

Abstract
Plasmid pL32 from the Natto strain of Bacillus subtilis belongs to a group of low-copy-number plasmids in gram-positive bacteria that replicate via a theta mechanism of replication. We studied the DNA region encoding the replication protein, RepN, of pLS32, and obtained the following results. Transcription of the repN gene starts 167 nucleotides upstream from the translational start site of repN. The copy number of repN-coding plasmid pHDCS2, in which the repN gene was placed downstream of the IPTG (isopropyl-1-thio-β-d-galactopyranoside)-inducible Pspac promoter, was increased 100 fold by the addition of IPTG. Histidine-tagged RepN bound to a specific region in the repN gene containing five 22-bp tandem repeats (iterons) with partial mismatches, as shown by gel retardation and foot printing analyses. Sequence alterations in the first three iterons resulted in an increase in plasmid copy number, whereas those in either the forth or fifth iteron resulted in the failure of plasmid replication. The iterons expressed various degrees of incompatibility with an incoming repN-driven replicon pSEQ243, with the first three showing the strongest incompatibility. Finally, by using a plasmid, pHDMAEC21, carrying the sequence alterations in all the five iterons in repN and thus unable to replicate but encoding intact RepN, the region necessary for replication was confined to a 96-bp sequence spanning the 3′-terminal half of the fourth iteron to an A+T-rich region located downstream of the fifth iteron. From these results, we conclude that the iterons in repN are involved in both the control of plasmid copy number and incompatibility, and we suggest that the binding of RepN to the last two iterons triggers replication by melting the A+T-rich DNA sequence.
Circular plasmids can be grouped into two classes by the mode of replication. One group replicates via a rolling-circle intermediate, while the other uses the theta-type intermediate. In the case of plasmids from natural isolates of Bacillus subtilis, it has been demonstrated that the sizes of the plasmids belonging to the first group are small, while those belonging to the second group are large (37). The small plasmids of B. subtilis are further grouped into seven classes based on their sizes and restriction patterns and show a replication function related to that of pC194 derived from Staphylococcus aureus (26). It remains unknown why the only pC194-type replicon is found in B. subtilis among rolling-circle replicons with different types of replication functions (13, 18, 20). On the other hand, plasmids that replicate via a theta mechanism of replication are classified into six groups (36); in addition, a recently reported plasmid, pBS72, carries a replicon of a new type (37). The large plasmids found in natural isolates of B. subtilis show diverse modes of theta replication, as exemplified by pLS20 (25), pLS32 (36), and pBS72 (37). Whereas the replication functions are unique for pLS20 and pBS72, several plasmids, including pLS32 and their relatives (see below), show amino acid sequence similarities in the replication initiation proteins (Rep), suggesting that they replicate in similar mechanisms.
The low-copy-number plasmid pLS32 used in this study was originally isolated from the Natto strain of Bacillus B. subtilis and replicates via a theta mechanism without the need for DnaA and DNA polymerase I (14, 35, 36). The unique feature of this plasmid is that it can support replication of the entire chromosome of B. subtilis and create a subgenome when it is placed in a chromosomal DNA region surrounded by direct repeats (14, 17). Large plasmids that have been isolated from a variety of bacterial genera, including Lactococcus, Lactobacillus, Staphylococcus, and Enterococcus (10, 19), carry rep genes that specify Rep proteins with amino acid sequence homologies among them. A common feature of this group of plasmids and pLS32 is that they appear to contain the replication initiation origin, ori, in the coding sequence of the rep gene, and this was verified experimentally for pLS32, staphylococcal plasmid pSX267, and Enterococcus faecalis plasmid pAD1 (11, 12, 36). It has also been shown for this group of plasmids that there are tandem (iterons) and/or inverted repeat sequences in the rep genes. Recently, the tandem repeats in pAD1 and in staphylococcal plasmid pSK41 were shown to be the targets of the Rep proteins of those plasmids (11, 23).
The amino acid sequences of the RepN family proteins are more homologous in the N-terminal regions than those in the C-terminal regions, and the middle regions show the least homology (10). Recently, Francia and coworkers made a striking observation that a spontaneous deletion removing a 105-nucleotide sequence (corresponding to 35 codons) in the repA gene of pAD1 bordered by two identical 31-bp direct repeats does not affect the replication ability of the plasmid (11). The nucleotide sequences in the direct repeats in the repA gene encode the same amino acids except for one, and this structural feature is found in all the plasmids belonging to this group (10). By analogy with the above observation, it can be assumed that a similar deletion would also lead to functional plasmids in this family of plasmids.
The replication origin (ori) regions of low-copy-number plasmids from both gram-positive and -negative organisms are characterized by the presence of iterons (10, 15). It has been shown for many low-copy-number Escherichia coli plasmids such as P1, F, R1, RK2, R6K, and pSC101 that iterons not only are essential for replication but also are key elements controlling plasmid replication (8).
In this paper, we report the binding of a histidine-tagged replication initiation protein of pLS32 (RepN) to a DNA sequence containing five direct repeats (iterons) with various degrees of sequence mismatches. We also show the results including incompatibility shown by the iterons, the effects of sequence alterations of the iterons on the plasmid copy number, and an attempt to locate the replication origin, oriN. From the data of these analyses, we present a model of pLS32 replication, in which RepN binds to the iterons and causes melting of the A+T-rich region located downstream of the iterons.
MATERIALS AND METHODS
Materials.
Synthetic oligonucleotides were commercially prepared by Espec Oligo Service Co. (Table 1).
TABLE 1.
Primers used in this studya
| Primer | Nucleotide sequence |
|---|---|
| RepNBio2 | 5′-XCCGAAGCAACACCTGATATA-3′ |
| 6634Bio | 5′-XGATGACCTTTTAATTAAAGCAGAGG-3′ |
| RepN3 | 5′-GTAAGCTTCGTTTATGAGTTCGTAAAACG-3′ |
| 6683Bio | 5′-XCTCTTGTTAACAATTTGTTAACAAGATTATTCTC-3′ |
| RepN5 | 5′-GTAGATCTATGAGTAAATATTTCACAGCTAATAAC-3′ |
| RepN4 | 5′-AAGGATCCTTAATTATCTAACCAATTATAAA-3′ |
| EC6655 | 5′-GTGAATTCGAGGAAACAGCCAAAAACATAG-3′ |
| CL6770 | 5′-GTATCGATCTCTTGATGGTTCAATTTGAA-3′ |
| EC7047 | 5′-GTGAATTCGTATGGAAATATTGATTTCAAAGGG-3′ |
