

Abstract
We have determined the effect of cisplatin–DNA damage on the ability of the DNA-dependent protein kinase (DNA-PK) to interact with duplex DNA molecules in vitro. The Ku DNA binding subunits of DNA-PK display a reduced ability to translocate on duplex DNA containing cisplatin–DNA adducts compared to control, undamaged duplex DNA. The decreased rates of translocation resulted in a decrease in the association of the p460 catalytic subunit of DNA-PK (DNA-PKcs) with the Ku–DNA complex. In addition to a decrease in DNA-PKcs association, the DNA-PKcs that is bound with Ku at a DNA end containing cisplatin–DNA adducts has a reduced catalytic rate compared to heterotrimeric DNA-PK assembled on undamaged DNA. The position of the cisplatin–DNA lesion from the terminus also effects kinase activation, with maximal inhibition occurring when the lesion is closer to the terminus. These results are consistent with a model for DNA-PK activation where the Ku dimer translocates away from the DNA terminus and facilitates the association of DNA-PKcs which interacts with both Ku and DNA resulting in kinase activation. The presence of cisplatin adducts decreases the ability to translocate away from the terminus and results in the formation of inactive kinase complexes at the DNA terminus. The results are discussed with respect to the ability of cisplatin to sensitize cells to DNA damage induced by ionizing radiation and the ability to repair DNA double-strand breaks.
INTRODUCTION
The DNA-dependent protein kinase (DNA-PK) is a heterotrimeric protein required for DNA double-strand break (DSB) repair via the non-homologous end joining pathway (NHEJ). DNA-PK associates with DNA ends via interactions with the Ku DNA binding subunits in addition to direct interactions with DNA (1). Ku is a heterodimer of 70 and 80 kDa subunits that has DNA end binding activity, which is required for DSB repair (2). Cells harboring mutations in any of the DNA-PK subunits or treated with compounds that inhibit DNA-PK kinase activity display hypersensitivity to ionizing radiation (IR). The interaction of the Ku subunits of DNA-PK have been extensively studied with respect to their DNA binding activity (3). Ku binds preferentially to DNA ends and discontinuities, nicks and gaps for example. A photocrosslinking method was employed to assess the interaction of Ku with DNA in an orientation-specific fashion. The results demonstrated that the Ku70 subunit was positioned closer to the DNA end while the 80 kDa subunit was positioned more proximal on a short (28 bp) DNA molecule (4). The interaction of Ku on longer DNA molecules has also been studied and reveals that DNA end-binding is followed by ATP-independent translocation of the Ku dimer to internal positions of the duplex DNA (5–7). The translocation of Ku to more distal regions has been observed on short DNA substrates in the presence of the catalytic subunit of DNA-PK (DNA-PKcs), and photocrosslinking experiments demonstrate that upon binding of DNA-PKcs, the Ku70/Ku80 complex moved further into the duplex DNA (8). These results are also consistent with the translocation of Ku subunits away from the DNA terminus to allow a direct interaction of the ssDNA ends with the DNA-PKcs to result in activation of the protein kinase activity (9).
Cisplatin is a DNA-damaging agent that forms coordinate covalent DNA adducts. The formation of cisplatin–DNA adducts results in cell cycle arrest, inhibition of DNA replication and transcription, and eventually apoptosis. Cisplatin is very effective in treating certain types of cancer including testicular and ovarian. In addition, recent clinical trials have demonstrated that cisplatin combined with radiotherapy provides for a significantly better prognosis when compared with radiation treatment alone (10). The mechanism of this interaction is not well understood. Cisplatin has been demonstrated to sensitize cells to IR in tissue culture model systems, again the specific mechanism of sensitization was not defined (11,12). We have previously demonstrated that cisplatin-damaged DNA is effectively bound by Ku, but failed to activate DNA-PK phosphorylation activity in vitro (13). Further analysis revealed that on short DNA substrates there was little effect on the affinity of Ku for cisplatin damaged DNA by determination of the equilibrium dissociation constant Kd while the majority of the inhibition was attributed to a decreased kcat (14). To further investigate the mechanism of inhibition of DNA-PK by cisplatin-damaged DNA, we have focused on the translocation activity of Ku and the ability to form a heterotrimeric DNA complex on duplex DNA substrates containing cisplatin–DNA lesions.
MATERIALS AND METHODS
Materials
Unlabeled nucleotides were from Life Technologies (Gaithersburg, MD) and radiolabeled nucleotides were from NEN Life Sciences (Boston, MA). Sequenase v.2.0 was from United States Biochemical (Cleveland, OH). Mung Bean Nuclease, HaeIII, EcoRI and PvuII were from Life Technologies. Synthetic oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA). ImmunoPure Streptavidin was purchased from Pierce (Rockford, IL) and was reconstituted to a concentration of 1 mg/ml in 20 mM potassium phosphate, pH 8.0. Cisplatin was purchased from Sigma Chemical Co. (St Louis, MO). All other reagents were purchased from standard suppliers. The DNA-PK holoenzyme and the Ku heterodimer purified free of DNA-PKcs were purified from HeLa cells as described previously (13).
