HIV-1 reverse transcription in vivo. Since a complete tRNA molecule was not required to complement the defective virus, it may be possible to use this information to design small RNAs that would compete with tRNALys3 in primer selection and disrupt HIV reverse transcription without interfering with the natural cellular function of the tRNA in protein synthesis. Further delineation of the important elements of the tRNA required for primer selection will aid in the understanding of the interaction between the tRNA and U5-PBS of the virus, which will facilitate the development of new therapeutics designed to inhibit HIV replication.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Steve Harvey, Dr Lesley Dupuy, Nathan Kelly and James Buescher for helpful comments and Tricia Elgavish for mutant design assistance. C.D.M. thanks Mark A. Richardson for helpful discussions. We are grateful to Dr Stephen Hajduk and Chad Barker for providing the synthetic RNA molecules. The pseudovirus culture was carried out in the UAB AIDS Center virus core facility (AI-27767). This work was supported by grants (AI34749 and GM56544) to C.D.M.
REFERENCES
- 1.Mak J. and Kleiman,L. (1997) J. Virol., 71, 8087–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das A.T., Klaver,B. and Berkhout,B. (1995) J. Virol., 69, 3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X., Mak,J., Arts,E.J., Gu,Z., Kleiman,L., Wainberg,M.A. and Parniak,M.A. (1994) J. Virol., 68, 6198–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wakefield J.K., Wolf,A.G. and Morrow,C.D. (1995) J. Virol., 69, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang S.M., Wakefield,J.K. and Morrow,C.D. (1996) Virology, 222, 401–414. [DOI] [PubMed] [Google Scholar]
- 6.Wakefield J.K., Kang,S.M. and Morrow,C.D. (1996) J. Virol., 70, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang S.M., Zhang,Z. and Morrow,C.D. (1997) J. Virol., 71, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang S.M. and Morrow,C.D. (1999) J. Virol., 73, 1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang S.M., Zhang,Z. and Morrow,C.D. (1999) Virology, 257, 95–105. [DOI] [PubMed] [Google Scholar]
- 10.Brulé F., Bec,G., Keith,G., Le Grice,S.F., Roques,B.P., Ehresmann,B., Ehresmann,C. and Marquet,R. (2000) Nucleic Acids Res., 28, 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isel C., Ehresmann,C., Keith,G., Ehresmann,B. and Marquet,R. (1995) J. Mol. Biol., 247, 236–250. [DOI] [PubMed] [Google Scholar]
- 12.Isel C., Keith,G., Ehresmann,B., Ehresmann,C. and Marquet,R. (1998) Nucleic Acids Res., 26, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isel C., Westhof,E., Massire,C., Le Grice,S.F., Ehresmann,B., Ehresmann,C. and Marquet,R. (1999) EMBO J., 18, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oude Essink B.B., Das,A.T. and Berkhout,B. (1995) J. Biol. Chem., 270, 23867–23874. [DOI] [PubMed] [Google Scholar]
- 15.Friant S., Heyman,T., Bystrom,A.S., Wilhelm,M. and Wilhelm,F.X. (1998) Mol. Cell. Biol., 18, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keeney J.B., Chapman,K.B., Lauermann,V., Voytas,D.F., Astrom,S.U., von Pawel-Rammingen,U., Bystrom,A. and Boeke,J.D. (1995) Mol. Cell. Biol., 15, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lund A.H., Duch,M., Lovmand,J., Jorgensen,P. and Pedersen,F.S. (1997) J. Virol., 71, 1191–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu Q. and Morrow,C.D. (1999) Virology, 254, 160–168. [DOI] [PubMed] [Google Scholar]
- 19.Wakefield J.K., Rhim,H. and Morrow,C.D. (1994) J. Virol., 68, 1605–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robertus J.D., Ladner,J.E., Finch,J.T., Rhodes,D., Brown,R.S., Clark,B.F. and Klug,A. (1974) Nature, 250, 546–551. [DOI] [PubMed] [Google Scholar]
- 21.Burns J.C., Friedmann,T., Driever,W., Burrascano,M. and Yee,J.K. (1993) Proc. Natl Acad. Sci. USA, 90, 8033–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sampson J.R. and Uhlenbeck,O.C. (1988) Proc. Natl Acad. Sci. USA, 85, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z., Yu,Q., Kang,S.M., Buescher,J. and Morrow,C.D. (1998) J. Virol., 72, 5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaeger J.A., Turner,D.H. and Zuker,M. (1990) Methods Enzymol., 183, 281–306. [DOI] [PubMed] [Google Scholar]
- 25.Zuker M. (1989) Methods Enzymol., 180, 262–288. [DOI] [PubMed] [Google Scholar]
- 26.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 27.Arts E.J., Li,X., Gu,Z., Kleiman,L., Parniak,M.A. and Wainberg,M.A. (1994) J. Biol. Chem., 269, 14672–14680. [PubMed] [Google Scholar]
- 28.Schimmel P., Giege,R., Moras,D. and Yokoyama,S. (1993) Proc. Natl Acad. Sci. USA, 90, 8763–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennell J.C., Wang,H. and Lambowitz,A.M. (1994) Mol. Cell. Biol., 14, 3094–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S.H., Suddath,F.L., Quigley,G.J., McPherson,A., Sussman,J.L., Wang,A.H., Seeman,N.C. and Rich,A. (1974) Science, 185, 435–440. [DOI] [PubMed] [Google Scholar]
- 31.Ladner J.E., Jack,A., Robertus,J.D., Brown,R.S., Rhodes,D., Clark,B.F. and Klug,A. (1975) Proc. Natl Acad. Sci. USA, 72, 4414–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wohrl B.M., Ehresmann,B., Keith,G. and Le Grice,S.F. (1993) J. Biol. Chem., 268, 13617–13624. [PubMed] [Google Scholar]
- 33.Sarih-Cottin L., Bordier,B., Musier-Forsyth,K., Andreola,M.L., Barr,P.J. and Litvak,S. (1992) J. Mol. Biol., 226, 1–6. [DOI] [PubMed] [Google Scholar]
- 34.Thrall S.H., Reinstein,J., Wohrl,B.M. and Goody,R.S. (1996) Biochemistry, 35, 4609–4618. [DOI] [PubMed] [Google Scholar]