

Abstract
Substituting thermodynamically favorable ethanol oxidation reaction (EOR) for oxygen evolution reaction (OER) engenders high‐efficiency hydrogen production and generates high value‐added products as well. However, the main obstacles have been the low activity and the absence of an explicit catalytic mechanism. Herein, a heterostructure composed of amorphous vanadium oxide and crystalline nickel nitride (VOx−Ni3N) is developed. The heterostructure immensely boosts the EOR process, achieving the current density of 50 mA cm−2 at the low potential of 1.38 V versus reversible hydrogen electrode (RHE), far surpassing the sluggish OER (1.65 V vs RHE). Electrochemical impedance spectroscopy indicates that the as‐fabricated heterostructure can promote the adsorption of OH− and the generation of the reactive species (O*). Theoretical calculations further outline the dual polarization of the Ni site at the interface, specifically the asymmetric charge redistribution (interfacial polarization) and in‐plane polarization. Consequently, the dual polarization modulates the d‐band center, which in turn regulates the adsorption/desorption strength of key reaction intermediates, thereby facilitating the entire EOR process. Moreover, a VOx−Ni3N‐based electrolyzer, coupling hydrogen evolution reaction (HER) and EOR, attains 50 mA cm−2 at a low cell voltage of ≈1.5 V. This work thus paves the way for creating dual polarization through interface engineering toward broad catalysis.
Keywords: dual polarization, ethanol oxidation reaction, interface engineering, nickel nitride, vanadium oxide
A meticulously engineered heterostructure has been developed, integrating amorphous vanadium oxide with crystalline nickel nitride (VOx−Ni3N), featuring a well‐defined Ni─O─V interface. This dual polarization at the Ni site within the interface significantly enhances the efficiency of the ethanol oxidation reaction (EOR). Furthermore, an electrolyzer configured for EOR‐coupled hydrogen production has been constructed and successfully powered by a commercial dry battery.
1. Introduction
With increasing energy and environmental concerns, electrocatalytic water splitting for hydrogen production has become a prominent topic in recent years.[ 1 , 2 , 3 , 4 ] Nevertheless, the sluggish kinetics of the anodic oxygen evolution reaction (OER) limits the efficiency of the overall water splitting.[ 5 , 6 , 7 , 8 ] One promising approach is utilizing the thermodynamically more favorable ethanol oxidation reaction (EOR) in place of the OER, which shows great potential as an alternative to conventional electrolytic water splitting for hydrogen production.[ 9 , 10 ]
In previous studies, transition metal‐based nanomaterials have been proven to be efficient for EOR, particularly nickel and cobalt‐based compounds, such as NiO,[ 11 ] Ni(OH)2,[ 12 ] Co3O4,[ 13 , 14 ] Co3S4,[ 15 ] and so on. The catalytic activity of the mentioned catalyst is primarily attributed to the high‐valence metals. However, a higher applied potential (>1.35 V vs RHE) is required to achieve the specific valence state, which is far surpassing the EOR thermodynamic potential (<0.5 V vs RHE).[ 16 , 17 ] As a result, the higher activation potential of transition metal‐based chalcogenides limits the efficiency in EOR application. Therefore, it is crucial to explore efficient EOR electrocatalysts with lower activation potential and further clarify their reaction mechanisms.
Transition metal nitrides (TMNs), due to their various electrocatalytic properties and excellent corrosion resistance, are widely used in various catalysis.[ 18 , 19 ] However, according to the Sabatier principle, the monoactive species could not favor the adsorption and activation of the involved intermediates. Thus, it remains challenging to regulate multi‐step proton‐coupled electron transfer.[ 20 , 21 ] Given this, Song et al. synthesized defective carbon–CoP nanoparticle hybrids and applied them in oxygen catalysis.[ 22 ] It revealed that the interfacial charge polarization, with the electrons gathering at the defective carbon surface, facilitates the activation of various oxygen‐involved intermediates. Ma's group reported that the built‐in interfacial polarization between BP nanoflakes and Nb2C nanosheets triggered high‐efficiency electrochemical nitrate reduction.[ 23 ] Theoretical calculations demonstrated that the enhanced NO3 −RR performance of the BP/Nb2C composite is attributed to the presence of polarization effects between BP nanoflakes and Nb2C nanosheets, leading to a high electron distribution at the interface of the BP/Nb2C composite. Therefore, interface engineering in heterostructures, especially the interfacial polarization, is an effective means to maneuver the active sites in close proximity to synergistically improve the intrinsic activity of catalysts and thus enhance the catalytic performance.
Herein, we reported on the synthesis of a VOx−Ni3N heterostructure, specifically integrating amorphous VOx with crystalline Ni3N. Dual polarization, including the in‐plane polarization and interfacial polarization between the hybrid phase, is demonstrated by detailed characterization. As a result, the VOx−Ni3N delivers superior EOR and HER catalytic activity and durability in alkaline ethanol media. By assembling an ethanol electrolyzer using VOx−Ni3N as a bifunctional electrocatalyst, a current density of 50 mA cm−2 could be achieved at a voltage of ≈1.5 V, comparable to the charge from a commercial dry battery. Consequently, high‐value‐added acetic acid could be produced at a cost‐effective route. Moreover, in‐situ electrochemical impedance spectroscopy and theoretical calculation analyses reveal the catalytic mechanism. The asymmetric charge distribution induced by dual polarization synergistically facilitates the coupling of Oads and ethanol at the VOx−Ni3N interface, thus boosting the catalytic performance. Therefore, creating dual−polarization through interface engineering could effectively promote multi‐step proton‐coupled electron transfer during heterogeneous catalysis beyond the Sabatier principle.