| RepN1 | 5′-GTAAGCTTAAAGGAGCAACAAAAATGAG-3′ |
| DEGS5 | 5′-GGATTCCAAAGTGCTGGATTC-3′ |
| DEGU3 | 5′-TCCATGATCACAACATCAGGATG-3′ |
| MU66765 | 5′-GATAAAAACTGGATGGGACAGAACGAGCTGTCAAGTAATGAACGGATAGG-3′ |
| MU66763 | 5′-CCTATCCGTTCATTACTTGACAGCTCGTTCTGTCCCATCCAGTTTTTATC-3′ |
| MU67035 | 5′-CTTTCCAGTAATGAACGAATTGGACAGAACGAGCTGTCAGGGAAAGTTCAAATTGA-3′ |
| MU67033 | 5′-TCAATTTGAACTTTCCCTGACAGCTCGTTCTGTCCAATTCGTTCATTACTGGAAAG-3′ |
| MU67475 | 5′-AGTTCAAATTGAACCATCAAGGGAAGTAAAGCTAAACCATCCGGATGGTTCAAAAAG-3′ |
| MU67473 | 5′-CTTTTTGAACCATCCGGATGGTTTAGCTTTACTTCCCTTGATGGTTCAATTTGAACT-3′ |
| DMU667651 | 5′-GATAAAAACTGGATGGGACAGAACGAGCTGTCAAGTAATGAACGAATTGGA-3′ |
| DMU667631 | 5′-TCCAATTCGTTCATTACTTGACAGCTCGTTCTGTCCCATCCAGTTTTTATC-3′ |
| MU67295 | 5′-GAACGAGCTGTCAGGGAAGGTGCAGATAGAGCCCTCAAGAGAGGTCAAATTG-3′ |
| MU67293 | 5′-CAATTTGACCTCTCTTGAGGGCTCTATCTGCACCTTCCCTGACAGCTCGTTC-3′ |
| MU67745 | 5′-CAAATTGAACCATCCGGACGGGAGCAAGAGGACCACTAATAATAATAACTCT-3′ |
| MU67743 | 5′-AGAGTTATTATTATTAGTGGTCCTCTTGCTCCCGTCCGGATGGTTCAATTTG-3′ |
| MMU7475 | 5′-GGTGCAGATAGAGCCCTCAAGGGAAGTAAAGCTAAACCATCCGGATGGTTCAAAAAG-3′ |
| MMU7473 | 5′-CTTTTTGAACCATCCGGATGGTTTAGCTTTACTTCCCTTGAGGGCTCTATCTGCACC-3′ |
| EC6241 | 5′-GTGAATTCAAAGGAGCAACAAAAATGAG-3′ |
| CL7119 | 5′-GTATCGATTAATTATCTAACCAATTATAAA-3′ |
| CL6827 | 5′-GTATCGATAGAGTTATTATTATTAGTGGTTG-3′ |
| CL6778 | 5′-GTATCGATTTGACCTCTCTTGATGGT-3′ |
| CL6741 | 5′-GTATCGATGGAAAGTTCATTTTGACCTA-3′ |
| CL6726 | 5′-GTATCGATACCTATCCGTTCATTACTGGAA-3′ |
| CL6702 | 5′-GTATCGATCATTTTGACCCATCCAGTTT-3′ |
| CL6771 | 5′-GTATCGATCTCTCTTGATGGTTCAATTTG-3′ |
| EC6684 | 5′-AGTGAATTCTGGATGGGTCAAAATGAACTTTCCAGT-3′ |
| EC6705 | 5′-GTGAATTCCAGTAATGAACGGATAGGT-3′ |
| EC6736 | 5′-GTGAATTCTTTCCGGGAAAGTTCAAATTG-3′ |
| EC6760 | 5′-AGTGAATTCCATCAAGAGAGGTCAAAT-3′ |
| CL6883 | 5′-AGTATCGATCTCTTGTTAACAATTTGTTAACAAGATTATTCTC-3′ |
| EC6867411 | 5′-AATTCGGATGGGTCAAAATGAACTTTCCAGTAATGAACGGATAGGTCAAAATGAACTTTCC-3′ |
| CL7416861 | 5′-CGGGAAAGTTCATTTTGACCTATCCGTTCATTACTGGAAAGTTCATTTTGACCCATCCG-3′ |
| EC6718S | 5′-AATTCGGATAGGTCAAAATGAACTTTCCGGGAAAGTTCAAATTGAACCATCA-3′ |
| CL6718S | 5′-CGTGATGGTTCAATTTGAACTTTCCCGGAAAGTTCATTTTGACCTATCCG-3′ |
| EC6736S | 5′-AATTCTTTCCGGGAAAGTTCAAATTGAACCATCAAGAGAGGTCAAATTGAACCATCC-3′ |
| CL6736S | 5′-CGGGATGGTTCAATTTGACCTCTCTTGATGGTTCAATTTGAACTTTCCCGGAAAG-3′ |
| EC686741M | 5′-AATTCGGATGGGACAGAACGAGCTGTCAAGTAATGAACGAATTGGACAGAACGAGCTGTCA-3′ |
| CL7416861M | 5′-CGTGACAGCTCGTTCTGTCCAATTCGTTCATTACTTGACAGCTCGTTCTGTCCCATCCG-3′ |
| EC6767 | 5′-AGTGAATTCGAGAGGTCAAATTGAACCATCCGGA-3′ |
| EC6772 | 5′-AGTGAATTCGTCAAATTGAACCATCCGGATG-3′ |
| REC6984 | 5′-AGTGAATTCACATCTGGGGTTTTCTCTACTTCG-3′ |
| REC6883 | 5′-AGTGAATTCTCTTGTTAACAATTTGTTAACAAGATTATTCTC-3′ |
| REC6851 | 5′-AGTTGAATTCTCATTATTATTAAAGTTATTTTTAATAGAG-3′ |
| REC6824 | 5′-AGTTGAATTCGAGTTATTATTATTAGTGGTTCTTTTTG-3′ |
X, biotin attached to the nucleotide at the 5′ end.
Plasmids and plasmid construction.
Plasmids used in this study are shown in Fig. 1 and Table 2. Plasmid pSEQ243 carries a 1.5-kb, repN-containing EcoRI-HindIII fragment that was isolated from pBET131, blunt ended, and inserted into the blunt-ended SalI and XbaI sites of pUKM504 (36). Plasmid pBECS21 was made by insertion of a BamHI fragment containing the neomycin resistance (Nmr) gene from pBEST509 into pHDCS2.
FIG. 1.

Structure of the plasmids used in this study. Expression of the repN and mutant repN genes are under the control of the spac promoter induced by IPTG. The dotted arrow in pHDMAE21 indicates the mutant repN gene containing sequence alterations in all five iterons. Abbreviations: Ac, AccI; Ba, BamHI; Bn, BanIII; Bp, BspEI; Bs, BstXI; Ec, EcoRI; Hi, HindIII; Ps, PstI; Sm, SmaI; Xb, XbaI.
TABLE 2.
Bacterial strains and plasmids
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| B. subtilis | ||
| CU741 | trpC2 leuC7 | 39 |
| RECT741 | trpC2 leuC7 recA::tet | This study |
| SSM190 | trpC2 leuC7 recA::tet amyE::[cat] | This study |
| BEST4173 | Carries repN at the proB locus | 17 |
| ISHI11 | trpC2 leuC7 recA::tet amyE::[repN 6241-7119 cat] | This study |
| ISHI12 | trpC2 leuC7 recA::tet amyE::[repN 6241-6827 cat] | This study |
| ISHI13 | trpC2 leuC7 recA::tet amyE::[repN 6241-6778 cat] | This study |
| ISHI14 | trpC2 leuC7 recA::tet amyE::[repN 6241-6741 cat] | This study |
| ISHI15 | trpC2 leuC7 recA::tet amyE::[repN 6241-6726 cat] | This study |
| ISHI16 | trpC2 leuC7 recA::tet amyE::[repN 6241-6702 cat] | This study |
| ISHI21 | trpC2 leuC7 recA::tet amyE::[repN 6684-6884 cat] | This study |
| ISHI22 | trpC2 leuC7 recA::tet amyE::[repN 6705-6884 cat] | This study |
| ISHI23 | trpC2 leuC7 recA::tet amyE::[repN 66736-6884 cat] | This study |
| ISHI24 | trpC2 leuC7 recA::tet amyE::[repN 6760-6884 cat] | This study |
| ISHI31 | trpC2 leuC7 recA::tet amyE::[repN 6686-6741 cat] | This study |
| ISHI32 | trpC2 leuC7 recA::tet amyE::[repN 6718-6765 cat] | This study |
| ISHI33 | trpC2 leuC7 recA::tet amyE::[repN 6736-6778 cat] | This study |
| ISHI34 | trpC2 leuC7 recA::tet amyE::[repN 6686-6741 cat]; repN sequence altered | This study |
| E. coli | ||
| JM103 | Δlac-pro thi rpsL supE sbcB hsdR4 F′ [traD36 proAB+lacIqlacZΔM15] | 41 |
| XL1 | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 lac F′ [proAB+lacIqlacZ DM15 Tn10] | 5 |
| Plasmids | ||
| pUKM504 | pUC18 carrying neo in the bla gene | 28 |
| pHDCS2 | Carries repN under control of the Pspac promoter | 36 |
| pSEQ243 | pUKM504 carrying repN between the EcoRI and HindIII sites | 36 |
| pMU63 | pSEQ243 derivative carrying sequence alterations in IT1 | This study |
| pMU75 | pSEQ243 derivative carrying sequence alterations in IT2 | This study |
| pDMU1 | pSEQ243 derivative carrying sequence alterations in IT1 and IT2 | This study |
| pMT34 | pSEQ243 derivative carrying sequence alterations in IT1, IT2, and IT3 | This study |
| pQDM42 | pSEQ243 derivative carrying sequence alterations in IT1, IT2, and IT5 | This study |
| pQMT11 | pSEQ243 derivative carrying sequence alterations in IT1, IT2, IT3, and IT4 | This study |
| pQMT51 | pSEQ243 derivative carrying sequence alterations in IT1, IT2, IT3, and IT5 | This study |
| pSMU19 | pSEQ243 derivative carrying sequence alterations in IT4 | This study |
| pSEM2129 | pSEQ243 derivative carrying sequence alterations in IT5 | This study |
| pMAE5 | pSEQ243 derivative carrying sequence alterations in IT1, IT2, IT3, IT4, and IT5 | This study |
| pHDMAE21 | pHDCS2 derivative carrying sequence alterations in IT1, IT2, IT3, IT4, and IT5 | This study |
| pQE8 | Expression vector with the T5 promoter | QIAGEN |
| pQErepN | pQE8 carrying repN under control of the T5 promoter | This study |
| ptrpBG1 | Integration vector at the amyE locus | 33 |
| pUH101 | pBR322 carrying cat at the EcoRV site | 34 |
| pBEST4B | pGEM2 carrying at the BamHI site a cat-containing Sau3 A fragment from pUH101 | M. Itaya, unpublished data |
| pBEST509 | pGEM2 derivative carrying Nmr | M. Itaya, unpublished data |
| pBECS21 | pHDCS2 derivative carrying the Nmr gene from pBEST509 at the BamHI site | This study |