DNA substrates
The 119 bp DNA was prepared by digesting pBS+ DNA (Strategene) with EcoRI and PvuII for 5 h at 37°C. The 119 bp fragment (767–882) was gel-purified by electrophoresis on a 2% agarose gel. The restriction fragment was purified from the agarose gel using the QIAquick Gel Extraction Kit (Qiagen Inc., Chatsworth, CA), radiolabeled by extension using Sequenase v.2.0, [α-32P]dATP (3000 Ci/mmol) and 100 µM dTTP in 50 mM Tris–Cl, pH 7.5, 5 mM DTT and 10 mM magnesium acetate which yielded a duplex 119 bp DNA substrate. Specific activity was determined, and the 119 bp DNA was mock-treated or treated with cisplatin at a variety of concentrations as described previously (13,15).
The 115 bp duplex DNA containing a 5′-biotin modification was prepared by PCR amplification of M13mp18 ssDNA. A 5′-biotinylated sense primer (TCTGGTAAACGAGGG- TTATGATAGTG) and an antisense primer (GGCTCTAGACAGTTGAGATTTAGGAATACCACATTCA) were used in amplification reactions containing 40 pmol of each primer, 20 mM Tris–Cl, pH 8.4, 50 mM KCl, 2 mM MgCl2, 0.4 mM dNTP, 20 µCi [α-32P]dCTP, 0.125 fmol of M13mp18 DNA and 2.5 U AmpliTaq DNA polymerase (Life Technologies). Reactions were cycled at 94°C for 1 min, 55°C for 2 min and 72°C for 0.5 min for 25 cycles followed by a 7 min incubation at 72°C for elongation. The specific activity of the DNA product was calculated and the DNA was gel purified on a native 6% polyacrylamide gel, eluted and quantified by the Hoechst 33258 fluorescence dye binding assay. Cisplatin treatment of DNA was performed as described previously (13,15).
Preparation of the 25 and 30 bp DNA substrates containing the single site-specific cisplatin lesions were constructed as described previously (14,16). The 41 bp DNA substrate was constructed by similar methods using the following oligonucleotides: AK1 (TCATTACTACTCACTCTGTCGGCCATCGCTCTCTATTCCC) and AK1.2 (GGGGAATAGAGAGCGATGGCCGACAGAGTGAGTAGTAATGA). The 120 bp DNA substrate was prepared by ligation of a series of oligonucleotides as we have described previously (17).
Electrophoretic mobility shift assays (EMSAs)
Equilibrium DNA binding assays were performed using an undamaged or cisplatin damaged 119 bp duplex DNA as described previously (13,15). Briefly, 50 fmol of DNA was incubated with the indicated amount of Ku for 30 min on ice. The samples were then separated on 8% native polyacrylamide gels at room temperature. EMSA analysis to measure the kinetics of Ku binding was performed in a final volume of 20 µl containing 40 fmol of undamaged or cisplatin damaged 115 bp, 5′-biotin modified DNA. The DNA was preincubated with 2 µg of streptavidin. Reactions were initiated by the addition of Ku (570 fmol) at various time intervals. Reactions were terminated by the addition of ice-cold buffer and immediately loaded onto a precooled 4% native gel such that the total incubation times ranged from 1 to 45 min. Electrophoresis was performed for 2 h at 200 V. Gels were dried under vacuum, exposed to X-ray film and radioactivity was quantified by PhosphorImager analysis using ImageQuant software in volume integration mode (Molecular Dynamics, CA). Global analysis was performed on the quantified time course data using Pro-K software (Applied Photophysics). Rate constants were determined for each Ku loading step assuming a single irreversible step for each binding event. EMSA analysis to measure DNA-PK binding to double-strand DNA was performed as described previously (1). Briefly, reactions were performed under reduced ionic strength to allow stable binding of the DNA-PKcs to the DNA terminus containing Ku.
Kinase assays
DNA-PK assays were performed as described previously (14). Reactions contained 40 mM HEPES, pH 7.5, 50 mM KCl, 8 mM MgCl2, 1 mM DTT, 2.5% glycerol, 0.2 mM EGTA, 400 µM concentration of the synthetic p53-based peptide substrate (EPPLSQEAFADLWKK) and 125 µM [γ-32P]ATP (30 Ci/mmol) in a 20 µl final volume. Reactions were incubated at 37°C for 15 min, terminated with a final concentration of 15% acetic acid and spotted on Whatman P81 filter paper. Filters were washed 5 × 5 min with 15% acetic acid, once briefly in 100% methanol and radioactivity quantified by PhosphorImager analysis.
