


Abstract
DNA ligases are grouped into two families, ATP-dependent and NAD-dependent, according to the cofactor required for their activity. A surprising capability of both kinds of ligases to complement for one another in vivo has been observed. Bacillus subtilis harbours one NAD-dependent ligase, YerG, and two ATP-dependent ligases, YkoU and YoqV, this last one being encoded by the 134 kb lysogenic bacteriophage SPβ and consisting of a single adenylation domain typical of ATP-dependent ligases. Because the genetics of ligases in B.subtilis had not been studied previously, the genes encoding for one ligase of each kind, yerG and yoqV, were investigated. We found that the yerG gene was essential in B.subtilis. This suggests that none of the ATP-dependent ligases was able to complement the yerG defect. In addition, the ATP-dependent ligase encoded by yoqV, when cloned on a plasmid under appropriate expression signals, was unable to rescue a yerG mutant strain. The two B.subtilis ligase genes yerG and yoqV were also introduced in an Escherichia coli strain encoding a thermosensitive ligase (ligts), and whereas yoqV did not complement the ligts defects, yerG fully complemented the growth and UV sensitivity defects of the lig mutant. We propose to rename the yerG and yoqV genes of B.subtilis ligA and ligB respectively.
INTRODUCTION
DNA ligases catalyse the joining of breaks in duplex DNA during DNA replication, repair and recombination. They are grouped into two families, ATP-dependent DNA ligases and NAD-dependent ligases, according to the cofactor required for their activity. Apart from a common KXDG motif, these two families share few amino acid similarities. However, the reactions catalysed by the two classes of DNA ligase are identical, except for the fact that the AMP group is derived from two different cofactors. This implies that first there is covalent attachment of AMP to the lysine residue of the KXDG motif, followed by its transfer to the 5′-phosphoryl group at a nicked site of duplex DNA. The AMP group is finally then released from the adenylated DNA as the phosphodiester bond is formed. The crystal structure of the 41 kDa ATP-dependent ligase of bacteriophage T7 has been solved (1), as well as the adenylation domain of the NAD-dependent DNA ligase of Bacillus stearothermophilus (2). This enabled Wigley and colleagues (2) to make a structural alignment of the adenylation domain of the two proteins, which prove to have a similar fold and to share several critical residues. More recently the three-dimensional structure of the complete 76 kDa NAD-dependent ligase of Thermus filiformis has been reported (3).
NAD-dependent ligases are of a uniform size (~70 kDa) and reveal extensive amino acid sequence conservation throughout the entire protein. ATP-dependent ligases are more diverse in size, but share a common ligase domain, ∼270 residues in length, possibly flanked by different domains. Until recently, the common view was that NAD-dependent ligases are present in eubacteria only, whereas ATP-dependent ligases are common to all other living organisms, including bacteriophages, archeabacteria, viruses and eukaryotes. However, a different picture has emerged with the rising number of complete bacterial genomes that have been sequenced. Among the 17 complete genomes of eubacteria presently available, all contain at least one gene homologous to the NAD-dependent ligase family, but three contain one or several copies of genes homologous to the ATP-dependent ligase family as well: Bacillus subtilis, Mycobacterium tuberculosis and Haemophilus influenzae (Table 1).
Table 1. Distribution of DNA ligases in B.subtilis, H.influenzae and M.tuberculosis.
| Bacterium species | NAD-dependent ligase (length in amino acids) | ATP-dependent ligase (length in amino acids) |
|---|---|---|
| B.subtilis | YerG (668) | YoqV (270) |
| YkoUa (611) | ||
| M.tuberculosis | LigA (691) | LigB (507) |
| LigC (358) | ||
| O28549 (759) | ||
| H.influenzae | DNLJ (679) | DNL1 (268) |
aThis protein is not mentionned as a putative ligase in data banks, however the Pfam algorithm for protein alignment includes it in its ATP-dependent ligase family. Indeed all conserved motives among ATP-dependent ligase are present in this protein (to see the alignment: http//www.sanger.ac.uk/Software/Pfam).
In two eubacterial species, Escherichia coli and Salmonella typhimurium, the NAD-dependent ligase gene, encoded by the lig gene, is known to be essential (4–6). However in the three species where more than one ligase gene is present, nothing is known as to whether any of the genes are essential. This question is addressed here with regard to B.subtilis. The B.subtilis genome harbours one NAD-dependent ligase-like gene, yerG, and two ATP-dependent ligase-like genes, yoqV and ykoU. The yoqV gene is encoded by the 134 kb lysogenic bacteriophage SPβ, but ykoU has no apparent bacteriophage origin. YoqV is a small protein composed of 270 amino acids, which consist of a single domain, the adenylation domain typical of ATP-dependent ligases. The YkoU protein is 611 amino acids in length, and its adenylation domain encompasses the first 292 residues. Of these two, only the smaller one, YoqV, was studied here in detail. Both ATP-dependent ligase genes are dispensable, because a strain cured of the SPβ prophage is viable, as well as a ykoU mutant, which has been constructed in the frame of a systematic analysis of unknown genes of B.subtilis (Micado database: http://locus.jouy.inra.fr/cgi-bin/genmic/madbase_home.pl; strain constructed in the laboratory of Dr K. Devine).