| pMAEC11, -12 | pHDMAE21 carrying the repN region between nt 6705 and 6986 at the EcoRI site | This study |
| pMAEC21 | pHDMAE21 carrying the repN region between nt 6736 and 6986 at the EcoRI site | This study |
| pMAEC39 | pHDMAE21 carrying the repN region between nt 6760 and 6986 at the EcoRI site | This study |
| pMAEC42 | pHDMAE21 carrying the repN region between nt 6760 and 6885 at the EcoRI site | This study |
| pMAEC51 | pHDMAE21 carrying the repN region between nt 6767 and 6885 at the EcoRI site | This study |
| pMAEC60 | pHDMAE21 carrying the repN region between nt 6772 and 6885 at the EcoRI site | This study |
| pMAEC541 | pHDMAE21 carrying the repN region between nt 6786 and 6885 at the EcoRI site | This study |
| pMAEC395 | pHDMAE21 carrying the repN region between nt 6760 and 6867 at the EcoRI site | This study |
| pMAEC111 | pHDMAE21 carrying the repN region between nt 6705 and 6851 at the EcoRI site | This study |
| pMAEC824 | pHDMAE21 carrying the repN region between nt 6705 and 6824 at the EcoRI site | This study |
The general strategy to construct pSEQ243 derivatives that carry mutations in the iterons was as follows. First, various combinations of PCR primers containing sequence changes in the iterons were annealed to plasmid pSEQ243, used as a template, and extended by PfuTurbo DNA polymerase according to the procedure described in the Quik Change site-directed mutagenesis kit (Stratagene), and the reaction products were transformed into Escherichia coli XL1. The PCR conditions were as follows: heating at 95°C for 30 s, 55°C for 1 min, and 68°C for 11 min 12 s. Sequence alterations in the repN gene in the resultant plasmids were confirmed by sequence determination. Second, to avoid possible sequence alterations in a region other than the repN sequence, the 1.5-kb EcoRI-HindIII fragments containing the altered repN-coding region were excised from agarose gels and cloned between the EcoRI and HindIII sites of pUKM504. Construction of pSEQ243 derivatives carrying multiple mutations in the iterons was carried out either by following the procedure described above using specific primers or by restriction and ligation of pSEQ243 derivatives. Plasmids pMU63, pMU75, and pSMU19 carrying single mutations in iteron 1 (IT1), IT2, and IT4, respectively, were constructed by using primer pairs MU66765 and MU66763, MU67035 and MU67033, and MU67475 and MU67473, respectively. Likewise, plasmids pDMU1, pMT34, pQDM42, and pQMT11 carrying multiple iteron mutations were constructed by using the sets of the following primer pairs and template: DMU667651 plus DMU667631 and pMU75, MU67295 plus MU67293 and pDMU1, MU67745 plus MU67743 and pDMU1, and MMU7475 plus MMU7473 and pMT34, respectively. Plasmids pQMT51, pSEM2129, and pMAE5 were constructed by exchanging the 447-bp BspEI-EcoRI fragment containing the mutant IT5 (Fig. 1) between plasmids pMT34 and pQDM42, pSEQ243 and pQDM42, and pQMT11 and pSEM2129, respectively. Plasmid pHDMAE21 (Fig. 1) was constructed by ligation of the 0.61-kb AccI-SmaI fragment containing the five altered iterons derived from pMAE5 with pHDCS2 (Fig. 1) from which the same C-terminal repN region had been removed by BanIII digestion, followed by blunting with T4 DNA polymerase and subsequent AccI digestion.
Plasmid pQErepN was constructed by insertion of a PCR fragment between the BamHI and HindIII sites of pQE8 that had been prepared by using primers RepN5 and RepN3 and digested with both BglII and HindIII.
The pMAEC series plasmids, except for pMAEC541 and pMAEC395, were constructed in E. coli JM103 by ligation of EcoRI-treated PCR fragments with pMAEC21 that had been cleaved with EcoRI and treated with alkaline phosphatase. The following primers were used as follows: for pMAEC11 and 12, EC6705 and REC6984; for pMAEC21, EC6736 and REC6984; for pMAEC39, EC6760 and REC6984; for pMAEC42, EC6760 and REC6883; for pMAEC51, EC6767 and REC6883; for pMAEC60, EC6772 and REC6883; for pMAEC111, EC6705 and REC6851; and for pMAEC824, EC6705 and REC6824. Plasmid pMAEC541 was constructed as follows. First, pMAEC60 was cut with BspEI, blunt ended with Klenow DNA polymerase I, and further cleaved with HindIII. The smaller BspEI (blunted)-HindIII fragment (390 bp) contains a DNA region from nucleotides (nt) 6786 to 6885 linked to the EcoRI site upstream of the Pspac promoter (42) of pHDMAE21 (Fig. 1). Second, pHDMAE21 was digested with EcoRI, blunt ended, and further digested with HindIII. After agarose gel electrophoresis, the larger fragment was isolated and ligated with the 390-bp fragment, resulting in pMAEC541. Plasmid pMAEC395 was constructed by ligation of the smaller fragment of the HpaI and PstI digestion of pMAEC39 with the larger fragment of the EcoRI- and PstI-cleaved product of pMAEC21, whose EcoRI site had been blunt ended with Klenow DNA polymerase I.
Bacterial strains, strain construction, and medium.
The bacterial strains used in this study are listed in Table 2. Strains ISHI11 to ISHI24, carrying various regions of repN in the chromosomal amyE locus, were constructed as follows. First, the regions to be studied were amplified by PCR with primers carrying EcoRI and ClaI sites and cloned between the EcoRI and ClaI sites of ptrpBGI (33). Second, the ptrpBGI derivatives were linearized by ScaI treatment and transformed into strain CU741 to chloramphenicol resistance (Cmr). Third, the resultant strains were made deficient in recA by transformation with DNA from strain RECT741. Strain SSM190 was constructed similarly, except that the region between the EcoRI and ClaI sites of ptrpBG1 was deleted by cleavage with the restriction enzymes, followed by blunting and ligation. For constructs ISHI31 through ISHI34, synthetic oligonucleotide pairs were annealed and cloned at the same EcoRI and ClaI sites in ptrpBG1, and the resultant plasmids were transformed into CU741 as described above. Oligonucleotides used were EC6241 and CL7119 for ISHI11, EC6241 and CL6827 for IGHI12, EC6241 and CL6778 for ISHI13, EC6241 and CL6741 for ISHI14, EC6241 and CL6726 for ISHI15, EC6241 and CL6702 for ISHI16, EC6684 and CL6883 for ISHI21, EC6705 and CL6883 for ISHI22, EC6736 and CL6883 for ISHI23, EC6760 and CL6883 for ISHI24, EC6867411 and CL7416861 for ISHI31, EC6718S and CL6718S for ISHI32, EC6736S and CL6736S for ISHI33, and EC686741 M and CL7416861 M for ISHI34.
Both E. coli and B. subtilis were grown in Luria-Bertani (LB) medium or LB plates (32).
Primer extension analysis.
Primer extension was performed with an AMV RT cDNA Synthesis kit obtained from Life Sciences, Inc. The reaction mixture contained 10 μg of RNA and a biotinylated primer, RepNBio2 (nt 6230 to 6207). The reaction product was run on a sequencing gel, together with sequencing ladders prepared by using the same primer and pSEQ243 as a template. RNA was isolated as described previously (43).
Purification of histidine (His)-tagged RepN.