2. Results and Discussion
2.1. Material Characterization
The VOx−Ni3N heterostructure was synthesized by sequential hydrothermal reaction and annealing process, as schematically illustrated in Figure 1a. The V doped nickel layered double hydroxide (Ni–V LDH) precursor was prepared through a hydrothermal approach. Then, VOx−Ni3N heterostructure was obtained by annealing the precursor at 400 °C in a NH3 atmosphere. The crystalline Ni3N phase in VOx−Ni3N heterostructure, indexed to PDF#10–0280, is confirmed by the X‐ray diffraction (XRD) pattern,[ 23 ] while no other diffraction signals are observed. It suggests that vanadium might present in an amorphous state since no diffraction peak attributed to vanadium‐related structure is observed (Figure S1, Supporting Information).
Figure 1.
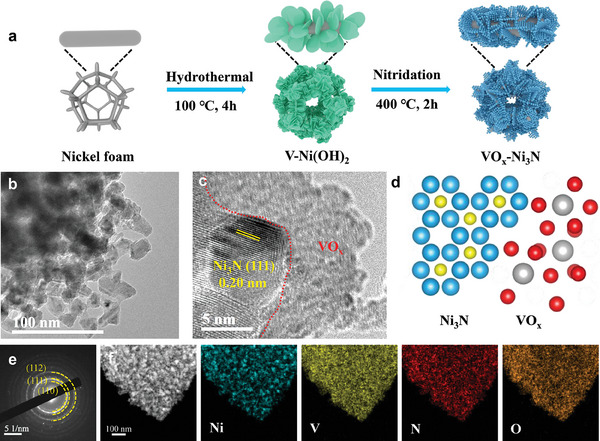
Structural characterization of the materials: a) Schematic of catalyst synthesis. TEM b) and HRTEM c) images of the VOx−Ni3N. d) Schematic atomic structure of VOx−Ni3N. e) Selected area electron diffractogram (SAED) and f) Scanning transmission electron microscopy (STEM) image and the corresponding elemental mapping.
The morphology of the obtained VOx−Ni3N heterostructure was characterized by field‐emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM). As shown in Figure S2 (Supporting Information), the Ni–V LDH is depicted with nanosheets that are well‐aligned and self‐assembled into a spherical structure. The nanosheet shape is inherited with rough and porous surfaces for VOx−Ni3N heterostructure after annealing. From the typical TEM image (Figure 1b), it is observed that the nanosheets are composed of interconnected nanoparticles, which might be caused by the phase transformation upon the annealing process. The lattice spacing of 0.20 nm corresponds to the (111) crystal plane of Ni3N (Figure 1c), consistent with selected area electron diffractogram (SAED) signatures in Figure 1e, as the schematic diagram shown in Figure 1d. Moreover, the amorphous phase at the edge of crystalline Ni3N can be identified. Given the elemental mapping profiles in Figure 1f, the margin of the entire heterostructure is composed by vanadium and oxygen. Thus, the observed amorphous phase can be attributed to VOx, which keeps a non‐crystalline state due to the relatively lower annealing temperature. Therefore, amorphous vanadium oxide coupled with crystalline nickel nitride can be demonstrated for the obtained VOx−Ni3N heterostructure.
X−ray photoelectron spectroscopy (XPS) was employed to investigate the electronic structure of the VOx−Ni3N heterostructure. In the Ni 2p spectra (Figure 2a), the peak pairs 852.2/869.5, 855.3/872.8, 856.1/873.8, and 860.6/878.9 eV can be well assigned to the binding energies of Ni─Ni, Ni─N, and Ni─O, and Sat. peaks.[ 24 , 25 ] Compared with that of the bare Ni3N, a positive shift of 0.3 eV in binding energies is detected, indicating the enhanced electron delocalization of the Ni3N after V is introduced. Meanwhile, Ni─O signals increase accompanied by a decrease in Ni─N intensity. The lifted Ni─O signature is potentially attributed to Ni3N bonded VOx species, different from surface oxidation. The migration of delocalized electrons from Ni3N to VOx at the interface is indicative of interfacial polarization. Then, a novel Ni─O─V interface is deduced within the VOx−Ni3N heterostructure.[ 26 ] As a result, a prominent charge redistribution occurs within the heterostructure, especially the Ni─O─V interface.
Figure 2.
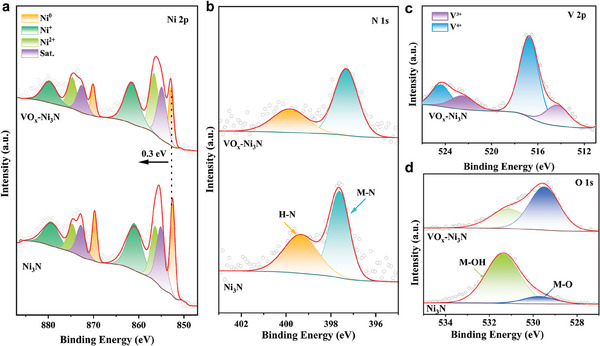
XPS spectra of Ni3N and VOx−Ni3N: Ni 2p a), N 1s (b). c) V 2p XPS spectrum of VOx−Ni3N and O 1s spectra of Ni3N and VOx–Ni3N (d).