Various studies have shown that heterologous complementation is possible between ATP-dependent and NAD-dependent enzymes. Among examples of ATP-dependent ligases able to complement bacterial ligase mutants, it has been shown that the bacteriophage T4 ligase is able to rescue a lig mutant of S.typhimurium (4), and the catalytic domain of human ligase I restores the thermoresistance of an E.coli strain encoding a thermosensitive ligase (ligts) (7). Conversely, the NAD-dependent ligase of E.coli restores the thermoresistance of a Saccharomyces cerevisiae strain encoding a thermosensitive ligase (cdc9ts) (8).
Because of the high degree of complementation between both classes of enzymes reported in the literature, it was not possible to tell a priori whether the three ligase genes of the B.subtilis genome would have overlapping functions, or whether one of them was essential for viability. We therefore decided to investigate whether the NAD-dependent ligase encoding gene, yerG, was essential or dispensable in B.subtilis. We show here that this gene is essential in B.subtilis. In addition, we found that the ATP-dependent ligase encoded by yoqV, when cloned on a plasmid under appropriate expression signals, is unable to rescue a yerG mutant strain. This indicates that the two ligases cannot substitute for one another in B.subtilis. The two B.subtilis ligase genes yerG and yoqV were also introduced in an E.coli ligts strain, and we found that whereas yoqV does not complement the ligts defects, yerG fully complements the growth and UV sensitivity defects of the lig mutant.
MATERIALS AND METHODS
Strains and plasmids
Strains and plasmids are listed in Table 2. Bacillus subtilis and E.coli cells were grown on LB medium unless otherwise stated. When needed, antibiotics were added to a final concentration of 5 µg/ml for kanamycin and 0.5 µg/ml for erythromycin for B.subtilis, and 100 µg/ml ampicillin for E.coli. The chemically defined medium has been described elsewhere (9). Constructions were performed as described below. The integrity of the sequence of all DNA fragments generated by PCR was verified by automated dideoxysequencing.
Table 2. Strains and plasmids.
| Name | Relevant genotype, description | Reference, source | |
|---|---|---|---|
| Strains | JJC75a | lig7ts | B.Michel |
| 168 | trpC2 | C.Anagnostopoulos | |
| HVS610 | 168 insb pMAP149 in yerH | This work | |
| HVS609 | 168 ins pMAP52 in yerG | This work | |
| HVS611 | 168 ins pMAP59 in yerG, with plasmid pMAP141 | This work | |
| Plasmids | pDG148 | pBR322-pUB110 KanR lacI Pspac | (15) |
| pMUTIN2 | pBR322 lacZ lacI EmR Pspac | (16) | |
| pMAP52 | pMUTIN2 Pspac:(nt –20c to 275 of yerG) | This work | |
| pMAP59 | pMUTIN2MCS Pspac:(nt 330–666 of yerG) | This work | |
| pMAP65 | pUB110 KanR lacI | (9) | |
| pMAP138 | pDG148 Pspac:yoqV | This work | |
| pMAP140 | pDG148 Pspac:yerG | This work | |
| pMAP149 | pMUTIN2MCS Pspac:(nt 540–854 of yerH) | This work | |
| pIL253 | EmR | (17) | |
| pMAP139 | pMAP138Δ(pBR322 lacI) from AatII to PstI, KanR | This work | |
| pMAP141 | pMAP140Δ(pBR322 lacI) from AatII to PstI, KanR | This work |
aAB1157 background, hsdR thr+ pro+.
bins indicates the insertion of a given plasmid in the B.subtilis chromosome.
cGene numbering starts at the start codon.
yerG transcriptional fusion strain HVS609. To fuse yerG to an inducible promoter, plasmid pMUTIN2 was used. This is a pBR322 derivative containing a Pspac IPTG inducible promoter upstream of a multiple cloning site flanked by BamHI and EcoRI. The promoter is preceded by two transcription terminators. Further downstream on the same plasmid are the lacI gene and a selectable erythromycin resistance marker (EmR) for B.subtilis. A DNA fragment starting 20 bp upstream of the yerG start codon, so as to include a ribosome binding site 296 bp in length, and flanked by artificially introduced EcoRI and BamHI sites, was generated by PCR and cloned into pMUTIN2. The resulting plasmid, pMAP52, contained a transcriptional fusion between Pspac and the first few hundred nucleotides of yerG. To generate the fusion between Pspac and the complete ORF of yerG, plasmid pMAP52 was introduced by single crossing over into the chromosome of B.subtilis wild-type 168 strain. The resulting strain, HVS609 (Fig. 1), harbors a single copy of the yerG gene under the control of the Pspac promoter.
