


Abstract
Cyclin E, a G1 cyclin, is overexpressed and present in low molecular weight (LMW) isoforms in breast cancer cells and tumor tissues. In this study we have examined the possibility that the shortened mRNA splice variants could give rise to tumor-specific cyclin E LMW proteins. We used the Splice Capture method to identify, enumerate and isolate known spliced mRNAs and to look for previously undetected mRNA forms of cyclin E that might be translated into the LMW proteins. We show that a new splice variant of cyclin E found in tumor cells isolated by the Splice Capture strategy, named Δ48, activates CDK2 more robustly than full-length cyclin E when assayed from transiently transfected cells with the natural substrate GST-Rb. We also found the Splice Capture method to be superior to the conventional RNase protection assay in analyzing the cyclin E mRNA present in normal and tumor cells. Splice Capture enumerated the relative abundance of known forms of cyclin E mRNA and easily discovered new splice variants in both normal and tumor cells. We conclude that the abundance of cyclin E splice variants in cells may represent a novel form of regulation of cyclin E, and if translated they show altered substrate specificity compared to the full length form of cyclin E.
INTRODUCTION
Cell division is a precisely regulated process driven unidirec-tionally through key regulatory checkpoints by the timely expression of cyclins. The cyclins are synthesized and then destroyed, modulating the activity of cyclin-dependent protein kinases (CDKs). In contrast to cyclins, the CDKs remain at a constant level through the cell cycle (1). Active cyclin–CDK complexes phosphorylate substrates essential for commitment of the cell to pass through checkpoint barriers. The precisely timed proteolysis of cyclins is the most effective form of regulating CDK activity, ensuring that the cells move irreversibly through the cell cycle (2).
Cyclin E binds to and activates CDK2, initiating the processes required for the G1 to S phase transition (3). In dividing cells the expression of cyclin E increases to a maximum at the G1–S transition. The ability to enter S phase requires cyclin E or cells will arrest at S phase (3), defining the important role substrates phosphorylated by cyclin E–CDK2 have in promoting DNA synthesis. Cyclin E expression in tumor cells is different from normal cells. In tumor cells cyclin E is overexpressed, correlating with poor prognosis (reviewed in 4). This protein expression is constitutive rather than cell cycle regulated (5–7) and in addition to the typical 50 kDa protein, a ladder pattern of low molecular weight (LMW) isoforms is evident when examined by immunoblotting (8,9). These LMW forms are of interest to us since they occur only in tumor tissues or tumor cell lines. The tumor-specific pattern of expression of cyclin E is a complex mixture of alternative translation start sites, post-translational proteolysis and post-translational covalent modifications. Cyclin E pre-mRNA splicing may also be involved in the generation of its LMW forms specific to the tumor phenotype. In this report we describe the discovery of three new splice variants of cyclin E, examine their distribution between normal and tumor cell lines and analyze their functionality in transfection assays.
Six splice variants have been identified to date, including the 45 kDa form originally considered the wild-type (10,11). A form 15 amino acids longer than the wild-type, coding for a 50 kDa protein, is termed EL and is the predominantly expressed cyclin E protein (3), with some minor translation from the methionyl residue at position 46. Translation from the methionine at position 16 does not occur and therefore the 45 kDa form is not made in tumor cells (D.C.Porter and K.Keyomarsi, manuscript in preparation). Other forms of cyclin E described include ES, where the cyclin box is absent and which is unable to activate CDK2 in vitro (12), a 9 bp in-frame deletion in the 5′ domain of the message termed Δ9 (5), a splice variant containing a 135 bp deletion downstream of the cyclin box known as ET (13) and, finally, a 148 bp deletion causing a C-terminal frameshift found in the 3′ domain of the cyclin E mRNA termed Δ148 (5). Cyclin E, EL, ET, Δ9 and Δ148 can all bind to and activate CDK2 in vitro using histone H1 as substrate (3,5,10,11,13). We have also observed that four N-terminal truncations, including one near the cyclin box, activate CDK2 (8). This suggests that subtle changes in the cyclin E protein resulting from splice variants are not intended to eliminate kinase activity.
In order to quantify how alternative splicing could generate the LMW forms of cyclin E observed only in tumor cells, we used Splice Capture to identify, enumerate and isolate splice variants of cyclin E. This strategy uses a combination of a reverse transcription (RT)–PCR library for ‘capture’ followed by a PCR product restriction map to ‘identify’ the new splice variants. This method is amenable to high throughput screening and has led to the discovery of three new splice variants and a previously unknown phenomenon where multiple splice variants occur in a single cyclin E mRNA. Alternative splicing of cyclin E pre-mRNA is an active process and appears to occur in both normal and tumor cells as a normal aspect of cyclin E expression. We have also carefully examined the cyclin E mRNA from normal and tumor cell lines using an RNase protection assay in order to quantitate mRNA levels of the major cyclin E transcripts. The RNase protection assay is also useful for detecting splice variants and the efficiency in that respect is compared to Splice Capture. Lastly, we show that one of the novel splice variants of cyclin E identified is biochemically active in transfected cells and, if translated, could account for one of the LMW forms of cyclin E found in tumor cells.