In the N 1s spectrum (Figure 2b) the peaks of 397.6 and 399.4 eV belong to the Ni─N and N─H bonds, respectively. Notably, the Ni─N bond undergoes a negative shift of 0.3 eV, indicating a weak Ni─N interaction in VOx−Ni3N.[ 27 ] This is quite consistent with the formation of the Ni─O─V interface, whilst diminishing the Ni─N species. Then, the in‐plane polarization of nickel sites within the VOx‐Ni3N heterostructure is evident from the emerging Ni─N species relative to the metallic nickel, especially the nickel sites at the surface. For vanadium, the hybrid valence of both 3+ and 4+ is confirmed in Figure 2c, as two sets of peaks are shown in the V 2p1/2 range. The prominent peak at 513.7 eV indicates the dominated V2O3 species in the amorphous VOx. The higher binding energy related to V4+ species might be attributed to the formed Ni─O─V interface, particularly the charge redistribution within it.[ 28 , 29 ] The shift of the corresponding M─O binding energy in O1s spectra also confirms the mixed metal–oxygen species (Figure 2d), consistent with the emerging Ni─O─V interface in the VOx−Ni3N heterostructure. Therefore, it can be concluded that introducing V into Ni3N encourages the formation of the Ni─O─V interface in the VOx−Ni3N heterostructure.
To further clarify the coordination environments and electronic structures of the VOx−Ni3N heterostructure, specifically the Ni─O─V interface, we performed X‐ray absorption fine structure (XAFS) analyses. As shown in Figure 3a, the X‐ray absorption near edge structure (XANES) spectra of Ni K‐edge indicate that the adsorption edges of both VOx−Ni3N and Ni3N are close to that of the Ni foil. Moreover, VOx−Ni3N presents a high‐energy shift of the pre‐edge feature, compared with Ni foil and Ni3N, suggesting an oxidation of Ni upon forming the Ni─O─V interface.[ 30 , 31 ] This is consistent with the binding energy shift in XPS spectra. Figure 3b shows the Fourier transform k2‐weighted extended X‐ray absorption fine structure (FT‐EXAFS) spectra (Table S1, Supporting Information). The prominent peak is located at ≈2.16 Å (uncorrected value) for VOx−Ni3N, assigned to the nitrogen‐bridged Ni─Ni scattering.[ 32 ] Besides, a scattering peak corresponding to Ni─N/O scattering is located at ≈1.50 Å. Notably, the Ni─N/O scattering increases upon the decrease of the Ni─Ni scattering for VOx−Ni3N when compared with that of bare Ni3N, indicating that the introduced V encourages the formation of the Ni─O─V interface.
Figure 3.
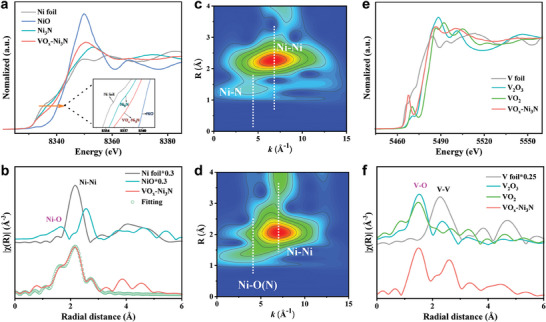
a) Ni K−edge XANES spectra of VOx−Ni3N, Ni3N, NiO, and Ni foil. b) Fourier transforms of the EXAFS (FT−EXAFS) spectra of VOx−Ni3N, Ni3N, NiO, and Ni foil. WT−EXAFS contour plots of the c) Ni3N and d) VOx−Ni3N. e) V K−edge XANES spectra of VOx−Ni3N, VO2, V2O3 and V foil. f) FT−EXAFS spectra of VOx−Ni3N, VO2, V2O3 and V foil.
The wavelet‐transformed (WT) of Ni K‐edge EXAFS oscillation was employed to illustrate the variation in the coordination structure at high k‐space resolution. Figure 3c,d provide the WT contour spectra of Ni3N and VOx−Ni3N, respectively. They both exhibit intensity maxima at ≈7 Å−1, attributed to the prominent Ni─Ni scattering due to the Ni3N principle.[ 33 ] The same intensity maximum also suggests that V stays in a separate phase, rather than insertion into the Ni3N framework. For Ni─N/O scattering at 4.4 Å−1, the enhanced intensity of Ni─N/O scattering for VOx−Ni3N further confirms the VOx‐mediated Ni─O─V interface. The electronic structure of V in VOx−Ni3N was probed from the V K‐edge XAFS spectra (Figure 3e). The absorption edge of V in VOx−Ni3N is comparable with V2O3. Meanwhile, V mainly coordinates with oxygen in the first scattering shell, consistent with standard vanadium oxides (Figure 3f). The subtle differences observed in the pre‐edge and white‐line intensity imply that the valence state of V in VOx−Ni3N is somewhat higher than that in V2O3, not entirely +3. The emerging Ni─O─V interface in VOx−Ni3N further encourages the charge transfer and results in an increased valence of V. Therefore, the Ni─O─V interface in VOx−Ni3N is clearly identified, exhibiting prominent charge redistribution accompanied by interfacial polarization.