JM103 carrying pQErepN was grown to early log phase, treated with isopropyl-1-thio-β-d-galactopyranoside (IPTG; 2 mM), and further incubated for 4 h. Purification of the His-tagged RepN protein was done according to the procedure provided from QIAGEN, except that proteins were eluted from a Ni2+-nitrilotriacetic acid silica column with a gradient of imidazole from 0 mM to 300 mM. The final preparation was at least 80% pure as judged by densitometric scanning of a sodium dodecyl sulfate-polyacrylamide gel after electrophoresis.
Binding of His-tagged RepN protein to DNA.
The reaction mixture contained 20 mM HEPES (pH 7.6), 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM dithiothreitol, 2% (wt/vol) Tween 20, 30 mM KCl, and 0.9 μg of either λ DNA or poly(dI-dC) in a total volume of 15 μl. DNA bands were detected in two ways. One method used SYBR green staining after agarose gel electrophoresis in a NuSieve 3:1 gel (4%; BMA Co.), while the other used the CSPD {disodium 3-(4-methoxyspiro[1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan]-4-yl)phenylphosphate}based chemiluminescence method (Roche Diagnostics) by the procedure recommended by the manufacturer.
Footprinting analysis.
The DNA fragments from nt 6634 to 7170 and from nt 6883 to 6256 biotin labeled at their 5′ ends were prepared by using primers 6634Bio and RepN3 (for the coding strand), and 6883Bio and RepN5 (for the noncoding strand), respectively. Footprinting was performed according to the procedure described in the SureTrack Footprinting Kit (Amersham Pharmacia Biotech, Inc.). Sequencing ladders prepared by using primer 6634Bio or 6883Bio were run alongside the lanes. Only the nucleotide numbers are shown (see Fig. 5).
FIG. 5.

DNase I footprinting analysis of the binding region of His-tagged RepN (A) and alignment of the nucleotide sequences protected from the nuclease digestion (B). (A) DNA fragments labeled with biotin at either nt 6634 or nt 6883 were subjected to footprinting analysis as described in Materials and Methods. The reaction mixture contained 0, 0.3, 0.9, and 2.7 μg of His-tagged RepN in a total volume of 45 μl. The triangles above the panels show the increment of His-tagged RepN. The filled and open arrowheads to the right of each panel show the bands decreased and increased in intensity, respec-tively, with increasing amounts of His-tagged RepN. The bars and arrows indicate the protected regions from DNase I and iterons, respectively. In the left panel, two X-ray films with different exposure time were joined for better clarity. (B) The iterons depicted by the large arrow at the bottom are arranged so that their 22 nucleotides show maximum homology. The numbers indicate the positions of the first and last nucleotides of the sequences. The boxes and small arrows show identical nucleotides among the five iterons and inverted repeat sequences in each iteron, respectively. The last two nucleotides shown in italics depict part of the A+T-rich sequence downstream from the iterons (see Fig. 2).
Quantification of DNA.
The copy number of pHDCS2 (see Fig. 6) was determined as follows. The cells of strain CU741 carrying pHDCS2 were grown overnight at 37°C in LB medium containing Cm (5 μg/ml) and transferred at 2% inoculation to the same medium with various concentrations of IPTG. Total DNA was isolated from the cells at stationary phase by the method of Saitoh and Miura (31) and further purified by the Wizard Plus SV Minipreps (Promega) according to the manufacturer's recommendation, except that the alkaline treatment step was omitted. The number of plasmid copies per chromosome was calculated from the relative concentration of a repN region in pHDCS2 to a pyrR region in the chromosome, which was determined by real-time PCR. Strain BEST4173 in which the entire repN is inserted at the proB locus was used as the standard cell carrying one copy of repN per chromosome. The PCR was carried out in a Smart Cycler System (Cepheid) by using a SYBR Premix Ex Taq real-time PCR kit (Takara) with primers EC6655 and REC6984 for the repN region and primers PYRR3 and PYR322 for the pyrR region in the chromosome. The fraction of plasmid-harboring cells in the stationary-phase population was estimated by spreading the cells on LB plates just before collection and testing 100 colonies obtained after overnight growth for resistance to Cm.
FIG. 6.

Amplification of the copy number of pHDCS2 by overexpression of the repN gene. Experimental procedures are described in Materials and Methods. Lane 1, λ DNA digested with HindIII; BamHI digests of total DNA isolated from the cells grown in the presence of IPTG at concentrations of 0 mM (lane 2); 0.008 mM (lane 3); 0.04 mM (lane 4); 0.2 mM (lane 5); 1.0 mM (lane 6).
For the determination of the relative copy numbers of the pSEQ243 derivatives (Table 3), total DNA was isolated from the plasmid-carrying cells, cleaved with both HindIII and XbaI, and electrophoresed in a 1% agarose gel. The enzyme treatment gave rise to 1.5-kb and 2.3-kb fragments containing the entire repN and chromosomal degS-degU genes, respectively. The DNA bands were transferred to a nylon membrane and detected by Southern hybridization with digoxigenin-labeled PCR fragments prepared for the repN and chromosomal degS-degU regions. The PCR fragments for probing were prepared with a PCR DIG Probe Synthesis kit (Roche Applied Science) with primer pairs RepN1 and RepN4 for the repN region and DEGS5 and DEGU3 for the degS-degU region. The intensity of the repN region was divided by that of the degS-degU region for each sample, and the values obtained for the mutant plasmids were further divided by that of pSEQ243 to calculate the relative abundance of the plasmids. The fluorescent bands were quantified by Atto LightCapture type AE-6961 (Atto Bioscience & Technology).
TABLE 3.
Properties of pSEQ243 and its repN mutants
| Plasmid | Mutation site(s) | Replicationa | Abundanceb |
|---|---|---|---|
| pSEQ243 | No | Yes | 1.0 |
| pMU63 | IT1 | Yes | 2.9 |
| pDMU1 | IT1, IT2 | Yes | 11.2 |
| pMT34 | IT1, IT2, IT3 | Yes | 2.0 |
| pQMT11 | IT1, IT2, IT3, IT4 | No | |
| pQMT51 | IT1, IT2, IT3, IT5 | No | |
| pSMU19 | IT4 | No | |
| pSEM2129 | IT5 | No |
Transformation of CU741 to Nmr.
The amounts of the plasmids in strain CU741 relative to that of pSEQ243 were estimated as described in Materials and Methods.
Transformation.
B. subtilis cells were made competent as described previously (29), and transformants were selected on LB plates containing appropriate antibiotics. The concentrations of the antibiotics used were 15 μg/ml for Nm and 5 μg/ml for Cm.
Sequence determination.
The nucleotide sequences of the DNA regions derived from PCR were confirmed by using an ABI Prism sequencer 377.
RESULTS AND DISCUSSION
Transcription initiation of repN.
We have shown previously that the DNA region between the HincII and BglI sites located upstream from the repN-coding region (Fig. 2) is required for replication of a repN-containing plasmid (36), which suggests that a controlling element(s) for repN transcription is located between the two restriction sites. In concert with this observation, we found that transcription starts at nt 6089 located between the two restriction sites (Fig. 3). Putative −35 and −10 sequences could be assigned to the hexamers from nt 6053 to 6058 and from nt 6079 to 6084, respectively, which would be recognized by σA-containing RNA polymerase. Although the distance between the two regions seems somewhat longer than that found in the consensus sequence, examples similar to this situation have been reported previously (16). The distance between the start sites of transcription and translation of repN is 167 bp (Fig. 2). We note in this respect that the transcription of the rep gene of pSK41 starts 240 bp upstream of the rep-coding sequence and that its expression appears to be regulated by antisense RNA whose transcriptional start site is located 156 bp upstream from the translational start site (23). It remains to be studied whether such regulatory system is also present in repN regulation.
FIG. 2.

Nucleotide sequence of repN and its flanking regions. The nucleotides are numbered according to the previous report (36), and the dots at the top are placed at every 10 nucleotides. The encircled region shows the repN-coding sequence. The promoter and SD sequences for repN are underlined. The asterisk and triangles depict the transcriptional start site and restriction cleavage sites, respectively. The arrows show IT1 through IT5, and the nucleotides shown above the iterons are those introduced by mutation. The open reading frame before the repN gene terminates at the TAA codon located between nt 5893 and 5895. The nucleotides in italics indicate those in the A+T-rich sequence, which we defined as the region containing A+T in ≥80% within a window of 10 consecutive nucleotides after IT5. Abbreviations: Ac, AccI; Bg, BglI; Bp, BspEI; Bs, BstXI; Ec, EcoRI; Hc, HincII; Hi, HindIII; Hp, HpaI. The nucleotide sequence of repN and its vicinity was deposited in the GSDB, DDBJ, EMBL, and NCBI nucleotide sequence databases under the accession number D49467 (36).