MATERIALS AND METHODS
Materials, cell lines, culture conditions and transfections
Serum was purchased from Hyclone Laboratories (Logan, UT) and cell culture medium from Life Technologies (Grand Island, NY). All other chemicals used were reagent grade. The culture conditions for the normal 76N cell strain, immortalized MCF-10A cell line and breast cancer MCF-7, MDA-MB-157, MDA-MB-231 and MDA-MB-436 cell lines have been described previously (6). The 76N-E6 cell line (a gift from Dr V. Band, Tufts Medical Institute, Boston, MA) was immortalized and cultured as described previously (14,15). All cells were cultured and treated at 37°C in a humidified incubator containing 6.5% CO2 and maintained free of mycoplasma as determined by Hoechst staining (16). Cells were transfected using FuGENE Transfection Reagent (Roche, Indianapolis, IN) as per the manufacturer’s recommendations. Briefly, cells were plated at 5 × 105 cells/100 mm plate. Forty-eight hours following the initial plating of cells, 1 ml of serum-free medium containing 10 µl of the FuGENE Transfection Reagent mixed with 5 µg of DNA was added to each plate. Twenty-four hours following addition of the DNA/FuGENE mix, cells were harvested and subjected to western blot or kinase assay as described below.
Western blotting, immunoprecipitation and H1 kinase analysis
Cell lysates were prepared and subjected to western blot analysis as previously described (17). Briefly, 50 µg of protein from each condition was electrophoresed in each lane of a 10% SDS–PAGE gel and transferred to Immobilon P overnight at 4°C at 35 mV constant voltage. The blots were blocked overnight at 4°C in Blotto (5% non-fat dry milk in 20 mM Tris, 137 mM NaCl, 0.25% Tween, pH 7.6). After six 10 min washes in TBST (20 mM Tris, 137 mM NaCl, 0.05% Tween, pH 7.6) the blots were incubated in primary antibodies for 3 h. Primary antibodies used were cyclin E HE-12 monoclonal (Santa Cruz Biochemicals, Santa Cruz, CA) at 1 µg/ml and anti-FLAG polyclonal antibody (Santa Cruz Biotechnology) at 0.25 µg/ml. All dilutions were made in Blotto. Following primary antibody incubation the blots were washed and incubated with the appropriate goat anti-mouse or anti-rabbit horseradish peroxidase conjugate at a dilution of 1:5000 in Blotto for 1 h and finally washed and developed with the Renaissance chemiluminesence system as directed by the manufacturer (NEN Life Sciences, Boston, MA).
For immunoprecipitation a polyclonal antibody to cyclin E (A. Koff) or to FLAG (Santa Cruz Biotechnology) was incubated with protein A–Sepharose and chemically cross-linked to the protein A–Sepharose with dimethyl pimelimidate dihydrochloride (Pierce) according to a published method (18). The cross-linked antibody–protein A–Sepharose was incubated with 200 µg total cell extract, washed, boiled in reducing SDS sample buffer and analyzed on a 13% SDS–PAGE gel, followed by western blot analysis with anti-cyclin E antibody.
For histone H1 or GST-Rb kinase assay 250 µg cell extract were used per immunoprecipitation with polyclonal antibody to FLAG in lysis buffer containing 50 mM Tris buffer, pH 7.5, 250 mM NaCl, 0.1% NP-40, 25 µg/ml leupeptin, 25 µg/ml aprotinin, 10 µg/ml pepstatin, 1 mM benzamidine, 10 µg/ml soybean trypsin inhibitor, 0.5 mM PMSF, 50 mM NaF and 0.5 mM sodium orthovanadate. The protein/antibody mixture was incubated with protein A–Sepharose for 1 h and the immunoprecipitates were then washed twice with lysis buffer and four times with kinase buffer (50 mM Tris–HCl, pH 7.5, 250 mM NaCl, 10 mM MgCl2, 1 mM DTT and 0.1 mg/ml BSA). The immunoprecipitates were then incubated with kinase assay buffer containing 60 µM cold ATP and 5 µCi [32P]ATP in a final volume of 50 µl at 37°C for 30 min. The products of the reaction were then analyzed on a 13% SDS–PAGE gel. The gels were then stained, destained, dried and exposed to X-ray film. For quantitation the protein bands corresponding to histone H1 or GST-Rb were excised and the radioactivity of each band was measured by Cerenkov counting.
RT–PCR library formation and PCR analysis
Total RNA from normal and tumor cell lines was prepared as described (19) and pelleted through CsCl. mRNA was prepared by binding to oligo(dT) attached to magnetic beads using a kit designed for this purpose (Promega). The mRNA was reverse transcribed with Superscript Reverse Transcriptase (Gibco BRL) and oligo(dT) as primer. The RT product was PCR amplified with the oligonucleotide primers UFCE (CTCCTCGAGATCATGCCG AGGGAGCGCAGGGA) and CE1247 (AGCAAACGCACGCCTCCGCTGCAACA) using 2 µl of RT product and Taq polymerase in the buffer provided, for 35 cycles of 94°C for 30 s, 70°C for 60 s and 60°C for 72 s. The PCR products were ligated into the T/A cloning vector pCRII with T4 ligase (Promega), transformed into DH10B, plated onto ampicillin and colonies selected by nylon filter lift using a 32P-labeled (Random Primer Kit; Boehringer Mannheim) cyclin EL probe. Plasmids were prepared from positive colonies and used as templates for a second PCR reaction using the same oligonucleotides as above. The PCR reaction was performed with 10 ng template using Taq polymerase in the buffer provided, for 18 cycles of 95°C for 30 s, 65°C for 20 s and 72°C for 30 s. The PCR product was then digested with the restriction enzyme Sau3AI and desalted on a G-50 Sephadex spin column. The restriction digest was separated on a 5% PAGE gel with TAE buffer and the restriction fragments visualized with ethidium bromide. Plasmids representing each variation in restriction pattern were selected and the inserts were sequenced to confirm identity. RT–PCR (as above) products from cDNA derived from the cell lines MCF-10A, 76N, MDA-MB-157 and MDA-MB-436 were gel purified and used as template in four separate PCR reactions using oligonucleotides specific for four of the known splice variants. The oligonucleotides used were: for EL, UFCE and CE1247 (see above); for Δ48, C3FOR (TCATGCCGAGGGAGCGCAGGGAGT) and CE1247; for Δ97, C31FOR (CGCAGGGAGCGCAGGGAGCGGAT) and CE1247; for ET, UFCE and ETREV (CCAGGACACAGAGATCCAACAGCTTC). The PCR reactions were performed with 10 ng template using Taq polymerase in the buffer provided, for 18 cycles of 95°C for 30 s and 80°C for 120 s for each splice variant except the ET form, where the PCR conditions were performed with 10 ng template using Taq polymerase in the buffer provided, for 18 cycles of 95°C for 30 s and 75°C for 120 s.