2.2. Electrocatalytic Performance
The EOR performance of the catalysts was evaluated using a standard three‐electrode system. The test conditions were first optimized by choosing various electrolytes (Figure S3, Supporting Information). The current densities achieved by VOx−Ni3N after the addition of 0.05, 0.1, 0.25, 0.5, and 1 m ethanol (EtOH)to 1 m KOH were 5, 23, 32, 22, and 14 mA cm−2, respectively, at a voltage of 1.4 V versus reversible hydrogen electrode (VRHE). Hence, the electrolyte condition was maintained as 1 m KOH with 0.25 M EtOH. Notably, VOx−Ni3N delivers the desired catalytic activity, outperforming nickel‐based hydroxides, oxides, and nickel metals (Figure 4a). A potential of merely 1.4 V is required to perform a current density of 50 mA cm−2, which is far superior than that of V−Ni (1.45 V), V−NiO (1.46), and V−Ni(OH)2 (1.47 V). According to the electrochemical impedance spectroscopy (EIS) plots in Figure 4b and Table S2 (Supporting Information), the VOx−Ni3N shows the smallest charge transfer resistance (Rct), indicating the kinetic charge transfer during the EOR process. Also, the durability of the VOx−Ni3N toward EOR is demonstrated by the steady current density observed at a fixed potential, as depicted in Figure 4c.
Figure 4.
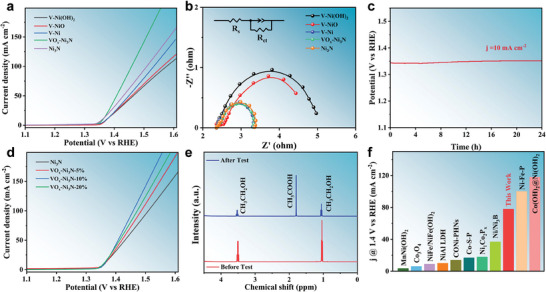
The EOR electrocatalytic properties of the VOx−Ni3N catalyst in 1 m KOH with 0.25 m EtOH electrolyte (without iR compensation): a) Linear sweep voltammetry (LSV) curves of V−Ni(OH)2, V−NiO, V−Ni and VOx−Ni3N. b) EIS curves. The inset is the equivalent circuit. c) Constant potential V–t curves of VOx−Ni3N. d) LSV curves of Ni3N, VOx−Ni3N−5%, VOx−Ni3N−10% and VOx−Ni3N−20%. e) 1H NMR measurements of the product after the reaction. f) Comparison of EOR performance with other transition metal−based catalyst histograms.
The dependence of catalytic performance on the amount of introduced V was also evaluated in Figure 4d and Table S3 (Supporting Information). The findings suggest that incorporating 10% V into VOx−Ni3N yields the best catalytic performance. Meanwhile, VOx−Ni3N−10% (with 10% vanadium content) exhibits the lowest Tafel slope and charge transfer resistance (Rct), as depicted in Figure S4 (Supporting Information). This suggests that the specific incorporation of vanadium significantly facilitates the electron transfer rate, which in turn optimizes the electrocatalytic activity for EOR. Besides, the electrochemically active surface area (ECSA) of the catalysts is measured to further evaluate the effect of surface area on the electrochemical performance (Figure S5, Supporting Information). Upon the incorporation of V, the ECSA exhibits a slight reduction compared to the pristine Ni3N. According to the ECSA normalized catalytic activity, VOx−Ni3N−10% behaves the optimal EOR performance, consistent with the result in Figure 4d. This result indicates that the ECSA is not the primary factor responsible for the observed variations in catalytic performance. Moreover, the lower VOx component will reduce the interface component, potentially hindering the catalytic process. Conversely, an elevated VOx content may lead to the encapsulation of Ni3N by amorphous VOx, which could compromise the availability of active nickel sites. Consequently, an intermediate loading content of ≈10% of vanadium is proposed to strike a balance between maintaining dual‐polarization and ensuring the exposure of the nickel species necessary for optimal catalytic activity. Thus, the enhanced catalytic activity of VOx−Ni3N is mainly attributed to the intrinsic activity of the embedded Ni–O–V interface within the VOx−Ni3N.
As shown in Figure 4e, the products before and after the reaction were evaluated by using 1H NMR spectroscopy. The chemical shifts ≈1.0 and 3.5 ppm before EOR correspond to the triple and quadruple peaks of the hydrogen proton in ethanol, respectively.[ 18 ] After EOR, the intensity of the two peaks decreases appreciably, indicating that most of the ethanol was consumed. Additionally, a new peak appears at 1.7 ppm, which is attributable to the methyl group adjacent to the carboxylate in potassium acetate.[ 34 ] Therefore, the EOR process follows a 4e− reaction pathway: *CH3CH2OH→*CH3CH2O →*CH3CHO→*CH3CO→*CH3COOH. This catalytic performance of VOx−Ni3N toward EOR is benchmarked against the state‐of‐the‐art nickel‐ and cobalt‐based catalysts (Figure 4f; Table S4, Supporting Information).
2.3. Catalytic Mechanism
To gain an insight into the catalytic mechanism of the Ni─O─V interface on the EOR process over VOx−Ni3N, Operando electrochemical impedance spectroscopy was employed to investigate the potential‐dependent interfacial behavior.[ 35 , 36 ] Figure 5a,b describe the Bode plots of V─Ni(OH)2 and VOx−Ni3N. Before 1.35 V, there is only a high‐frequency interface with a very weak charge transfer.[ 37 ] After 1.35 V, there is a sudden decrease in the phase angle, suggesting that the catalyst initiates its reaction. The interfacial response of V─Ni(OH)2 in the high‐frequency region indicates that the dominant reaction process is the dehydrogenation of Ni(OH)2 species into Ni(OH)O species. In contrast, the interfacial response of VOx−Ni3N occurs in the low‐frequency region, indicating that the reaction occurs at the catalyst surface, involving adsorbed species such as OH* or O*.
Figure 5.