RESULTS
We have demonstrated previously that on short duplex DNA molecules 25 bp in length, the affinity of the Ku subunits of DNA-PK is not dramatically altered when the DNA contains various cisplatin DNA adducts (14). On longer DNA substrates, 44 and 75 bp in length, again the affinity was similar to undamaged DNA, but we did observe a change in the distribution of Ku–DNA complexes on DNA substrates damaged with cisplatin (13). To examine this effect further, we have prepared a DNA substrate capable of binding multiple Ku dimers and have addressed the effect of cisplatin on the formation of these multimeric protein–DNA complexes. A 119 bp DNA was purified, 5′ labeled with 32P and treated with various concentrations of cisplatin. Cisplatin-damaged DNAs were then employed in EMSAs with purified Ku protein free from the DNA-PKcs. Increasing concentrations of Ku were added and the reactions were incubated for 30 min on ice. The reaction products were separated by native PAGE. The results of one such experiment are presented in Figure 1 and demonstrate that multiple DNA–Ku complexes are observed. At the lowest Ku concentration (lane 2), the predominant product is a single Ku bound to the DNA and a small amount of a 2Ku–DNA complex, which likely represents a Ku dimer bound to each DNA terminus of a single DNA molecule. At the highest Ku concentration, the slowest migrating complex observed is consistent with the complex containing five Ku dimers bound to a single DNA substrate (lane 4). Upon the introduction of cisplatin–DNA adducts (lanes 5–12), a clear change in the product distribution was observed. A single Ku–DNA complex is only observed on the undamaged DNA at the lowest level of Ku (lane 2), while this complex can be observed at the intermediate concentration of Ku (lanes 7 and 11) for both of the cisplatin-damaged DNA substrates. Comparing the intermediate levels of Ku, the predominant product is 3Ku–DNA using undamaged DNA, while at the lowest level of cisplatin an equal amount of the 2Ku–DNA and 3Ku–DNA complexes are observed. At the highest level of cisplatin damage, the 2Ku DNA complex predominates. In reactions performed with the highest level of Ku (lanes 4, 8 and 12), the 5Ku–DNA complex represents >90% of the products with undamaged DNA. The 4Ku–DNA complex and the 3Ku–DNA complex represent the majority of the products observed at the low and high level of cisplatin–DNA damage, respectively. These data demonstrate that the presence of cisplatin–DNA adducts results in a decrease in the number of Ku molecules that are bound to a single DNA substrate.
Figure 1.

The effect of cisplatin–DNA adducts on the binding of Ku to a duplex 119 bp DNA. The undamaged and cisplatin damaged 119 bp DNA substrates were prepared and purified as described in Materials and Methods. Binding reactions contained 50 fmol of DNA substrate and 0, 16.6, 66.6 and 266.6 fmol of Ku. Reactions were performed with either undamaged DNA (lanes 1–4) or DNA treated at a D/N of 0.1 (lanes 5–8) or 1.0 (lanes 9–12). Ku–DNA complexes were separated from unbound DNA by 4% native PAGE, visualized by autoradiography and quantified by PhosphorImager analysis using ImageQuant software in volume integration mode. The location of free DNA and the multimeric Ku–DNA complexes are depicted on the left.
Kinetics of Ku binding to undamaged DNA
To further investigate the mechanism of how the different Ku DNA complexes are formed on DNA damaged with cisplatin, we performed a series of kinetic assays. To accurately determine the kinetics of the formation of multiple Ku–DNA complexes, we sought to limit Ku binding to a single terminus of the linear duplex DNA fragment. We employed a system established previously that has been demonstrated to limit Ku binding to a single DNA end by blocking the opposite end with a streptavidin–biotin complex (4,8). To prepare a duplex DNA of sufficient length with a 5′-biotin, we employed PCR amplification of M13mp18 ssDNA using two primers, one of which was synthesized with a 5′-biotin modification. The resulting 115 bp DNA substrate was internally labeled with 32P during the amplification reaction. The amount of streptavidin required to bind 100% of the input DNA was determined as well as the amount of Ku required to form multiple complexes on the 115 bp blocked DNA substrate (data not shown). Time course assays were performed as described in Materials and Methods and the results of one such experiment using an undamaged DNA substrate are shown in Figure 2A. To increase the resolution of the Ku–DNA complexes, electrophoresis was performed until the streptavidin–DNA complex migrated to the bottom of the gel. The data were quantified by PhosphorImager analysis calculating the percentage of each product in each lane. This approach allowed us to control for variations in gel loading and the results are presented in Figure 2B. The results demonstrate that at the earliest time point taken (1 min), we were unable to observe a product with a single Ku molecule bound to the DNA substrate. At lower Ku concentrations (data not shown), we were able to detect the single Ku–DNA complex but the formation of the multimeric complexes was diminished. The 2Ku–DNA complex could be observed at both the 1 and 2 min time points and was undetectable after 10 min. The 3Ku–DNA complex predominated at these early time points and decreased within 20 min, after which it was no longer detectable. The decrease in the 2Ku–DNA and 3Ku–DNA complexes was accompanied by an increase in the formation of the 4Ku–DNA complex and at the later time points only the 4Ku–DNA and 5Ku–DNA complexes were observed.