RNase protection assay
32P-labeled antisense RNA probes were prepared with a Maxiscript kit from Ambion according to the manufacturer’s directions using T7 RNA polymerase, 1 µg plasmid template and [32P]CTP. After probe synthesis the template DNA was digested with RNase-free DNase and gel purified. DNA template was prepared for probes 1–5 using the oligonucleotide primers shown below. The oligonucleotides were used for PCR to create T7 RNA polymerase templates of ∼300 bp segments of cyclin E cDNA with a T7 promoter on the bottom strand. The same conditions were used as for plasmid amplification in the RT–PCR library construction. The PCR products were ligated into pCRII vector, transformed into DH10B, colonies selected and plasmids in the same orientation were prepared as described. The plasmids were linearized with HindIII prior to use as T7 polymerase template. The RNase protection assay was performed with an RPA II kit from Ambion. The streamlined procedure was as follows. Total RNA from various cell lines was prepared as described for RT–PCR libraries (19). Then 10 µg total RNA in 10 µl were hybridized to 5 µl of 32P-labeled antisense probe. The probe was also hybridized to serially diluted full-length cyclin E sense RNA as a standard curve. A full-length sense RNA was prepared using the Maxiscript kit from Ambion, quantitated by ethidium staining in comparison to a known standard RNA. After hybridization for 15 h the samples were digested with RNase, precipitated and separated on a 5% TBE polyacrylamide gel. The gel was dried, exposed to film and the radioactive bands cut from the gel and Cerenkov c.p.m. measured. The probes used were as follows: probe p1B, L1 (GGGATGCGAAGGAGCGGGACA) and 271+T7 (GGATCCTAATACGACTCACTATAGGGAGGATTTGCCCAGCTCAGTACAGG); probe p3B, 184 (ACACCTGACAAAGAAGATGATGAC) and 523+T7 (GGATCCTAATACGACTCACTATAGGGAGGTAAAGATGAAATCCCAATAAG); probe p4H, 484 (ATGGCGACACAAGAAAATGTTGTA) and 823+T7 (GGATCCTAATACGACTCACTATAGGGAGGATAAGGAAATTCAAGGCAGTC); probe p11B, 784 (CAGATTGCAGAGCTGTTGGATCTC) and 1247+T7 (GGATCCTAATACGACTCACTATAGGGAGGAGCAAACGCACGCCTCCGCTGCAACA); probe p7B, L1 (GGGATGCGAAGGAGCGGGACA) and 523+T7 (GGATCCTAATACGACTCACTATAGGGAGGTAAAGATGAAATCCCAATAAG).
RESULTS
Cyclin E LMW forms are predominant in breast cancer cells
The characteristic pattern of cyclin E found in tumor cells and tumor tissues includes a number of LMW forms. The forms represent N-terminal truncations since the monoclonal antibody (HE12) used to detect them on a western blot is directed toward a C-terminal peptide epitope (residues 318–338) (20). The mass of these LMW proteins calculated from SDS–PAGE (45–33 kDa) suggests that they contain an intact cyclin box domain and should bind and activate the cyclin E kinase partner CDK2 (8). Figure 1 shows normal and tumor cell extracts and immunoprecipitation of CDK2 from the same extracts, analyzed by cyclin E western blot, indicating which forms of cyclin E bind to and immunoprecipitate with CDK2. The western blot of total tumor cell extract in Figure 1, lane 3, is an example of the full complement of LMW forms that are typically found in tumor cells and tissues. Many of the LMW forms of cyclin E from tumor cells immunoprecipitate with CDK2 (Fig. 1, lane 4) yet these are barely detectable in normal cells (lanes 1 and 2). In normal cells the 50 kDa form is the predominant cyclin E binding to CDK2 (lanes 7 and 8). We have recently shown that in vitro synthesized truncations of cyclin E which bracket the molecular weight range of those found in tumor cells are able to bind and activate CDK2 (8).
Figure 1.
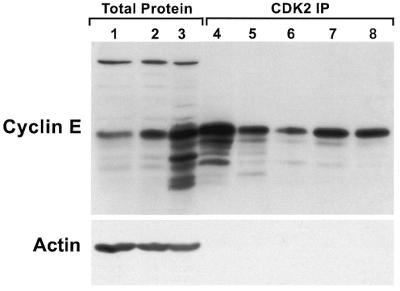
Cyclin E LMW forms bind to and immunoprecipitate with CDK2. Protein was extracted from the 76N normal (lanes 1 and 7), MCF-10A normal (lanes 2 and 8), MDA-MB-157 cancer (lanes 3 and 4), MDA-MB-436 cancer (lane 5) and MCF-7 cancer (lane 6) cell lines. For western blotting 50 µg total protein was loaded (lanes 1–3). Analysis with anti-actin antibody reveals equal loading of total protein. For CDK2 immunoprecipitation (lanes 4–8) 200 µg total cell extract were incubated with a polyclonal antibody to CDK2 cross-linked to protein A–Sepharose. The immune complexes were washed, boiled in reducing sample buffer and western immunoblotted alongside the total cell extracts.