FIG. 3.
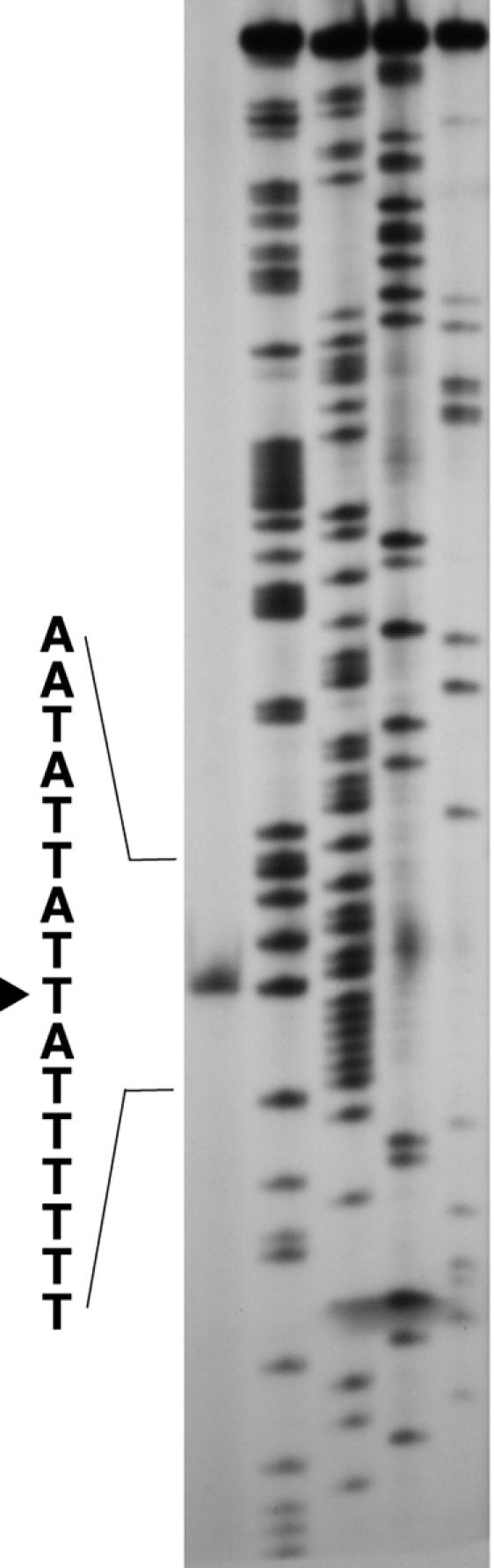
Determination of the transcriptional start site of repN by primer extension. The arrowhead indicates the nucleotide at which transcription initiates. The experimental conditions used are described in Materials and Methods.
Binding of His-tagged RepN to a DNA region containing direct repeats in repN.
Rep proteins are known to start replication by binding to the replication origin regions (15). To test whether RepN also binds to a specific region in the repN-coding sequence, we prepared a His-tagged RepN protein and incubated it with the restriction fragments generated by digestion of the 1.5-kb EcoRI-HindIII fragment with AccI, BstXI, and HpaI (Fig. 4). It should be noted that the His-tagged RepN used is functional in vivo, since a recombinant plasmid made between pQErepN from which His-tagged RepN was prepared (see Materials and Methods), and a Cmr marker DNA conferred resistance on B. subtilis cells to this antibiotic (data not shown). It was shown that the 189-bp fragment between the AccI and BstXI sites was missing in the agarose gel (Fig. 4). In a separate experiment, it was shown that the mobility of the 445-bp BstXI-HindIII fragment obtained without HpaI digestion did not change upon incubation with His-tagged RepN (data not shown), indicating that the HpaI sites are not in the RepN-binding site. From these results we presumed that His-tagged RepN bound to a region within the 189-bp fragment or to a region encompassing the AccI or BstXI site. We reasoned that if any of the two restriction sites was within the RepN-binding site, it would be the BstXI site, since this site is within repeats (Fig. 2).
FIG. 4.

Binding of His-tagged RepN to a specific region in the repN gene as shown by agarose gel electrophoresis. Approximately 120 ng of the 1.5-kb EcoRI-HindIII fragment (nt 5730 to 7228) isolated from pSEQ243 was digested with the three restriction enzymes shown. The DNA fragments thus obtained and 0.9 μg of λ DNA were incubated with 0.8 μg of the His-tagged RepN protein, and analyzed by agarose gel electrophoresis. The experimental conditions are described in Materials and Methods. The numbers above and under the horizontal line indicate the distances between the restriction sites and their positions, respectively. The restriction map is not drawn to scale. Lanes: 1, size markers (φX174 DNA digested with HaeIII); 2, a digest of the 1.5-kb fragment with the three restriction enzymes; 3, the 1.5-kb DNA digest incubated with His-tagged RepN.
To study the RepN-binding sequence more precisely, we performed footprinting analysis using DNase I. As shown in Fig. 5A, the DNA regions extending from nt 6685 to 6810 and from nt 6685 to 6809 for the coding and noncoding strands, respectively, were protected from DNase I digestion. Previously, we reported that this region contains several tandem and inverted repeat sequences (36). Since pLS32 is a low-copy-number plasmid that uses a theta mechanism of replication, and many low-copy-number plasmids of E. coli (for example, P1, F, R1 and R6K) carry several tandem repeats (iterons) that are involved in replication, we reexamined whether the tandem and inverted repeat sequences could be arranged into a simple array of iterons. As a result, we identified five tandem repeats consisting of 22 nt by allowing various degrees of sequence mismatches (Fig. 2 and 5B). We noted that each iteron contained a partial inverted repeat and that there was a perfect inverted repeat of 11 nt between the 3′ half of IT4 and the 5′ half of IT5 (Fig. 5B), although the functional significance remains unknown.
Copy number control by RepN.
We next investigated whether the copy number of a repN-driven plasmid is affected by RepN protein levels. To do this, we constructed pHDCS2 in which the repN gene is placed downstream of the Pspac promoter that is inducible by the addition of IPTG. After the B. subtilis CU741 cells carrying pHDCS2 (Fig. 1) were grown in the presence of various concentration of IPTG, total DNA was isolated, cut with BamHI which cleaves pHDCS2 at a unique site, and subjected to agarose gel electrophoresis. As shown in Fig. 6, the plasmid content was found to be increased with increasing concentrations of IPTG. Quantification by reverse transcription-PCR revealed that the copy numbers of pHDCS2 was 0.45 and 400 per chromosome in the cells grown without and with IPTG (at 0.2 mM and 1.0 mM), respectively. It was noted that only 13% of the cells were Cmr in the cell population grown without IPTG, indicating that pHDCS2 are segregationally unstable. Taking the fraction of the Cmr cells into consideration, we estimated the copy number of the plasmid to be 3 to 4 per chromosome. On the other hand, all the cells cultured with IPTG (100 colonies tested) were resistant to Cm. These results show that overexpression of repN by the addition of IPTG at 0.2 mM or more results in an increase in the copy number by 100 fold.
It has been demonstrated that the copy number of iteron-containing E. coli plasmids such as P1, F, and RK2 does not increase even when the Rep protein levels were increased (7, 24, 30). This is explained by the handcuffing model, in which plasmids pair at the iterons through interactions between Rep proteins and thus are incapable of replication (1, 7, 9, 24, 30). Apparently, this mechanism does not apply for the replication machinery in pLS32.
Effect of iteron mutations on plasmid replication and copy number.
To investigate the role of the iterons in replication, we introduced sequence changes in the five iterons of pSEQ243 (Fig. 2) individually or in combinations as described in Materials and Methods and tested whether the mutations affected the replication ability of the host plasmid. The nucleotide sequence changes were introduced so that the amino acid sequence of RepN was not changed. It was found that the mutations in the first three tandem repetitions did not affect the transforming ability of the plasmids (pMU63, pDMU1, and pMT34) (Table 3), but further addition of sequence changes in either IT4 (pQMT11) or IT5 (pQMT51) abolished the transforming activity (Table 3). It was also found that the mutation in IT4 (pSMU19) or IT5 (pSEM2129) alone led to the inactivation of the transforming ability (Table 3). It is known that plasmids have to be multimers to transform B. subtilis strains (6). To avoid conformational variations among the plasmid preparations used above, we also performed transformation with plasmids that had been cleaved at a unique BamHI site and ligated, but identical results were obtained (data not shown). These results indicate that at least both IT4 and IT5 are essential for plasmid replication, although it is not known at present which nucleotides in the altered sequences are responsible for the functional loss of plasmid replication.