Figure 2.

Kinetic analysis of Ku binding to an undamaged 115 bp DNA. (A) The undamaged 5′-biotin modified 115 bp substrate was prepared and purified as described in Materials and Methods. Kinetic assays were performed with 40 fmol of DNA prebound to streptavidin (lane 1) and the reactions initiated by the addition of Ku protein (570 fmol, lanes 2–9). Reactions were incubated for the indicated times and the products were separated using 4% native PAGE visualized by autoradiography and quantified by PhosphorImager analysis using ImageQuant software in volume integration mode. The location of the SA–DNA complex and the multimeric Ku–DNA complexes are depicted on the left. (B) Quantification of the time course for Ku loading to unplatinated 115 bp duplex DNA substrate. The percentage of each Ku–DNA complex in each lane was plotted versus time. The 2Ku–DNA complex is denoted by triangles, the 3Ku–DNA complex by inverted triangles, the 4Ku–DNA complex by squares and the 5Ku–DNA complex by open circles.
Kinetics of Ku binding to cisplatin-damaged DNA
To assess the effect of cisplatin on the kinetics of Ku binding, the 5′ biotinylated 32P-labeled 115 bp DNA duplex was treated with cisplatin at a drug to nucleotide ratio of 0.1. The duplex DNA was gel purified and used in kinetic analyses as described above. Preliminary experiments were also performed demonstrating that cisplatin damage did not effect the streptavidin binding to the DNA substrates. The results from a typical experiment are presented in Figure 3. The most obvious difference is that a single Ku–DNA complex was easily observable at the initial time points and decreased to undetectable levels after 20 min. The 2Ku–DNA complex was also present at the initial time points and decreased over the entire time period of the assay: 45 min. The 3Ku–DNA complex increased to a maximum of >50% of the total products at 15 min and decreased over the remainder of the assay. The concentration of the 4Ku–DNA complex, initially 10–15% of the reaction products, increased over the entire time course to represent >60% of the reaction products at 45 min. There was no detectable 5Ku–DNA complex in the reaction performed with cisplatin-damaged DNA compared to the undamaged DNA where it represented >20% of the products at 45 min (Fig. 2A).
Figure 3.

Kinetic analysis of Ku binding to a cisplatin damaged 115 bp DNA. (A) The cisplatin damaged 5′-biotin modified 115 bp substrate was prepared and purified as described in Materials and Methods. Kinetic assays were performed with 40 fmol of DNA prebound to streptavidin (lane 1) and the reactions initiated by the addition of Ku protein (570 fmol, lanes 2–9). Reactions were incubated for the indicated times and the products were separated using 4% native PAGE, visualized by autoradiography and quantified by PhosphorImager analysis using ImageQuant software in volume integration mode. The location of the SA–DNA complex and the multimeric Ku–DNA complexes are depicted on the left. (B) Quantification of the time course for Ku loading to cisplatin damaged 115 bp duplex DNA substrate. The percentage of each Ku–DNA complex in each lane was plotted versus time. The 1Ku–DNA complex is denoted by circles, the 2Ku–DNA complex by triangles, the 3Ku–DNA complex by inverted triangles and the 4Ku–DNA complex by squares.
The data obtained in Figures 2B and 3B were subject to global analysis using a model for Ku binding that assumes a single irreversible binding event. Under the conditions of the assay with limiting DNA and Ku binding restricted to one end of the DNA, the limiting step in binding a second Ku molecule is the movement of the first bound Ku away from the terminus to more proximal positions (Fig. 4). Once the terminus is free, the second Ku dimer binds very rapidly. Using this simple model, the calculated rate constants for Ku translocation are presented in Table 1. The k3, k4 and k5 rate constants were determined for the undamaged DNA while the k2, k3 and k4 were determined for the cisplatin-damaged DNA. The k1 and k2 constants were not determined for the undamaged DNA as the 2Ku–DNA complex was the first observable product. The k5 was not determined for the cisplatin-damaged DNA as the 5Ku–DNA complex was not detectable over the time course of the assay. Based on these analyses, the k3 and k4 can be directly compared and reveal that the rate of Ku translocation is 15–20 times faster on undamaged compared to the cisplatin-damaged DNA substrate. The decrease in rates from k2 to k3 to k4 are consistent with a model of irreversible binding to the DNA where the limiting step in the reaction is movement away from the DNA terminus to allow binding of the next Ku molecule.
Figure 4.