Novel cyclin E splice variants in normal and tumor cells
To identify the cyclin E forms found in tumor cells we examined the possibility that shortened mRNA splice variants could give rise to the cyclin E LMW proteins. We used an RT–PCR-based assay to enumerate the relative abundance of the known forms of cyclin E mRNA and search for new splice variants (Fig. 2). For these studies poly(A)+ mRNA from the 76N normal cell strain and MDA-MB-157 tumor cell line was prepared and used to create RT–PCR plasmid libraries in Escherichia coli. Thirty-one tumor and 33 normal cell line derived clones containing cyclin E cDNAs were picked from the libraries and plasmids isolated. The plasmids were used as templates in a PCR reaction using the oligonucleotides from the RT–PCR step, amplifying only the cDNA sequences without interference from the plasmid sequence. The PCR products were digested with a single restriction enzyme, Sau3AI, separated by electrophoresis on a 5% acrylamide gel and stained with ethidium bromide. This strategy distinguishes the various known cyclin E splice variants in each cell line examined (Fig. 2). A clone of each splice variant of cyclin E was also sequenced to confirm our restriction mapping accuracy. Figure 2A shows the restriction digest of representative clones found in normal and tumor cells. Figure 2B indicates the frequency of each cDNA variant in normal and tumor cells. The results of this library analysis reveal that the EL form is the dominant mRNA in normal and tumor cells. Three previously discovered splice variants were detected, including the Δ9, ET and ES forms. There are novel splice variants found in the cell lines in addition to the known forms. The novel cyclin E splice variants identified were: Δ48, with 48 bp lost from position 22 to 69; IN3, with 190 bp from intron 3 which was not spliced (21); Δ97, with 97 bp lost from position 24 to 120. While Δ48 can be translated into protein products (see Fig. 5) the others could not, due to frameshifts (an unspliced variant IN3 and the Δ97 variant). In addition to these novel splice variants, we also identified cDNAs containing more than one splice variation in a single cDNA. An example is shown in Figure 2A, lane 3, where a Δ9 splice variant and the ES splice variant are present in the same cDNA. With this method, Splice Capture, we found seven of 64 examples where more than one splice variation occurred in a single cDNA.
Figure 2.

RT–PCR library analysis shows an equal representation of cyclin EL and its splice variants in normal and tumor cells. RT–PCR was performed on normal (76N) and tumor (MDA-MB-157) mRNA using 3′ and 5′ flanking oligonucleotides to the cyclin EL cDNA sequence. The PCR products representing a sample of the cyclin E mRNA species were then cloned into the PCR vector, transformed into E.coli, plated, filter lifted, probed with the 32P-labeled cyclin E probe, positive colonies picked, plasmids prepared, PCR performed again to create a clean product for restriction digest with Sau3AI, electrophoresed on 5% acrylamide gels and stained with ethidium bromide. (A) The gel represents the different cyclin E splice variants observed in both normal and tumor cells and the identities were confirmed by sequencing. MW, 100 bp ladder; lane 1, pLXSN-EL plasmid containing the EL form of cyclin E; lane 2, EL, cDNA identical to pLXSN-EL; lane 3, Δ9 + ES, cDNA containing the Δ9 splice variant in combination with the ES splice variant; lane 4, ET, with 135 bp lost from position 706 to 840 (gccctt…gcagag); lane 5, Δ48, with 48 bp lost from position 22 to 69 (cgggat…gcggag); lane 6, Δ9; lane 7, IN3, with 190 bp of genomic sequence from intron 3 which failed to be spliced (bases 1999–2190 of the genomic sequence, accgttGTGAGT…TGTTAGtttttg) (21); lane 8, Δ97, with 97 bp lost from position 24 to 120 (ggatgc…ttgcag). The numbers to the right of the gel identify the length in bp of the Sau3AI restriction fragments from the EL cDNA PCR product. (B) The occurrence of each individual variation is shown in normal (76N) and tumor (MDA-MB-157) cells. The number in parentheses indicates the total number of clones examined for each cell line. The ET and Δ48 variants contain an open reading frame and could be translated into a cyclin E protein. IN3 and Δ97 contain a frameshift precluding complete translation from the M1 start. Some cDNAs contained more than one splice variation and these were counted individually.
Figure 5.
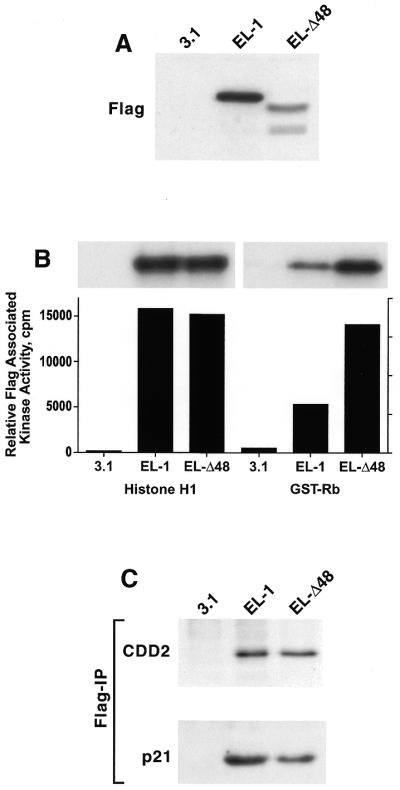
The Δ48 cyclin E splice variant is functionally active. 76N-E6 cells were transiently transfected with cyclin EL–FLAG, Δ48–FLAG or the vector backbone (pCDNA3.1) using the FuGENE reagent. Twenty-four hours following transfection cells were harvested and cell lysates were prepared and subjected to (A) western blot analysis, (B) FLAG kinase analysis and (C) FLAG immune complex formation. For western blot analysis 50 µg protein extract from each condition were analyzed by western blotting with the anti-FLAG antibody. The blot was developed with chemiluminescence reagents. For FLAG kinase activity equal amounts of protein (250 µg) from cell lysates were prepared for each condition and immunoprecipitated with anti-FLAG antibody (polyclonal) coupled to protein A beads, using histone H1 or GST-Rb as substrate. For each condition we show the resulting autoradiogram of the histone H1 and GST-Rb SDS–PAGE and the quantitation of the histone H1 and GST-Rb associated kinase activities by Cerenkov counting. For immunoprecipitation followed by western blot analysis equal amounts of protein (250 µg) from cell lysates prepared for each condition were immunoprecipitated with anti-FLAG (polyclonal) antibody coupled to protein A beads and the immunoprecipitates were subjected to western blot analysis with CDK2 and p21 antibodies.