We realized in our routine work that the mutant plasmids pMU63, pDMU1, and pMT34 were obtained in larger amounts than the wild-type plasmid, pSEQ243, from B. subtilis cells. This prompted us to estimate the quantities of the mutant plasmids relative to that of pSEQ243 in the cell. The results showed that the copy number of pMU63 carrying the mutant IT1 was increased 2.9 fold compared to that of pSEQ243, whereasthe presence of both the IT1 and IT2 mutations in plasmid pDMU1 caused a further increase in copy number, amounting to 11.2 fold (Table 3). On the other hand, there was a 2.0-fold increase when the three iterons were mutated simultaneously (pMT3). It appears from these results that the first three iterons are involved in negative control of the plasmid copy number, with the first two together exerting a strong effect on replication efficiency.
Binding of RepN to DNA regions containing mutant iterons.
To examine a correlation between plasmid replication and the binding of RepN to the iterons, we performed a gel shift assay using His-tagged RepN and PCR-amplified 493-bp (nt 6634 to 7119) DNA fragments that contained different numbers of the wild-type and mutant iterons. Incubation of the His-tagged RepN protein with a PCR fragment carrying the five wild-type iterons caused a shift in the DNA band to an upper, broad region (Fig.7Aa). The DNA bands were also shifted to the upper region, although less extensively, when they carried mutations in IT1, both IT1 and IT2, and the three iterons IT1, IT2, and IT3 together (Fig. 7Ab to d). On the other hand, no shift band was detected with DNA fragments carrying additional mutations in IT4 and IT5 (Fig. 7Ae and f), indicating that the His-tagged RepN binds to a DNA region containing both intact IT4 and IT5.
FIG. 7.

Binding of His-tagged RepN to DNA fragments carrying different numbers of wild-type and mutant iterons (IT) (A) and to a DNA fragment containing the first three iterons (B). (A) Fifteen nanograms of the PCR fragments (nt 6634 to 7119) prepared with primers RepN4 and 6634Bio and mutant plasmids as templates were incubated with His-tagged RepN, followed by agarose gel electrophoresis as described in Materials and Methods. The wild-type and mutant iterons are represented by horizontal bars without and with a cross, respectively, and numbered from the 5′ ends. The arrow and arrowhead indicate the 486-bp fragment and λ DNA, respectively. The templates used were pSEQ243 (a), pMU63 (b), pDMU1 (c), pMT34 (d), pQMT11 (e), and pMAE5 (f). The triangles above the panels show the increment of His-tagged RepN. The amounts of His-tagged RepN added to each set of experiment were 0, 0.4 and 0.8 μg from left to right. The leftmost lane shows the bands of HaeIII-digested φX174 DNA used as size markers. The data shown were obtained in the same set of experiment, but electrophoresis was performed in a different gel in the same gel electrophoresis apparatus. (B) Digoxigenin-labeled DNA fragments from nt 6655 to 6771 containing IT1, IT2, and IT3 (left) and from nt 7047 to 7169 without a iteron (right) were obtained by using primer pairs EC6655 plus CL6771 and EC7047 plus RepN3, respectively, and pSEQ243 as a template. Ten nanograms of the PCR products was incubated as described in Materials and Methods and subjected to polyacrylamide gel electrophoresis. DNA bands were detected by the chemiluminescent detection method using CSPD as described in Materials and Methods. The labeled DNA fragments were prepared with digoxigenin-dideoxyUTP and terminal transferase using a DIG Gel Shift Kit obtained from Roche Diagnostics. The amounts of His-tagged RepN added were 0, 0.4, 0.6, 0.8, and 1.0 μg (from left to right).
The inability of pSMU19 and pSEM2129 to transform B. subtilis cells (Table 3) could have been caused by the inability of RepN to bind IT1 through IT3 without intact IT4 and IT5. It was found, however, that His-tagged RepN bound to a PCR fragment containing only iterons IT1 through IT3 (Fig. 7B, left), indicating that the protein can bind to IT1 through IT3 independent of IT4 or IT5. In the same experimental condition, the His-tagged RepN protein did not bind to a PCR fragment with a similar size that derived from a C-terminal part of repN (nt 7047 to 7169) (Fig. 7B, right). These results show that the binding of RepN to IT4 and IT5 but not to IT1 through IT3 is necessary for replication. We note that the banding patterns are different when DNA fragments with different number of iterons were used. This could be due to differences in overall conformations of the DNA-protein complexes or to a result of the binding of different number of His-tagged RepN molecules to various iterons. Anomalous banding patterns similar to those reported here were observed for the complexes between the wild-type and mutant sequences of plasmid P1 DNA and its Rep protein (4).
Incompatibility directed by iterons.
Two different plasmids with the same mode of replication or sharing the common partitioning system cannot coexist in the same cell, which is called incompatibility (inc) (for reviews, see references 2, 15, and 27). For some E. coli plasmids, incompatibility is exerted by a specific DNA region containing iterons (24, 30, 38). In an attempt to locate the replication origin (oriN) of pLS32, we tried to transform pHDCS2-carrying CU741 cells with derivatives of pSEQ243 that carry various regions of repN, but no transformant was obtained (data not shown). We presumed that this was due to strong incompatibility, and this notion prompted us to identify the sequences responsible for this activity. To locate the inc region, we inserted various regions of repN in the lowest possible copy number state, i.e., at the amyE locus in the chromosome, and tested whether the constructed strains could accommodate pSEQ243 specifying Nmr. Transformants thus obtained were grouped in three categories on the basis of colony morphology. In the cases where no transformant was observed on selection plates, the DNA region was classified as showing strong incompatibility, whereas when minute transformant colonies were formed but the cells in them could not form colonies upon restreaking on a fresh plate within 24 h, we termed the phenomenon intermediate incompatibility. When colonies similar in size to or slightly smaller than those of the transformants of strain SSM190 (a control strain with no repN sequence) were obtained, we designated these cells as showing no incompatibility. The three groups are designated as ++, +, and − for strong, intermediate, and no incompatibility, respectively (Fig. 8A). The DNA region from nt 6241 to 7119 containing the entire repN region showed strong incompatibility toward the incoming pSEQ243 (construct ISHI11) (Fig. 8A). Deletions extending from the 3′ end (nt 7119) to nt 6741 (ISHI12, -13, and -14) did not affect the strong inc property, whereas those up to nt 6726 (ISHI15) and nt 6702 (ISHI16) showed intermediate and no inc activities, respectively. Likewise, deletions from the 5′ end (nt 6241) to nt 6684 (ISHI21) or nt 6705 (ISHI22) showed strong inc activity, but a further deletion to nt 6736 (ISHI23) resulted in intermediate incompatibility. A deletion removing the first three iterons (ISHI24) did not show an inc activity. It appears from these results that the simultaneous presence of both IT1 and IT2 caused stronger incompatibility. We then examined the effect of two consecutive iterons on incompatibility. Combinations of IT1 and IT2 (ISHI31) and of IT2 and IT3 (ISHI32) showed strong and intermediate incompatibilities, respectively, whereas neither the combination of IT3 and IT4 (ISHI33) nor IT4 and IT5 (ISHI24) exhibited incompatibility. Thus, the intermediate activity shown by IT3, IT4, and IT5 together as observed in ISH23 suggests a cumulative effect of the iterons. The sequence alterations in both IT1 and IT2 (ISHI34) that are contained in pDMU1 abolished the inc activity, indicating that those iterons are indeed involved in incompatibility. In a separate experiment, we found that IT1 (nt 6686 to 6708) alone in ISH35 but not IT2 alone (nt 6719 to 6741) had a weak but significant inc activity (data not shown). These results together with the result of ISHI15 show that IT1 has the strongest inc activity among the five iterons. The difference in the inc activities in ISHI15 and ISHI35 might be due to the presence of extra nucleotides in ISHI15.
FIG. 8.

Incompatibility (A) and replication ability (B) shown by various regions of repN. The coding region of repN depicted in the box is from nt 6256 to 7116 with iterons (arrows) located from nt 6686 to nt 6810. The map is not drawn to scale. The small boxes indicate the A+T-rich region in the repN-coding region as defined in the legend to Fig. 2. (A) The host strains (ISHI11-ISHI34) carrying the respective regions at the amyE locus were made competent, and transformed with pSEQ243. ++, +, and − indicate strong, intermediate, and no incompatibility activities, respectively (for definitions, see the text). (B) Replication ability of pHDMAE21 carrying the DNA regions shown. + and − indicate the colony forming abilities of the plasmids on Cm-containing plates when transformed into CU741.