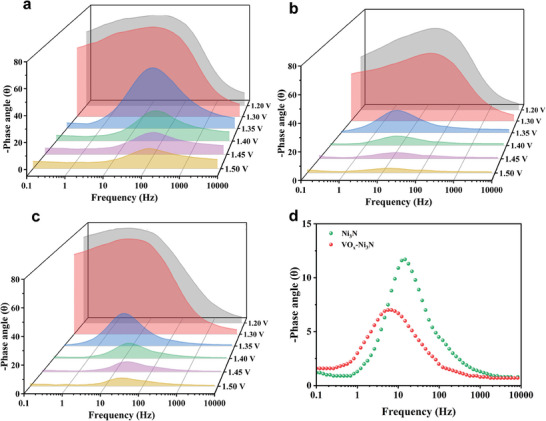
The operand electrochemical impedance spectroscopy characterization: Bode plots of a) V─Ni(OH)2, b) VOx−Ni3N, and c) Ni3N electrode for EOR in different potentials. d) Bode phase plots of VOx−Ni3N and Ni3N at 1.4 V RHE.
Bode plots were utilized for various ethanol concentrations to determine the primary adsorbed species on the catalyst. As illustrated in Figure S6 (Supporting Information), the pre‐oxidation reactions, specifically the adsorption of OH−, are consistently maintained between 1.2 to 1.3 V, even with the escalation of ethanol concentrations. Notably, there is no discernible reduction in potential below 1.35 V. This observation points to the low reactivity of OH* adsorption during EOR, thereby suggesting that the dominant active species in the process is more likely to be O*.[ 38 ] Besides, at low ethanol concentrations, the generated O* species are not immediately utilized. Whereafter, an accumulation of O* occurs, resulting in the formation of an oxide layer on the catalyst's surface as the potential rises. As a result, OER begins competing with the EOR, consequently diminishing the selectivity of the EOR. According to Figure S6c–d (Supporting Information), the phase angle frequency exhibits a positive shift with an increase in ethanol concentration, suggesting that an excess of ethanol impedes the adsorption of active species. This interference with adsorption is suggested to be the cause of the observed decline in EOR activity. The variation in phase angle values between the phase peaks of 1.2 and 1.3 V represents the strength of OH− adsorption before the EOR. In comparison to Ni3N (Figure 5c), the greater phase angle difference observed for VOx−Ni3N suggests more robust adsorption of OH−. The phase angles of the two samples were also further compared at a voltage of 1.4 VRHE. It is found that the phase peaks of VOx−Ni3N at the low‐frequency interface show smaller phase angles and displacements (Figure 5d), This observation suggests a kinetic deprotonation process of the oxygen‐containing intermediate OH* during the EOR process over VOx−Ni3N.[ 39 , 40 ]
Density Functional Theory (DFT) calculations have provided insights into the catalytic activity for EOR over the Ni─O─V interface. It is well understood that heterogeneous components possess distinct work functions, which naturally give rise to an internal electric field (IEF).[ 41 ] The charge difference density distribution, as depicted in Figure 6a, indicates that the built‐in IEF encourages a pronounced charge redistribution at the Ni─O─V interface. The delocalized electron further promotes the polarization of the Ni─O─V interface, thereby facilitating the transfer of electrons at the interface.[ 42 ] Figure 6b provides the calculation of the Bader charge value for the Ni site on pristine Ni3N and VOx−Ni3N. The average Bader charge value of Ni on Ni3N was calculated to be −0.20 e, signifying electrons transfer from Ni to N. This electron transfer suggests the occurrence of spontaneous in‐plane lattice polarization, where Ni atoms lose electrons into positively charged, and N atoms gain electrons into negatively charged. In the case of the Ni─O─V interface in VOx−Ni3N, the Bader charge value of −0.36 e for Ni indicates a significant electron transfer from Ni3N to VOx. This interfacial polarization is instrumental in leading to an increased valence state of Ni at the Ni─O─V interface. This charge transfer is in alignment with the electronegativity order: O (3.44) > N(3.04) > Ni (1.91) > V (1.63). As a result, the dual polarization at the Ni─O─V interface, particularly in‐plane polarization, and IEF−induced interfacial polarization, plays a crucial role in modulating the electronic properties of the Ni site, especially the increase in the d‐band center of Ni.
Figure 6.
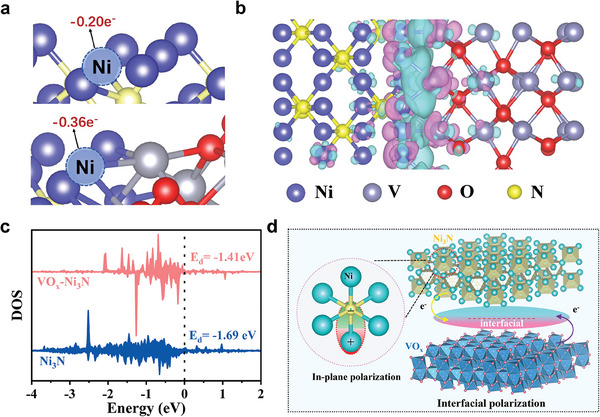
DFT calculation results of the HER process on the catalyst surfaces: a) Differential charge density of VOx−Ni3N heterostructure. Cyan and pink indicate charge accumulation and consumption, respectively. b) Bader charge analysis of Ni3N and VOx−Ni3N. c) Density of states (DOS) of Ni−d orbitals in Ni3N and VOx−Ni3N. d) Schematic of dual polarization in VOx−Ni3N.