The type or frequency of the splice variants relative to the EL form does not differ significantly between normal and tumor cell lines except for Δ48 (IN3 and Δ97 both contain a frameshift at the 5′-end; see Fig. 2 legend). The Δ48 variation, which represents 10% of the total mRNA coding for cyclin E, would be translated into a cyclin E molecule of predicted mass 44.8 kDa, making it a candidate for one of the LMW forms of cyclin E (see Figs 1 and 5). Furthermore, the Splice Capture analysis shows that the most abundant cyclin E mRNA codes for the full-length (EL) form in both normal and tumor cells, since ∼50% of all cyclin E mRNA codes for the EL form. The next most common splice variant is Δ9, occurring in six of 33 normal mRNAs and seven of 31 tumor mRNAs.
Appearance of alternatively spliced forms of cyclin E in normal and tumor cells
We compared the effectiveness of Splice Capture in characterizing the mRNA present in normal and tumor cells to the RNase protection analysis of cyclin E mRNA (Fig. 3). RNase protection analysis is quantitative and more sensitive than northern blotting and detects small sequence variations, including splice variants.
Figure 3.

Expression of cyclin E mRNA in breast cancer cell lines. (A) Schematic representation of the anitsense RNA probes within the cyclin E cDNA used to quantitate cyclin E mRNAs by RNase protection assay in normal (76N and MCF-10A) and tumor (MCF-7, MDA-MB-157, MDA-MB-231, MDA-MB-436 and ZR75T) cells. (B) RNase protection assay with antisense probe 3B in normal and tumor cells. The numbers represent copies of mRNA per cell as quantitated after gel electrophoresis by Cerenkov counts of the protected 32P-labeled antisense RNA probe (± SD). (C) Four overlapping antisense RNA probes were used to quantitate the amount of mRNA and look for the presence of splice variants in MDA-MB-157 cells. (D) Autoradiograph of an RNase protection assay using RNA from a variety of cell lines and the antisense RNA probe 7B. Probe 7B extends the range of protected mRNA (see Fig. 2A) and detects the presence of a smaller protected fragment, possibly due to a low abundance splice variant. This pattern was similar in all cell lines examined.
Figure 3A schematically depicts the full-length cyclin E target mRNA and the relative locations of the RNA probes 1B, 3B, 4H, 11B and 7B complementary to the mRNA sequence. We used probe 3B to quantitate cyclin E mRNA levels in seven human breast cell lines (two normal and five tumor) (Fig. 3B) and three additional overlapping antisense probes to examine the entire coding domain of cyclin E in the tumor cell line MDA-MB-157 (Fig. 3C). When probes mismatch the target mRNA they also serve as a method to scan for splice variants (Fig. 3D). The results of the quantitation (Fig. 3B) show that there are between two and three copies of cyclin E mRNA per cell in asynchronous normal breast epithelial cells (76N) and an immortalized breast cell line (MCF-10A). Breast tumor cell lines exhibited a range of cyclin E mRNA expression from as low as 1 copy/cell (MCF-7 and ZR75T) to 4 copies/cell (MDA-MB-231) to 8 copies/cell (MDA-MB-436) to 43 copies/cell in MDA-MB-157. Human β-actin was quantitated by RNase protection analysis in each preparation of total RNA as a control and was present consistently at ∼3000 copies/cell regardless of cell type.
When the cyclin E mRNA from the tumor cell line MDA-MB-157 was quantitated using four overlapping probes scanning the full-length coding sequence, one RNase protection analysis probe specific for bases 190–523 is over-represented 4-fold when compared to the rest of the mRNA (Fig. 3C). This part of the mRNA includes the cyclin box coding domain of the cyclin E protein. When a longer probe (p7B) was used in the RNase protection analysis (bases 25–523, spanning three known splice variants) two distinct protected fragments were detected, one representing the full-length mRNA and a second weaker protected fragment ∼50–100 bases shorter (Fig. 3D). This shorter cyclin E could represent the cyclin ES mRNA or a new splice variant containing only a short segment of the known coding sequence for cyclin E. This form could escape detection by Splice Capture if it lacks the 5′ sequence needed in the PCR step. The data presented also indicate that both normal and tumor cells express the alternatively spliced variants of cyclin E as detected by RNase protection analysis.