Figure 1.

Chromosomal structure of wild-type strain 168 and mutant strains. Strain 168 is shown at the top of the figure. In strain HVS609, plasmid pMAP52 is integrated in yerG, so as to fuse yerG to the Pspac promoter. In strain HVS610, plasmid pMAP149 is disrupting the yerH gene.
yerH mutant strain HVS610. An internal fragment of the yerH gene, 350 bp in length, was generated by PCR and cloned between the EcoRI and BamHI sites of pMUTIN2 to give plasmid pMAP149. To disrupt yerH, plasmid pMAP149 was introduced by a single crossing over into the chromosome of B.subtilis to give strain HVS610 (Fig. 1).
Plasmid pMAP59. To disrupt the yerG gene, a 314 bp internal segment of yerG was generated by PCR and cloned between the EcoRI and BamHI sites of pMUTIN2 to give plasmid pMAP59.
Plasmids pMAP138 to 141. To clone yoqV under strong expression signals, plasmid pDG148 was used as a vector. It is a pBR322-pUB110 shuttle vector, which replicates both in E.coli and B.subtilis. It contains a strong promoter active both in B.subtilis and E.coli and a canonical ribosome binding site immediately upstream of an XbaI site followed by a SalI site. A DNA fragment containing the yoqV ORF flanked by artificially introduced XbaI and SalI sites was generated by PCR and cloned into pDG148 in E.coli. The resulting plasmid, pMAP138, was used for complementation tests in E.coli. For B.subtilis a smaller derivative, pMAP139, deleted for the pBR322 sequences and the lacI gene, was made by an AatII–PstI deletion of pMAP138. Using the same strategy, the yerG ORF was cloned into pDG148 to give pMAP140, and a smaller derivative devoid of pBR322 and lacI sequences, pMAP141, was obtained by an AatII–PstI deletion of pMAP140.
Adenylation test
Bacillus subtilis and E.coli crude extracts were prepared in the following way. A 100 ml overnight culture was centrifuged, and the pellet stored at –80°C. To lyse cells, the frozen pellet was resuspended in 1 ml of 50 mM Tris pH 7.5, 5 mM DTT, 1 mM EDTA, 2 mg/ml lysozyme, complemented with a cocktail of protease inhibitors (Roche). The suspension was incubated for 20 min in ice. Sonication was applied for 5 s using 1 s pulse/9 s pause cycles, with a small probe (Vibracell apparatus). The soluble fraction (2 ml) was collected after a 30 min ultracentrifugation at 100 000 g, its concentration was ~4 mg/ml of total protein.
To test the adenylation activity of crude extracts, 4 µl of extract was incubated in buffer A (50 mM Tris pH 7.5, 10 mM MgCl2, 2 mM DTT, 0.1 mM ATP), in a final volume of 20 µl, in the presence of 5 µCi of [α-32P]ATP, for 15 min at 20°C (the ratio between cold- and radiolabelled ATP was critical for optimal detection of the adenylation activity in crude extracts). Proteins were then denatured in a SDS buffer and separated by polyacrylamide gel electrophoresis (PAGE). The gel was fixed, colored and dried prior to exposition. Radioactivity was detected using a Storm apparatus (Molecular Dynamics).
UV sensitivity test
Exponentially growing cells were diluted serially and plated on LB. Plates were irradiated at a fluence of 2 J/m2/s for different times, and then incubated for growth at 30°C (for strain JJC75) or 42°C for the other two strains.
RESULTS
yerG is an essential gene
The NAD-dependent ligase gene is located at 55° on the B.subtilis chromosome, as the third gene of a putative operon encoding pcrB, pcrA, yerG and yerH (Fig. 1). Among these four genes, only pcrA, encoding a DNA helicase of the SF1 family, has been characterised and found to be an essential gene (9). The pcrB and yerH genes are unknown and do not match with any known protein from other organisms.
A yerG mutant strain was constructed to examine whether the encoded NAD-dependent ligase enzyme is essential in vivo. To disrupt the yerG gene, B.subtilis was transformed with pMAP59, a non-replicative plasmid containing an internal fragment of the gene. No transformants were obtained on rich media, nor on chemically defined media.
The impossibility of disrupting yerG could result from a polar effect of the insertion on the downstream gene of the operon (yerH), or from the requirement of yerG for viability. To test for the first alternative, a yerH mutant was constructed. As for the yerG gene, a non-replicative plasmid containing an internal fragment of yerH was used to transform B.subtilis. Transformants were obtained, which contained the appropriate gene disruption, and the resulting strain (strain HVS610, Fig. 1) did not show any growth defect. We concluded therefore that yerH was not essential, and in turn, that the impossibility to disrupt yerG was likely to be due to its requirement for viability.