The results with the inc activities conferred by IT1, IT2, and IT3 (see, for example, ISHI13, -14, -15, -31, and -34), and those with the increase in the copy number of the plasmids containing mutations in those iterons (Table 3) show an inverse relationship between the inc activity and copy number, suggesting that the iterons are involved in the negative control of pLS32 replication. On the other hand, the replication efficiency was shown to depend on the concentration of RepN (Fig. 6). These results suggested that the incompatibility was caused by competition for RepN between the iterons embedded in the chromosome and the replication machinery on the RepN-driven plasmid pSEQ243. To test this possibility, we constructed pBECS21, a pHDCS2 derivative containing an Nmr gene. When the plasmid was transformed into ISHI12 and selected for Cm and Nm resistance on LB plates with and without 20 μM IPTG, transformants were obtained only on IPTG-containing plates. When the ISHI12 transformants and a control strain, SSM190 (without the iterons) carrying pBECS21, were streaked on LB plates containing various concentrations of IPTG, the former formed colonies only on plates containing IPTG at concentrations of 3.1 μM or more, whereas the latter formed colonies at all the concentrations tested (Table 4). These results suggest that higher concentrations of RepN in the cell overrode the incompatibility exerted by the iterons in the chromosome. It should be noted that when the same IT-containing DNA fragment present in ISH12 was placed on a multicopy plasmid, pUB110, and used for the host for pHDCS2, no transformant was obtained (data not shown), apparently indicating the multicopy effect of the inc-coding DNA region.
TABLE 4.
Effect of overproduction of RepN in pBECS21 on incompatibility exerted by the repN iteronsa
| IPTG (μM) | Strain
|
|
|---|---|---|
| ISHI12 (pBECS21)b | SSM190 (pBECS21) | |
| 0 | − | + |
| 1.6 | − | + |
| 3.1 | + | + |
| 12.5 | + | + |
| 50 | + | + |
Transformants of strains ISHI12 and SSM190 with pBECS21 were selected on LB plates containing Cm (5 μg/ml), Nm (15 μg/ml), and IPTG (20 μM); colonies formed were streaked on the same plates containing various concentrations of IPTG.
+ and −, colony formation and no colony formation, respectively.
In the case of the E. coli F plasmid, a sequence containing two out of the five iterons in the incC region showed strong incompatibility activity (38). In contrast to this and the incompatibility shown the repN iterons, two entire replicons could be maintained in the same cell in the case of plasmids pAD1, pSX267, and pSK41 that are grouped in the same family with pLS32 (11, 12, 23). Apparently the strong incompatibility shown by the repN iterons is an intrinsic nature of pLS32 and may be explained by competition for RepN between iterons on two replicons. This is in concert with the titration model presented for an iteron-containing broad-host-range plasmid R1162, in which an increase in the concentration of the cognate replication initiation protein RepC results in an increase in plasmid copy number and overcomes incompatibility caused by iterons (21).
Localization of replication initiation (oriN) region.
To localize the replication origin (oriN) we used plasmid pHDMAE21, apHDCS2 derivative in which all five iteron sequences have been changed by replacement with synonymous codons (Fig. 1; Table 2). This plasmid cannot replicate in B. subtilis, but specifies the intact RepN protein from the altered repN gene under the control of the Pspac promoter. We expected, therefore, that if a DNA sequence containing oriN is placed in pHDMAE21, it would initiate replication with a supply of RepN from the mutant repN gene on the same plasmid.
Various DNA regions in the repN gene were amplified by PCR and cloned in the EcoRI site of pHDMAE21 using E. coli JM103. The resultant recombinant plasmids were examined for the ability to transform B. subtilis cells to Cmr. Since pHDMAE21 is a derivative of pHDCS2 that can replicate without the addition of IPTG (see above), selection plates for transformants did not contain the drug. Plasmids pMAEC11 and pMAEC12 carrying the insert from nt 6705 to 6986 in different orientations could generate transformant colonies (Fig. 8B) whose appearances were indistinguishable. Restriction enzyme analysis showed that the plasmids obtained from the B. subtilis transformants were indistinguishable from those obtained from the E. coli hosts. These results indicate that oriN is functional in both orientations. The results shown below were those obtained by using plasmids containing the inserts in the same direction as that of the repN gene in pHDMAE21.
Plasmids with deletions from nt 6705 to 6771 gave transformants, but a further deletion to nt 6785 (pMAEC541), which removed IT4 but left IT5 intact, abolished the transforming activity (Fig. 8B). It should be noted that the transformant colonies with plasmid pMAEC60 were slightly smaller than those obtained with the plasmids carrying smaller deletions (pMAEC11 through pMAEC51), suggesting that the replication ability is partly affected by the deletion up to nt 6771, the sixth nucleotide in IT4 (Fig. 2). On the other hand, deletions from the 3′ end to nt 6868 (pMAEC39, pMAEC42, and pMAEC395) did not affect the transforming activity. However, the colonies obtained with pMAEC395 were smaller than those transformed with the other two plasmids carrying the same sequence at the 5′ ends. A further deletion to nt 6852 (pMAEC111) or nt 6824 (pMAEC824) resulted in inactivation of the transforming activity. The 3′ ends of pMAEC111 and pMAEC824 are within the A+T-rich sequence (Fig. 8B) (for the definition of the A+T-rich sequence, see the legend to Fig. 2), suggesting that this region is important for replication. From these results, we conclude that the region consisting of most of the 3′ end sequence of IT4, IT5, and the downstream A+T-rich sequence constitutes a core region for plasmid replication. The results that the deletion plasmids pMAEC60 and pMAEC395 gave rise to smaller colonies suggest that the replication efficiency is gradually lost by deletion from both ends. Thus, although definite boundaries for the oriN activity are not clear from these studies, it is possible that a minimum of 96 bp from nt 6772 to 6867 can serve as the replication origin of pLS32.
The repN gene in pHDMAE21 is under the control of the Pspac promoter as described above. When IPTG was added to the selection plates at a concentration of 0.1 mM to amplify RepN, transformation with pMAEC111 and pMAEC824 but not with pMAEC541 gave rise to small colonies (data not shown). Upon restreaking, they could form colonies on the same selective plates but not on plates without IPTG. These results suggest that the 5′ end of the core sequence is strictly required for replication perhaps for the binding of RepN but that the requirement of the A+T-rich sequence is rather relaxed and compensated for by a large supply of the RepN protein.
In E. coli plasmid R6K, one of the replication origins, ori-γ, contains seven iterons; removal of one does not affect replication, but a deletion of two reduces the efficiency of replication, and further deletion results in a failure of plasmid replication (22). In contrast, all five iterons are required in vivo in the case of P1 replication (40). It appears from these observations that iterons affect different levels of functional roles, i.e., negative and positive control of replication, the balance of which possibly determines the final copy number in the cell. By analogy with the model of replication of various plasmids and the E. coli chromosome presented by Bramhill and Kornberg (3), we suggest that the replication of pLS32 initiates by binding of RepN to IT4 and IT5 and the ensuing melting of the A+T-rich region. This would provide the structure for the subsequent steps in DNA elongation. During replication, the copy number may be maintained low by IT1 through IT3, probably because if the copy number of a large plasmid like pLS32 increased it would impose a metabolic burden on the cell.
Acknowledgments
We thank N. Kogiso and E. Kawachi for technical assistance.
This work was supported by a Grant-in-aid for Scientific Research (B) from the Ministry of Education, Science, Sports and Culture of Japan.