RT–PCR detects novel splice variants in both normal and tumor cell lines
From our screening for splice variants it appeared that the expression of some mRNA species reflected a bias between normal and tumor cells. For example, we found three cases of the Δ48 form of cyclin E mRNA which only occurred in tumor cells. In order to explore the normal and tumor expression of this and other new splice variants we identified in this study in a larger sample size, we used unique oligonucleotide primers designed to PCR amplify a single specific splice variant form of cyclin E cDNA. Selected splice variants were examined by RT–PCR using cDNA made from normal and tumor cell lines. The Δ9 splice variant described previously exists in both normal and tumor cells as shown by RT–PCR (5). Figure 4 shows that the Δ48, Δ97 and ET splice variant-specific oligonucleotides are able to PCR amplify a product in two normal and two tumor human breast cell lines. Figure 4A shows the results from the control PCR amplification of the EL form. The oligonucleotides used for the EL form flank all described splice variants and will amplify all cyclin E cDNAs we have described. The PCR amplification of the EL form shows that the only detectable product is full length, confirming that in a mixed population of cDNAs the predominant form is EL in normal and tumor cell lines. Figure 4A and B shows that mRNA (converted to cDNA) for Δ48, Δ97 and ET, respectively, are also present in the MCF-10A (normal/immortal), 76N (normal), MDA-MB-157 (tumor) and MDA-MB-436 (tumor) cell lines. This demonstrates that the large variety of spliced forms of cyclin E found in both normal and tumor cells reflect an ongoing natural process of splicing largely unaffected by the tumor phenotype.
Figure 4.
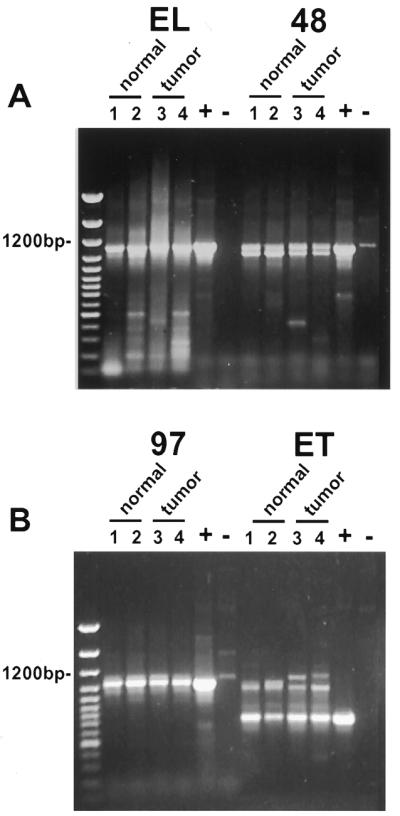
General expression of novel splice variants of cyclin E in normal and tumor breast cells. RT–PCR amplification of the cyclin E coding sequence from normal and tumor-derived breast epithelial cell lines using oligonucleotides UFCE and CE1247 for EL, C3FOR and CE1247 for Δ48, C31FOR and CE1247 for Δ97 and UFCE and ETREV for ET. The cell lines used were as follows: lane 1, MCF-10A; lane 2, 76N; lane 3, MDA-MB-157; lane 4, MDA-MB-436. Normal cells are represented in lanes 1 and 2 and tumor-derived cell lines in lanes 3 and 4. + indicates a positive control using vectors with the appropriate splice variant cDNA inserts as template. – indicates a negative control using the EL vector as template. The negative control for EL was an empty vector. The first lane of each gel shows the 100 bp ladder molecular weight size markers. Note that the only difference observed between normal and tumor cells was the detection of slower migrating ET primed RT–PCR products which were tumor specific and distinguishable from the ET products based on size.
The Δ48 splice variant can give rise to an activated LMW form of cyclin E
We next investigated whether the Δ48 cyclin E protein product is functional and biochemically active in mammary epithelial cells. For these experiments we engineered two cyclin E constructs with the FLAG epitope sequence at the C-terminus. These constructs included cyclin EL–FLAG (described previously; 8) and Δ48–FLAG. Immortalized 76N-E6 mammary epithelial cells were used in transient transfection assays using these two constructs (Fig. 5). The endogenous level of cyclin E in this cell line is very low (data not shown), making it an ideal system to examine the biochemical effects of overexpression of epitope-tagged Δ48 cyclin E. Following transfection of 76N-E6 with the indicated cyclin E–FLAG constructs, expression and activity of the resultant protein products were assessed using western blot analysis with anti-FLAG antibody (Fig. 5A), histone H1 and GST-Rb phosphorylation (Fig. 5B) and immune complex formation with CDK2 (Fig. 5C), respectively. Histone H1 and GST-Rb were used as substrates for active cyclin E–CDK2 complexes in immunoprecipitates prepared with an antibody to FLAG (Fig. 5B). This analysis revealed that while both cyclin EL–FLAG and Δ48–FLAG stimulate CDK2 equally well to phosphorylate the substrate histone H1, the expressed Δ48–FLAG construct transiently activates CDK2 with greater activity toward the substrate GST-Rb. (Fig. 5B). The kinase activity associated with each construct was proportional to the amount of FLAG protein product expressed with the exception of the higher activity associated with Δ48–FLAG and GST-Rb phosphorylation (Fig. 5A) (e.g. compare the expression versus associated kinase activities of EL to Δ48). Immune complex formation of cyclin E–FLAG to CDK2 reveals that both cyclin EL and Δ48 bind equally well to CDK2 (Fig. 5C). An explanation for the higher Δ48–FLAG-dependent kinase activity against GST-Rb is the reduced amount of the CDK2 inhibitor p21 present in Δ48–FLAG immune complexes (Fig. 5C). We conclude that the novel Δ48 splice variant of cyclin E stimulates a greater kinase activity towards GST-Rb than the full-length cyclin EL–FLAG.
DISCUSSION
The array of low molecular weight forms of cyclin E with established prognostic value (9,22–26) arise from a range of cellular activities, including partial proteolysis, alternative translation starts and post-translational modification. The crucial role of cyclin E in normal cell division and the importance of the LMW forms in breast tumor prognosis invites the exploration of how the LMW forms of cyclin E arise and what impact these isoforms have on the cell cycle. In this paper we have explored the role of splicing in the creation of tumor-specific LMW forms. More precisely, whether cyclin E pre-mRNA is spliced differently in normal and tumor cells and whether such a difference could account for the dominant forms of cyclin E detected by immunoblot of tumor cell extracts.