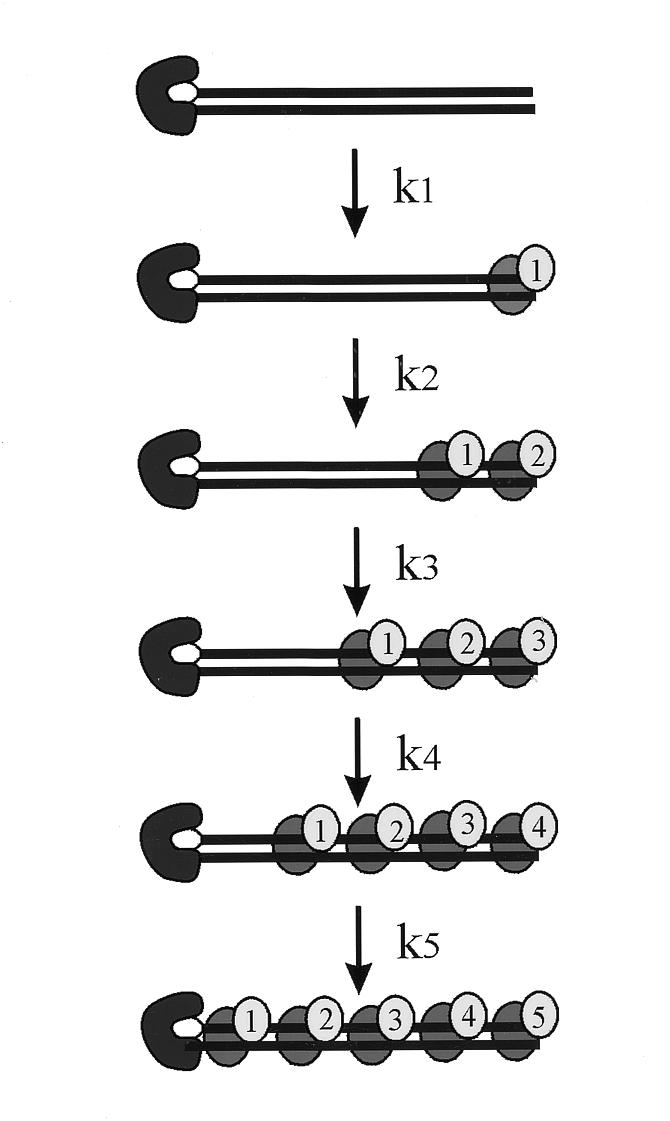
Model for multiple Ku binding events. The data from Figures 2B and 3B were fit using global analysis to a model where each binding event is a single irreversible reaction. The DNA substrate is depicted with the biotin–streptavidin complex blocking the left terminus. The Ku dimer is depicted by grey ovals. The conditions of the assay were designed such that the limiting step in the pathway is translocation of Ku away from the terminus to allow a subsequent Ku dimer to bind.
Table 1. Rate constants for Ku bindinga.
| Reaction step | –Pt | +Pt | –Pt/+Pt |
|---|---|---|---|
| k1 | ND | ND | – |
| k2 | ND | 162 | – |
| k3 | 2.5 | 0.125 | 20 |
| k4 | 0.21 | 0.0137 | 15 |
| k5 | ND | ND | – |
aThe values obtained represent unimolecular rate constants presented in min–1.
DNA-PK binding to undamaged and cisplatin-damaged DNA
Having determined the effect of cisplatin on Ku movement in reactions without DNA-PKcs, we sought to determine if in fact the association of DNA-PK was inhibited by cisplatin-damaged DNA. For these experiments a 59 bp duplex DNA was employed and was modified to contain six cisplatin 1,2-d(GpG) adducts at defined locations. The optimal cisplatin treatment conditions were determined empirically to assure that 100% of the substrates were platinated at each site. The substrate was confirmed by exonuclease digestion mapping of the substrate (data not shown). EMSA conditions were modified to enable detection of the association of the heterotrimeric DNA-PK with the duplex DNA substrate (1). The results of the experiment are presented in Figure 5. Reactions were performed with the undamaged control DNA and as the concentration of DNA-PK was increased a complex migrating just below the well of the gel was observed (lanes 3–5). This complex is not observed in reactions performed with Ku purified free from DNA-PKcs (lane 2). Identical reactions were performed with cisplatin-damaged DNA (lanes 6–10). A DNA-PK–DNA complex was observed but the results show a decrease in the amount of this complex formed compared to that observed with undamaged DNA. Quantification of the data demonstrate that DNA-PKcs can bind to cisplatin-damaged DNA with the Ku–DNA complex but the amount of DNA-PK was decreased to ~35% of that observed with the undamaged DNA. These results are consistent with a decrease in the ability of Ku to translocate on the cisplatin-damaged DNA such that the association of DNA-PKcs with the DNA terminus and the Ku dimer is inhibited.
Figure 5.

DNA-PKcs binding to a 59 bp cisplatin-damaged duplex DNA. Undamaged and cisplatin-damaged duplex 59 bp DNA substrates were prepared and purified as described in Materials and Methods. Binding assays were performed with 50 fmol of undamaged DNA (lanes 1–5) or the same DNA containing six cisplatin d(GpG) adducts (lanes 6–10). Reactions contained buffer alone (lanes 1 and 6), purified Ku (lanes 2 and 7) or 40, 80 or 160 fmol of the heterotrimeric DNA-PK (lanes 3–5 and 8–10). Reaction products were separated by 8% native PAGE, visualized by autoradiography and quantified by PhosphorImager analysis using ImageQuant software in volume integration mode as described in Materials and Methods. The locations of free DNA and the multimeric Ku–DNA and DNA-PK–DNA complexes are depicted on the right.