REFERENCES
- 1.Abeles, A. L., and S. J. Austin. 1991. Antiparallel plasmid-plasmid pairing may control P1 plasmid replication. Proc. Natl. Acad. Sci. USA 88:9011-9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin, S. J., and K. Nordstrom. 1990. Partition-mediated incompatibility of bacterial plasmids. Cell 60:351-354. [DOI] [PubMed] [Google Scholar]
- 3.Bramhill, D., and A. Kornberg. 1988. A model for initiation at origins of DNA replication. Cell 54:915-918. [DOI] [PubMed] [Google Scholar]
- 4.Brendler, T. G., A. L. Abeles, L. D. Reaves, and S. J. Austin. 1997. The iteron bases and spacers of the P1 replication origin contain information that specifies the formation of a complex structure involved in initiation. Mol. Microbiol. 23:559-567. [DOI] [PubMed] [Google Scholar]
- 5.Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. Xl1-blue: a high efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection. BioTechniques 5:376-379. [Google Scholar]
- 6.Canosi, U., A. Iglesias, and T. A. Trautner. 1981. Plasmid transformation in Bacillus subtilis: effects of insertion of Bacillus subtilis DNA into plasmid pC194. Mol. Gen. Genet. 181:434-440. [DOI] [PubMed] [Google Scholar]
- 7.Chattoraj, D. K. 2000. Control of plasmid DNA replication by iterons: no longer paradoxical. Mol. Microbiol. 37:467-476. [DOI] [PubMed] [Google Scholar]
- 8.del Solar, G., R. Giraldo, M. J. Ruiz-Echevarria, M. Espinosa, and R. Diaz-Orejas. 1998. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 62:434-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durland, R. H., and D. R. Helinski. 1990. Replication of the broad-host-range plasmid RK2: direct measurement of intracellular concentrations of the essential TrfA replication proteins and their effect on plasmid copy number. J. Bacteriol. 172:3849-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Firth, N., S. Apisiridej, T. Berg, B. A. O'Rourke, S. Curnock, K. G. Dyke, and R.A. Skurray. 2000. Replication of staphylococcal multiresistance plasmids. J. Bacteriol. 182:2170-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francia, M. V., S. Fujimoto, P. Tille, K. E. Weaver, and D. B. Clewell. 2004. Replication of Enterococcus faecalis pheromone-responding plasmid pAD1: location of the minimal replicon and oriV site and repA involvement in initiation of replication. J. Bacteriol. 186:5003-5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gehring, M., F. Gotz, and R. Bruckner. 1996. Sequence and analysis of the replication region of the Staphylococcus xylosus plasmid pSX267. Gene 182:117-122. [DOI] [PubMed] [Google Scholar]
- 13.Gruss, A., and S. D. Ehrlich. 1989. The family of highly interrelated single-stranded deoxyribonucleic acid plasmids. Microbiol. Rev. 53:231-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassan, A. K., S. Moriya, M. Ogura, T. Tanaka, F. Kawamura, and N. Ogasawara. 1997. Suppression of initiation defects of chromosome replication in Bacillus subtilis dnaA and oriC-deleted mutants by integration of a plasmid replicon into the chromosomes. J. Bacteriol. 179:2494-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helinski, D. R., A. E. Toukdarian, and R. P. Novick. 1996. Replication control and other stable maintenance mechanisms of plasmids, p. 2295-2324. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umberger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 16.Helmann, J. D. 1995. Compilation and analysis of Bacillus subtilis σA-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA. Nucleic Acids Res. 23:2351-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itaya, M., and T. Tanaka. 1997. Experimental surgery to create subgenomes of Bacillus subtilis 168. Proc. Natl. Acad. Sci. USA 94:5378-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janniere, L., A. Gruss, and S. D. Ehrlich. 1993. Plasmids, p. 625-644. In A. L. Sonenshein, J. A. Hoch, and R. Losick (ed.), Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. ASM Press, Washington, D.C.
- 19.Kearney, K., G. F. Fitzgerald, and J. F. Seegers. 2000. Identification and characterization of an active plasmid partition mechanism for the novel Lactococcus lactis plasmid pCI2000. J. Bacteriol. 182:30-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan, S. A. 1997. Rolling-circle replication of bacterial plasmids. Microbiol. Mol. Biol. Rev. 61:442-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim, K., and R. J. Meyer. 1985. Copy number of the broad host-range plasmid R1162 is determined by the amounts of essential plasmid-encoded proteins. J. Mol. Biol. 185:755-767. [DOI] [PubMed] [Google Scholar]
- 22.Kolter, R., and D. R. Helinski. 1978. Activity of the replication terminus of plasmid R6K in hybrid replicons in Escherichia coli. J. Mol. Biol. 124:425-441. [DOI] [PubMed] [Google Scholar]
- 23.Kwong, S. M., R. A. Skurray, and N. Firth. 2004. Staphylococcus aureus multiresistance plasmid pSK41: analysis of the replication region, initiator protein binding and antisense RNA regulation. Mol. Microbiol. 51:497-509. [DOI] [PubMed] [Google Scholar]
- 24.McEachern, M. J., M. A. Bott, P. A. Tooker, and D. R. Helinski. 1989. Negative control of plasmid R6K replication: possible role of intermolecular coupling of replication origins. Proc. Natl. Acad. Sci. USA 86:7942-7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meijer, W. J., A. J. de Boer, S. van Tongeren, G. Venema, and S. Bron. 1995. Characterization of the replication region of the Bacillus subtilis plasmid pLS20: a novel type of replicon. Nucleic Acids Res. 23:3214-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meijer, W. J., G. B. Wisman, P. Terpstra, P. B. Thorsted, C. M. Thomas, S. Holsappel, G. Venema, and S. Bron. 1998. Rolling-circle plasmids from Bacillus subtilis: complete nucleotide sequences and analyses of genes of pTA1015, pTA1040, pTA1050 and pTA1060, and comparisons with related plasmids from gram-positive bacteria. FEMS Microbiol. Rev. 21:337-368. [DOI] [PubMed] [Google Scholar]
- 27.Nordstrom, K. 1990. Control of plasmid replication: how do DNA iterons set replication frequency? Cell 63:1121-1124. [DOI] [PubMed] [Google Scholar]
- 28.Ogura, M., and T. Tanaka. 1996. Transcription of Bacillus subtilis degR is σD dependent and suppressed by multicopy proB through σD. J. Bacteriol. 178:216-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogura, M., and T. Tanaka. 1997. Transcription of Bacillus subtilis ComK negatively regulates degR gene expression. Mol. Gen. Genet. 254:156-165. [DOI] [PubMed] [Google Scholar]
- 30.Pal, S. K., and D. K. Chattoraj. 1988. P1 plasmid replication: initiator sequestration is inadequate to explain control by initiator-binding sites. J. Bacteriol. 170:3554-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saito, H., and K. Miura. 1963. Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim. Biophys. Acta 72:619-629. [PubMed] [Google Scholar]
- 32.Sambrook, J., E. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory PRess, Cold Spring Harbor, N.Y.
- 33.Shimotsu, H., and D. J. Henner. 1986. Construction of a single-copy integration vector and its use in analysis of regulation of the trp operon of Bacillus subtilis. Gene 43:85-94. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka. T., and M. Kawata. 1988. Cloning and characterization of Bacillus subtilis iep, which has positive and negative effects on production of extracellular proteases. J. Bacteriol. 170:3593-3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka, T., and T. Koshikawa. 1977. Isolation and characterization of four types of plasmids from Bacillus subtilis (natto). J. Bacteriol. 131:699-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka, T., and M. Ogura. 1998. A novel Bacillus natto plasmid pLS32 capable of replication in Bacillus subtilis. FEBS Lett. 422:243-246. [DOI] [PubMed] [Google Scholar]
- 37.Titok, M. A., J. Chapuis, Y. V. Selezneva, A. V. Lagodich, V. A. Prokulevich, S. D. Ehrlich, and L. Janniere. 2003. Bacillus subtilis soil isolates: plasmid replicon analysis and construction of a new theta-replicating vector. Plasmid 49:53-62. [DOI] [PubMed] [Google Scholar]
- 38.Tolun, A., and D. R. Helinski. 1981. Direct repeats of the F plasmid incC region express F incompatibility. Cell 24:687-694. [DOI] [PubMed] [Google Scholar]
- 39.Ward, J. B. Jr., and S. A. Zahler. 1973. Genetic studies of leucine biosynthesis in Bacillus subtilis. J. Bacteriol. 116:719-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickner, S., J. Hoskins, D. Chattoraj, and K. McKenney. 1990. Deletion analysis of the mini-P1 plasmid origin of replication and the role of Escherichia coli DnaA protein. J. Biol. Chem. 265:11622-11627. [PubMed] [Google Scholar]
- 41.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-109. [DOI] [PubMed] [Google Scholar]
- 42.Yansura, D. G., and D. J. Henner. 1984. Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 81:439-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida, K.-I., K. Kobayashi, Y. Miwa, C.-M. Kang, M. Matsunaga, H. Yamaguchi, S. Tojo, M. Yamamoto, R. Nishi, N. Ogasawara, T. Nakayama, and Y. Fujita. 2001. Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 29:683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]