To confirm this conclusion, strain HVS609 was constructed, in which yerG was fused to an IPTG inducible promoter. We investigated whether strain HVS609 was able to grow in the absence of IPTG. Almost no sign of lethality was observed after approximately 30 generations of growth in the absence of inducer. This could be due to a low concentration of LacI in the cell (only one copy of the lacI gene had been inserted into the chromosome, together with the Pspac–yerG fusion), allowing leaky yerG expression. When plasmid pMAP65, a pUB110 derivative encoding LacI, was introduced into strain HVS609, arrest of growth was observed in the absence of inducer, as shown in Figure 2. After 2 h, the number of viable cells ceased to increase but optical density continued to increase for one additional hour, as a result of cell filamentation, before leveling off. Therefore, strain HVS609 containing pMAP65 is IPTG-dependent for growth. This confirms that yerG is essential.
Figure 2.

Growth curves of strain HVS609 harbouring plasmid pMAP65. Colony forming units (cfu, left) and optical density (OD650, right) of the strain grown either in the presence (squares) or absence (circles) of IPTG are reported as a function of time.
Overexpression of the yoqV encoded ATP-dependent ligase does not rescue a yerG mutation
Two potential ATP-dependent ligases are encoded by the B.subtilis genome, ykoU and yoqV, neither of which is essential. Since yerG is essential in a wild-type strain of B.subtilis, this suggests that neither ATP-dependent ligase can complement yerG activity. Because the yoqV gene belongs to SPβ prophage, we reasoned that its transcription was likely to be repressed in the chromosome. The possibility existed therefore that the ATP-dependent ligase once fused to appropriate expression signals would be able to complement a yerG mutation. To test this possibility, a plasmid was constructed, pMAP139, in which the yoqV ORF was cloned into a pUB110 derivative, under strong expression signals, including the Pspac promoter and a canonical ribosome binding site. As a control, the yerG gene was cloned similarly on the pUB110 derivative, to give plasmid pMAP141.
In order to investigate whether this new strain, in which the yoqV ATP-dependent ligase was constitutively expressed, could survive in the absence of the yerG encoded NAD-dependent ligase, an attempt at disrupting yerG with the non-replicative plasmid pMAP59 (see first paragraph) was repeated. Table 3, lane 1, shows the proportion of transformants in which the yerG gene was effectively interrupted in various B.subtilis strains.
Table 3. A yerG, but not a yoqV overproducing strain allows the selection of B.subtilis transformants harbouring a yerG disruption.
| Proportion of disrupted yerG clonesa among transformantsb of B.subtilis 168 strain containing | |||
|---|---|---|---|
| Transforming DNA | no plasmid | pMAP141 (yerG) | pMAP139 (yoqV) |
| Plasmid DNA pMAP59 (disrupts yerG) | N.R. | 5/15 | 0/2 |
| Total DNA of strain | |||
| HVS611 (yerG disrupted) | 0/48 | 18/18 | 0/18 |
aThe proportion of transformants effectively containing a yerG disruption but not resulting from a congression is indicated (see text for details).
bCompetent cells of the indicated strains were transformed to EmR with the indicated DNA. The three strains tested had a similar level of competence, estimated by transformation with the plasmid pIL253 (between 1.3 × 105 and 3.7 × 105 transformants per µg of DNA were obtained).
N.R., not relevant (no transformants obtained).
In the B.subtilis strain harbouring the yerG encoding plasmid pMAP141, only a few transformants were obtained (50/µg of plasmid DNA). This was expected and resulted from the reduced homology between recipient and transforming DNA: plasmid pMAP59 shares a 337 bp homology with the yerG gene, and also a 293 bp homology with both plasmids pMAP139 and pMAP141 in a segment encompassing the Pspac promoter. To distinguish between the transformants in which pMAP59 had recombined with the chromosomal copy of yerG, and those in which pMAP59 had recombined with the resident plasmid pMAP141, total DNA was extracted from 15 transformants. Five of the 15 clones contained a disrupted copy of yerG (Table 3, lane 1) as determined by Southern blot hybridisation (not shown). The other 10 clones contained a recombinant plasmid between pMAP141 and pMAP59. From this analysis we concluded that it is possible to construct a yerG disruption in a B.subtilis strain containing a plasmidic copy of the gene. The resulting strain was named HVS611.