Kinase activity on undamaged and cisplatin-damaged DNA substrates
If the DNA-PKcs that is bound to the cisplatin-damaged DNA terminus is active for phosphorylation, then we would expect to observe ~35% of the kinase activity relative to the undamaged control. Interestingly, under identical conditions used to measure DNA-PK binding in the EMSA analysis, the kinase activity is inhibited by >95% (Fig. 6). These results suggest that in addition to a decrease in the association of DNA-PKcs with the DNA ends of cisplatin-damaged DNA, the DNA-PK that is bound is in an inactive conformation such that there is a reduced catalytic efficiency. These results are consistent with our initial mechanistic findings that there was a reduced kcat for phosphorylation, while the overall affinity of the Ku subunits for DNA is largely unaffected by cisplatin damage(14).
Figure 6.

Kinase activation using a 59 bp DNA substrate with multiple cisplatin adducts. Phosphorylation reactions were performed as described in Materials and Methods. Kinase activity was determined in the presence of undamaged (open bar) or cisplatin-damaged (closed bars) duplex 59 bp DNA substrates. The data are presented as the percent of activity obtained compared to the undamaged control DNA. The activity obtained with the undamaged DNA at each concentration of protein was defined as 100%. Reactions were initially performed at a DNA-PK–DNA ratio of 0.16:1 and then using increasing protein:DNA ratios of 0.8:1, 1.64:1 and 3.28:1, similar to reaction conditions in Figure 5.
Length of cisplatin–DNA damage from the terminus alters the degree of DNA-PK inhibition
If the ability of the Ku subunits to translocate or move away from a DNA terminus is required for activation of DNA-PK, then the position of the adduct from the terminus should influence the degree of inhibition. Specifically, if the adduct is far enough from the terminus such that Ku can freely translocate the required distance to allow DNA-PKcs to bind and be activated, then no inhibition should be observed. To address this question, a series of DNA substrates were prepared with site-specific cisplatin–DNA adducts at defined distances from the DNA termini. To facilitate the direct comparison of these DNA substrates, the adducts were placed an equal distance from each end on a given DNA molecule and the total length of the DNA was varied to allow for the different distances between the DNA termini and the adduct. Duplex substrates of size 30, 41 and 120 bp were prepared with centrally located cisplatin adducts such that the distances of the lesion from the termini was 14, 20 and 60 bases, respectively. As others have reported, we also observed a size dependence on the ability to activate DNA-PK. The specific activity of DNA-PK using the shorter DNA substrates, 30 bp, was ~40% of that observed for the longer DNA substrates, 40 and 120 bp in length (data not shown). To determine the effect of the cisplatin lesion on kinase activation, the DNA-PK activity obtained with each cisplatin-damaged DNA substrate is presented as a percentage of the activity obtained with the undamaged control of identical sequence and length. For comparison, we have determined previously that the kinase activity using a 25 bp duplex containing a single 1,2-d(GpG) cisplatin lesion was reduced by ~80% compared to the undamaged control (14). The data obtained with the new DNA substrates are presented in Figure 7 and demonstrate that with the 30 bp DNA the presence of the cisplatin lesion 14 bp from each termini resulted in a 50% decrease in kinase activity, less than that observed with the 25 bp DNA. The trend continued for the 41 bp DNA where we observed only 20% inhibition for this DNA where the adduct was 20 bp from each termini. Interestingly, this degree of inhibition is similar to the result we observed previously using a 25 bp DNA containing a 1,2-d(GpG) lesion at one terminus such that the opposite end was 22 bp from the lesion (14). Results obtained with the 120 bp DNA substrate revealed no statistically significant difference between the undamaged and cisplatin-damaged DNA.
Figure 7.

Kinase activation using DNA substrates with single, site-specific 1,2-d(GpG) cisplatin adducts located varying distances from the DNA termini. Phosphorylation assays were performed as described in Materials and Methods. DNA-PK (160 fmol) was incubated in the presence of unplatinated or platinated duplex 30, 41 and 120 bp DNA substrates (1 pmol). The data are presented as the percent kinase activity obtained with the cisplatin-damaged DNA compared to the undamaged counterparts. The activity with each undamaged DNA was defined as 100%.