The documented splice variants (3,5,12) demonstrate a propensity for the cyclin E primary transcript to be spliced into many final mRNA forms. Utilizing the Splice Capture library screening process we have found three new splice variations of cyclin E and show evidence that combinations of splice variants exist in populations of mRNA from both normal and tumor cells. Most of these variants occur at a low frequency, however, the ET and Δ9 forms are shown to have a surprisingly high frequency. The Δ9 form of cyclin E may have a specific function since it codes for an active cyclin E and represents 20% of cyclin E mRNA in both normal and tumor cells. For example, we have recently shown that Δ9 splicing removes a peptide sequence in a domain critical for N-terminal proteolysis by a nuclear elastase-like activity (D.C.Porter and K.Keyomarsi, manuscript in preparation). The abundance of splice variants includes a number of frameshifts not translated into protein and proteins with missing blocks of amino acid sequence. We suspect that the Δ9 form of cyclin E may represent the focus of a cell cycle-regulated splicing. The purpose of this variation could be to alter its susceptibility to proteolysis or affect cyclin E–CDK2 substrate specificity. Clearly, the splicing fidelity of cyclin E is less than perfect, perhaps due to the extra demands of programmed alternative splicing. Much of splicing activity, including Δ9, Δ48, Δ97 and IN3, is focused around the exon 2–3 and 3–4 splice sites (21). Cyclin E may even have feedback regulation involving splicing since the ET form is cell cycle regulated (13).
The profusion of cyclin E splice variants in cells may represent a novel form of regulation of cyclin E, affecting its functionality. For example, in addition to proteolytic stability, substrate specificity may be altered by splicing. We have shown that the Δ48 splice variant promotes greater kinase activity toward GST-Rb (Fig. 5), a substrate whose phosphorylation state regulates E2F activity. This change in activity toward GST-Rb may be modulated by the reduced binding of p21 to the Δ48–CDK2 complex. As more substrates for cyclin E–CDK2 are discovered, the role of splice variants in substrate selection will be clearer.
Splicing may also alter the subcellular or subnuclear localization of cyclin E. A change in localization of cyclin E could have profound affects on the ability of cyclin E–CDK2 to phosphorylate substrates. Specifically, cyclin E associates with and phosphorylates SAP155, a component of the SF3 splicing factor (20). Only the phosphorylated form of SAP155 is found in functioning spliceosomes (27). However, the association of SAP155 and cyclin E is not cell cycle dependent, yet cyclin E expression and activity is cell cycle regulated, peaking at G1/S. If phosphorylation of SAP155 by cyclin E modulates the splicing selectivity of cyclin E, then cyclin E pre-mRNA may be spliced differently as cyclin E–CDK2 kinase activity increases during the G1 to S transition, effecting a feedback form of regulation. In addition, the cyclin E subcellular localization has not been fully characterized. The active spliceosome has a characteristic speckled appearance in the nucleus (28) and the spliceosome association previously reported would suggest that cyclin E could have a specialized localization determined by its protein structure. If the protein structure of cyclin E is varied by mRNA splicing then another potential form of feedback regulation might occur. There is a precedent for this possibility as cyclins B1 and B2 have different patterns of subcellular localization through mitosis, which presumably affects the function of MPF (29,30). While splice variants of cyclin B have not been described, the notion of subcellular localization which affects cyclin function is important.
It is not clear whether all the cyclin E splice variants give rise to protein products. Our data shows that only 50% of the cyclin E mRNA is of the EL 50 kDa form. The other forms, particularly those in the correct reading frame, must play an as yet undiscovered role in regulating the cell cycle. These mRNAs certainly may be translated into protein in tumor, but not normal, cells and contribute to the pattern of LMW forms seen on immunoblots. Furthermore, the translated protein products arising from these splice variants are also functionally active (see Fig. 5). In addition to the translated splice variants, we have also identified mRNAs that are frameshifted and cannot be translated into functional cyclin E. We suspect that the abundance of frameshifted forms reflects the error rate of an active splicing process used to generate functionally diverse forms of cyclin E. Aberrant transcripts have been described for tumor suppressor genes which, like cyclin E, do not differ in normal and tumor cell frequency (31). Lastly, there may also be non-catalytic functions of cyclin E; for example the ES form does not bind to CDK2 or appear to have any kinase activation activity either in vitro or in vivo (12).
Equally important, we show that using Splice Capture we can isolate novel splice variants of a specific gene, in this case cyclin E, and examine its functionality in cells. The Δ48 splice variant form of cyclin E identified and isolated by the Splice Capture strategy was used to examine the biochemical activity of this novel splice variant of cyclin E in human mammary cells. Our results (Fig. 5) suggest that the protein product of the Δ48 splice variation, which comprises 10% of the total cyclin E mRNA, has an altered substrate specificity, as it phosphorylates GST-Rb more effectively than the full-length form of cyclin E, and defines a new aspect of cyclin E regulation.
In summary, we have identified several novel splice variants of cyclin E using Splice Capture. One splice variant is significantly altered in its ability to select a substrate and to bind p21. This method is a powerful tool for comparing the relative abundance of known splice variants and for generating cloned sequences for evaluation by transfection. These novel splice variants of cyclin E are generally expressed in normal and tumor cells and may provide a novel form of regulation of cyclin E in mammalian cells. Deregulation of the splicing of cyclin E could contribute to the alteration of cyclin E and generation of the LMW forms of cyclin E detected in tumor cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Mr Christopher Danes for excellent technical assistance. We also gratefully acknowledge the use of Wadsworth Center’s Molecular Genetics, Tissue Culture and Photography/Graphics core facilities. This research was supported in part by grant DAMD-17-94-J-4081 from the US AMRAA and by grant no. R29-CA666062 from the National Cancer Institute (both to K.K.).