In the B.subtilis strain harbouring the yoqV encoding plasmid pMAP139, transformation with plasmid pMAP59 yielded two transformants only, and none of these two transformants contained a disrupted copy of yerG. Rather, both of them had a mixture of two plasmids, the resident pMAP139, and a 14 kb plasmid recombinant between pMAP139 and pMAP59. The low transformation efficiency, and the absence of transformants harbouring a yerG disruption, suggested that a yerG mutation was not tolerated in a strain expressing yoqV. However, given the low number of transformants, the possibility that such a disruption is indeed tolerated could not be completely excluded.
To increase the chances to get a yerG disruption in strain 168 harbouring pMAP139, another attempt at disrupting yerG was made. Instead of pMAP59 as a source of DNA, total DNA of strain HVS611, in which pMAP59 is already integrated into yerG, was used (Table 3, lane 2). In such a case, the homology between resident and transforming DNA is greatly increased, and higher transformation frequencies are expected. However, because the total DNA of strain HVS611 also contains pMAP141 plasmid DNA, the possibility exists to obtain ‘false positive transformants’, resulting from a congression, i.e. the simultaneous acquisition of two independent markers, yerG::EmR and plasmid pMAP141. In strain 168 harbouring the yerG encoding plasmid, a high yield of transformants (3.5 × 104/µg of DNA) were obtained as expected, and of among 18 clones analysed, all contained an interrupted copy of yerG (Table 3, lane 2) as determined by a PCR analysis. With strain 168, 10 times less transformants (2 × 103/µg of DNA) were obtained, and among 48 clones analysed, all were resistant to kanamycin, the marker of plasmid pMAP141. This showed that these transformants were ‘false positives’. In strain 168 harbouring pMAP139, 6 × 103 transformants per µg of DNA were obtained, a result similar to strain 168. This suggested that all transformants also resulted from a congression of yerG::EmR and pMAP141 DNA. Unlike recipient strain 168, the strain containing plasmid pMAP139 was already resistant to kanamycin and as such congression could not be tested phenotypically. To check for the presence of plasmid pMAP141 in the transformants, total DNA of 18 clones was extracted, and the plasmid and chromosomal DNA was analysed by Southern blotting. In all clones, plasmid pMAP141 was found effectively, either alone or in combination with plasmid pMAP139. Therefore a proportion of 0 among 18 clones contained a yerG disruption (Table 3, lane 2).
In conclusion, when plasmid pMAP141 was present in the recipient wild-type strain of B.subtilis, the chromosomal copy of yerG could be disrupted. However, when the yoqV encoding plasmid pMAP139 was present, no transformants containing an disrupted copy of yerG were obtained.
To verify that the ATP-dependent ligase cloned into pMAP139 was active, an adenylation test was performed on B.subtilis crude extracts. The first step of the ligation process consists of the covalent attachment of AMP to the lysine residue of the ligase, which can be visualised easily on a protein gel if the AMP is radiolabelled. Extracts treated with RNase were incubated in the presence of [α-32P]ATP for 15 min at room temperature and analysed by denaturing PAGE. As shown in Figure 3, lanes 4 and 5, a radioactive signal was detected in samples extracted from the YoqV-containing strain, and was absent in the B.subtilis wild-type extract. This signal corresponded to a protein of ~30 kDa, which is close to the expected size of 31 kDa for YoqV. Therefore the YoqV ligase was present and active in the strain containing plasmid pMAP139. The absence of any signal in crude extracts of B.subtilis wild-type extracts suggests that neither the YoqV nor the YkoU ATP-dependent ligases were expressed at a detectable level in this strain.
Figure 3.
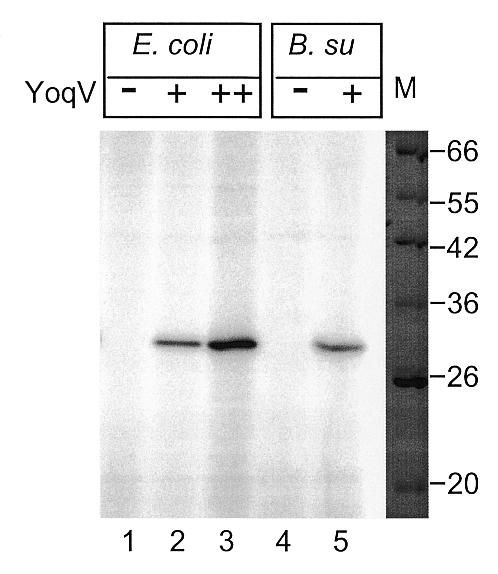
Presence of an adenylate activity specific of E.coli and B.subtilis crude extracts from strains containing a plasmid overproducing YoqV. Crude extracts of E.coli strains JM105 (lane 1), JM105 + pMAP138 grown without IPTG (lane 2), JM105 + pMAP138 grown with IPTG (lane 3), and B.subtilis strains 168 (lane 4) and 168 + pMAP139 (lane 5), were incubated with radiolabelled ATP, separated by PAGE, stained with Coomassie blue (right, only the marker lane is shown), and exposed (left). Lane M, molecular weight markers, relevant sizes are indicated in kDa.