DISCUSSION
The multifunctional Ku protein is a required component of the NHEJ DSB repair machinery. The protein displays fairly unique properties with respect to its interaction with DNA. How these DNA binding properties function to process DNA DSBs have only been loosely defined. The DNA end-binding activity of Ku has been postulated to bring DNA ends together to allow their ligation, and in vitro biochemical analyses of NHEJ bear out the requirement for Ku (18). Another indication of Ku promoting the association of DNA ends comes from experiments demonstrating that Ku can transfer between two DNA molecules containing at least 4-base complementary ends (19). The proposed model is that the annealing of the termini allow Ku to bridge the two DNAs. Ku also has specific interactions with DNA-PKcs that facilitate the association of DNA-PKcs with DNA termini. A study of the interaction of DNA-PKcs with a variety of DNA substrates in the absence of Ku led to a model in which DNA-PKcs promotes the association of two DNA ends that may lead to ligation (20). DNA-PKcs activation was observed on duplex DNAs as short as 12 bp with 5-base single-stranded overhangs on each end (20). Analyses of the DNA-PK holoenzyme on short duplex DNA substrates revealed that the minimum DNA length that supports kinase activation in the presence of Ku is 26 bp with non-productive complexes being formed on shorter DNA molecules (21). Another role of the Ku protein could be to generate single-stranded DNA ends that would facilitate the interaction of DNA-PKcs with the DNA molecule. This limited strand separation activity is consistent with the reported helicase activity of Ku and that the helicase activity is extremely inefficient. Atomic force microscopy has also been used to demonstrate the association of DNA-PK at DNA termini and the DNA joining activity (22). This report also observed another interesting aspect of Ku binding to DNA: the formation of multimeric Ku–DNA complexes.
One of the many interesting activities of Ku is the proposed ability to slide or translocate on duplex DNA molecules in an ATP-independent manner (5,6). The translocation of Ku has been observed in the absence of DNA-PKcs and more recently, DNA-PKcs was demonstrated to promote Ku movement away from a DNA terminus to more internal positions (8). A recent report has demonstrated that kinase activity is required for the DNA-PK holoenzyme to vacate the DNA end, which allows other proteins access to the DNA ends (23). However, the proteins employed were bacterial polymerases and exonucleases and while they were inhibited by the presence of the DNA-PK holoenzyme, the possibility exists that the activities required for NHEJ might behave differently. A general model for Ku translocation can be envisioned where the movement of Ku away from a DNA terminus is via a second molecule, DNA-PKcs or another Ku, acting to displace the bound Ku protein from the DNA terminus promoting the association with an internal stretch of bases on the duplex DNA. The data we obtained in the kinetic analyses support this model as the rates for Ku movement decrease as the number of Ku bound molecules increases. That is, the energy required to move the Ku molecules is greater with more Ku bound. The kinetic data are also consistent with a model where random movement of Ku to internal position results in the ability of a free DNA end to which another Ku dimer or DNA-PKcs could bind. The presence of cisplatin adducts on the DNA substrate resulted in a dramatically reduced rate of Ku movement on the duplex DNA. The adducts do not appear to be a complete block to Ku translocation but simply serve to reduce the rate at which the protein moves or slides along the duplex. The decreased rate results in decreased association of DNA-PKcs in addition to the formation of inactive DNA-PK complexes at the termini. The association of DNA-PK at DNA termini of DSBs is a required step in the repair by the NHEJ pathway. The ability of cisplatin lesions to inhibit DNA-PK activity in vitro and potentially the NHEJ catalyzed repair of DSBs is consistent with the ability of cisplatin to sensitize cells to killing by treatment with IR. This possibility would require the presence of compound DNA lesions, essentially positioning a DNA DSB in close proximity to a cisplatin lesion. The likelihood of this occurring, if both processes are random, is exceedingly small. The possibility exists that DNA containing cisplatin lesions results in the preferential formation of IR-induced DNA damage in the vicinity of the cisplatin–DNA adducts. The exact mechanism of how cisplatin achieves its sensitization activity is not known. Our in vitro analyses support a mechanism involving decreased DSB repair via the NHEJ pathway by cisplatin–DNA lesions inhibiting the kinase activity of DNA-PK.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by NIH award CA64374 and CA82741 (to J.J.T.).