According to the d‐band center (εd) theory, the lower energy level of the d‐band center indicates a higher occupancy of the antibonding states, which correlates with weaker adsorption toward intermediates.[ 43 ] As the density of state depicted in Figure 6c, the εd value of VOx−Ni3N is −1.41 eV, higher than that of pristine Ni3N (−1.69 eV). As illustrated in Figure 6d, the dual polarization at the Ni─O─V interface could effectively optimize the adsorption of key intermediates, such as OH−, and kinetically promote the formation of reactive species O*.[ 44 ]
Furthermore, we calculated the Gibbs free energy required for the EOR process (Figure 7 ). For the key step of the formation of reactive O*, an uphill energy of 1.1 eV is required for Ni3N, while this energy drops to 0.57 eV for Ni─O─V interface‐mediated VOx−Ni3N. The calculated result is quite consistent with the in situ electrochemical impedance analyses. Moreover, the Ni sites in the Ni─O─V interface were tuned for favorable adsorption for all involved intermediates. A more energetic desorption (0.95 eV) of acetic acid over the Ni─O─V interface is demonstrated in comparison to pristine Ni3N (1.86 eV). Therefore, DFT calculation clarifies the dual polarization within the Ni─O─V interface in VOx−Ni3N heterostructure, which substantially encourages the EOR process thermodynamically.
Figure 7.
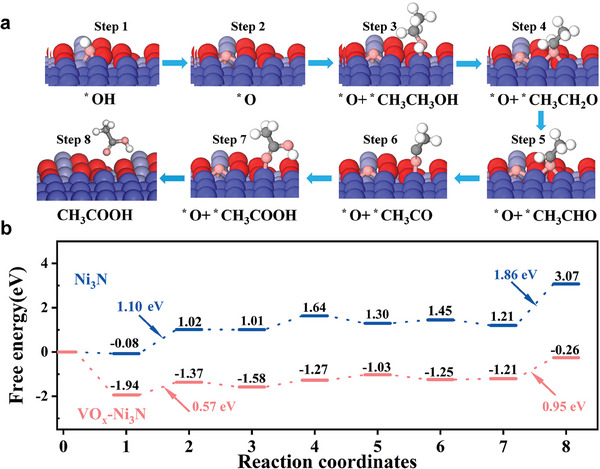
DFT calculation for the reaction of ethanol on Ni3N and VOx−Ni3N. a) The reaction coordinates (a) and the corresponding Gibbs free energies b) during the EOR process. The blue, cyan, red, grey, and white balls represent nickel, vanadium, oxygen, carbon, and hydrogen, respectively.
2.4. Hybrid Electrolyzer
In a further study, it was revealed that a proper Ni─O─V interface could also significantly enhance the HER catalytic activity. As shown in Figure 8a, VOx−Ni3N exhibits an excellent HER catalytic performance in alkaline conditions. Specifically, when the proportion of vanadium is 20% (VOx−Ni3N−20%), the required overpotential is 60.3 mV. This value is significantly lower than that of Ni3N (117.6 mV), VOx−Ni3N−10% (101.4 mV), and VOx−Ni3N−30% (81.7 mV). The superior hydrogen evolution reaction (HER) performance exhibited by VOx−Ni3N−20% as opposed to VOx−Ni3N−10% can likely be attributed to the increased presence of VOx‐induced Ni–O species, which are essential for water dissociation in the alkaline HER process The corresponding Tafel and impedance analyses (Figure S7a−b, Supporting Information) signify that the dual polarization within the Ni─O─V interface could synergistically promote the Volmer step and the sequential hydrogen production.[ 45 ] Besides, the desired durability of VOx−Ni3N for alkaline HER is demonstrated as 24 h of steady catalysis at 10 mA cm−2 (Figure S7c, Supporting Information).
Figure 8.

HER performance in 1 m KOH + 0.25 m EtOH electrolyte (without iR compensation): a) LSV curves of Ni3N, VOx−Ni3N−10%, VOx−Ni3N−20% and VOx−Ni3N−30%. b) schematic diagram of the HER coupled EOR hybrid electrolysis. c) LSV curves of the hybrid and conventional hydrolysis, where VOx−Ni3N−10% was used as the anode and VOx−Ni3N−20% as the cathode. d) V−t diagrams of the hybrid hydrolysis.
Due to the outstanding bifunctional application of VOx−Ni3N for both EOR and HER, a hybrid electrochemical cell was established (Figure 8b). The anodic chamber contains KOH and ethanol at the concentrations of 1 and 0.25 m, respectively; while the cathodic chamber only contains KOH at the concentration of 1 m. Figure 8c,d demonstrate that the system offers outstanding catalytic performance and favorable durability in ethanol oxidation coupled hydrogen production. At a current density of 50 mA cm−2, a cell voltage of ≈1.5 V is required, which is 300 mV lower than overall water splitting. Meanwhile, the performance is significantly superior to many mixed electrolytic water catalyst systems with non−precious metals (Tables S5−S6, Supporting Information). Therefore, creating dual‐polarization by precisely engineering an interface within heterostructure is of great significance toward broad catalysis.
3. Conclusion
In summary, we have meticulously crafted a heterostructure that integrates amorphous vanadium oxide with crystalline nickel nitride (VOx−Ni3N), featuring a well‐defined Ni─O─V interface. The catalyst demonstrates high‐efficiency ethanol oxidation into acetic acid, specifically delivering a current density of 50 mA cm−2 at 1.38 VRHE, significantly surpassing pristine Ni3N. Various characterization techniques and theoretical calculations elucidate the dual polarization, particularly the interfacial polarization induced by the internal electric field and the in‐plane polarization within the Ni─O─V interface, which encourages the modification of electronic structure, thereby facilitating the EOR process. Given the superior catalytic performance of VOx−Ni3N toward alkaline hydrogen evolution, an EOR‐coupled‐HER electrolyzer was established. A commercial dry battery (1.5 V) is capable of driving the electrolyzer to deliver a current density of 50 mA cm−2, far surpassing the traditional overall water−splitting electrolyzer both in energy conservation and the production of value‐added products. This strategy of precisely engineering interface in heterostructure is applicable to a broad range of applications.