The yoqV ligase does not complement a ligts mutant of E.coli, but the yerG ligase does
In E.coli, the C-terminal part of the human ATP-dependent ligase I is able to restore the thermoresistance to a ligts strain (7), and in S.typhimurium the bacteriophage T4 ATP-dependent ligase, which contains only the minimal adenylation domain, like the yoqV gene product, allows growth of the NAD-dependent ligase disrupted strain (4). We therefore investigated whether YoqV was able to complement a ligts strain of E.coli. For this, plasmid pMAP138, a hybrid composed of pBR322, lacI and pMAP139, was constructed. The plasmid was transformed into strain JJC75, containing the lig7 ts allele, and the resulting strain was grown at various temperatures. Plates incubated at 42 and 30°C are shown in Figure 4. As with the ligts, the strain harbouring plasmid pMAP138 did not grow at 42 or 37°C (not shown), both in the presence and absence of IPTG. To further confirm this defect, cells were grown exponentially at 30°C and the plating efficiency at 42°C relative to 30°C was determined. Only 10–5 survivors were found at 42°C with the ligts strain, with or without pMAP138. This shows that the B.subtilis ATP-dependent ligase does not complement the growth defect of the ligts strain.
Figure 4.
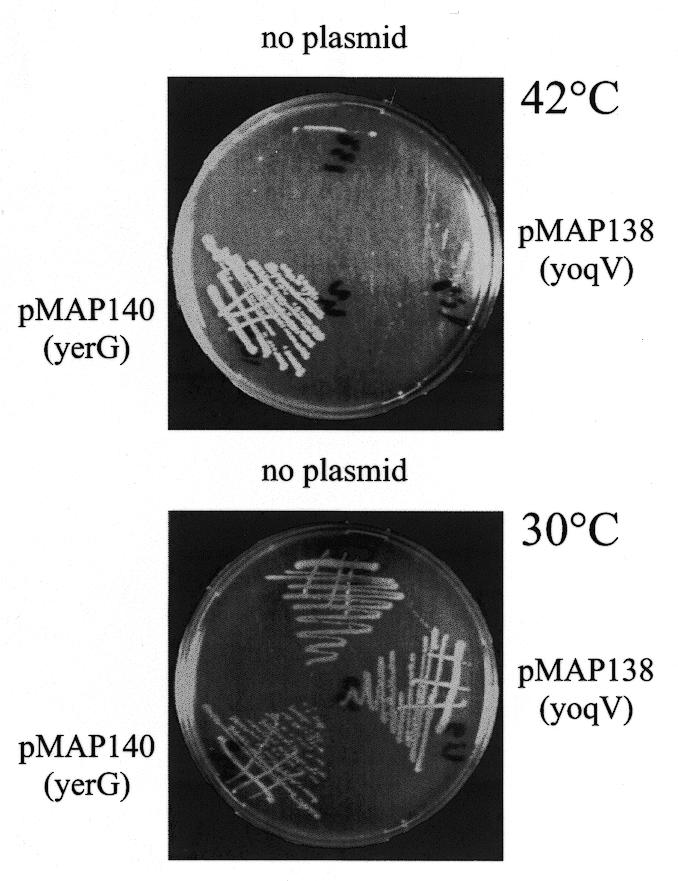
yerG, but not yoqV, restores thermoresistance to the E.coli ligts strain. Petri plates showing streaks on LB of the E.coli ligts strain harbouring no plasmid (no plasmid), plasmid pMAP138 [pMAP138 (yoqV)] or pMAP140 [pMAP140 (yerG)], grown either at 42 (top) or 30°C (bottom).
To verify that the ATP-dependent ligase cloned into pMAP138 was active, an adenylation test was performed on E.coli ligts crude extracts (Fig. 3, lanes 1–3). A radioactive signal was detected in samples extracted from the YoqV containing strain, and was absent in the extract that did not contain YoqV. It was 10-fold more intense in the sample in which expression of yoqV had been derepressed upon addition of IPTG. This showed that the ATP-dependent ligase encoded by yoqV was active in E.coli crude extracts.
Similar heterologous complementation experiments were carried out in parallel with the yerG encoded NAD ligase. Plasmid pMAP140, a hybrid of pBR322, lacI and pMAP141, which provides IPTG-dependent expression of the B.subtilis NAD-dependent ligase in E.coli, was constructed and introduced in the ligts background. In contrast to the yoqV experiment, this strain did form individual colonies at 42°C (Fig. 4), and it plated at an efficiency close to 100% at this temperature, even in the absence of IPTG. This shows that the B.subtilis NAD-dependent ligase complements the growth defect of the ligts strain.