REFERENCES
- 1.Sheaff R.J. (1997) Methods Enzymol., 283, 173–193. [DOI] [PubMed] [Google Scholar]
- 2.Pavletich N.P. (1999) J. Mol. Biol., 287, 821–828. [DOI] [PubMed] [Google Scholar]
- 3.Ohtsubo M., Theodoras,A.M., Schumacher,J., Roberts,J.M. and Pagano,M. (1995) Mol. Cell. Biol., 15, 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steeg P.S. and Zhou,Q. (1998) Breast Cancer Res. Treat., 52, 17–28. [DOI] [PubMed] [Google Scholar]
- 5.Keyomarsi K., Conte,D., Toyofuku,W. and Fox,M.P. (1995) Oncogene, 11, 941–950. [PubMed] [Google Scholar]
- 6.Gray-Bablin J., Zalvide,J., Fox,M.P., Knickerbocker,C.J., DeCaprio,J.A. and Keyomarsi,K. (1996) Proc. Natl Acad. Sci. USA, 93, 15215–15220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sgambato A., Zhang,Y.J., Arber,N., Hibshoosh,H., Doki,Y., Ciaparrone,M., Santella,R., Cittadini,A. and Weinstein,I.B. (1997) Clin. Cancer Res., 3, 1879–1887. [PubMed] [Google Scholar]
- 8.Harwell R.M., Porter,D.C., Danes,C. and Keyomarsi,K. (2000) Cancer Res., 60, 481–489. [PubMed] [Google Scholar]
- 9.Keyomarsi K., O’Leary,N., Molnar,G., Lees,E., Fingert,H.J. and Pardee,A.B. (1994) Cancer Res., 54, 380–385. [PubMed] [Google Scholar]
- 10.Koff A., Cross,F., Fisher,A., Schumacher,J., Leguellec,K., Philippe,M. and Roberts,J.M. (1991) Cell, 66, 1217–1228. [DOI] [PubMed] [Google Scholar]
- 11.Lew D.J., Dulic,V. and Reed,S.I. (1991) Cell, 66, 1197–1206. [DOI] [PubMed] [Google Scholar]
- 12.Sewing A., Ronicke,V., Burger,C., Funk,M. and Muller,R. (1994) J. Cell Sci., 107, 581–588. [DOI] [PubMed] [Google Scholar]
- 13.Mumberg D., Wick,M., Burger,C., Haas,K., Funk,M. and Muller,R. (1997) Nucleic Acids Res., 25, 2098–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Band V., DeCaprio,J.A., Delmolino,L., Kulesa,V. and Sager,R. (1991) J. Virol., 65, 6671–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Band V., Zajchowski,D., Kulesa,V. and Sager,R. (1990) Proc. Natl Acad. Sci. USA, 87, 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hessling J.J., Miller,S.E. and Levy,N.L. (1980) J. Immunol. Methods, 38, 315–324. [DOI] [PubMed] [Google Scholar]
- 17.Rao S., Lowe,M., Herliczek,T. and Keyomarsi,K. (1998) Oncogene, 17, 2393–2402. [DOI] [PubMed] [Google Scholar]
- 18.Newman R.A. (1987) Methods Enzymol., 150, 723–745. [DOI] [PubMed] [Google Scholar]
- 19.Chomczynski P. and Sacchi,N. (1987) Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 20.Seghezzi W., Chua,K., Shanahan,F., Gozani,O., Reed,R. and Lees,E. (1998) Mol. Cell. Biol., 18, 452–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geng Y., Eaton,E.N., Picon,M., Roberts,J.M., Lundberg,A.S., Gifford,A., Sardet,C. and Weinberg,R.A. (1996) Oncogene, 12, 1173–1180. [PubMed] [Google Scholar]
- 22.Keyomarsi K. and Herliczek,T. (1997) In Meijer,L., Guidet,S. and Philippe,M. (eds), Progress in Cell Cycle Research. Plenum Press, New York, NY, Vol. 3, pp. 171–191. [DOI] [PubMed]
- 23.Nielsen N.H., Arnerlov,C., Emdin,S.O. and Landberg,G. (1996) Br. J. Cancer, 74, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang A., Yoshimi,N., Suzui,M., Yamauchi,A., Tarao,M. and Mori,H. (1996) J. Cancer Res. Clin. Oncol., 122, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scuderi R., Palucka,K.A., Pokrovskaja,K., Bjorkholm,M., Wiman,K.G. and Pisa,P. (1996) Blood, 8, 3360–3367. [PubMed] [Google Scholar]
- 26.Donnellan R. and Chetty,R. (1999) FASEB J., 13, 773–780. [DOI] [PubMed] [Google Scholar]
- 27.Wang C., Chua,K., Seghezzi,W., Lees,E., Gozani,O. and Reed,R. (1998) Genes Dev., 12, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sleeman J.E. and Lamond,A.I. (1999) Curr. Biol., 9, 1065–1074. [DOI] [PubMed] [Google Scholar]
- 29.Jackman M., Firth,M. and Pines,J. (1995) EMBO J., 14, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagting A., Jackman,M., Simpson,K. and Pines,J. (1999) Curr. Biol., 9, 680–689. [DOI] [PubMed] [Google Scholar]
- 31.Wang N.M., Tsai,C.H., Yeh,K.T., Chen,S.J. and Chang,J.G. (1999) Int. J. Mol. Med., 4, 249–252. [PubMed] [Google Scholar]