Conflict of Interest
There is no conflict of interest to declare.
Author Contributions
M.Z., B.J., W.K., and Y.C. performed the experimental investigation. A.C. and X.Z. performed the theoretical investigations. M.Z. wrote the manuscript. F.L. and X.Z. revised the manuscript. F.L., X. W., and X.Z. supervised this work.
Supporting information
Supporting Information
Acknowledgements
This work is supported by the National Natural Science Foundation of China (52372097 and 52202310), the Youth Hundred Talents Program of Yangzhou University, the Qinglan Project and the Top‐talent Program of Yangzhou University, and the Innovation Technology Platform Project (YZ2020268, SSX2023000032) jointly built by Yangzhou City and Yangzhou University.
Zhou M., Jin B., Kong W., Chen A., Chen Y., Zhang X., Lu F., Wang X., Zeng X., Dual Polarization of Ni Sites at VOx−Ni3N Interface Boosts Ethanol Oxidation Reaction. Adv. Sci. 2024, 11, 2407473. 10.1002/advs.202407473
Contributor Information
Fei Lu, Email: feilu@yzu.edu.cn.
Xi Wang, Email: xiwang@bjtu.edu.cn.
Xianghua Zeng, Email: xhzeng@yzu.edu.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Navarro R. M., Peña M. A., Fierro J. L. G., Chem. Rev. 2007, 107, 3952. [DOI] [PubMed] [Google Scholar]
- 2. Dincer I., Int. J. Hydrog. Energy. 2012, 37, 1954. [Google Scholar]
- 3. Wang X., Zheng Y., Sheng W., Xu Z. J., Jaroniec M., Qiao S. Z., Mater. Today 2020, 36, 125. [Google Scholar]
- 4. Zou X., Zhang Y., Chem. Soc. Rev. 2015, 44, 5148. [DOI] [PubMed] [Google Scholar]
- 5. Gautam J., Liu Y., Gu J., Ma Z., Zha J., Dahal B., Zhang L. N., Chishti A. N., Ni L., Diao G., Wei Y., Adv. Funct. Mater. 2021, 31, 2106147. [Google Scholar]
- 6. Gong C., Zhao L., Li D., He X., Chen H., Du X., Wang D., Fang W., Zeng X., Li W., Chem. Eng. J. 2023, 464, 143124. [Google Scholar]
- 7. Li X., Xiao L., Zhou L., Xu Q., Weng J., Xu J., Liu B., Angew. Chem., Int. Ed. 2020, 59, 21106. [DOI] [PubMed] [Google Scholar]
- 8. Li X., Deng C., Kong Y., Huo Q., Mi L., Sun J., Cao J., Shao J., Chen X., Zhou W., Lv M., Chai X., Yang H., Hu Q., He C., Angew. Chem., Int. Ed. 2023, 62, e202309732. [DOI] [PubMed] [Google Scholar]
- 9. Arshad F., Haq T. U., Hussain I., Sher F., ACS App. Energy Mater. 2021, 4, 2342. [Google Scholar]
- 10. Luo H., Barrio J., Sunny N., Li A., Steier L., Shah N., Stephens I. E. L., Titirici M. M., Adv. Energy Mater. 2021, 11, 2101180. [Google Scholar]
- 11. Chen W., Xie C., Wang Y., Zou Y., Dong C. L., Huang Y. C., Xiao Z., Wei Z., Du S., Chen C., Zhou B., Ma J., Wang S., Chem 2020, 6, 2974. [Google Scholar]
- 12. Li Z., Ning S., Xu J., Zhu J., Yuan Z., Wu Y., Chen J., Xie F., Jin Y., Wang N., Meng H., Sun S., Energy Environ. Sci. 2022, 15, 5300. [Google Scholar]
- 13. Dai L., Qin Q., Zhao X., Xu C., Hu C., Mo S., Wang Y. O., Lin S., Tang Z., Zheng N., ACS Cent. Sci. 2016, 2, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Q., Qi S., Huo Q., Zhao Y., Sun J., Chen X., Lv M., Zhou W., Feng C., Chai X., Yang H., He C., J. Am. Chem. Soc. 2024, 146, 2967. [DOI] [PubMed] [Google Scholar]
- 15. Ding Y., Xue Q., Hong Q. L., Li F. M., Jiang Y. C., Li S. N., Chen Y., ACS Appl. Mater. Interface 2021, 13, 4026. [DOI] [PubMed] [Google Scholar]
- 16. Zheng J., Chen X., Zhong X., Li S., Liu T., Zhuang G., Li X., Deng S., Mei D., Wang J. G., Adv. Funct. Mater. 2017, 27, 1704169. [Google Scholar]
- 17. Chen G. F., Luo Y., Ding L. X., Wang H., ACS Catal. 2018, 8, 526. [Google Scholar]
- 18. Sun H., Tian C., Fan G., Qi J., Liu Z., Yan Z., Cheng F., Chen J., Li C. P., Du M., Adv. Funct. Mater. 2020, 30, 1910596. [Google Scholar]