REFERENCES
- 1.Hammarsten O. and Chu,G. (1998) DNA-Dependent Protein Kinase: DNA Binding and Activation in the Absence of Ku. Proc. Natl Acad. Sci.USA, 95, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin S. and Weaver,D.T. (1997) Double-Strand Break Repair by Ku70 Requires Heterodimerization With Ku80 and DNA Binding Functions. EMBO J., 16, 6874–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dynan W.S. and Yoo,S. (1998) Interaction of Ku Protein and DNA-Dependent Protein Kinase Catalytic Subunit With Nucleic Acids. Nucleic Acids Res., 26, 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo S., Kimzey,A. and Dynan,W.S. (1999) Photocross-Linking of an Oriented DNA Repair Complex — Ku Bound at a Single DNA End. J. Biol. Chem., 274, 20034–20039. [DOI] [PubMed] [Google Scholar]
- 5.de Vries E.G., van Driel,W., Bergsma,W.G., Arnberg,A.C. and van der Vliet,P.C. (1989) HeLa Nuclear Protein Recognizing DNA Termini and Translocating on DNA Forming a Regular DNA-Multimeric Protein Complex. J. Mol. Biol., 208, 65–78. [DOI] [PubMed] [Google Scholar]
- 6.Paillard S. and Strauss,F. (1991) Analysis of the Mechanism of Interaction of Simian Ku Protein With DNA. Nucleic Acids Res., 19, 5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stuiver M.H., Coenjaerts,F.E. and van der Vliet,P.C. (1990) The Autoantigen Ku Is Indistinguishable From NF IV, a Protein Forming Multimeric Protein-DNA Complexes. J. Exp. Med., 172, 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo S. and Dynan,W.S. (1999) Geometry of a Complex Formed by Double Strand Break Repair Proteins at a Single DNA End: Recruitment of DNA-PKcs Induces Inward Translocation of Ku Protein. Nucleic Acids Res., 27, 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leuther K.K., Hammarsten,O., Kornberg,R.D. and Chu,G. (1999) Structure of DNA-Dependent Protein Kinase: Implications for Its Regulation by DNA. EMBO J., 18, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keys H.M. (1999) Cisplatin, Radiation, and Adjuvant Hysterectomy Compared With Radiation and Adjuvant Hysterectomy for Bulky Stage IB Cervical Carcinoma. N. Engl. J. Med., 340, 1144–1153. [DOI] [PubMed] [Google Scholar]
- 11.Dolling J.A., Boreham,D.R., Brown,D.L., Mitchel,R.E.J. and Raaphorst,G.P. (1998) Modulation of Radiation-Induced Strand Break Repair by Cisplatin in Mammalian Cells. Int. J. Radiat. Biol., 74, 61–69. [DOI] [PubMed] [Google Scholar]
- 12.Raaphorst G.P., Wang,G., Stewart,D. and Ng,C.E. (1996) Concomitant Low Dose-Rate Irradiation and Cisplatin Treatment in Ovarian Carcinoma Cell Lines Sensitive and Resistant to Cisplatin Treatment. Int. J. Radiat. Biol., 69, 623–631. [DOI] [PubMed] [Google Scholar]
- 13.Turchi J.J. and Henkels,K. (1996) Human Ku Autoantigen Binds Cisplatin-Damaged DNA but Fails to Stimulate Human DNA-Activated Protein Kinase. J. Biol. Chem., 271, 13861–13867. [DOI] [PubMed] [Google Scholar]
- 14.Turchi J.J., Patrick,S.M. and Henkels,K.M. (1997) Mechanism of DNA-Dependent Protein Kinase Inhibition by Cis-Diamminedichloroplatinum(II)-Damaged DNA. Biochemistry, 36, 7586–7593. [DOI] [PubMed] [Google Scholar]
- 15.Turchi J.J., Li,M. and Henkels,K.M. (1996) Cisplatin-DNA Binding Specificity of Calf High-Mobility Group 1 Protein. Biochemistry, 35, 2992–3000. [DOI] [PubMed] [Google Scholar]
- 16.Patrick S.M. and Turchi,J.J. (1999) Replication Protein A (RPA) Binding to Duplex Cisplatin-Damaged DNA Is Mediated Through the Generation of Single-Stranded DNA. J. Biol. Chem., 274, 14972–14978. [DOI] [PubMed] [Google Scholar]
- 17.Hermanson I.L. and Turchi,J.J. (2000) Overexpression and Purification of Human XPA Using a Baculovirus Expression System. Protein Expr. Purif., 19, 1–11. [DOI] [PubMed] [Google Scholar]
- 18.Baumann P. and West,S.C. (1998) DNA End-Joining Catalyzed by Human Cell-Free Extracts. Proc. Natl Acad. Sci. USA, 95, 14066–14070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bliss T.M. and Lane,D.P. (1997) Ku Selectively Transfers Between DNA Molecules With Homologous Ends. J. Biol. Chem., 272, 5765–5773. [DOI] [PubMed] [Google Scholar]
- 20.Hammarsten O., DeFazio,L.G. and Chu,G. (2000) Activation of DNA-Dependent Protein Kinase by Single-Stranded DNA Ends. J. Biol. Chem., 275, 1541–1550. [DOI] [PubMed] [Google Scholar]
- 21.West R., Yaneva,M. and Lieber,M. (1998) Productive and Nonproductive Complexes of Ku and DNA-Dependent Protein Kinase at DNA Termini. Mol. Cell. Biol., 18, 5908–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pang D., Yoo,S., Dynan,W.S., Jung,M. and Dritschilo,A. (1997) Ku Proteins Join DNA Fragments As Shown by Atomic Force Microscopy. Cancer Res., 57, 1412–1415. [PubMed] [Google Scholar]
- 23.Calsou P., Frit,P., Humbert,O., Muller,C. and Chen,D.J. (1999) The DNA-Dependent Protein Kinase Catalytic Activity Regulates DNA End Processing by Means of Ku Entry into DNA. J. Biol. Chem., 274, 7848–7856. [DOI] [PubMed] [Google Scholar]