Because the YerG ligase was able to restore growth of the ligts strain at 42°C, its ability to complement the UV sensitivity of the ts strain was further investigated. Whereas the ligts strain was UV sensitive at 30°C (Fig. 5, circles), this defect disappeared in the presence of yerG, even at 42°C (triangles), and the strain became as UV resistant as the original wild-type strain (squares). In conclusion, the yerG gene product allowed the full restoration of the growth and UV resistance defects of the ligts strain.
Figure 5.

UV sensitivity of wild-type E.coli strain JJC40 grown at 42°C (squares), its ligts derivative JJC75 grown at 30°C (circles) and JJC75 harbouring the yerG encoding plasmid pMAP140, grown at 42°C (triangles). The fraction of surviving clones is reported as a function of the UV dose.
DISCUSSION
In B.subtilis, we have shown that the yerG-encoded NAD-dependent ligase, which we propose to rename ligA, is essential for growth. This was somewhat unexpected, given the presence of two other potential ligase genes in its chromosome. These two other ligases, ykoU and yoqV, belong to the family of ATP-dependent ligases. Observations have shown that some ATP-dependent ligases, although originating from organisms completely unrelated to bacteria, such as the T4 bacteriophage and Homo sapiens, were able to restore the viability of a S.typhimurium or a E.coli lig mutant (4,7), therefore suggesting a surprisingly broad range of activity and compatibility for these enzymes.
Because the yoqV gene is part of the SPβ prophage, the possibility existed that the absence of complementation was simply due to a lack of a sufficient level of expression. To increase the expression of the yoqV encoded ATP-dependent ligase, which we propose to rename ligB, this gene was cloned on a plasmid under appropriate expression signals. Even in this case, the yerG gene could not be disrupted. To further investigate the activity of yoqV, the gene was cloned in E.coli and its ability to complement the growth defect of the lig7ts strain was tested. Again, at 42°C yoqV failed to restore growth of the ligts mutant. To exclude the possibility that the ligase encoded by yoqV was in fact an inactive enzyme, its adenylation activity in B.subtilis and E.coli crude extracts was tested and found to be present. It appears therefore that, in B.subtilis as well as in E.coli, not all ATP-dependent ligases provide sufficient activity for maintaining alive bacterial cells devoid of their native NAD-dependent ligase.
In cases where a complementation has been reported, it should be noted that not all phenotypes were restored. Neither the Chlorella virus ATP-dependent ligase nor the E.coli NAD-dependent ligase, although restoring growth of a S.cerevisiae cdc9Δ strain, restored complete UV resistance or MMS resistance (8). It has been suggested that DNA ligases perform different functions, some of which are generic and can be achieved by an NAD-dependent ligase as well as by an ATP-dependent ligase, the others being enzyme specific. The ligation of Okasaki fragments during DNA replication was suggested to be a generic, as well as an essential function. Here we report, however, that the SPβ ATP-dependent ligase is not able to provide this essential function, whichever it may be. What properties of the bacteriophage T4 ATP-dependent ligase, which are not present in the SPβ ligase, make it a good substitute to the bona fide ligase of S.typhimurium? Both enzymes are small in size and do not contain additional domains supposedly involved in protein–protein interactions, so it is tempting to speculate that the difference lies in the intrinsic enzymatic activities of these proteins, one having a higher specific activity, or a higher stability than the other. As a point of comparison, the adenylation activity of YoqV in B.subtilis crude extracts was equivalent to the signal obtained with 0.25 U of commercial T4 ligase mixed with a wild-type B.subtilis crude extract (not shown). Reported to the total amount of protein, and converted into the units as defined by Modrich and Lehman (10), it corresponded to 4 U/mg of protein. The NAD-dependent ligase of E.coli gives between 0.4 and 1.7 U/mg of protein, and this amount was reported to be much more than actually needed in the cell (11). Therefore, in terms of quantity and adenylation activity, YoqV does not appear to be defective. A reasonable possibility is that the second step of the ligation reaction, the transfer of the AMP moiety to the DNA and subsequent ligation, is inefficient in YoqV.
The NAD-dependent ligase of B.subtilis is able to complement a ligts strain of E.coli. This is interesting in view of the growing evidence that DNA ligases of a large size, apart from their ligase domain, possess several other domains that may be involved in protein–protein interactions. For instance, the mammalian DNA ligase I has been shown to bind to PCNA (12), and mammalian DNA ligase IV to XRCC4 (13,14). No interaction has yet been reported for the bacterial NAD-dependent ligases, but it has been suggested that their BRCT domain may serve this function (3). If a NAD-dependent ligase from a distantly related bacterium like B.subtilis (the two proteins share 49% of amino acid identity) is able to complement the ligase defect in E.coli, this suggests either that this interaction is conserved between the two organisms, or that the B.subtilis protein does not need to interact with any E.coli protein to work efficiently.