- 19. Zhai P., Wang C., Zhao Y., Zhang Y., Gao J., Sun L., Hou J., Nat. Commun. 2023, 14, 1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bai Y., Zhang W., Zhang Z., Zhou J., Wang X., Wang C., Huang W., Jiang J., Xiong Y., J. Am. Chem. Soc. 2014, 136, 14650. [DOI] [PubMed] [Google Scholar]
- 21. Hu Q., Zhou W., Qi S., Huo Q., Li X., Lv M., Chen X., Feng C., Yu J., Chai X., Yang H., He C., Nat. Sustain. 2024, 7, 442. [Google Scholar]
- 22. Lin Y., Yang L., Zhang Y., Jiang H., Xiao Z., Wu C., Zhang G., Jiang J., Song L., Adv. Energy Mater. 2018, 8, 1703623. [Google Scholar]
- 23. Wang S., Song C., Cai Y., Li Y., Jiang P., Li H., Yu B., Ma T., Adv. Energy Mater. 2023, 13, 2301136. [Google Scholar]
- 24. Wang Z., Xu L., Huang F., Qu L., Li J., Owusu K. A., Liu Z., Lin Z., Xiang B., Liu X., Zhao K., Liao X., Yang W., Cheng Y. B., Mai L., Adv. Energy Mater. 2019, 9, 1900390. [Google Scholar]
- 25. Li R. Q., Liu Q., Zhou Y., Lu M., Hou J., Qu K., Zhu Y., Fontaine O., J. Mater. Chem. A. 2021, 9, 4159. [Google Scholar]
- 26. Chen X. H., Li X., Wu L. L., Fu H. C., Luo J., Shen L., Zhang Q., Lei J. L., Luo H. Q., Li N. B., J. Mater. Chem. A. 2021, 9, 11563. [Google Scholar]
- 27. Yang M., Zhao M., Yuan J., Luo J., Zhang J., Lu Z., Chen D., Fu X., Wang L., Liu C., Small 2022, 18, 2106554. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Q., Liu B., Li L., Ji Y., Wang C., Zhang L., Su Z., Small 2021, 17, 2005769. [DOI] [PubMed] [Google Scholar]
- 29. Zhou M., Li H., Long A., Zhou B., Lu F., Zhang F., Zhan F., Zhang Z., Xie W., Zeng X., Yi D., Wang X., Adv. Energy Mater. 2021, 11, 2101789. [Google Scholar]
- 30. Wang T., Wang M., Yang H., Xu M., Zuo C., Feng K., Xie M., Deng J., Zhong J., Zhou W., Cheng T., Li Y., Energy Environ. Sci. 2019, 12, 3522. [Google Scholar]
- 31. Wu T., Song E., Zhang S., Luo M., Zhao C., Zhao W., Liu J., Huang F., Adv. Mater. 2022, 34, 2108505. [DOI] [PubMed] [Google Scholar]
- 32. Liu H., He Q., Jiang H., Lin Y., Zhang Y., Habib M., Chen S., Song L., Angew. Chem., Int. Ed. 2017, 11, 11574. [DOI] [PubMed] [Google Scholar]
- 33. Li S., Wang S., He J., Li K., Xu Y., Wang M., Zhao S., Wang Y., Li X., Zhong X., Wang J., Angew. Chem., Int. Ed. 2023, 62, e202306553. [DOI] [PubMed] [Google Scholar]
- 34. Zhao X., Dai L., Qin Q., Pei F., Hu C., Zheng N., Small 2017, 13, 1602970. [DOI] [PubMed] [Google Scholar]
- 35. Lu Y., Liu T., Dong C. L., Yang C., Zhou L., Huang Y. C., Li Y., Zhou B., Zou Y., Wang S., Adv. Mater. 2022, 34, 2107185. [DOI] [PubMed] [Google Scholar]
- 36. Wang H. Y., Hung S. F., Chen H. Y., Chan T. S., Chen H. M., Liu B., J. Am. Chem. Soc. 2016, 138, 36.26710084 [Google Scholar]
- 37. Lyons M. E. G., Brandon M. P., J. Electroanal. Chem. 2009, 631, 62. [Google Scholar]
- 38. Tao H. B., Xu Y., Huang X., Chen J., Pei L., Zhang J., Chen J. G., Liu B., Joule 2019, 3, 1498. [Google Scholar]
- 39. Palmas S., Ferrara F., Vacca A., Mascia M., Polcaro A. M., Electrochim. Acta. 2007, 53, 400. [Google Scholar]
- 40. Zhao T., Shen X., Wang Y., Hocking R. K., Li Y., Rong C., Dastafkan K., Su Z., Zhao C., Adv. Funct. Mater. 2021, 31, 2100614. [Google Scholar]
- 41. Chen L., Wang H. Y., Tian W. W., Wang L., Sun M. L., Ren J. T., Yuan Z. Y., Small 2023, 20, 2307252. [Google Scholar]
- 42. Zhang Y., Guo F., Di J., Wang K., Li M. M. J., Dai J., She Y., Xia J., Li H., Nano‐Micro Lett. 2024, 16, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang N., Ning S., Yu X., Chen D., Li Z., Xu J., Meng H., Zhao D., Li L., Liu Q., Lu B., Chen S., Appl. Catal., B 2022, 302, 120838. [Google Scholar]
- 44. Zhao B., Liu J., Wang X., Xu C., Sui P., Feng R., Wang L., Zhang J., Luo J. L., Fu X. Z., Nano Energy 2021, 80, 105530. [Google Scholar]
- 45. Zhou P., Zhai G., Lv X., Liu Y., Wang Z., Wang P., Zheng Z., Cheng H., Dai Y., Huang B., Appl. Catal., B 2020, 283, 119590. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.