On a practical level, the observations reported here encourage the search for antibiotics directed specifically against NAD-dependent ligases, since these enzymes prove to be essential for bacterial growth and specific for this reign. Although some bacteria like B.subtilis, M.tuberculosis and H.influenzae, harbour more than one ligase gene, the NAD-dependent ligase gene is always unique, and at least in the case of B.subtilis it proves to be essential.
Acknowledgments
ACKNOWLEDGEMENTS
M.-A.P. wishes to thank B. Michel for the gift of strain JJC75, and E. d’Alençon, S. Marsin and C. Bruand for critical reading of the manuscript.
REFERENCES
- 1.Subramanya H.S., Bird,L.E., Brannigan,J.A. and Wigley,D.B. (1996) Crystal structure of a DExx box DNA helicase. Nature, 384, 379–383. [DOI] [PubMed] [Google Scholar]
- 2.Singleton M.R., Hakansson,K., Timson,D.J. and Wigley,D.B. (1999) Structure of the adenylation domain of an NAD+-dependent DNA ligase. Structure Fold Des., 7, 35–42. [DOI] [PubMed] [Google Scholar]
- 3.Lee J.Y., Chang,C., Song,H.K., Moon,J., Yang,J.K., Kim,H.K., Kwon,S.T. and Suh,S.W. (2000) Crystal structure of NAD(+)-dependent DNA ligase: modular architecture and functional implications. EMBO J., 19, 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park U.E., Olivera,B.M., Hughes,K.T., Roth,J.R. and Hillyard,D.R. (1989) DNA ligase and the pyridine nucleotide cycle in Salmonella typhimurium. J. Bacteriol., 171, 2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konrad E.B., Modrich,P. and Lehman,I.R. (1973) Genetic and enzymatic characterization of a conditional lethal mutant of Escherichia coli K12 with a temperature-sensitive DNA ligase. J. Mol. Biol., 77, 519–529. [DOI] [PubMed] [Google Scholar]
- 6.Gottesman M.M., Hicks,M.L. and Gellert,M. (1973) Genetics and function of DNA ligase in Escherichia coli. J. Mol. Biol., 77, 531–547. [DOI] [PubMed] [Google Scholar]
- 7.Kodama K., Barnes,D.E. and Lindahl,T. (1991) In vitro mutagenesis and functional expression in Escherichia coli of a cDNA encoding the catalytic domain of human DNA ligase I. Nucleic Acids Res., 19, 6093–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sriskanda V., Schwer,B., Ho,C.K. and Shuman,S. (1999) Mutational analysis of Escherichia coli DNA ligase identifies amino acids required for nick-ligation in vitro and for in vivo complementation of the growth of yeast cells deleted for CDC9 and LIG4. Nucleic Acids Res., 27, 3953–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petit M.A., Dervyn,E., Rose,M., Entian,K.D., McGovern,S., Ehrlich,S.D. and Bruand,C. (1998) PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol. Microbiol., 29, 261–273. [DOI] [PubMed] [Google Scholar]
- 10.Modrich P. and Lehman,I.R. (1970) Enzymatic joining of polynucleotides. IX. A simple and rapid assay of polynucleotide joining (ligase) activity by measurement of circle formation from linear deoxyadenylate-deoxythymidylate copolymer. J. Biol. Chem., 245, 3626–3631. [PubMed] [Google Scholar]
- 11.Gellert M. and Bullock,M.L. (1970) DNA ligase mutants of Escherichia coli. Proc. Natl Acad. Sci. USA, 67, 1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin D.S., Bai,W., Yao,N., O’Donnell,M. and Tomkinson,A.E. (1997) An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc. Natl Acad. Sci. USA, 94, 12863–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grawunder U., Zimmer,D. and Leiber,M.R. (1998) DNA ligase IV binds to XRCC4 via a motif located between rather than within its BRCT domains. Curr. Biol., 8, 873–876. [DOI] [PubMed] [Google Scholar]
- 14.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 15.Stragier P., Bonamy,C. and Karmazyn-Campelli,C. (1988) Processing of a sporulation sigma factor in Bacillus subtilis: how morphological structure could control gene expression. Cell, 52, 697–704. [DOI] [PubMed] [Google Scholar]
- 16.Vagner V., Dervyn,E. and Ehrlich,S.D. (1998) A vector for systematic gene inactivation in Bacillus subtilis. Microbiology, 144, 3097–3104. [DOI] [PubMed] [Google Scholar]
- 17.Simon D. and Chopin,A. (1988) Construction of a vector plasmid family and its use for molecular cloning in Streptococcus lactis. Biochimie, 70, 559–566. [DOI] [PubMed] [Google Scholar]